
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilylation reactions on nanoporous gold via homolytic Si–H activation of silanes†
Hongbo
Li‡
a,
Huifang
Guo‡
a,
Zhiwen
Li
b,
Cai
Wu
a,
Jing
Li
a,
Chunliang
Zhao
a,
Shuangxi
Guo
a,
Yi
Ding
*b,
Wei
He
 *a and
Yadong
Li
c
*a and
Yadong
Li
c
aTsinghua-Peking Joint Centers for Life Sciences, School of Pharmaceutical Sciences, Tsinghua University, Beijing 100084, China. E-mail: whe@mail.tsinghua.edu.cn
bTianjin Key Laboratory of Advanced Functional Porous Materials, Tianjin University of Technology, Tianjin 300384, China. E-mail: yding@tjut.edu.cn
cDepartment of Chemistry, Tsinghua University, Beijing 100084, China
First published on 30th April 2018
Abstract
Si–H bond activation is an important process implicated in many useful synthetic applications including silylation and transfer hydrogenation reactions. Herein we discovered homolytic activation of Si–H bonds on the surface of nanoporous gold (NPG), forming hydrogen radicals and [Au]–[Si] intermediates. By virtue of this new reactivity, we achieved highly selective mono and sequential alcoholysis of dihydrosilane. In addition, the amphiphilic nature of the [Au]–[Si] intermediate allows for a new bis-silylation reaction of cyclic ethers. The present work showcased that the surface reactivity of nanocatalysts may provide exciting opportunity for new reaction discovery.
Introduction
Catalytic activation of Si–H bonds provides rapid construction of silicon-containing functional molecules or materials, a process collectively referred to as “silylation reactions”.1–4 This also enables transfer hydrogenation reactions where the silanes serve as the hydrogen gas equivalent.5–8 A plethora of homogeneous metal catalysts including Pd,9 Ir,10–15 Rh,16–18 Ni,19,20 Ru,21–24 Co,25 and Fe26–30 salts had been discovered for these important processes. These homogeneous catalysts often activate silanes through oxidative addition or a σ-bond metathesis pathway, giving rise to metal hydride ([M]–H) intermediates.20,22,23,27 The metal hydride is known to be highly reactive towards unsaturated bonds, thus posing considerable limitations on the substrate scope.22,23,29There is an intriguing pattern in the past few decades that heterogeneous catalysts can display reactivity inaccessible by known homogeneous catalysts.31–33 For example, Porco and co-workers reported an elegant silver nanoparticle catalysed Diels–Alder cycloaddition that was not catalysed by homogeneous Lewis acids.34 Glorius and his colleagues discovered that Pd/C showed completely different regioselectivity in the arylation of benzothiophenes as compared to homogeneous Pd catalysts.35 Recently, we reported a catalytic deoxygenation reaction of aromatic epoxides on the Cu surface, affording alkenes with high cis isomer selectivity.36
We thus became curious about Si–H activation on the surface of heterogeneous metal catalysts.37–39 Importantly, the pioneering studies of Yamamoto,40 Jin,5 and Chen41 have shown that silane activation on NPG can lead to the efficient oxidation of silanes to silanols, as well as transfer semi-hydrogenation of alkynes. Herein, we disclose that NPG activates Si–H bonds through a homolytic pathway, forming hydrogen radicals and [Au]–[Si] intermediates. This new activation mode allows efficient monoalcoholysis of dihydrosilanes in a controlled (only one Si–H is activated) and chemoselective (hydrogen radicals tolerate many functional groups) manner. By tuning the reaction conditions, the second Si–H bond activation/alcoholysis can proceed to give unsymmetrical silaketals in a one-pot reaction. Moreover, the [Au]–[Si] intermediates could serve as both an electrophile (forming Si–O bonds) and nucleophile (forming C–Si bonds), resulting in the ring opening/bis-silylation reaction of a range of cyclic ethers.
Results and discussion
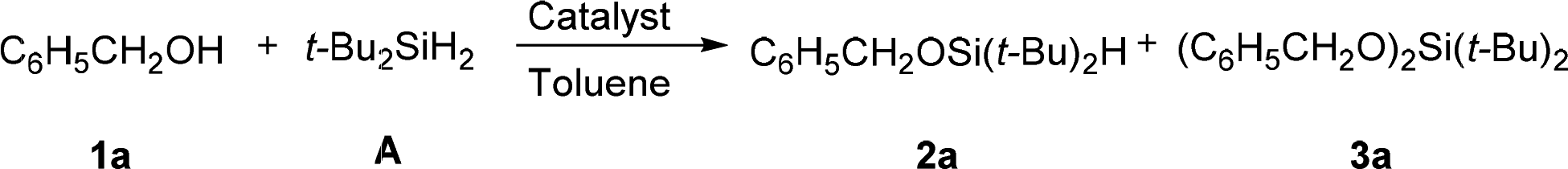
Monoalcoholysis of di-tert-butylsilane
Our investigation started with testing a number of heterogeneous catalysts in the monoalcoholysis of di-tert-butylsilane (A) with benzyl alcohol 1a (Table 1). Commercial Pd/C is the most reactive among the tested catalysts, however affording undesired disiloxane 3a in a high percentage. Au–Pd alloy nanoparticles and Au nanoparticles showed moderate activity but good selectivity. These observations suggested that Au is less reactive but more selective than Pd in the monoalcoholysis. When NPG was employed, the reaction rate was significantly improved, achieving 70% conversion in 12 h (Table 1, entry 5). A synthetically useful condition was obtained at 50 °C with 92% yield of 2a in 2 h. The turnover number (TON) and the turnover frequency (TOF) of the NPG under these conditions were calculated to be as high as 5377 and 2689 h−1, respectively (Table 1, entry 6). Such a high TOF and TON highlight the activity enhancement brought by the nanostructure of the NPG. In contrast, homogeneous Au salts,8 such as 0.5 ppm AuCl or AuCl3 solutions showed a very low conversion even at 50 °C (Table 1, entry 7), which is similar to the control reaction without any catalyst (Table 1, entry 8), suggesting that the homogenous Au salts did not contribute to the reaction.|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Conversionb | 2a | 3a |
| a Reaction conditions: 1a (1.0 mmol), t-Bu2SiH2 (A, 2.1 mmol) and catalyst (1 mol%) in toluene (5 mL) under nitrogen, 25 °C for 12 h. b Conversion monitored by NMR on the crude material. c Isolated yields. d Au–Pd alloy nanoparticles (Au 1.5‰, Pd 1‰, w/w) supported on activated carbon, 1 mol% based on metal. e 50 °C, 2 h. n.d. = not detected. | ||||
| 1 | Pd/C | 99% | 60% | 15% |
| 2 | AuPd/Cd | 79% | 30% | 11% |
| 3 | Au/Al2O3 | 25% | 21% | n.d. |
| 4 | Gold foil | 13% | 11% | n.d. |
| 5 | NPG | 70% | 66% | n.d. |
| 6e | NPG | 99% | 92% | n.d. |
| 7e | Au salt | 10% | 8% | n.d. |
| 8e | — | 10% | 8% | n.d. |
EPR study
With great curiosity we sought to understand the intrinsic selectivity of NPG by probing how the NPG catalyst activates the dihydrosilanes. To this end, we employed the electron paramagnetic resonance (EPR) spectroscopy using PBN as the spin trap (Fig. 1).42 Under our model reaction conditions, a persistent signal attributed to the PBN-H adduct was observed (Fig. 1a, aN = 15.26 G, aH(1) = aH(2) = 7.60 G).42,43 A similar radical was observed in the absence of the benzyl alcohol under otherwise identical conditions (Fig. 1b, aN = 15.00 G, aH(1) = aH(2) = 7.28 G). However, no signal was detected in the absence of the di-tert-butylsilane (Fig. 1c and d). Collectively, these results suggested that the radical originated from the di-tert-butylsilane but not the benzyl alcohol44 nor the solvent toluene. Interestingly, EPR of the silane alone in the toluene also gave weak radical signals (Fig. 1e), which is consistent with the spontaneous reaction without a catalyst (Table 1, entry 8). In contrast, the trapping of a silyl radical44 by PBN was not observed, suggesting the silyl moiety is still covalently linked to the gold surface ([Au]–[Si] species). Such a homolytic Si–H activation by the NPG is consistent with the earlier observations of Raffa,39 and is completely different from that by the homogeneous gold catalysts. In the latter cases, discrete [Au]–H was responsible for the catalytic activity and no [Au]–[Si] intermediate was ever proposed.45,46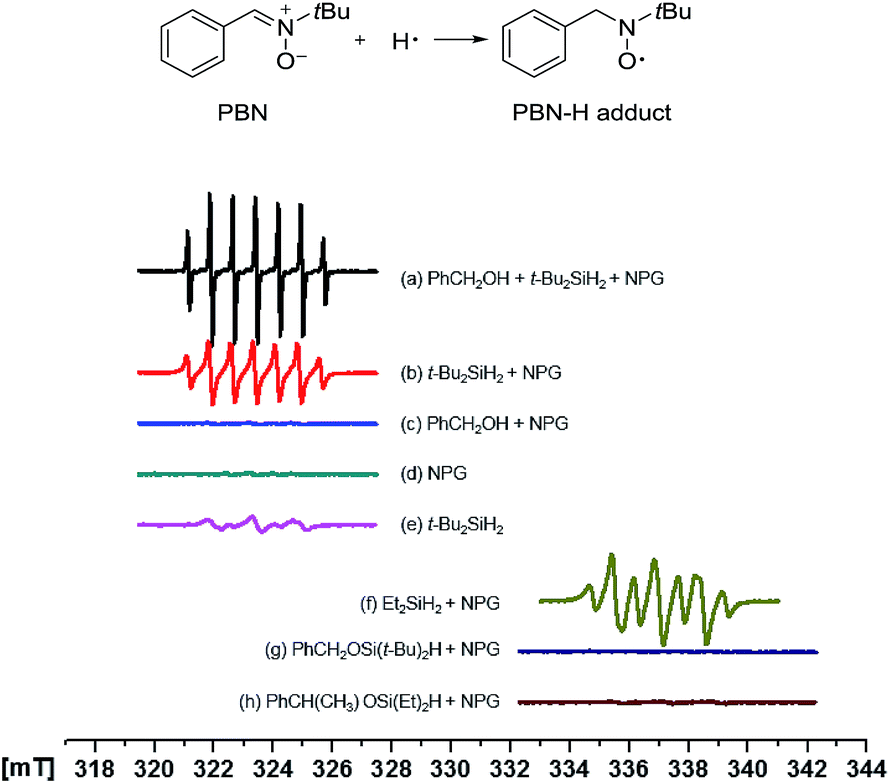 | ||
| Fig. 1 EPR spectra using PBN (0.02 mmol) as the spin trap. In respective tests, 0.5 mL toluene solvent was used. Other reagents if used: alcohol (0.15 mmol), silane (0.23 mmol), and NPG (0.015 mmol). Tests (a–e) were run at 0 °C after reaction at 50 °C for 5 min. Tests (f–h) were run at r.t. after reaction at r.t. for 10 min. | ||
Similar behaviour was also observed with the less hindered, more reactive diethylsilane (Fig. 1f, aN = 14.43 G, aH = 7.54 G). However, when two representative hydridosilyl ethers 2a (Fig. 1g) and 2r (Fig. 1h) were submitted to the EPR experiments, no PBN-H adduct was observed. These data suggested that the activation of the Si–H bonds in the hydridosilyl ethers by the NPG was much slower. The steric and electronic differences brought by the alkoxy substitutions might account for the retarded Si–H activation on the NPG surface. Altogether, it is clear that the selective activation of the first Si–H bond in dihydrosilane by NPG is the origin of the observed monoalcoholysis.
Scope of the NPG catalysed monoalcoholysis
Based on the observed selective homolytic Si–H activation of dihydrosilanes with NPG, we demonstrated the scope of the monoalcoholysis (Table 2). Primary (1a–1c, 1j–1o) and secondary (1d–1f) alcohols are shown to be compatible with the current method, affording the desired hydridosilyl ethers. Reactions of tertiary alcohols (1g, 1h) with di-tert-butylsilane were slow. However, using diethylsilane instead led to excellent isolated yields of the desired products (2g and 2h). Phenol was also successfully converted into silane 2i, a precursor for ortho-C–H alkenylation.47 Mono silylation of substrates bearing two hydroxyl groups could also be achieved by controlling the stoichiometry of the dihydrosilane. In the presence of 1.0 equiv. of the di-tert-butylsilane, mono silylation of α,ω-diol gave a high yield of the desired product (2m), which might suggest that the absorption of 1m onto the surface of the NPG is faster than that of 2m. Moreover, the primary hydroxyl was selectively silylated over the secondary hydroxyl (2n), and the benzyl alcohol was selectively silylated over the phenol (2o). The two cases indicate that the primary alcohols are more reactive than secondary alcohols and phenols under the current reaction conditions.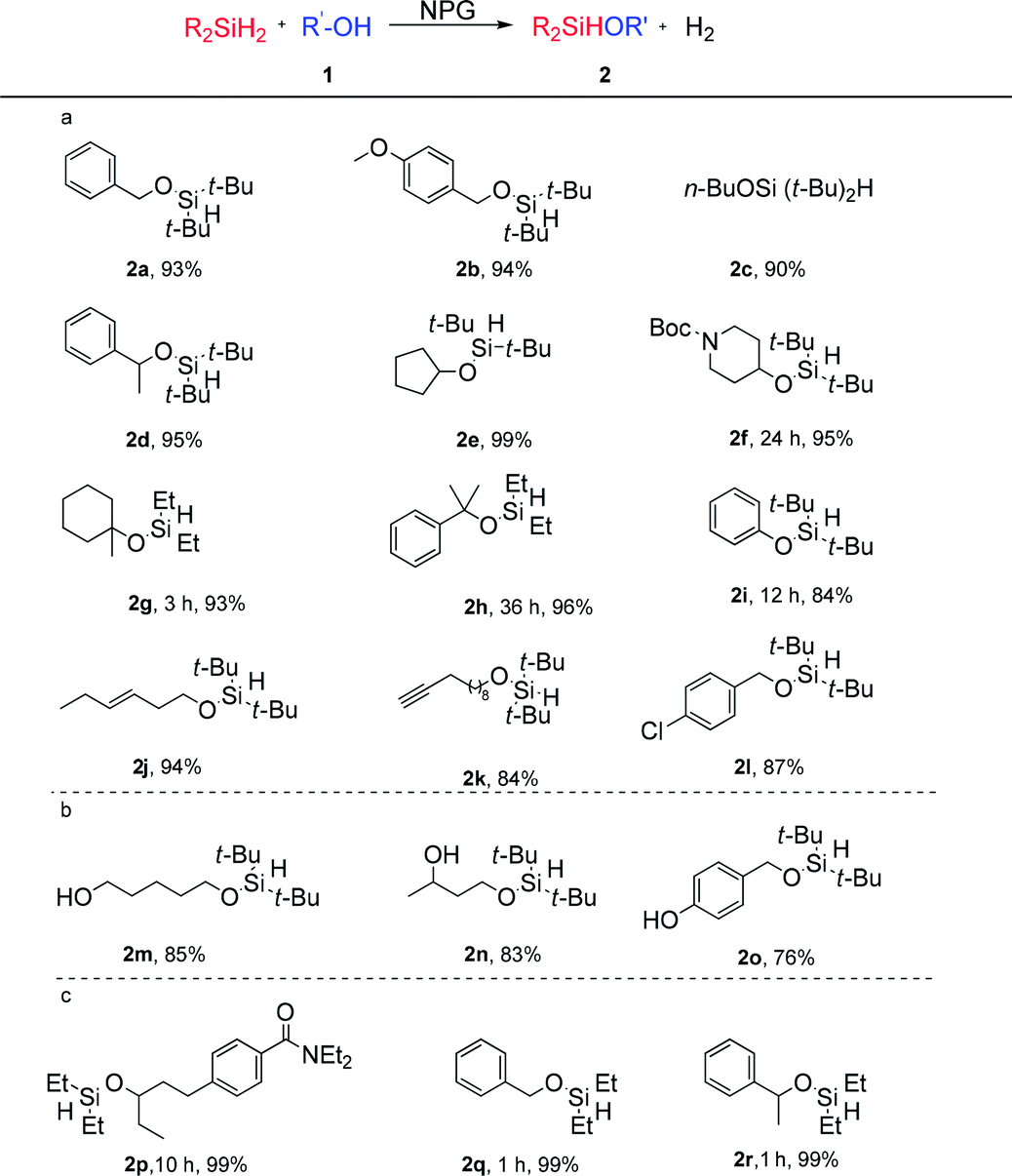
The current method showed excellent compatibility with a range of reducible functionalities, since the hydrogen radicals are much less reactive than the metal hydride. The internal alkene showed tolerance under the reaction conditions (2j). Surprisingly, the terminal alkyne was also intact (2k) despite the known reactivity of alkynes in NPG catalysis.5,48,49 The stability of the π bonds in the solvent acetone (see ESI 3.1†), the alkene (2j), the terminal alkyne (2k), the carbamate (2p) and the aryl halide (2l) towards the NPG is noteworthy.
The reactions between the less hindered diethylsilane (1.5 eq.) and less hindered alcohols (1p, 1q, and 1r) also gave exclusive monoalcoholysis products. These observations suggested that the chemoselectivity towards monoalcoholysis is dictated by the intrinsic reactivity of the NPG catalyst, and not by the steric hindrance of the dialkylsilanes or alcohols.
The overall selectivity of the NPG catalyst in the controlled monoalcoholysis of dialkylsilanes is unprecedented. There are a few reported catalysts for this purpose, for example, PdCl2,50 (PPh)3RhCl\(PPh)3RuCl2,51 tris(oxazolinyl)borato zinc hydride,52 and Ph3PCuH.53 However, these methods encounter one or more limitations including competitive disiloxane formation, hydrosilyation of π bonds and hydrogenolysis of aryl halide bonds. The efficient synthesis of compound 2p is a good example showing the advantage of our method, as the Hartwig group reported that the ruthenium complex Ru(PPh3)3Cl2 catalysed reaction suffered from hydrosilylation of the amide.12
Syntheses of unsymmetrical silaketals
We further developed efficient one-pot syntheses of unsymmetrical silaketals via sequential Si–H activations (Fig. 2). Unsymmetrical silaketals have been widely used in temporary tethering strategies.54 Their classic syntheses largely depended on multistep, stoichiometric displacement reactions of chlorosilanes. The Wilcox group has reported remarkable one-pot syntheses55,56 through sequential alcoholysis using two different readily available catalysts: a rhodium acetate dimer for the first Si–O formation and Pd/C55 or Mn(CO)5Br56 for the second Si–O formation. We discovered that using 1.1 equiv. diethylsilane, the first Si–H activation/alcoholysis could proceed smoothly to give the desired hydridosilyl ethers. Addition of a second alcohol 5 and then increasing the reaction temperature to 50 °C triggered the second Si–H activation/alcoholysis. The desired unsymmetrical silaketals were then obtained in good yields. Our method is complementary to the Wilcox protocol because: (1) a single catalyst is employed; (2) a less hindered, more reactive diethylsilyl linkage is used, while diisopropyl linkage was used in Wilcox reactions; (3) our system is compatible with alkene functionality without compromising the yields. Importantly, in both sets of reactions (Table 2 and Fig. 2), the NPG catalyst can be easily removed from the reaction and reused for a number of cycles (see ESI 3.3†). The products in Table 2 could be obtained in analytical purity by simple evaporation, which avoided the problem that these hydridosilyl ethers often decomposed rapidly during silica gel chromatography.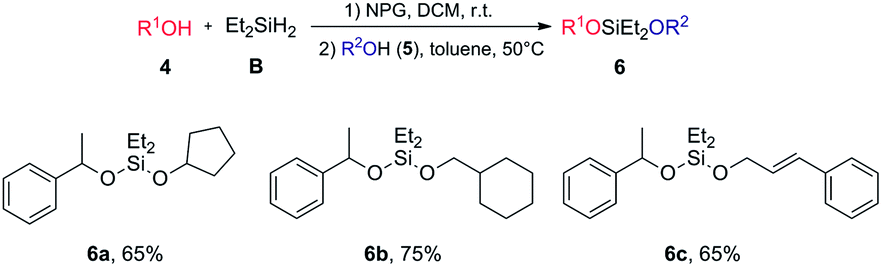 | ||
| Fig. 2 One-pot synthesis of unsymmetrical silaketals. | ||
Ring-opening bis-silylation reaction of cyclic ethers
We uncovered a surprising ring-opening bis-silylation reaction of cyclic ethers (Table 3). When di-tert-butylsilane was treated with epoxide 7a in the presence of 3 mol% NPG, an unexpected product was isolated in a 62% yield which was subsequently identified as bis-silicon 9a (Table 3, entry 1). Such a ring-opening/bis-silylation reaction was successfully extended to cyclic ethers of four, five and six membered rings (entries 2, 3 and 4). We discovered that at higher temperatures, the ratio of bis-silylation/hydrosilylation products could be improved, such that the bi-silicon compounds were obtained at synthetically useful yields in the range of 70–82%. The ring strain seems to dictate the reaction rate: smaller rings react faster and demand lower reaction temperature. Interestingly, the ratio of bis-silylated products increase as the ring size becomes bigger. In the case of 7d, only the bis-silylation products were obtained in a 82% yield.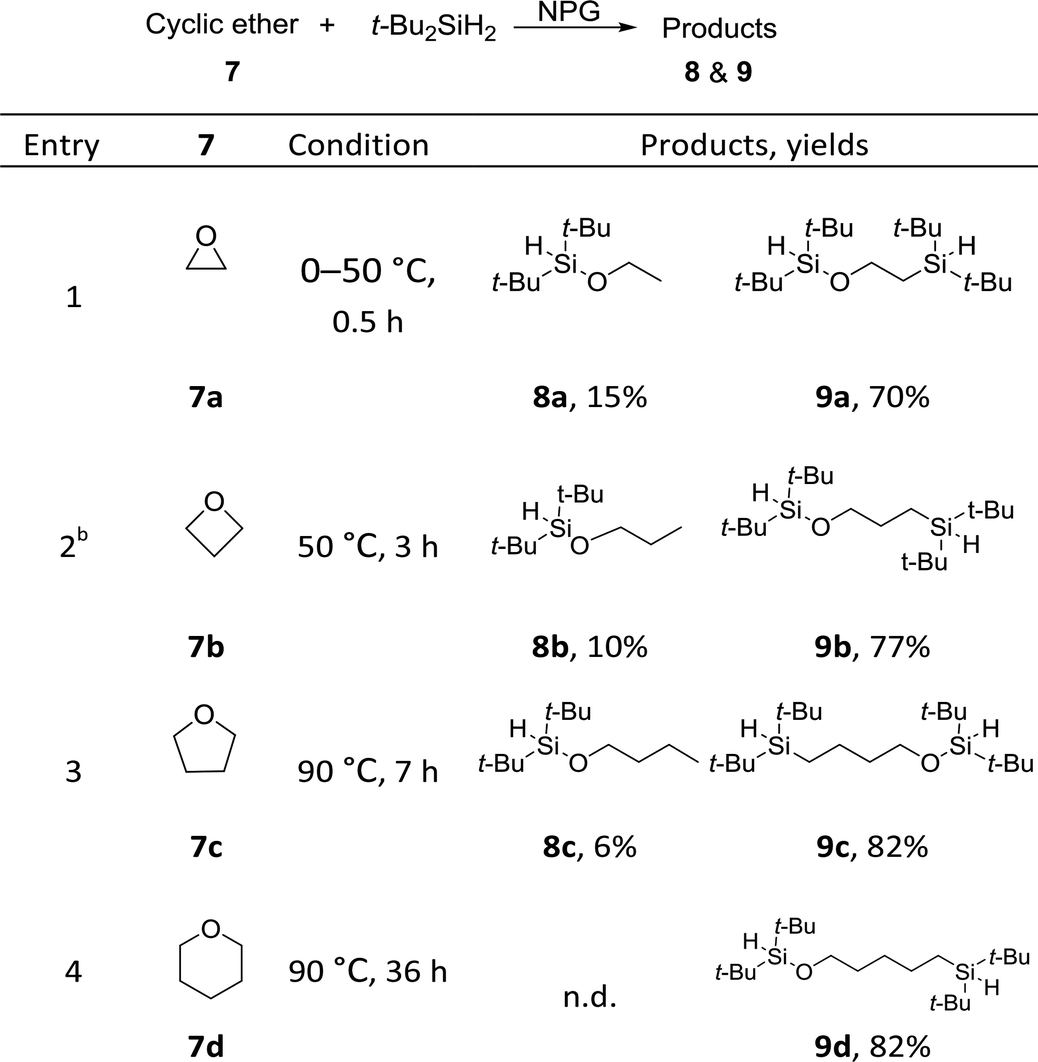
This represents the first examples of incorporating two silicon atoms into cyclic ethers, affording simultaneously an O–Si and a C–Si bond. In hindsight, the direct hydrosilylation of cyclic ethers57–60 had been reported in four papers, catalysed by a homogeneous iridium complex,57 dicobalt octacarbonyl,58 or boron Lewis acid.59,60 Stratakis and co-workers reported an elegant gold nanoparticle catalysed silaboration of oxetanes and unactivated epoxides. This interesting formal addition of Si–B was proposed to proceed through the oxidative insertion of Au into Si–B bonds, which is different than our radical pathway (vide infra).61 Others examples were limited to the ring-opening bromo- or iodo-silylation with Br- or I-silanes62 or its synthetic equivalents.63 Notably, our NPG catalysis provides a different bis-functionalization method, affording bis-silicons that could serve as useful building blocks in organic syntheses and materials chemistry. For example, the chemoselective O–Si cleavage of the bis-silicon 9c was successfully achieved under TBAF conditions, affording a terminal alcohol synthon (see ESI 5.1†). The versatile reactivity of the remaining silane bond also provides ample synthetic opportunities, including the preparation of silicone monomers.
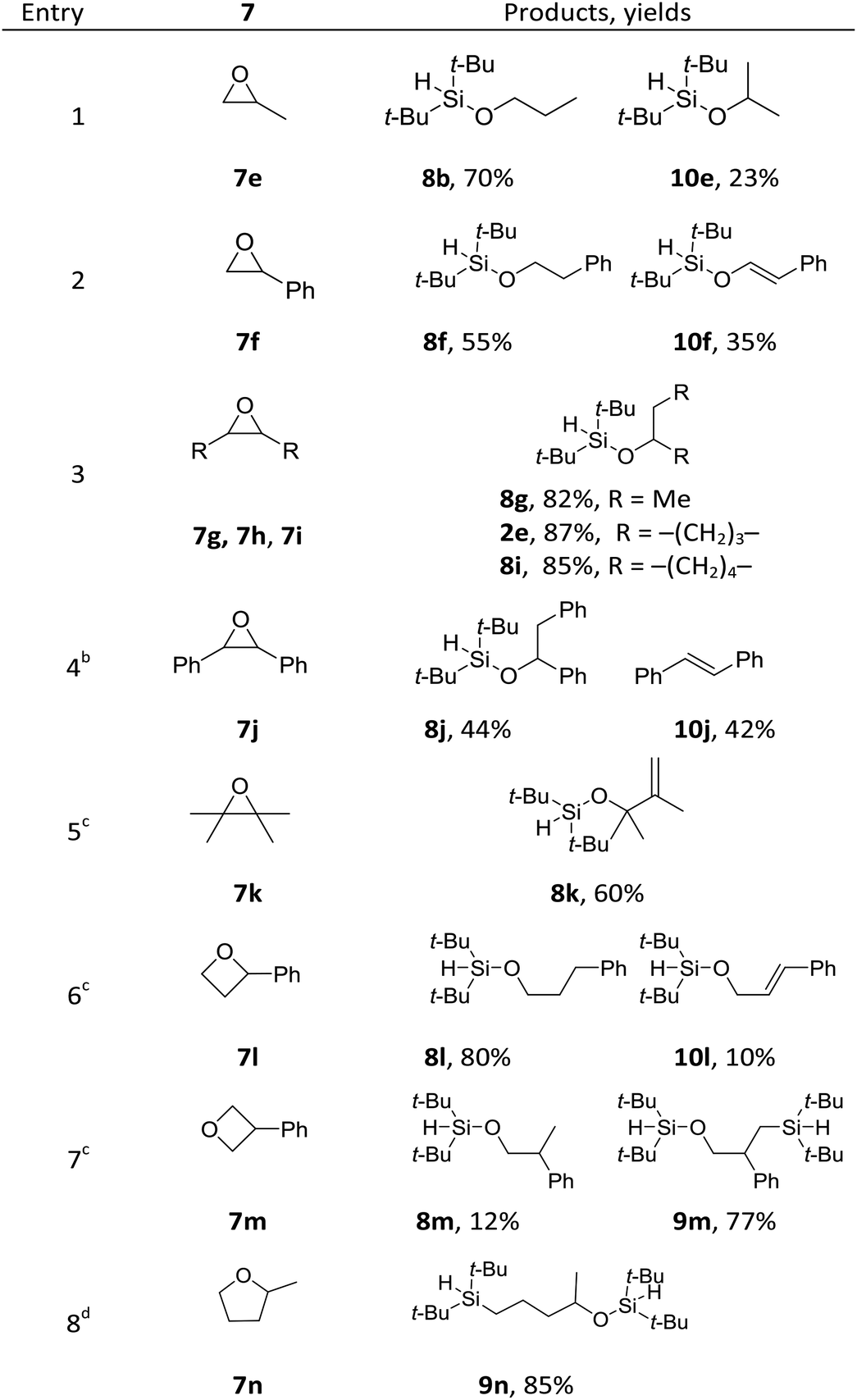
To further probe the reaction scope as well as to shed light on the mechanism of the reaction, substituted cyclic ethers were then studied (Table 4). Overall, the reactivity of the substituted cyclic ethers (mainly hydrosilyation) is very different than that of the unsubstituted ones (mainly bis-silylation). Three conspicuous trends could be summarized from the data: (1) the incorporation of a substituent on the carbon next to the oxygen atom shut down the bis-silylation (7e–7l), while a substituent further away resulted in bis-silylation (cf.7l and 7m). The hydrosilylation of 7g, 7h, 7i and 7l is not what we intended, but the yields are high. The bis-silyation of 7m and 7n is very efficient. (2) The ring opening took place preferentially at the more substituted carbon (cf.7e, 7f, and 7l), with the only exception in the case of 7n (vide infra); (3) when a quaternary carbon (7k) or a carbon connected with phenyl groups (7f, 7j and 7l) is present in the substrate, elimination products were observed. The elimination products (i.e., 10) alerted us to a possible elimination–hydrosilyation pathway. To this end, an independently synthesized (but-3-enyloxy)di-tert-butylsilane failed to produce the desired product 9c under the reaction conditions (see ESI†), which was consistent with the chemoselectivity shown in Table 2 (cf.2p) and suggested that the C–Si bond formation is unlikely to proceed through an elimination–hydrosilylation sequence.
| a Products and isolated yields of the NPG catalysed ring-opening silylation reaction. Conditions: unless otherwise specified, cyclic ether (3.0 mmol), t-Bu2SiH2 (6.6 mmol) and catalyst (3 mol%), under argon, in a sealed tube, 40 °C, 4 h. b Toluene 2 mL as the solvent, 70 °C, 5 h. c 70 °C, 5 h. d 90 °C, 15 h. |
|---|
|
A plausible mechanism of this cyclic ether ring opening/bis-silylation reaction is proposed (Fig. 3), which could explain our experimental observations. First, the Si–H bond of silane is activated on the surface of NPG, possibly in an oxidative-insertion manner, which gave the intermediate A. The Au–H bond is more susceptible to the PBN trapping than the Au–Si bond, so that only the hydrogen radical was observed. We observed hydrogen gas formation in the reaction, which is associated with the formation of species B from species A. The formations of hydrogen gas and Au–Si species of type A were also documented by the Stratakis group in their Au/TiO2 NP catalysed cis-1,2-disilylation of alkynes.64 We believe that at higher temperature the formation of hydrogen gas was more favourable, thus the ratio of B/A becomes bigger. A radical type C–O bond cleavage of the cyclic ether can be triggered either by the intermediate A or by the intermediate B, leaving the radical on the more substituted carbon (i.e., C). We proposed this radical was instantly trapped by the NPG surface, thus forming the intermediate D. The fate of the intermediate D diverges depending on the substrate structure. When it is a highly stable benzyl radical, it undergoes elimination to give alkene products (e.g.10f). It also could form the hydrosilylation products by a formal hydrogen shift on the surface of A, or form the bis-silylation product through a formal Si shift on the surface of B. The higher population of B at higher temperature explains the higher yields of bis-silyation products. This mechanism can also explain why most of the substituted cyclic ethers underwent hydrosilyation but not bis-silylation. The reason might lie in the steric difference. When there is a R group attached to the carbon linked to the Au surface, the Si shift is considerably more sterically demanding than the H shift. In the case of 7n, the successful formation of the bis-silyation product 9n is the result of both a higher reaction temperature and less steric repulsion. It should be noted that with a larger ring system, the organic compound depicted in intermediate D would have more flexibility to avoid repulsion with the Au surface, therefore rendering it possible to cleave the C–O bond in an opposite position. Such a mechanistic proposal is intriguing in two aspects: (1) the reaction selectivity is very much dependent on the surface reactivity of the catalyst; (2) the [Au]–[Si] species formally serves as both an electrophile and a nucleophile in this bis-silylation ring-opening reaction. Both aspects cannot be achieved by homogeneous catalysis.
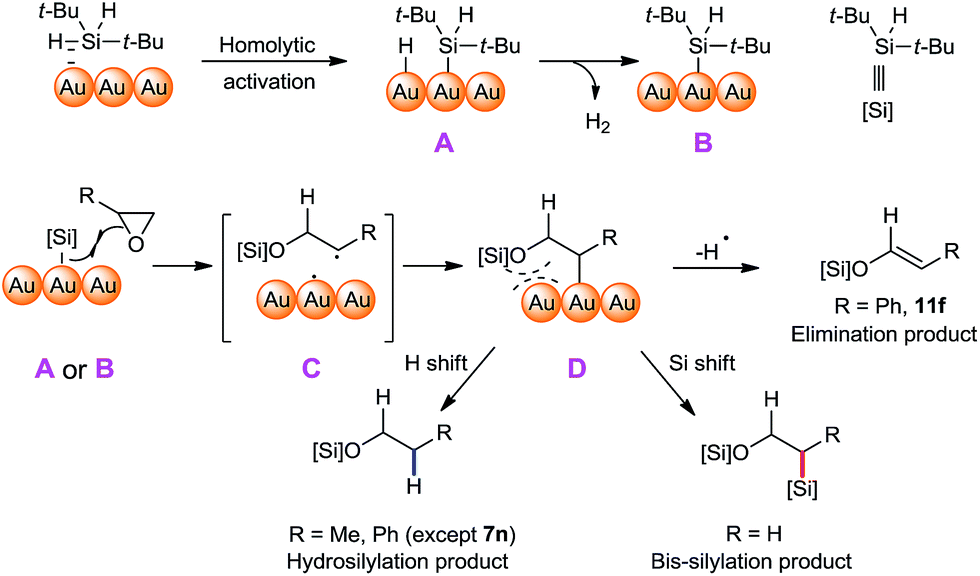 | ||
| Fig. 3 Proposed mechanism of the ring-opening reaction. | ||
Conclusions
In conclusion, by virtue of the NPG catalysed homolytic NPG activation of Si–H bonds, we realized three new silylation reactions: controlled monoalcoholysis of dihydrosilanes, one-pot formation of unsymmetrical silaketals and ring-opening bis-silylation of cyclic ethers. These reactions feature a readily available catalyst, operationally simple protocols and high efficiencies. Moreover, the high chemoselectivity and functional group compatibility achieved by this NPG catalysis are unprecedented in the literature. Together with the exotic amphiphilic reactivity of [Au]–[Si] intermediates, this study showcased the unique potential of heterogeneous catalysts in organic syntheses.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Key R&D Program of China (No. 2017YFA0505200) and NSFC (21625104 and 21521091).References
- B. Marciniec, Hydrosilylation: a comprehensive review on recent advances, Springer Science & Business Media, 2008 Search PubMed.
- S. E. Denmark and M. H. Ober, Aldrichimica Acta, 2003, 36, 75–85 CAS.
- C. Cheng and J. F. Hartwig, Chem. Rev., 2015, 115, 8946–8975 CrossRef CAS PubMed.
- S. Bähr and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 52–59 CrossRef PubMed.
- M. Yan, T. Jin, Y. Ishikawa, T. Minato, T. Fujita, L. Y. Chen, M. Bao, N. Asao, M. W. Chen and Y. Yamamoto, J. Am. Chem. Soc., 2012, 134, 17536–17542 CrossRef CAS PubMed.
- M. Yan, T. A. Jin, Q. Chen, H. E. Ho, T. Fujita, L. Y. Chen, M. Bao, M. W. Chen, N. Asao and Y. Yamamoto, Org. Lett., 2013, 15, 1484–1487 CrossRef CAS PubMed.
- L. Zamostna, M. Ahrens and T. Braun, J. Fluorine Chem., 2013, 155, 132–142 CrossRef CAS.
- H. F. Guo, X. L. Yan, Y. Zhi, Z. W. Li, C. Wu, C. L. Zhao, J. Wang, Z. X. Yu, Y. Ding, W. He and Y. D. Li, Nano Res., 2015, 8, 1365–1372 CrossRef CAS.
- Y. Sumida, T. Kato, S. Yoshida and T. Hosoya, Org. Lett., 2012, 14, 1552–1555 CrossRef CAS PubMed.
- T. A. Boebel and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 7534–7535 CrossRef CAS PubMed.
- E. M. Simmons and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 17092–17095 CrossRef CAS PubMed.
- E. M. Simmons and J. F. Hartwig, Nature, 2012, 483, 70–73 CrossRef CAS PubMed.
- D. Sieh and P. Burger, J. Am. Chem. Soc., 2013, 135, 3971–3982 CrossRef CAS PubMed.
- J. A. Muchnij, F. B. Kwaramba and R. J. Rahaim, Org. Lett., 2014, 16, 1330–1333 CrossRef CAS PubMed.
- Y. Hua, P. Asgari, T. Avullala and J. Jeon, J. Am. Chem. Soc., 2016, 138, 7982–7991 CrossRef CAS PubMed.
- S. J. O'Malley and J. L. Leighton, Angew. Chem., Int. Ed., 2001, 40, 2915–2917 CrossRef.
- J. T. Spletstoser, M. J. Zacuto and J. L. Leighton, Org. Lett., 2008, 10, 5593–5596 CrossRef CAS PubMed.
- T. Lee and J. F. Hartwig, Angew. Chem., Int. Ed., 2016, 55, 8723–8727 CrossRef CAS PubMed.
- T. Zell, T. Schaub, K. Radacki and U. Radius, Dalton Trans., 2011, 40, 1852–1854 RSC.
- I. Buslov, J. Becouse, S. Mazza, M. Montandonclerc and X. Hu, Angew. Chem., Int. Ed., 2015, 54, 14523 CrossRef CAS PubMed.
- F. Kakiuchi, K. Tsuchiya, M. Matsumoto, B. Mizushima and N. Chatani, J. Am. Chem. Soc., 2004, 126, 12792–12793 CrossRef CAS PubMed.
- S. T. Ding, L. J. Song, Y. Wang, X. H. Zhang, L. W. Chung, Y. D. Wu and J. W. Sun, Angew. Chem., Int. Ed., 2015, 54, 5632–5635 CrossRef CAS PubMed.
- S. Wübbolt and M. Oestreich, Angew. Chem., Int. Ed., 2015, 54, 15876–15879 CrossRef PubMed.
- W. Li, X. Huang and J. You, Org. Lett., 2016, 47, 666–668 CrossRef PubMed.
- C. Chen, M. B. Hecht, A. Kavara, W. W. Brennessel, B. Q. Mercado, D. J. Weix and P. L. Holland, J. Am. Chem. Soc., 2015, 137, 13244–13247 CrossRef CAS PubMed.
- H. Nishiyama and A. Furuta, Chem. Commun., 2007, 7, 760–762 RSC.
- J. Y. Wu, B. N. Stanzl and T. Ritter, J. Am. Chem. Soc., 2010, 132, 13214–13216 CrossRef CAS PubMed.
- D. J. Peng, Y. L. Zhang, X. Y. Du, L. Zhang, X. B. Leng, M. D. Walter and Z. Huang, J. Am. Chem. Soc., 2013, 135, 19154–19166 CrossRef CAS PubMed.
- J. H. Chen, B. Cheng, M. Y. Cao and Z. Lu, Angew. Chem., Int. Ed., 2015, 54, 4661–4664 CrossRef CAS PubMed.
- Z. Q. Zuo, L. Zhang, X. B. Leng and Z. Huang, Chem. Commun., 2015, 51, 5073–5076 RSC.
- M. Moreno-Manas and R. Pleixats, Acc. Chem. Res., 2003, 36, 638–643 CrossRef CAS PubMed.
- M. Pagliaro, V. Pandarus, R. Ciriminna, F. Beland and P. D. Cara, ChemCatChem, 2012, 4, 432–445 CrossRef CAS.
- F. Zaera, Chem. Soc. Rev., 2013, 42, 2746–2762 RSC.
- H. A. Cong, C. F. Becker, S. J. Elliott, M. W. Grinstaff and J. A. Porco, J. Am. Chem. Soc., 2010, 132, 7514–7518 CrossRef CAS PubMed.
- D. T. D. Tang, K. D. Collins and F. Glorius, J. Am. Chem. Soc., 2013, 135, 7450–7453 CrossRef CAS PubMed.
- B. Xiao, Z. Q. Niu, Y. G. Wang, W. Jia, J. Shang, L. Zhang, D. S. Wang, Y. Fu, J. Zeng, W. He, K. Wu, J. Li, J. L. Yang, L. Liu and Y. D. Li, J. Am. Chem. Soc., 2015, 137, 3791–3794 CrossRef CAS PubMed.
- Y. Zhang, X. Cui, F. Shi and Y. Deng, Chem. Rev., 2012, 112, 2467–2505 CrossRef CAS PubMed.
- M. Stratakis and H. Garcia, Chem. Rev., 2012, 112, 4469–4506 CrossRef CAS PubMed.
- P. Raffa, C. Evangelisti, G. Vitulli and P. Salvadori, Tetrahedron Lett., 2008, 49, 3221–3224 CrossRef CAS.
- N. Asao, Y. Ishikawa, N. Hatakeyama, Menggenbateer, Y. Yamamoto, M. W. Chen, W. Zhang and A. Inoue, Angew. Chem., Int. Ed., 2010, 49, 10093–10095 CrossRef CAS PubMed.
- T. Fujita, P. F. Guan, K. McKenna, X. Y. Lang, A. Hirata, L. Zhang, T. Tokunaga, S. Arai, Y. Yamamoto, N. Tanaka, Y. Ishikawa, N. Asao, Y. Yamamoto, J. Erlebacher and M. W. Chen, Nat. Mater., 2012, 11, 775–780 CrossRef CAS PubMed.
- M. Conte, H. Miyamura, S. Kobayashi and V. Chechik, J. Am. Chem. Soc., 2009, 131, 7189–7196 CrossRef CAS PubMed.
- E. G. Janzen, T. Kasai and K. Kuwata, Bull. Chem. Soc. Jpn., 1973, 46, 2061–2062 CrossRef CAS.
- J. Zeitouny and V. Jouikov, Phys. Chem. Chem. Phys., 2009, 11, 7161–7170 RSC.
- H. Ito, T. Saito, T. Miyahara, C. M. Zhong and M. Sawamura, Organometallics, 2009, 28, 4829–4840 CrossRef CAS.
- S. Labouille, A. Escalle-Lewis, Y. Jean, N. Mezailles and P. Le Floch, Chem.–Eur. J., 2011, 17, 2256–2265 CrossRef CAS PubMed.
- M. Mewald, J. A. Schiffner and M. Oestreich, Angew. Chem., Int. Ed., 2012, 51, 1763–1765 CrossRef CAS PubMed.
- A. M. Caporusso, L. A. Aronica, E. Schiavi, G. Martra, G. Vitulli and P. Salvadori, J. Organomet. Chem., 2005, 690, 1063–1066 CrossRef CAS.
- A. Corma, C. Gonzalez-Arellano, M. Iglesias and F. Sanchez, Angew. Chem., Int. Ed., 2007, 46, 7820–7822 CrossRef CAS PubMed.
- J. Ohshita, R. Taketsugu, Y. Nakahara and A. Kunai, J. Organomet. Chem., 2004, 689, 3258–3264 CrossRef CAS.
- R. J. P. Corriu and J. J. E. Moreau, J. Chem. Soc., Chem. Commun., 1973, 38–39 RSC.
- D. Mukherjee, R. R. Thompson, A. Ellern and A. D. Sadow, ACS Catal., 2011, 1, 698–702 CrossRef CAS.
- C. Lorenz and U. S. Schubert, Chem. Ber., 1995, 128, 1267–1269 CrossRef CAS.
- S. Bracegirdle and E. A. Anderson, Chem. Soc. Rev., 2010, 39, 4114–4129 RSC.
- C. N. Scott and C. S. Wilcox, Synthesis, 2004, 36, 2273–2276 Search PubMed.
- C. N. Scott and C. S. Wilcox, J. Org. Chem., 2010, 75, 253–256 CrossRef CAS PubMed.
- S. Park and M. Brookhart, Chem. Commun., 2011, 47, 3643–3645 RSC.
- K. T. Kang and W. P. Weber, Tetrahedron Lett., 1985, 26, 5753–5754 CrossRef CAS.
- J. B. Grande, D. B. Thompson, F. Gonzaga and M. A. Brook, Chem. Commun., 2010, 46, 4988–4990 RSC.
- M. Gevorgyan, S. Rubin, J. Benson, J.-X. Liu and Y. Yamamoto, J. Org. Chem., 2000, 65, 6179–6186 CrossRef PubMed.
- E. Vasilikogiannaki, A. Louka and M. Stratakis, Organometallics, 2016, 35, 3895–3902 CrossRef CAS.
- U. Kruerke, Chem. Ber., 1962, 95, 174–182 CrossRef.
- J. Ohshita, A. Iwata, F. Kanetani, A. Kunai, Y. Yamamoto and C. Matui, J. Org. Chem., 1999, 64, 8024–8026 CrossRef CAS.
- I. Saridakis, M. Kidonakis and M. Stratakis, ChemCatChem, 2018, 10, 980–983 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01427b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |