DOI:
10.1039/C8SC01537F
(Edge Article)
Chem. Sci., 2018,
9, 6041-6052
Selective oxymetalation of terminal alkynes via 6-endo cyclization: mechanistic investigation and application to the efficient synthesis of 4-substituted isocoumarins†
Received
4th April 2018
, Accepted 13th June 2018
First published on 15th June 2018
Abstract
The cyclization of heteroatom-containing alkynes with π acidic metal salts is an attractive method to prepare heterocycles because the starting materials are readily available and the organometallic compounds are useful synthetic intermediates. A new organometallic species in the heterocyclization provides an opportunity to synthesize heterocycles that are difficult to obtain. Herein, we describe a novel cyclic oxymetalation of 2-alkynylbenzoate with indium or gallium salts that proceeds with an unusual regioselectivity to give isocoumarins bearing a carbon–metal bond at the 4-position. This new type of metalated isocoumarin provided 3-unsubstituted isocoumarins that have seldom been investigated despite their important pharmacological properties. Indium and gallium salts showed high performance in the selective 6-endo cyclization of terminal alkynes while boron or other metals such as Al, Au, and Ag caused 5-exo cyclization or decomposition of terminal alkynes, respectively. The metalated isocoumarin and its reaction intermediate were unambiguously identified by X-ray crystallographic analysis. The theoretical calculation of potential energy profiles showed that oxyindation could proceed via 6-endo cyclization under thermodynamic control while previously reported oxyboration would give a 5-membered ring under kinetic control. The investigation of electrostatic potential maps suggested that the differences in the atomic characters of indium, boron and their ligands would contribute to such a regioselective switch. The metalated isocoumarins were applied to organic synthetic reactions. The halogenation of metalated isocoumarins proceeded to afford 4-halogenated isocoumarins bearing various functional groups. The palladium-catalyzed cross coupling of organometallic species with organic halides gave various 4-substituted isocoumarins. A formal total synthesis of oosponol, which exhibits strong antifungal activity, was accomplished.
Introduction
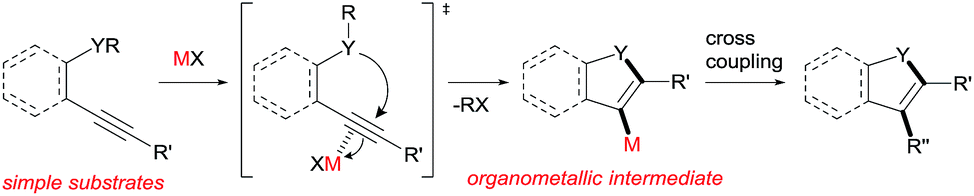
Heterocyclic compounds have attracted much attention in pharmaceutical chemistry as well as in photochemistry and also play a pivotal role as building blocks in organic synthetic transformation.1 Therefore, a novel efficient synthetic method for heterocyclic frameworks is highly desired in various fields of chemistry. Many well-established methods are available in the literature.2 The heterocyclization of ω-heteroatom-substituted alkynes using π acidic metal salts is undoubtedly a powerful strategy for the preparation of heterocycles (Scheme 1).3 This addition reaction uses readily available alkynes as a starting material. Furthermore, metal salt-mediated cyclization spontaneously forms a carbon–metal bond and a heterocyclic framework and produces organometallic intermediates leading to target heterocycles via appropriate synthetic reactions such as cross coupling. These features allowed us to directly access various substituted heterocycles from simple organic substrates.
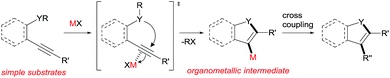 |
| | Scheme 1 Metal salt-mediated heterocyclization of ω-heteroatom-substituted alkynes. | |
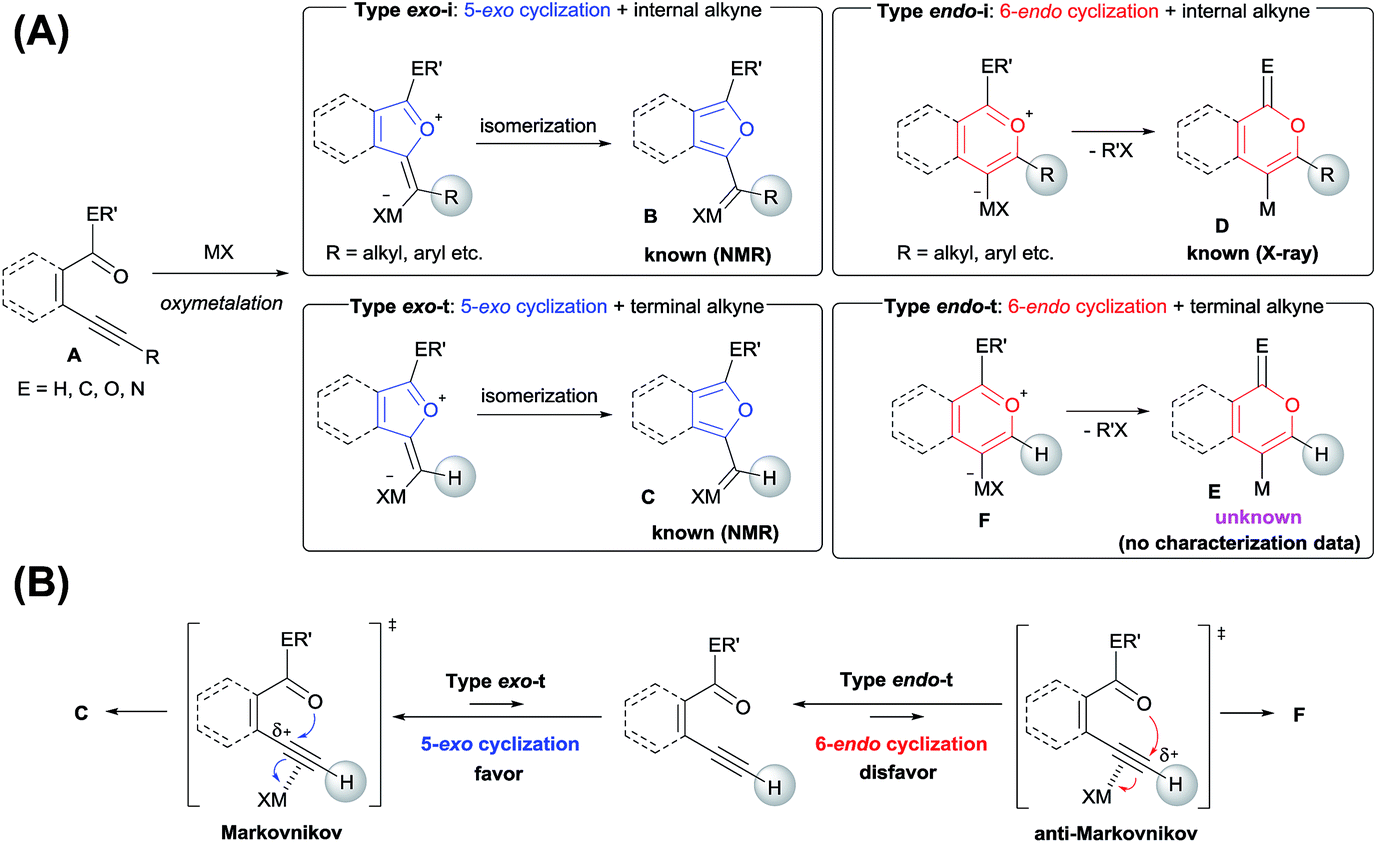
Various heteroatom-containing alkynes have been investigated for use in the synthesis of heterocyclic compounds using π acidic metal salts. Alkyne A includes a carbonyl moiety and is a feasible and beneficial substrate to obtain 5- or 6-membered oxacyclic alkenylmetals (Scheme 2A). When A is treated with a metal salt (MX), oxymetalation proceeds to afford heterocyclic compounds through either 5-exo or 6-endo cyclization (Type exo or endo). Considering that the structure of A bears either an internal (R = alkyl, aryl, etc.) or a terminal alkyne (R = H), oxymetalation can be distinguished by four types of reaction courses: Type exo-i, Type exo-t, Type endo-i, and Type endo-t. In Types exo-i and exo-t, the furan frameworks B and C have a metal carbenoid moiety and are obtained via the isomerization of zwitterion intermediates.4,5 On the other hand, Types endo-i and endo-t lead to 1H-isochromen derivatives D and Evia the elimination of R′X from zwitterion intermediates.6,7 Among these four types, only Type endo-t is kinetically unfavorable due to an unstable cationic transition state via an anti-Markovnikov addition manner (Scheme 2B). Furthermore, recent theoretical research about the regioselectivity of cyclization revealed that the nucleophilic cyclization of alkynes displays exo selectivity intrinsically.8 On the other hand, Lewis acidic metals can promote endo cyclization by decrease of the stereoelectronic penalty, but the exo cyclization was not disturbed, and, thus, the cyclization showed low selectivity.7,9 For the above reasons, there is no report of a preparation method for species Evia Type endo-t in contrast to the cases of Types exo-i, exo-t, and endo-i, for which target organometallic compounds (B–D) are well established.4b,5a,6c,6e If the reaction course of oxymetalation is realized from A to E in Type endo-t,10 various 6-membered heterocycles based on E, which have been difficult to obtain and remain unknown, should be synthesized. Therefore, the establishment of a strategy for Type endo-t is an important challenge in heterocyclic chemistry.
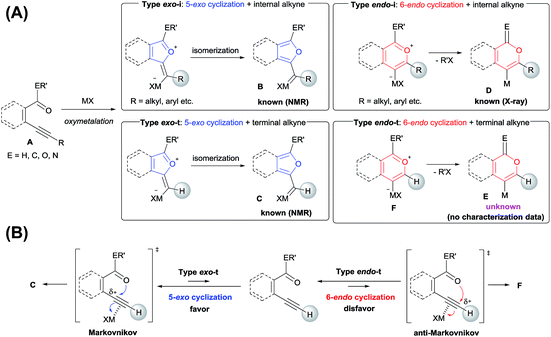 |
| | Scheme 2 (A) Four types of metalated heterocycles from the oxymetalation of alkyne A including a carbonyl moiety. (B) Comparison of the transition states of Type exo-t with Type endo-t. | |
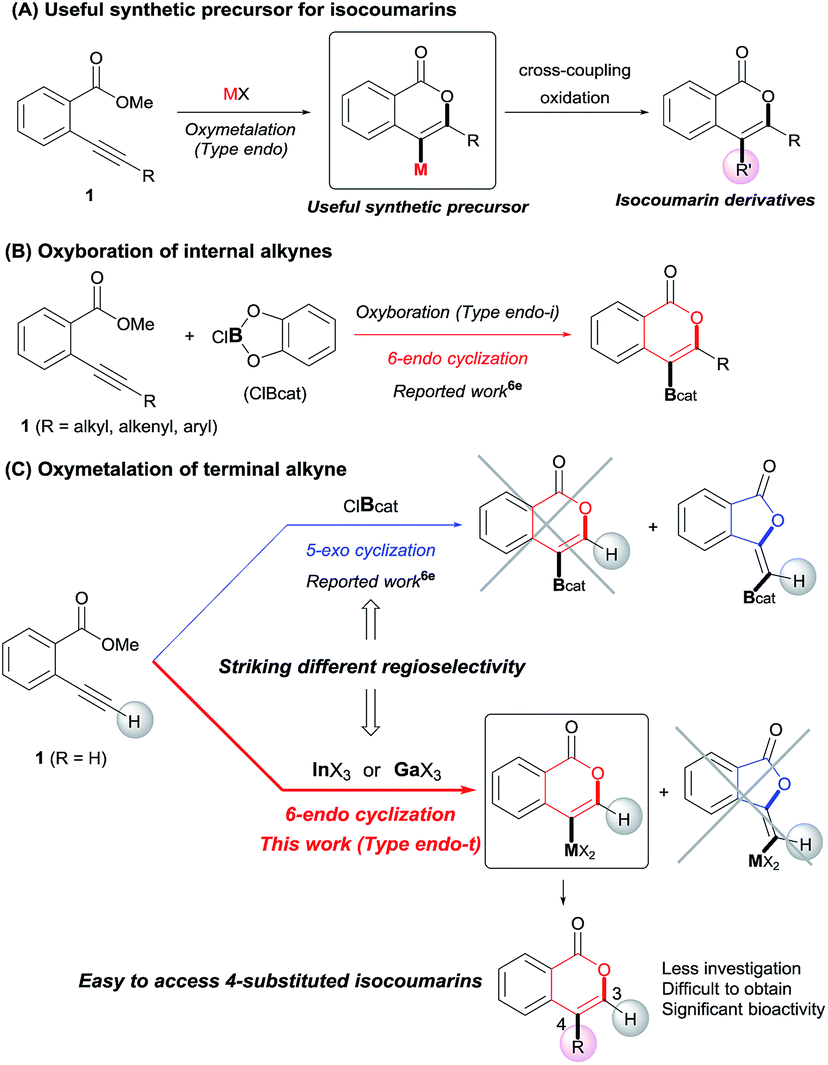
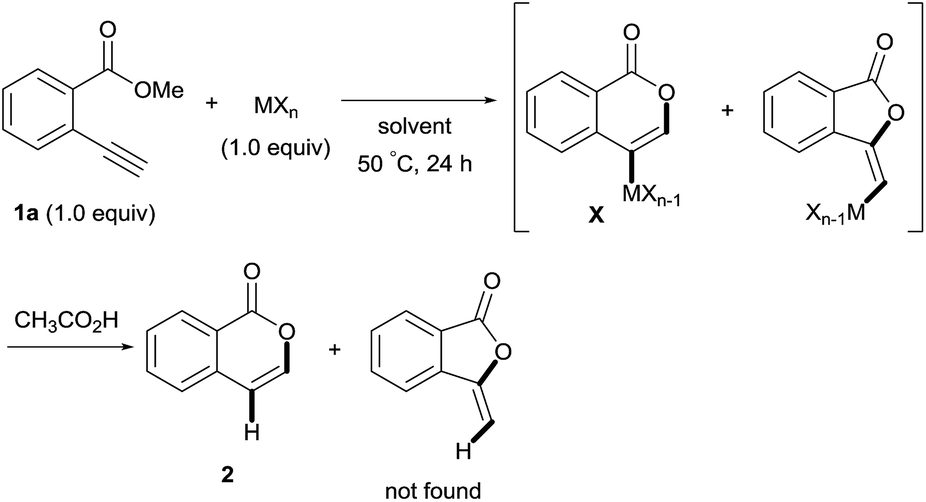
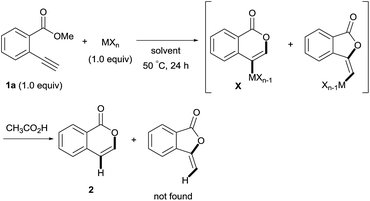
Isocoumarins are an important class of oxygen-containing heterocycles that exhibit a wide range of pharmacological properties.11 Thus, the development of their general synthetic method has attracted much attention. The reaction of Type endo would be a powerful tool for the synthesis of isocoumarins (Scheme 3A). In fact, reports have described the oxymetalation of 2-alkynylbenzoate 1 (R = alkyl, alkenyl, aryl) for Type endo-i and application to the synthesis of isocoumarins.6a,6b,6e Recently, Blum reported an excellent method for the construction of 4-borylated isocoumarins by the oxyboration of the internal alkynes 1 in the Type endo-i reaction course (Scheme 3B).6e However, terminal alkyne 1 (R = H) gave only a 5-exo cyclization product according to the Markovnikov rule (blue path in Scheme 3C).6e This result prompted us to explore the oxymetalation of the terminal alkynes 1 for Type endo-t. The oxymetalation of Type endo-t provides 3-unsubstituted and 4-substituted isocoumarins that are seldom investigated due to the lack of synthetic methods,12 and limited substituents have been introduced at the 4-position despite the well-known beneficial significant bioactivity characteristics such as antitumor,13 antiangiogenic,14 antifungal,15 and antibiotic.16
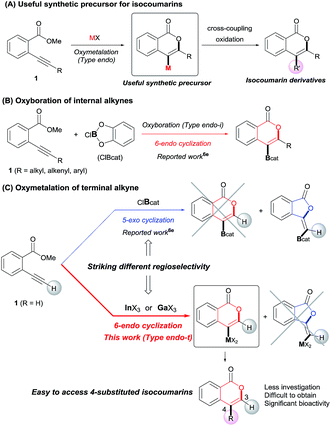 |
| | Scheme 3 (A) Oxymetalation of 2-alkynylbenzoate 1 followed by transformation for the construction of isocoumarins. (B) Previously reported oxyboration of internal alkynes to generate 4-borylated isocoumarins. (C) Oxymetalation of terminal alkynes, reported oxyboration for 5-membered compounds (blue path), and our developed oxymetalation for 6-membered compounds (red path). | |
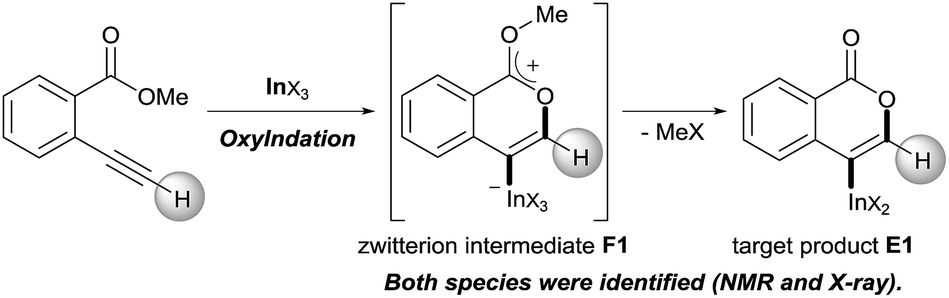
Our group developed the indium or gallium salt-mediated carbometalation of simple terminal alkynes with silyl ketene acetals by utilizing their high π electron affinity and moderate Lewis acidity.17 In this context, we investigated the Type endo-t reaction of 2-ethynylbenzoate using indium or gallium trihalides for the synthesis of corresponding metalated isocoumarins. In this report, we successfully achieved a 6-endo selective oxymetalation of terminal alkynes and fully characterized the target organometallic species E1via an NMR study and X-ray crystallographic analysis. Furthermore, the intermediate F1 was isolated, which revealed that oxymetalation proceeds via the zwitterion intermediate F1, and the elimination of the alkyl halide gives the target product E1 (Scheme 4). While benzopyrylium species such as F are known as highly reactive intermediates in the proposed catalytic oxymetalation cycle,10a,18 the isolation of species F is a challenging issue.10e,10f,19 To the best of our knowledge, F1 is the first example of a fully characterized benzopyrylium intermediate F. In addition, we fully disclosed the mechanism by combining experimental data and theoretical calculation. These mechanistic investigations were consistent with the achievement of isolation of the zwitterion intermediate and demonstrated that its stability is a crucial point in this remarkable cyclization regioselectivity.
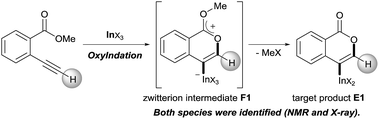 |
| | Scheme 4 Oxyindation of alkynes for the synthesis of an isocoumarin framework via a zwitterion intermediate. | |
Results and discussion
Optimization of reaction conditions
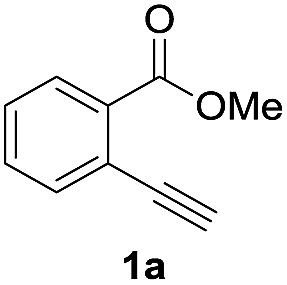
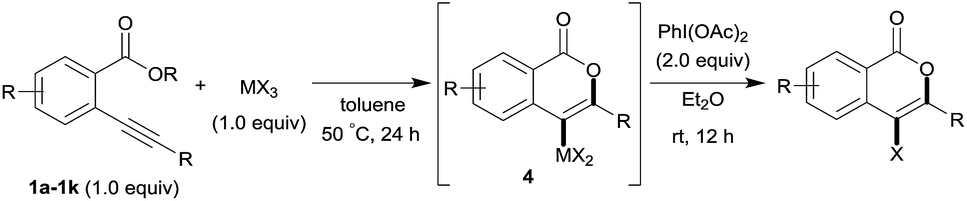
First, we examined the effect of Lewis acids on oxymetalation using methyl 2-ethynylbenzoate 1a (Table 1). The reaction of 1a with metal halides was carried out in toluene at 50 °C, and the reaction mixture was quenched with acetic acid. The reaction using InCl3 afforded the target isocoumarin 2via 6-endo cyclization, albeit in a low yield (entry 1). Gratifyingly, InBr3 and InI3 mediated oxymetalation smoothly proceeded in a 6-endo cyclization fashion to give 2 in high yields (entries 2 and 3). In these cases, the reaction mixture was quenched with deuterated acetic acid to afford 2 bearing deuterium at the 4-position. We did not observe an isocoumarin bearing deuterium at the 3-position which could be produced through the generation of indium acetylide20 followed by Lewis acid mediated cycloaddition. The reaction using InI3 showed a higher ratio of D/H than the case of InBr3. This result suggested the more efficient generation of the alkenylmetal intermediate X in the case of InI3. Gallium salts were also suitable for the 6-endo cyclization of 1a, and GaI3 gave a high yield (entries 4 and 5). On the other hand, typical Lewis acids such as AlCl3, AlI3, BBr3 and TiCl4 were ineffective (entries 6–9). Transition metal salts such as PdCl2, CuBr2 and FeBr3 provided no target product and resulted in a decomposition of 1a (entries 10–12). Alkynophilic π-acids such as gold and silver salts were subjected to the present cyclization. It was found that AuCl3, AuCl, AgOTf and AuCl/AgOTf resulted in low yields (entries 13–16). A decrease in yield was observed at lower temperature (entry 17). The solvent effect was examined on oxyindation using InI3. Dichloroethane as a solvent provided a good yield while chlorobenzene and hexane afforded only moderate yields (entries 18–20). The yields were appreciably decreased in CH3CN and THF (entries 21 and 22) probably because the coordination of these solvents to InI3 decreased the Lewis acidity. Finally, InI3 was the most effective Lewis acid, and, therefore, we chose entry 3 to represent the optimal conditions.
Table 1 Effect of Lewis acids on the oxymetalation of 2-ethynylbenzoate 1aa
|
|
| Entry |
MXn |
Solvent |
Yieldb of 2 (%) |
|
Reaction conditions: 1a (0.5 mmol), Lewis acid MXn (0.5 mmol), solvent (1 mL), 50 °C, 24 h.
The yield of 2 was determined by 1H NMR.
The reaction mixture was quenched with CH3CO2D (30 equiv., 5 min) and a subsequent addition of H2O (10 mL).
35 °C.
|
| 1 |
InCl3 |
Toluene |
13 |
| 2c |
InBr3 |
Toluene |
82 (77% D) |
| 3c |
InI3 |
Toluene |
79 (91% D) |
| 4 |
GaBr3 |
Toluene |
30 |
| 5 |
GaI3 |
Toluene |
77 |
| 6 |
AlCl3 |
Toluene |
0 |
| 7 |
AlI3 |
Toluene |
0 |
| 8 |
BBr3 |
Toluene |
0 |
| 9 |
TiCl4 |
Toluene |
0 |
| 10 |
PdCl2 |
Toluene |
0 |
| 11 |
CuBr2 |
Toluene |
0 |
| 12 |
FeBr3 |
Toluene |
0 |
| 13 |
AuCl3 |
Toluene |
7 |
| 14 |
AuCl |
Toluene |
5 |
| 15 |
AgOTf |
Toluene |
31 |
| 16 |
AuCl/AgOTf |
Toluene |
18 |
| 17d |
InI3 |
Toluene |
61 |
| 18 |
InI3 |
ClCH2CH2Cl |
78 |
| 19 |
InI3 |
ClC6H5 |
57 |
| 20 |
InI3 |
Hexane |
57 |
| 21 |
InI3 |
CH3CN |
17 |
| 22 |
InI3 |
THF |
0 |
Mechanistic investigation
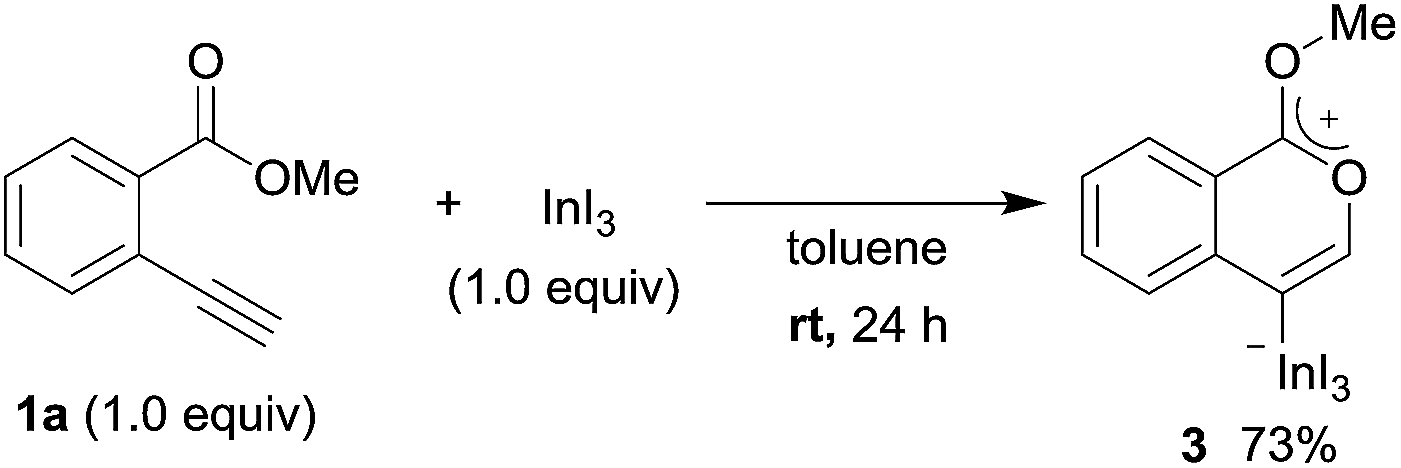
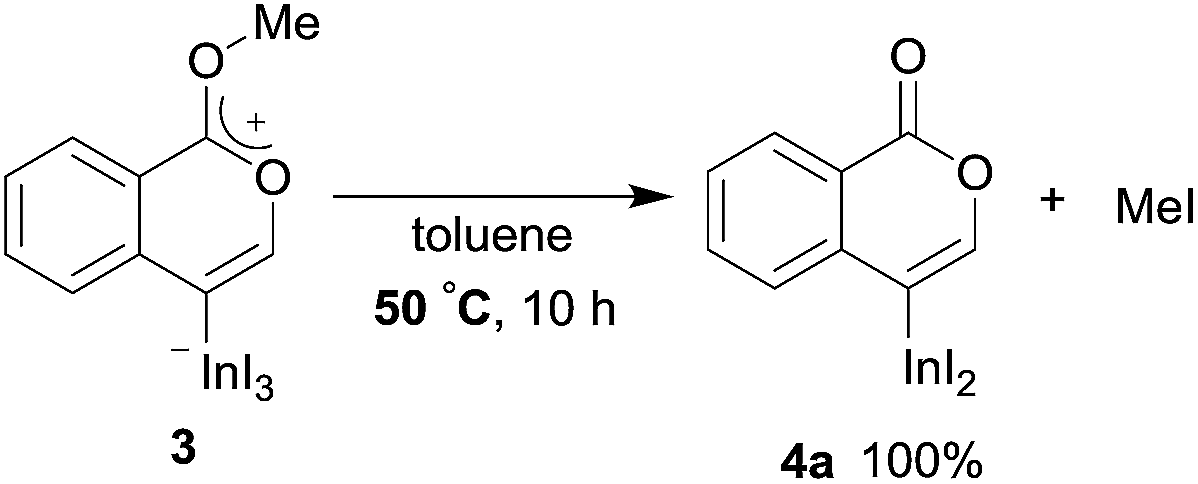
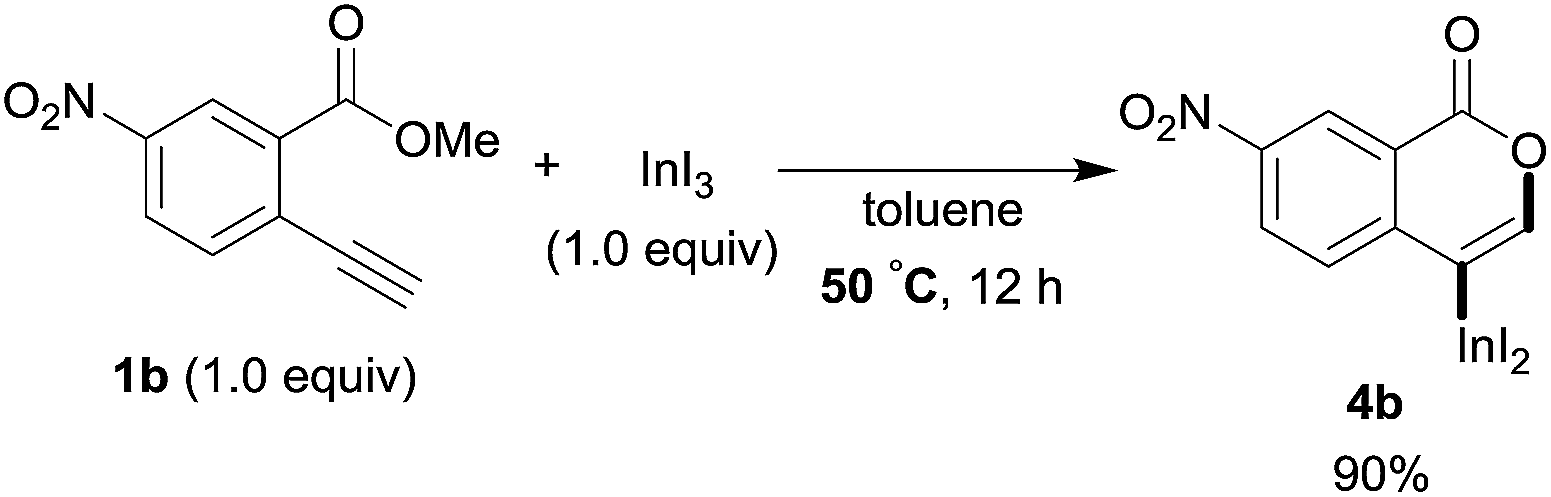
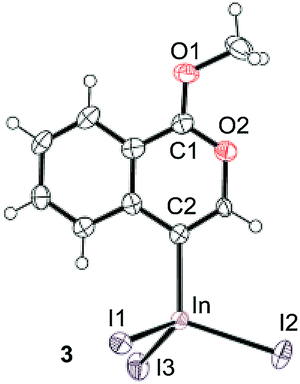
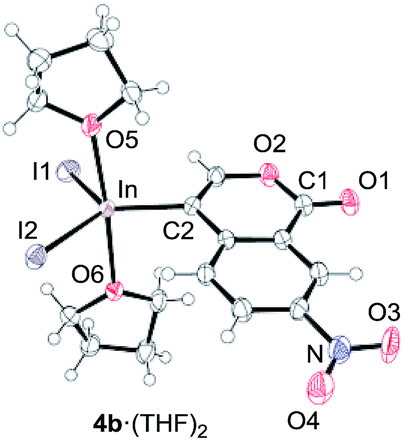
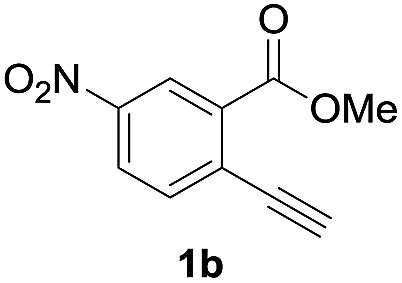
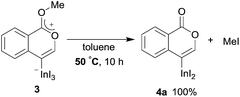
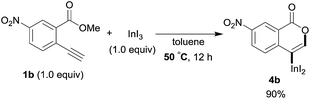
To gain insight into the reaction mechanism, we used 1H NMR spectroscopy to monitor the oxyindation. When 2-alkynylbenzoate 1a was mixed with InI3 in CDCl3 at −30 °C, no reaction occurred. At −5 °C, some amount of a new product was observed (see Fig. S1 and S2† in the ESI). At room temperature, a large amount of white precipitation was formed. This white solid was also obtained in the reaction of 1a with InI3 in toluene at room temperature (eqn (1)). X-ray crystallographic analysis revealed that the white solid was a 6-membered oxacyclic zwitterion 3 bearing a carbon–indium bond (Fig. 1). The bond lengths of the two carbon–oxygen bonds (C1–O1 = 1.267 Å and C1–O2 = 1.298 Å) in the zwitterion 3 existed between a C–O double bond (1.203 Å) and the single bond (1.377 Å) of a typical isocoumarin derivative,21 and, thus, the positive charge was delocalized in an ester moiety. The indium atom was coordinated to three iodines and showed a distorted tetrahedral structure with a formal negative charge. The formed zwitterionic alkenyl indium 3 was heated at 50 °C in toluene to give a neutral alkenylindium product 4a, quantitatively by the elimination of MeI (eqn (2)). Although a suitable single crystal of 4a for X-ray analysis was not obtained, we successfully conducted X-ray diffraction analysis of nitro-substituted alkenylindium 4b produced from 2-ethynyl-5-nitrobenzoate 1b (eqn (3) and Fig. 2). The bond lengths of C1–O1 (1.211 Å) and C1–O2 (1.367 Å) were similar to those of a reported isocoumarin framework.21 The indium complex 4b displayed trigonal bipyramidal coordination with two THF ligands in axial positions. These results indicated a two-step pathway including a fast cyclization and a slow elimination of MeI during the 6-endo oxyindation process from 1 to 4.| | 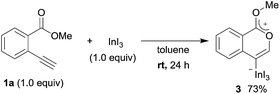 | (1) |
| |  | (2) |
| |  | (3) |
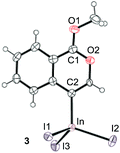 |
| | Fig. 1 The X-ray crystallographic structure of zwitterion intermediate 3 with the thermal ellipsoids shown at 50% probability (CCDC 1579824). Selected bond lengths (Å): C1–O1 = 1.267(9), C1–O2 = 1.298(11), C2–In = 2.171(7), In–I1 = 2.7392(8), In–I2 = 2.7219(8), and In–I3 = 2.6915(7). | |
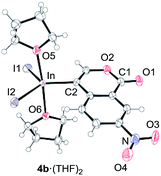 |
| | Fig. 2 The X-ray crystallographic structure of an isocoumarin including a carbon–indium bond at the 4-position, 4b·(THF)2, with the thermal ellipsoids shown at 50% probability (CCDC 1576342). Selected bond lengths (Å) and angles (deg): C1–O1 = 1.211(4), C1–O2 = 1.367(5), C2–In = 2.162(3), In–I1 = 2.7148(4), In–I2 = 2.7005(4), In–O5 = 2.318(3), In–O6 = 2.371(3), O5–In–O6 = 175.44(10), I1–In–C2 = 116.70(11), C2–In–I2 = 125.51(11), and I2–In–I1 = 117.621(12). | |
Theoretical calculation for oxyindation
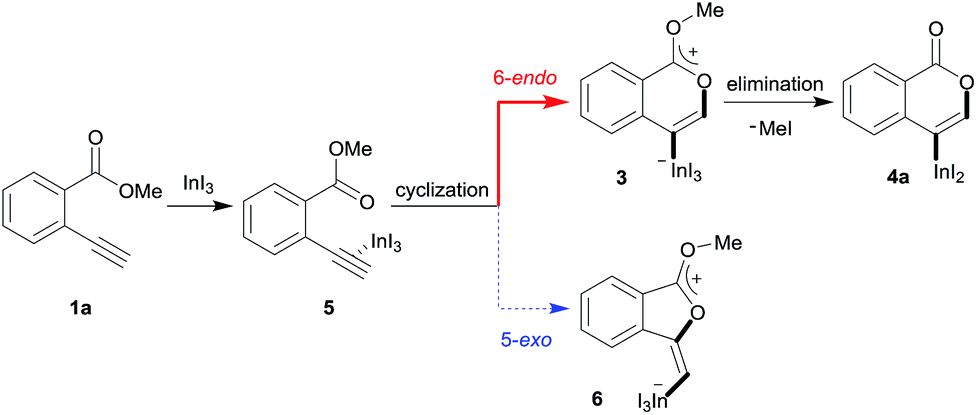
A mechanism for the formation of the target isocoumarin 4a using InI3 is proposed in Scheme 5, wherein InI3 is coordinated to the alkyne moiety in 5, oxyindation proceeds via 6-endo cyclization to give the zwitterion intermediate 3, and, finally, the elimination of MeI affords 4a.
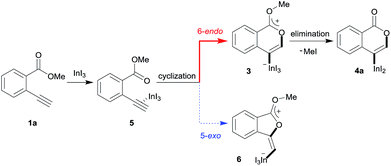 |
| | Scheme 5 A proposed mechanism for the formation of the isocoumarin 4a. | |
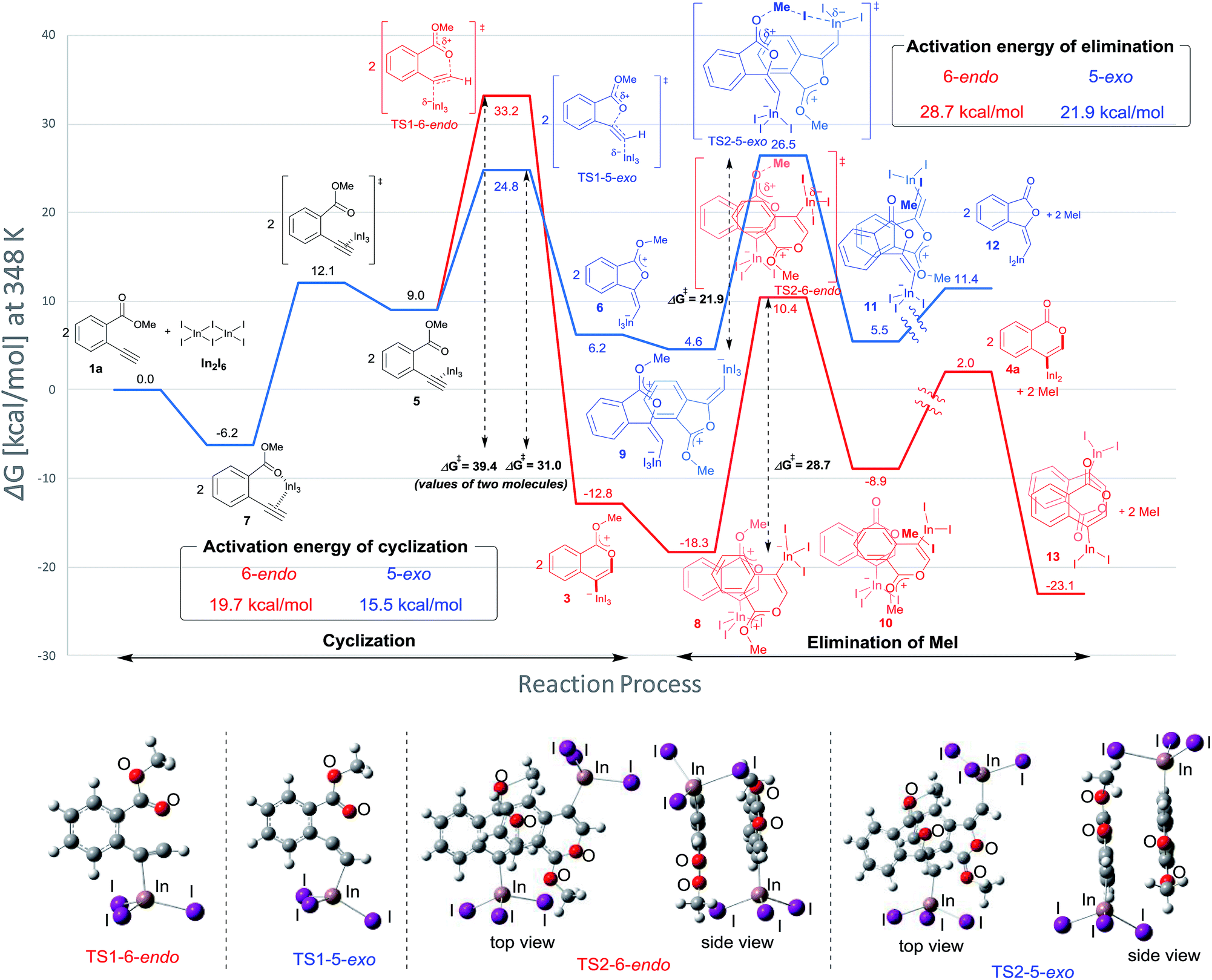
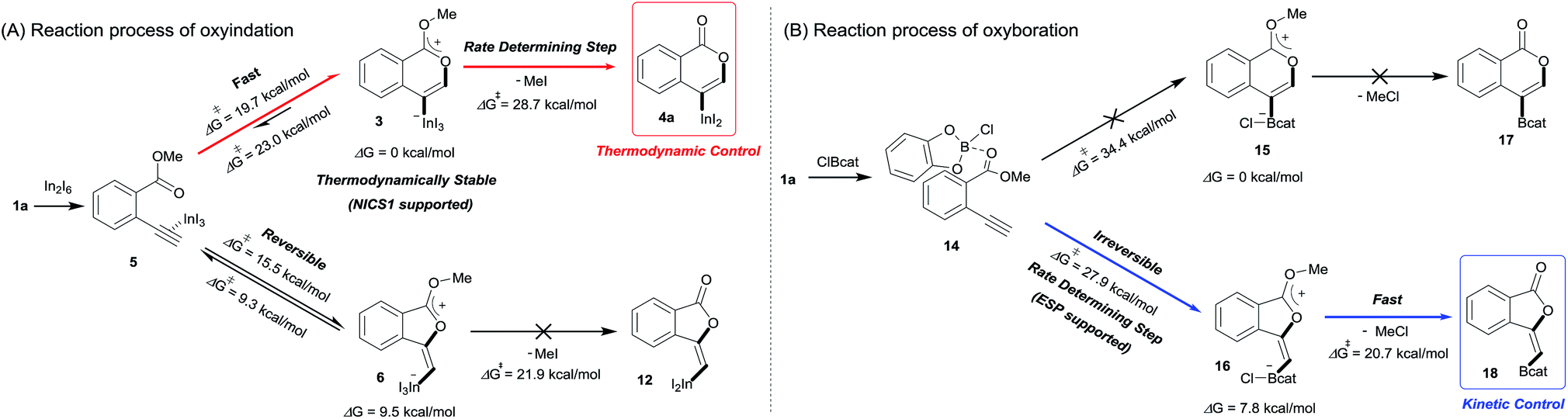
Density functional theory (DFT) calculations were performed to more thoroughly consider the reaction mechanism. The calculation of the potential energy profile for 6-endo cyclization (red) is shown in Fig. 3. We selected 1a and In2I6 as the starting materials because InI3 exists in a dimer fashion.22 The coordination of two 1a to In2I6 dissociates the aggregation of InI3 to give complex 7, in which InI3 is chelated with the alkyne moiety and carbonyl group of 1a (Fig. S3† in the ESI shows the detailed mechanism of generating the complex 7 from 1a and In2I6). The dissociation of the carbonyl oxygen atom generates complex 5, in which InI3 directly activates the alkyne moiety. In this pathway, the anti-addition of InI3 and the ester moiety to the alkyne moiety proceeds in a concerted mechanism to provide a stable 6-membered zwitterion intermediate 3. The elimination of MeI proceeds in an intermolecular fashion, because the intramolecular elimination of MeI requires a very unstable intermediate (Fig. S4† in the ESI shows the potential energy profile for the intramolecular elimination of MeI). Two zwitterions aggregate in a head-to-tail fashion to give complex 8, and then the elimination step starts from 8. The intermolecular nucleophilic substitution of the methyl group by I− proceeds in an SN2-mechanism to give complex 10 and MeI, and then a subsequent elimination of MeI affords the target product 4a.23 A carbonyl group of 4a coordinates to the indium atom of another 4a to give the stable dimeric product 13. The activation energy of the elimination step (8 to TS2-6-endo, 28.7 kcal mol−1) is much higher than that of the cyclization step (7 to TS1-6-endo, 19.7 kcal mol−1).24 Therefore, the elimination of MeI is a rate-determining step. We also calculated the 5-exo cyclization pathway (blue) to investigate the regioselectivity. This process proceeds via concerted cyclization, wherein the 5-membered zwitterion 6 is much more unstable than the 6-membered version 3. The intermolecular elimination of MeI takes place in an SN2-manner (9 to 11). The transition state (TS2-5-exo) shows the highest energy level, and it is even higher than the energy profile of the 6-endo cyclization process (red) due to the instability of the 5-membered zwitterion 6.
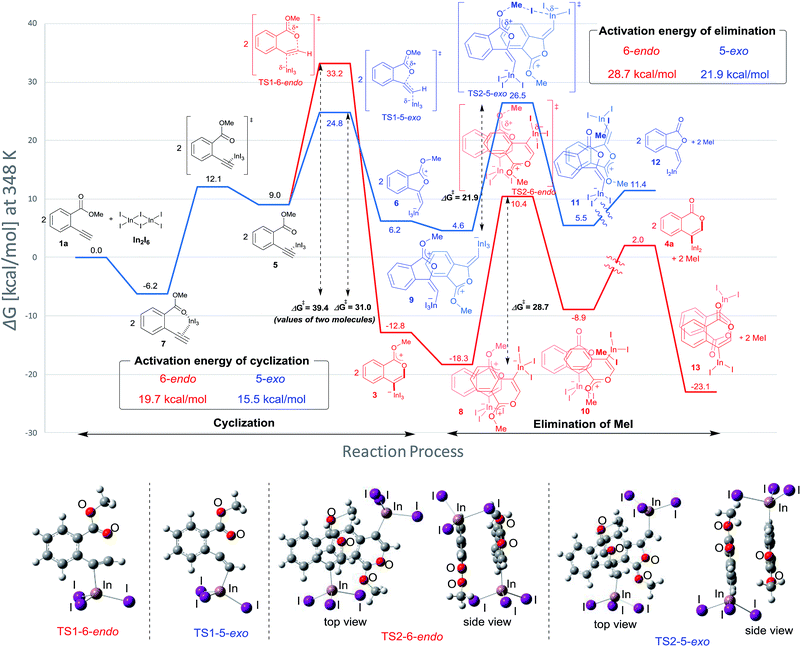 |
| | Fig. 3 The energy profiles of 6-endo and 5-exo oxyindations and 3D molecular structures of transition states. DFT calculation was performed using wB97XD/6-31+G (d,p) for C, H, and O and using DGDZVP for In and I. Solvation effect was introduced using the IEFPCM model, and toluene was used as a solvent. | |
In order to clarify the unique 6-endo cyclization selectivity of oxyindation, the energy profiles of the two cyclization manners were compared. The activation energy of 5-exo cyclization is lower (7 to TS1-5-exo, 15.5 kcal mol−1) than that of 6-endo cyclization (7 to TS1-6-endo, 19.7 kcal mol−1). However, 5-exo cyclization is reversible because the activation energy for the elimination of MeI (9 to TS2-5-exo, 21.9 kcal mol−1) is much higher than that of retro-cyclization (6 to TS1-5-exo, 9.3 kcal mol−1) due to the instability of the zwitterion 6. On the other hand, during 6-endo cyclization, both activation energies of elimination (8 to TS2-6-endo, 28.7 kcal mol−1) and retro-cyclization (3 to TS1-6-endo, 23.0 kcal mol−1) are high because the 6-membered zwitterion intermediate 3 is thermodynamically stable. This result indicates that 6-endo cyclization is irreversible and the most thermodynamically stable form of intermediate 8 is exclusively generated to provide the target product 4a, which is consistent with the successful isolation of the zwitterion intermediate 3 (Fig. 1). Therefore, oxyindation proceeds under thermodynamic control to afford the stable 6-membered product 4a. We also calculated an energy profile of InCl3-mediated oxyindation and found the same pathway with the case of InI3 (see Fig. S5† in the ESI). The activation energy of the elimination step in the case of InCl3 is higher than that of InI3 because of the low nucleophilicity of Cl−, and it caused much less reactivity of InCl3 (entry 1, Table 1).
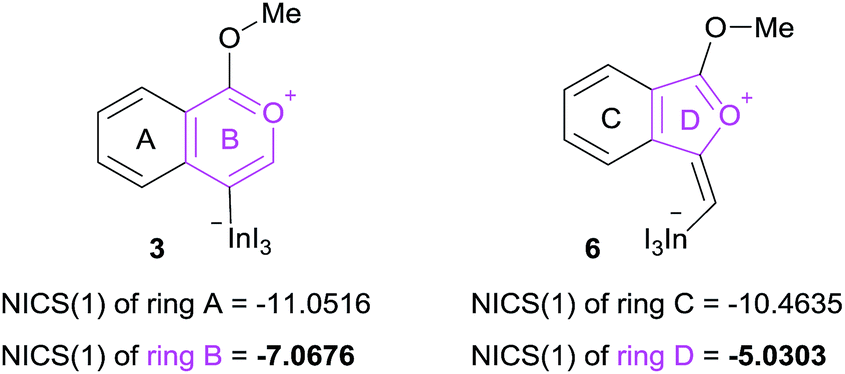
The remarkable regioselectivity of oxyindation is ascribed to the differences in stability between the 6-membered zwitterion 3 and the 5-membered 6. Zwitterion 3 is much more stable than 6, and this difference in stability originates from the aromaticity of these compounds, although ring strain is also a consideration. To verify this possibility, the aromaticity of zwitterions was evaluated via NICS(1)25 (Fig. 4), and the 6-membered compound 3 showed a higher level of aromaticity than 6.
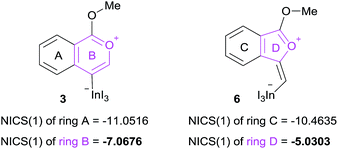 |
| | Fig. 4 NICS(1) values of 6-membered zwitterion 3 and 5-membered zwitterion 6. The aromaticity was calculated using B3LYP/6-31G (d,p) for C, H, and O and using DGDZVP for In and I for their optimized structures. | |
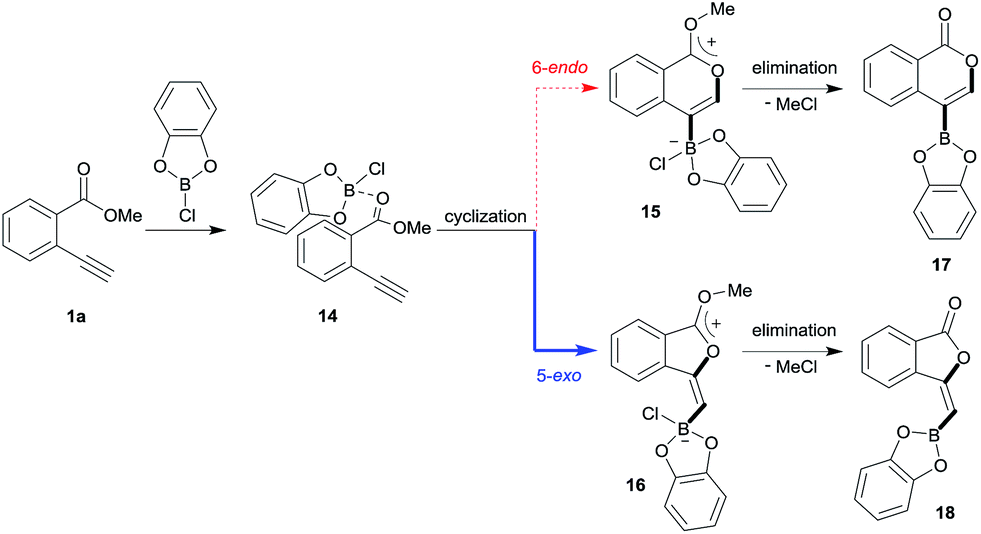
Theoretical calculation for oxyboration
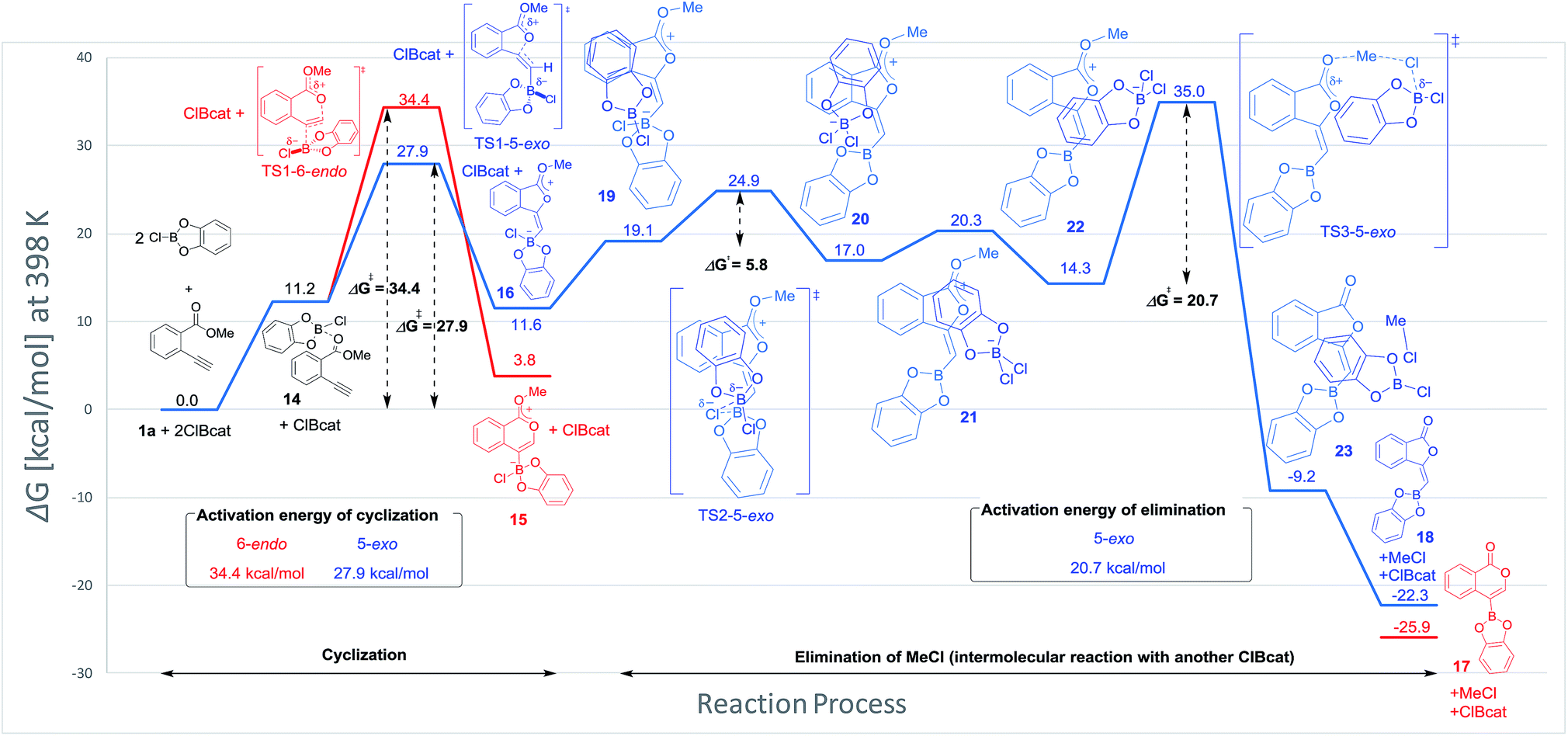
Blum and co-workers reported that the oxyboration of 1a using B-chlorocatecholborane (ClBcat) gave a 5-membered product6e rather than the 6-membered version (Scheme 6). ClBcat is coordinated to the carbonyl moiety of 1a. Then, oxyboration proceeds via 5-exo cyclization to give the zwitterion intermediate 16, and the elimination of MeCl gives the target product 18. We also performed DFT calculation of oxyboration to investigate the striking change in the regioselectivity between oxyboration and oxyindation. First, the calculation of oxyboration was performed for a similar oxyindation mechanism via concerted cyclization and SN2-type elimination of MeCl from aggregated zwitterion intermediates (see Fig. S6† in the ESI). We considered another possibility for the elimination step, because the recent theoretical investigation of ClBcat-mediated heterocyclization has shown other mechanisms,26 whereby the Me group is attacked either by dissociated chloride26a or by [Cl2Bcat]−.26b Thus, we considered these additional two plausible elimination steps assisted by either free Cl− or [Cl2Bcat]− (see Fig. S7† in the ESI and Fig. 5 and 6). The result of comparison between these three pathways showed that the most probable path was the use of [Cl2Bcat]− (details of the comparison are shown in the ESI†).
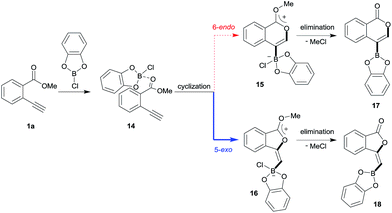 |
| | Scheme 6 A proposed mechanism for oxyboration. | |
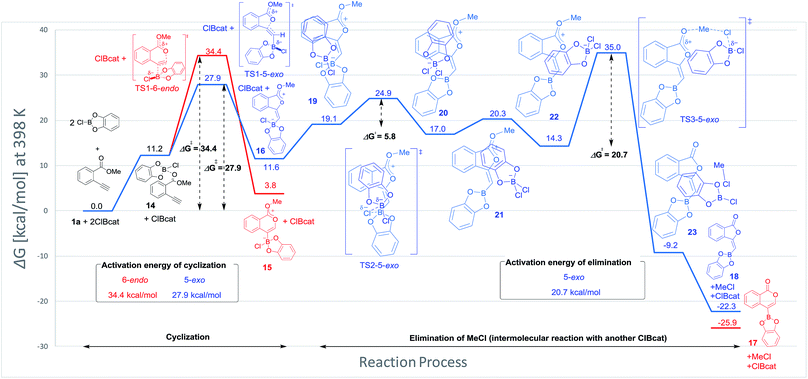 |
| | Fig. 5 The energy profiles of 5-exo and 6-endo oxyborations. DFT calculation was performed with wB97XD/6-31+G (d,p) for C, H, O, B, and Cl. Solvation effect was introduced using the IEFPCM model, and toluene was used as a solvent. | |
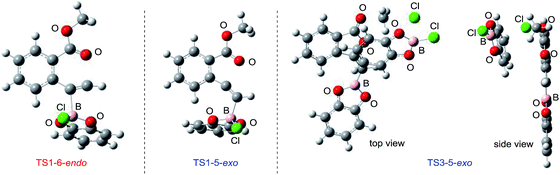 |
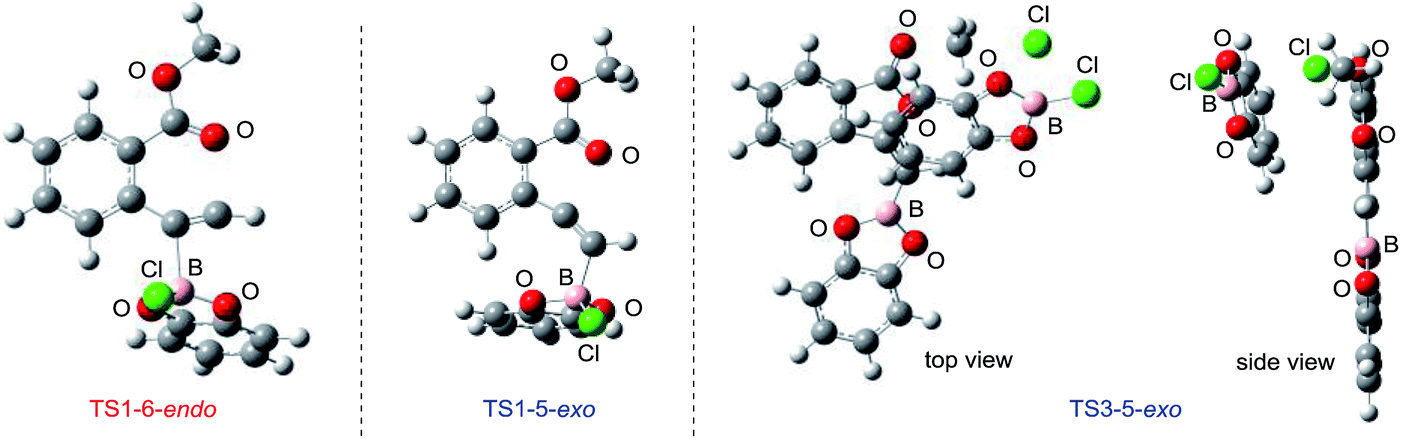
| | Fig. 6 3D molecular structures of transition states in oxyborations. | |
The total reaction profile of oxyboration is described in Fig. 5 and 6. In that profile, 5-exo cyclization from 1a and 2ClBcat to 16 has an activation energy (27.9 kcal mol−1) that is lower than that of 6-endo cyclization (1a and 2ClBcat to 15, 34.4 kcal mol−1). The chloride moiety of zwitterion 16 coordinates to another ClBcat to provide complex 19. The chloride transfer process (19 to 20) has a low energy barrier (5.8 kcal mol−1), and [Cl2Bcat]− is generated rapidly. Cl in [Cl2Bcat]− approaches the methyl group in the ester moiety (20 → 21 → 22), and an elimination of MeCl (22 to 23) in the SN2-mechanism occurs to give 5-membered product 18. The activation energy of the elimination of MeCl (22 to TS3-5-exo) is 20.7 kcal mol−1, which allows the elimination of MeCl to proceed smoothly to give the final product 18. The fast elimination step allows oxyboration to proceed under kinetic control to accomplish the 5-exo selective cyclization.
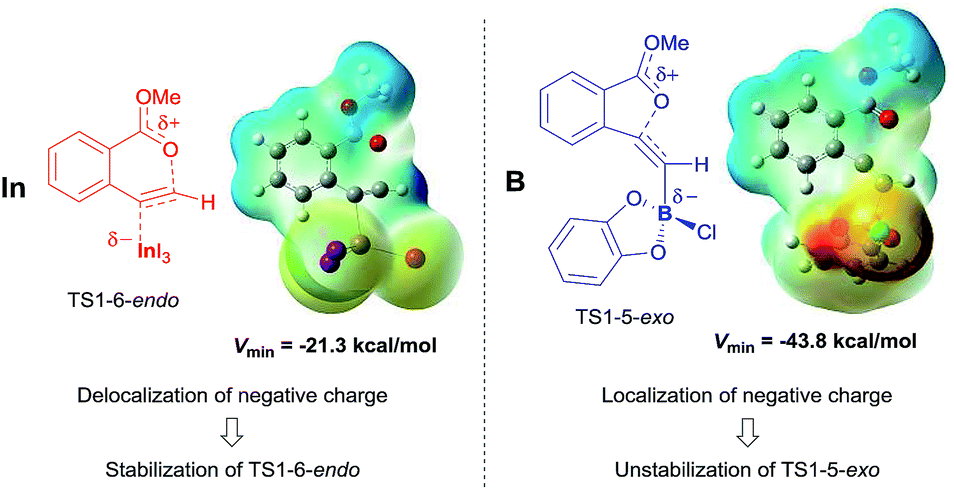
Comparing the transition state of the cyclization step in oxyindation with that in oxyboration based on an electrostatic potential map.
The significant difference between oxyindation and oxyboration was investigated because each showed a characteristic energy profile, particularly for the cyclization step. The energy barrier of cyclization in oxyindation (6-endo: 19.7 kcal mol−1, 5-exo: 15.5 kcal mol−1) is much lower than that of oxyboration (6-endo: 34.4 kcal mol−1, 5-exo: 27.9 kcal mol−1). Therefore, the electrostatic potential maps for the transition states of cyclization (TS1-6-endo and TS1-5-exo) were calculated (Fig. 7). The value of Vmin, which represents the most negative surface electrostatic potential, was investigated to evaluate the degree of localization for a negative charge.27 The Vmin of the organoindium species (left, in Fig. 7) was less negative than that of boron (right, in Fig. 7), which showed that the negative charge was delocalized in the transition state of oxyindation compared with oxyboration. The value of Vmax, which is the most positive surface electrostatic potential, was also calculated and was less affected by the differences in the metals (see Table S2† in the ESI). The polarizability of the indium, boron and heteroatoms binding to a metal explained these results. Indium and iodine atoms have large polarizability (αIn = 69 a.u. and αI = 35.1 a.u.),28 and the increasing negative charge in the TS1 of oxyindation was efficiently delocalized to stabilize the zwitterionic TS1-6-endo.29 On the other hand, boron, chlorine and oxygen atoms have smaller polarizability (αB = 20.5 a.u., αCl = 14.7 a.u., and αO = 6.04 a.u.)28 than indium and iodine atoms, so that TS1-5-exo becomes unstable due to the localization of a negative charge. The difference in the fundamental features between indium and boron atoms imparts a significant amount of influence on the regioselectivity of oxymetalation.
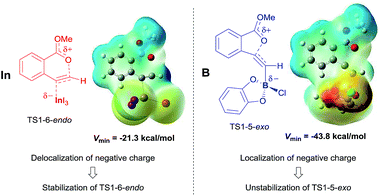 |
| | Fig. 7 Electrostatic potential maps were calculated on the 0.001 au isosurface of electron density for optimized structures of the transition states of oxyindation (left) and oxyboration (right). The potential is depicted by a color gradient from the most negative (red) to the most positive (blue) value (kcal mol−1). Vmin represents the most negative surface electrostatic potential. | |
Summary of DFT calculation
In oxyindation (Fig. 8A), the activation energy of 5-exo cyclization is much lower than that required for the elimination of MeI to lead to reversible 5-exo cyclization. Therefore, the thermodynamically stable 6-membered zwitterion 3 was selectively produced to accomplish the remarkable 6-endo selectivity. The elimination step from 3 is a rate-determining step that provides the target metalated isocoumarin 4a. On the other hand, the energy barrier for cyclization in oxyboration (Fig. 8B) is higher than that for the elimination of MeCl and the cyclization step is a rate-determining step, which leads to irreversible 5-exo cyclization to afford the 5-membered product 18 under kinetic control. Therefore, the activation energies of cyclization as well as elimination are important factors to determine the regioselectivity in cyclization.
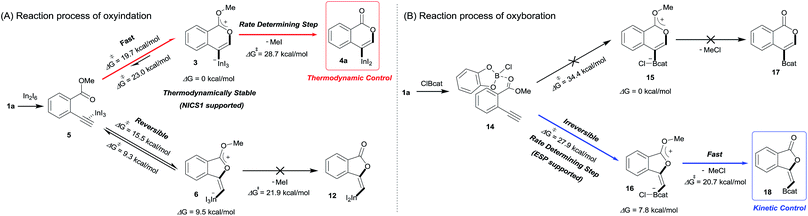 |
| | Fig. 8 The summarized results of DFT calculation. | |
Application to the synthesis of isocoumarin derivatives
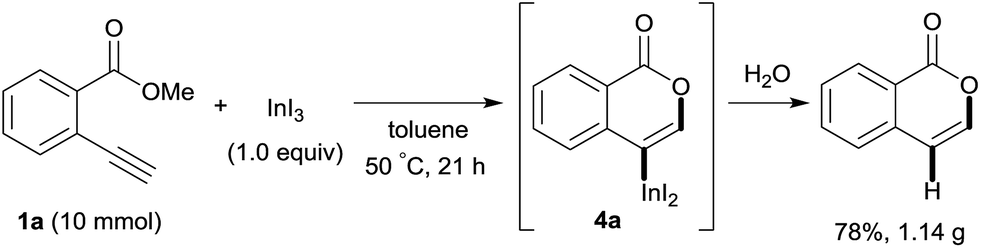
Our developed oxyindation was applied to the synthesis of isocoumarin derivatives. First, the gram-scale synthesis of an organoindium species was carried out. Methyl ester 1a (10 mmol) reacted with InI3 to give organoindium 4a, and 1.14 g of isocoumarin was isolated by the addition of H2O (Scheme 7).
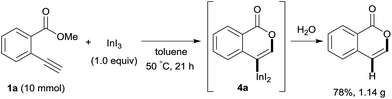 |
| | Scheme 7 Gram-scale synthesis of isocoumarin including a carbon–indium bond. | |
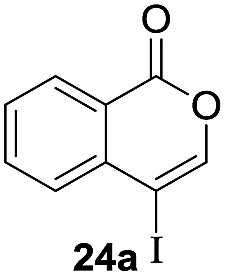
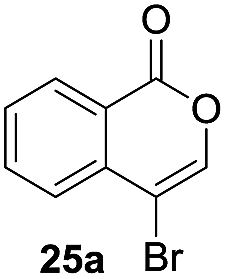
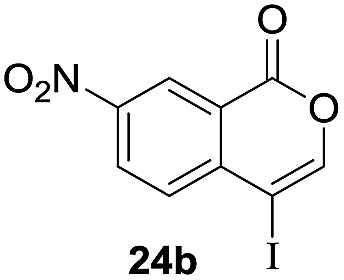
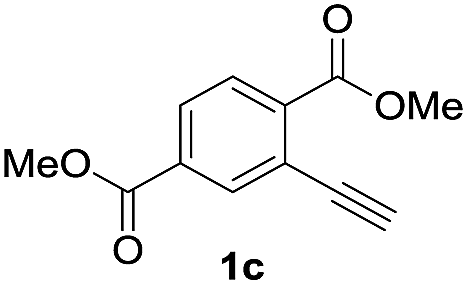
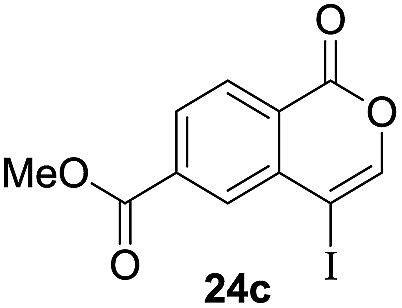
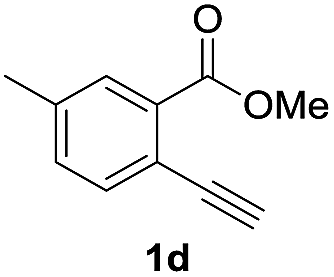
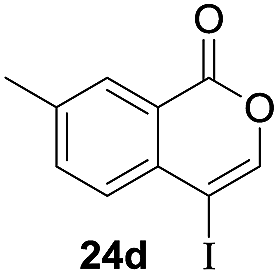
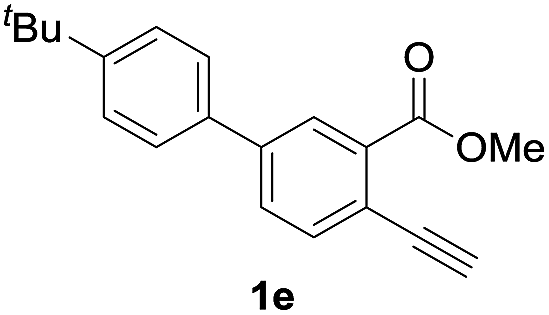
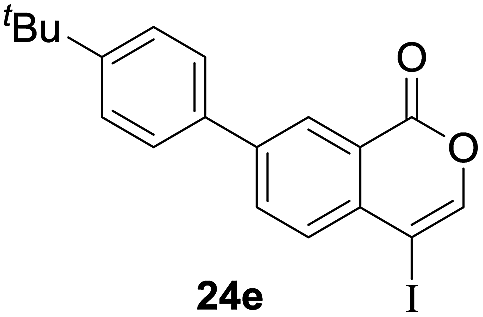
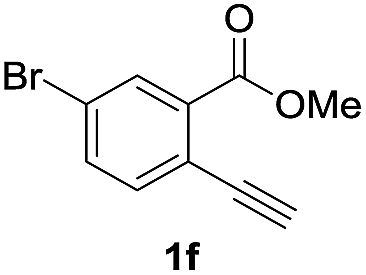
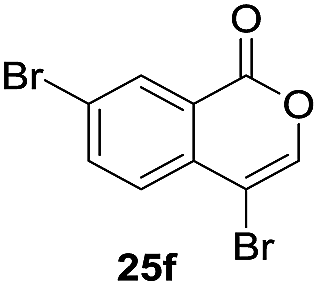
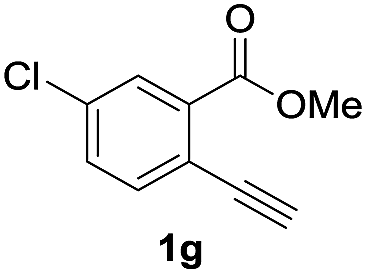
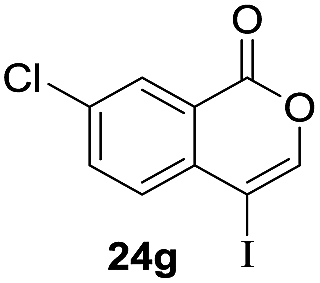
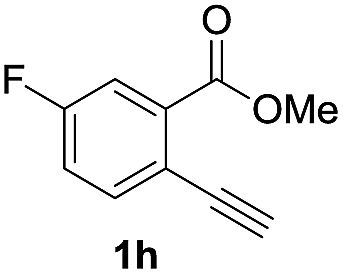
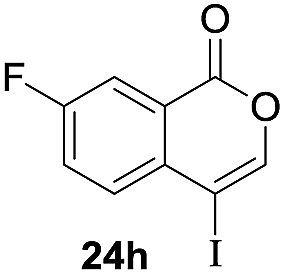
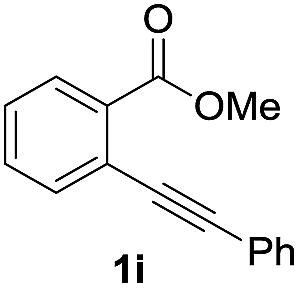
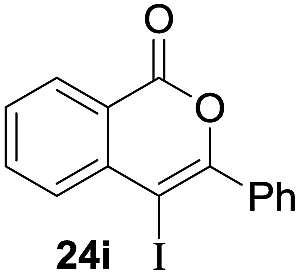
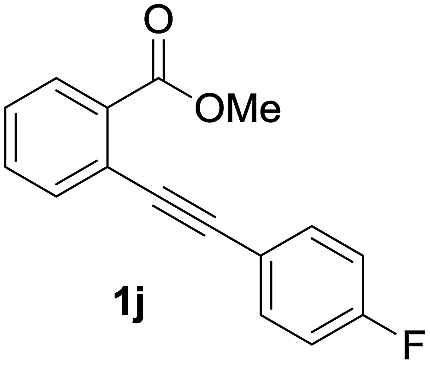
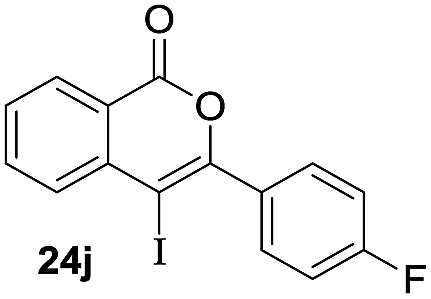
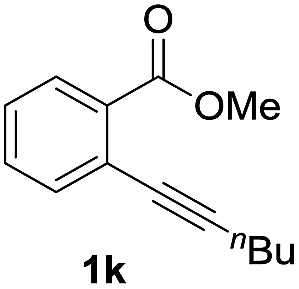
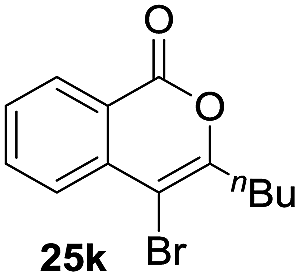
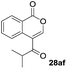
Next, the oxidation of the produced alkenylindium compounds was performed (Table 2). An oxyindation of 1a using InI3 was carried out, and the organoindium 4a was oxidized with PhI(OAc)2 in a one-pot procedure to give 4-iodoisocoumarin 24a (entry 1). Subjecting InBr3 to the oxidation reaction provided 4-bromoisocoumarin 25a in a high yield (entry 2). Therefore, various types of 2-alkynylbenzoates were surveyed in the sequential oxyindation/halogenation process to give 4-halogenated isocoumarins. Substrates with electron withdrawing groups such as nitro and carbonyl groups gave the target products 24b and 24c in high yields (entries 3 and 4). The structure of 24b was characterized by X-ray crystallographic analysis (see Fig. S11† in the ESI). Substrates with methyl or aryl groups efficiently afforded the target isocoumarins 24d and 24e (entries 5 and 6). Also, 2-alkynylbenzoates, including halogen moieties (Br, Cl and F), were suitable for this reaction system to give the isocoumarins 25f–24h in moderate yields (entries 7–9). The synthesis of isocoumarins from internal alkynes was also investigated. The optimization of the reaction conditions showed that gallium salts were more suitable than indium salts for the oxymetalation of an internal alkyne (see Table S3† in the ESI). Therefore, gallium salts were employed in the reactions of internal alkynes 1i–1k to provide the 3,4-disubstituted isocoumarins 24i, 24j and 25k (entries 10–12).
Table 2 Sequential oxymetalation/halogenation of various types of 2-alkynylbenzoate 1a
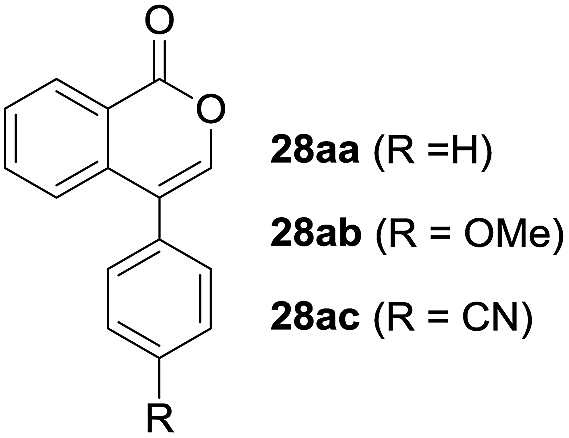
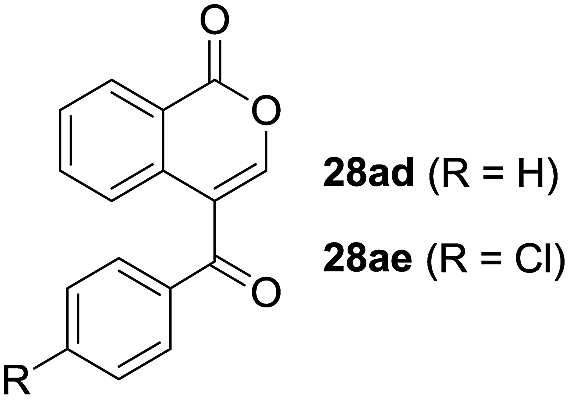
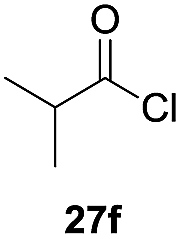
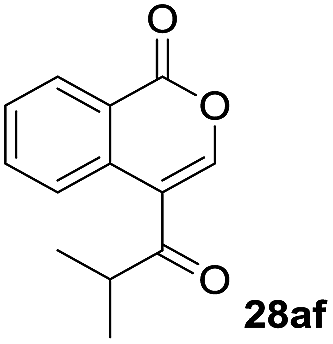
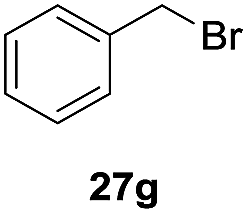
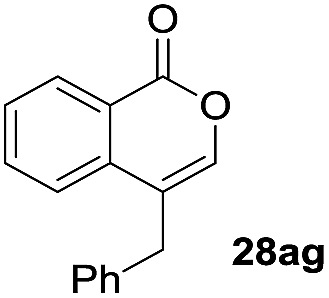
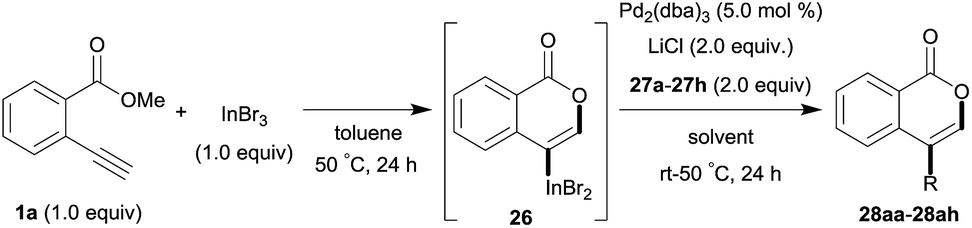
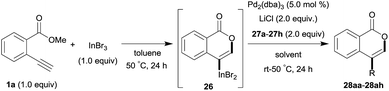
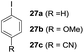
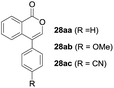
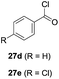
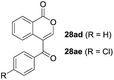
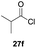
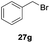
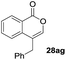
One-pot syntheses of 4-substituted isocoumarins were performed via oxyindation followed by a palladium-catalyzed cross-coupling reaction (Table 3).30 After the oxymetalation of 1a using InBr3, the addition of a palladium catalyst, lithium chloride, organic halides 27, and an additional solvent to the resultant toluene solution afforded the coupling product 28. Iodobenzene 27a and aryl iodides bearing an electron donating group 27b or an electron withdrawing group 27c were applicable to give the 4-arylisocoumarins 28aa–28ac in high yields (entry 1). Palladium-catalyzed cross coupling with acid chlorides also proceeded efficiently. Reactions using the benzoyl chloride derivatives 27d and 27e, as well as the alkanoyl chloride 27f, afforded the isocoumarins 28ad–28af with ketone moieties in good yields (entries 2 and 3). The structure of 28ae was characterized by X-ray crystallographic analysis (see Fig. S12† in the ESI). In this reaction system, alkyl halides such as benzyl bromide 27g and allyl bromide 27h were also suitable to give 4-alkylisocoumarins 28ag and 28ah, respectively (entries 4 and 5). Various types of 4-substituted isocoumarins were obtained from an isocoumarin that included a carbon–indium bond by utilizing palladium-catalyzed cross coupling.
Table 3 One-pot formation of 4-substituted isocoumarins by palladium-catalyzed cross coupling of organoindium species 26 with organic halides 27a
|
|
| Entry |
27
|
Target |
Yieldb (%) |
Basic reaction conditions of the first step: 1a (0.5 mmol), InBr3 (0.5 mmol), toluene (1 mL), 50 °C, 24 h. Second step: Pd2dba3 (0.025 mmol), LiCl (1.0 mmol), 27 (1.0 mmol), NMP (2.5 mL), 50 °C, 24 h.
Isolated yields.
HMPA (2.5 mL), rt, 24 h.
HMPA (2.5 mL), 50 °C, 24 h.
E/Z = 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. :10.
|
| 1 |
|
|
28aa: 8128ab: 7128ac: 72 |
| 2 |
|
|
28ad: 8228ae: 64 |
| 3c |
|
|
61 |
| 4d |
|
|
51 |
| 5d |
|
|
45e |
Formal total synthesis of oosponol
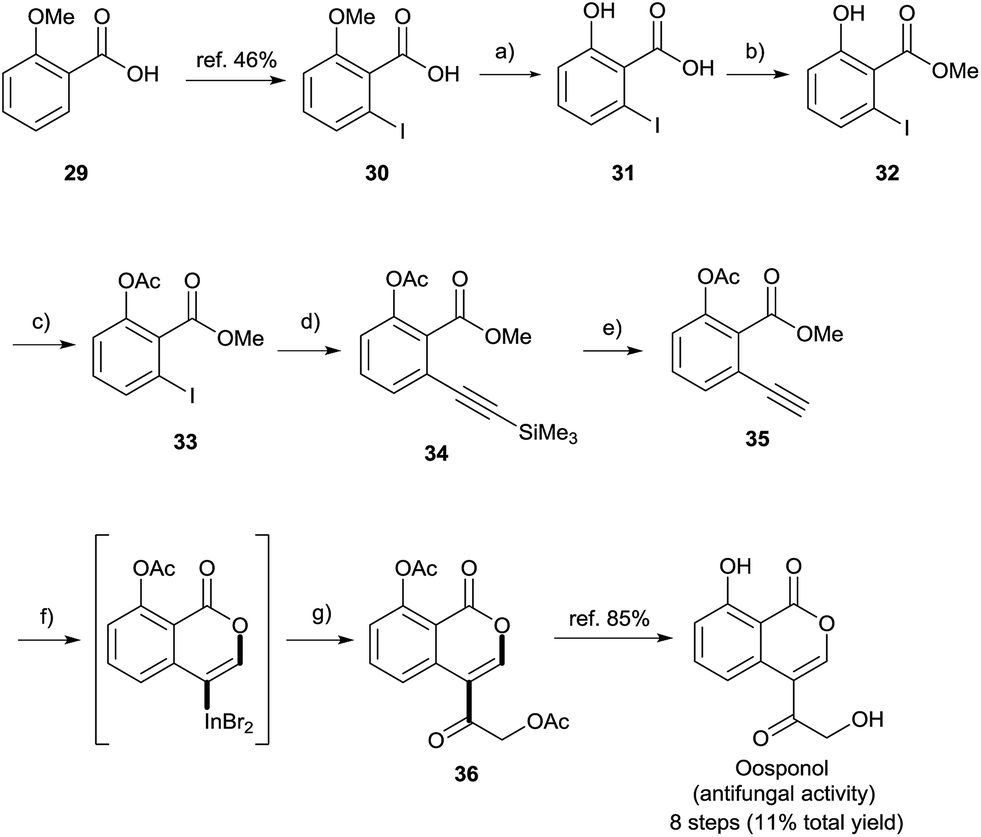
Finally, a formal total synthesis of oosponol, which exhibits strong antifungal activity,15 was conducted (Scheme 8). Firstly, the iodination of commercially available compound 29 proceeded via a method found in the literature.31 During the initial investigation, 30 was transformed into methyl 2-ethynyl-6-methoxybenzoate, and then we attempted the synthesis of the precursor of oosponol via oxyindation and cross-coupling, but the reaction returned a complicated mixture (see Scheme S1† in the ESI). Therefore, in another synthetic route, the OMe group of 29 was converted to an OAc group with less ability to donate electrons. The OMe moiety of 30 was completely deprotected with BBr3. The acid-catalyzed esterification and acetylation of the phenol moiety gave methyl 6-iodoacetylsalicylate 33 in a high yield. Sonogashira coupling followed by the removal of a silyl moiety afforded the desired 2-alkynylbenzoate derivative 35. The oxymetalation of 35 using InBr3 and sequential palladium-catalyzed cross coupling with acid chloride 27i produced the key intermediate 36, and the hydrolyzation of 36 yielded oosponol.16b Our method used a readily available starting material and gave a higher yield than previous studies.16b,32
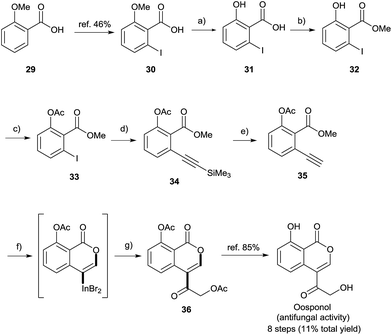 |
| | Scheme 8 Formal total synthesis of oosponol. Reagents and reaction conditions: (a) BBr3 (1 M in CH2Cl2, 2.0 equiv.), CH2Cl2, rt, 20 h, 100%. (b) H2SO4 (20 mol%), MeOH, reflux, 20 h, 87%. (c) AcCl (1.04 equiv.), pyridine (1.04 equiv.), acetone, rt, 14 h, 97%. (d) ethynyltrimethylsilane (1.1 equiv), PdCl2(PPh3)2 (2.0 mol%), CuI (20 mol%), NEt3, RT, 17 h, 100%. (e) 1 M KF aq. (1.65 equiv.), DMF, RT, 0.5 h, 76%. (f) InBr3 (1.0 equiv.), Toluene, 50 °C, 24 h. (g) Pd2dba3 (5.0 mol%), LiCl (2.0 equiv.), 2-(acetyloxy)acetyl chloride 27i (2.0 equiv.), HMPA, rt, 9 h, 44%. | |
Conclusions
We achieved the synthesis of isocoumarins bearing a metal–carbon bond at the 4-position via 6-endo selective oxymetalation of 2-alkynylbenzoate 1 (Type endo-t). Indium and gallium salts showed high activity for the oxymetalation of 2-ethynylbenzoate 1a. Both the metalated isocoumarin 4b and the zwitterion intermediate 3 were identified by X-ray crystallographic analysis. This is the first example of the isolation of the product E and the benzopyrylium intermediate F proposed in the mechanism of oxymetalation (Scheme 2A). The elimination of MeI from zwitterion 3 occurred under heating conditions to give the target product 4a, which means that the rate-determining step was the elimination step. DFT calculation suggested that thermodynamic control led to 6-endo selective oxyindation, while kinetic control led to 5-exo selective oxyboration. The 6-membered product proved much more stable than the 5-membered product due to a difference in the degree of aromatic stability. The investigation of the electrostatic potential of the transition state in the cyclization pathway suggested that a delocalization of negative charge by the large atomic radii of In and I atoms stabilizes the zwitterionic transition state. In contrast, the small atomic radii of B, Cl, and O atoms cause a localization of negative charge to destabilize the corresponding transition state. The difference in stability between the 6- and 5-membered zwitterions and the elemental character of InI3 both played important roles in the unique regioselectivity of oxymetalation and in the facile preparation of the E species.
These isocoumarins bearing a carbon–metal bond at the 4-position were applied to organic synthesis. Oxymetalation provided isocoumarins on a gram scale. The oxidation of organoindium or gallium species yielded various types of 4-halogenated isocoumarins. Palladium-catalyzed cross coupling with aryl iodide, acid chloride, and alkyl bromide gave a wide range of 4-substituted isocoumarins in a one-pot reaction. Therefore, the unprecedented regioselectivity of the present oxymetalation contributed to the synthesis of new types of isocoumarins. We accomplished a formal total synthesis of oosponol to demonstrate the utility of our reaction system.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP15H05848 in Middle Molecular Strategy, JP16K05719 and JP18H01977. Y. N. acknowledges support from the Frontier Research Base for Global Young Researchers, Osaka University, of the MEXT program and from Mitsui Chemicals Award in Synthetic Organic Chemistry. We thank Prof. Dr S. Blum (Department of Chemistry, University of California, Irvine) for helpful suggestions. We would like to thank Prof. Dr S. Akai (Graduate School of Pharmaceutical Science, Osaka University, Japan) for suggestions about the total synthesis of oosponol. Thanks are due to the Analytical Instrumentation Facility, Graduate School of Engineering, Osaka University, for assistance in obtaining the MS spectra.
Notes and references
-
(a) D. B. Kitchen, H. Decornez, J. R. Furr and J. Bajorath, Nat. Rev. Drug Discovery, 2004, 3, 935–949 CrossRef PubMed;
(b) R. E. Ziegert, J. Toräng, K. Knepper and S. Bräse, J. Comb. Chem., 2005, 7, 147–169 CrossRef PubMed;
(c) R. A. Ward and J. G. Kettle, J. Med. Chem., 2011, 54, 4670–4677 CrossRef PubMed;
(d) P. S. Mariano, Tetrahedron, 1983, 39, 3879–3983 CrossRef;
(e) N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef PubMed;
(f) B. H. Lipshutz, Chem. Rev., 1986, 86, 795–819 CrossRef;
(g) A. R. Katritzky, Chem. Rev., 1989, 89, 827–861 CrossRef;
(h) Y.-G. Zhou, Acc. Chem. Res., 2007, 40, 1357–1366 CrossRef PubMed.
-
(a) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef PubMed;
(b) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395–3442 CrossRef PubMed;
(c) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079–3159 CrossRef PubMed;
(d) A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199–2238 CrossRef PubMed;
(e) V. Nair, C. Rajesh, A. U. Vinod, S. Bindu, A. R. Sreekanth, J. S. Mathen and L. Balagopal, Acc. Chem. Res., 2003, 36, 899–907 CrossRef PubMed.
- For selective examples:
(a) J. Liu, X. Xie and Y. Liu, Chem. Commun., 2013, 49, 11794–11796 RSC;
(b) Y.-J. Feng, F.-Y. Tsai, S.-L. Huang, Y.-H. Liu and Y.-C. Lin, Eur. J. Inorg. Chem., 2014, 5406–5414 CrossRef;
(c) S. S. Racharlawar, D. Shankar, M. V. Karkhelikar, B. Sridhar and P. R. Likhar, J. Organomet. Chem., 2014, 757, 14–20 CrossRef;
(d) J. J. Hirner, D. J. Faizi and S. A. Blum, J. Am. Chem. Soc., 2014, 136, 4740–4745 CrossRef PubMed;
(e) D. Shankar, K. Jaipal, B. Sridhar, R. N. Chary, S. Prabhakar, L. Giribabu and P. R. Likhar, RSC Adv., 2015, 5, 20295–20301 RSC;
(f) E. Chong and S. A. Blum, J. Am. Chem. Soc., 2015, 137, 10144–10147 CrossRef PubMed;
(g) K. N. Tu, J. J. Hirner and S. A. Blum, Org. Lett., 2016, 18, 480–483 CrossRef PubMed;
(h) D. J. Faizi, N. A. Nava, M. Al-Amin and S. A. Blum, Org. Synth., 2016, 93, 228–244 CrossRef;
(i) D. J. Faizi, A. J. Davis, F. B. Meany and S. A. Blum, Angew. Chem., Int. Ed., 2016, 55, 14286–14290 CrossRef PubMed;
(j) A. J. Warner, A. Churn, J. S. McGough and M. J. Ingleson, Angew. Chem., Int. Ed., 2017, 56, 354–358 CrossRef PubMed;
(k) N. Chaisan, W. Kaewsri, C. Thongsornkleeb, J. Tummatorn and S. Ruchirawat, Tetrahedron Lett., 2018, 59, 675–680 CrossRef.
-
(a) Y. Kato, K. Miki, F. Nishino, K. Ohe and S. Uemura, Org. Lett., 2003, 5, 2619–2621 CrossRef PubMed;
(b) C. P. Casey, N. A. Strotman and I. A. Guzei, Organometallics, 2004, 23, 4121–4130 CrossRef;
(c) R. Vicente, J. González, L. Riesgo, J. González and L. A. López, Angew. Chem., Int. Ed., 2012, 51, 8063–8067 CrossRef PubMed;
(d) T. Murata, M. Murai, Y. Ikeda, K. Miki and K. Ohe, Org. Lett., 2012, 14, 2296–2299 CrossRef PubMed;
(e) Y. Xia, S. Qu, Q. Xiao, Z.-X. Wang, P. Qu, L. Chen, Z. Liu, L. Tian, Z. Huang, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2013, 135, 13502–13511 CrossRef PubMed;
(f) J.-M. Yang, Z.-Q. Li, M.-L. Li, Q. He, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2017, 139, 3784–3789 CrossRef PubMed.
-
(a) K. Miki, T. Yokoi, F. Nishino, K. Ohe and S. Uemura, J. Organomet. Chem., 2002, 645, 228–234 CrossRef;
(b) K. Miki, T. Yokoi, F. Nishino, Y. Kato, Y. Washitake, K. Ohe and S. Uemura, J. Org. Chem., 2004, 69, 1557–1564 CrossRef PubMed;
(c) K. Okamoto, M. Watanabe, A. Mashida, K. Miki and K. Ohe, Synlett, 2013, 24, 1541–1544 CrossRef.
-
(a) R. C. Larock and L. W. Harrison, J. Am. Chem. Soc., 1984, 106, 4218–4227 CrossRef;
(b) A. Nagarajan and T. R. Balasubramanian, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1987, 26B, 917–919 Search PubMed;
(c) Y. Zhu and B. Yu, Angew. Chem., Int. Ed., 2011, 50, 8329–8332 CrossRef PubMed;
(d) Y. Tang, J. Li, Y. Zhu, Y. Li and B. Yu, J. Am. Chem. Soc., 2013, 135, 18396–18405 CrossRef PubMed;
(e) D. J. Faizi, A. Issaian, A. J. Davis and S. A. Blum, J. Am. Chem. Soc., 2016, 138, 2126–2129 CrossRef PubMed;
(f) B. Akkachairin, J. Tummatorn, N. Supantanapong, P. Nimnual, C. Thongsornkleeb and S. Ruchirawat, J. Org. Chem., 2017, 82, 3727–3740 CrossRef PubMed.
- The metal species E was proposed as intermediate for oxymetalation/protonation sequential reactions, and these cyclic reactions showed low regioselectivity to afford mixture of 5- and 6-membered rings.
(a) E. Marchal, P. Uriac, B. Legouin, L. Toupet and P. v. d. Weghe, Tetrahedron, 2007, 63, 9979–9990 CrossRef;
(b) B. Y.-W. Man, A. Knuhtsen, M. J. Page and B. A. Messerle, Polyhedron, 2013, 61, 248–252 CrossRef.
-
(a) I. V. Alabugin, K. Gilmore and M. Manoharan, J. Am. Chem. Soc., 2011, 133, 12608–12623 CrossRef PubMed;
(b) K. Gilmore, R. K. Mohamed and I. V. Alabugin, WIREs Comput. Mol. Sci., 2016, 6, 487–514 CrossRef.
-
(a) I. V. Alabugin and K. Gilmore, Chem. Commun., 2013, 49, 11246–11250 RSC;
(b) P. W. Peterson, R. K. Mohamed and I. V. Alabugin, Eur. J. Org. Chem., 2013, 2505–2527 CrossRef;
(c) T. Yao and R. C. Larock, J. Org. Chem., 2003, 68, 5936–5942 CrossRef PubMed.
- Although some groups reported 6-endo cyclic oxymetalation using terminal alkynes, the formed 6-membered pyrylium intermediate F was trapped by a nucleophile to give not E but heterocyclic compounds or naphthalene derivatives, and F has not been isolated.
(a) N. Iwasawa, M. Shido and H. Kusama, J. Am. Chem. Soc., 2001, 123, 5814–5815 CrossRef PubMed;
(b) N. Asao, T. Nogami, K. Takahashi and Y. Yamamoto, J. Am. Chem. Soc., 2002, 124, 764–765 CrossRef PubMed;
(c) H. Kusama, H. Funami, J. Takaya and N. Iwasawa, Org. Lett., 2004, 6, 605–608 CrossRef PubMed;
(d) N. T. Patil and Y. Yamamoto, J. Org. Chem., 2004, 69, 5139–5142 CrossRef PubMed;
(e) H. Kusama, H. Funami, M. Shido, Y. Hara, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2005, 127, 2709–2716 CrossRef PubMed;
(f) H. Kusama, H. Funami and N. Iwasawa, Synthesis, 2007, 2014–2024 Search PubMed;
(g) R. Yanada, K. Hashimoto, R. Tokizane, Y. Miwa, H. Minami, K. Yanada, M. Ishikura and T. Takemoto, J. Org. Chem., 2008, 73, 5135–5138 CrossRef PubMed;
(h) X.-L. Fang, R.-Y. Tang, X.-G. Zhang, P. Zhong, C.-L. Deng and J.-H. Li, J. Organomet. Chem., 2011, 696, 352–356 CrossRef;
(i) S. Zhu, Z. Zhang, X. Huang, H. Jiang and Z. Guo, Chem.–Eur. J., 2013, 19, 4695–4700 CrossRef PubMed;
(j) G. Mariaule, G. Newsome, R. Y. Toullec, P. Belmont and V. Michelet, Org. Lett., 2014, 16, 4570–4573 CrossRef PubMed;
(k) J.-F. Cui, H.-M. Ko, K.-P. Shing, J.-R. Deng, N. C.-H. Lai and M.-K. Wong, Angew. Chem., Int. Ed., 2017, 56, 3074–3079 CrossRef PubMed.
-
(a) R. D. Barry, Chem. Rev., 1964, 64, 229–260 CrossRef;
(b)
R. A. Hill, in Progress in the Chemistry of Organic Natural Products, ed. W. Herz, 1986, vol. 49, pp. 1–78 Search PubMed;
(c) I.-U. Rahman, M. Arfan and G. A. Khan, J. Chem. Soc. Pak., 1998, 20, 76–87 Search PubMed;
(d) V. Rukachaisirikul, A. Rodglin, Y. Sukpondma, S. Phongpaichit, J. Buatong and J. Sakayaroj, J. Nat. Prod., 2012, 75, 853–858 CrossRef PubMed;
(e) M. E. Riveiro, A. Moglioni, R. Vazquez, N. Gomez, G. Facorro, L. Piehl, E. R. De Celis, C. Shayo and C. Davio, Bioorg. Med. Chem., 2008, 16, 2665–2675 CrossRef PubMed;
(f) L. M. Bedoya, E. Del Olmo, R. Sancho, B. Barboza, M. Beltrán, A. E. García-Cadenas, S. Sánchez-Palomino, J. L. López-Pérez, E. Muñoz, A. S. Feliciano and J. Alcamí, Bioorg. Med. Chem. Lett., 2006, 16, 4075–4079 CrossRef PubMed;
(g) N. Agata, H. Nogi, M. Milhollen, S. Kharbanda and D. Kufe, Cancer Res., 2004, 64, 8512–8516 CrossRef PubMed;
(h) T. Nakashima, S. Hirano, N. Agata, H. Kumagai, K. Isshiki, T. Yoshioka, M. Ishizuka, K. Maeda and T. Takeuchi, J. Antibiot., 1999, 52, 426–428 CrossRef PubMed;
(i) M. Yoshikawa, E. Harada, Y. Noritoh, K. Inoue, H. Matsuda, H. Shimoda, J. Yamahara and N. Murakami, Chem. Pharm. Bull., 1994, 42, 2225–2230 CrossRef PubMed;
(j) T. Furuta, Y. Fukuyama and Y. Asakawa, Phytochemistry, 1986, 25, 517–520 CrossRef.
-
(a) N. Panda, P. Mishra and I. Mattan, J. Org. Chem., 2016, 81, 1047–1056 CrossRef PubMed;
(b) K. Kobayashi, W. Miyatani and M. Kuroda, Helv. Chim. Acta, 2013, 96, 2173–2178 CrossRef;
(c) M. Lessi, T. Masini, L. Nucara, F. Bellina and R. Rossi, Adv. Synth. Catal., 2011, 353, 501–507 CrossRef;
(d) M. P. Pavan, M. Chakravarty and K. C. K. Swamy, Eur. J. Org. Chem., 2009, 5927–5940 CrossRef;
(e) Z. He and A. K. Yudin, Org. Lett., 2006, 8, 5829–5832 CrossRef PubMed;
(f) M. Chakravarty and K. C. K. Swamy, J. Org. Chem., 2006, 71, 9128–9138 CrossRef PubMed;
(g) S. Kim, G.-J. Fan, J. Lee, J. J. Lee and D. Kim, J. Org. Chem., 2002, 67, 3127–3130 CrossRef PubMed;
(h) B. A. Kowalczyk, Synthesis, 2000, 1113–1116 CrossRef;
(i) P. Babin and J. Dunoguès, Tetrahedron Lett., 1984, 25, 4389–4392 CrossRef;
(j) K. Beautement and J. M. Clough, Tetrahedron Lett., 1984, 25, 3025–3028 CrossRef;
(k) O. S. Wolfbeis, Liebigs Ann. Chem., 1981, 819–827 CrossRef.
- M. Kimura, I. Waki and M. Kokubo, Jpn. J. Pharmacol., 1978, 28, 693–697 CrossRef PubMed.
- J. H. Lee, Y. J. Park, H. S. Kim, Y. S. Hong, K.-W. Kim and J. J. Lee, J. Antibiot., 2001, 54, 463–466 CrossRef PubMed.
-
(a) J. Sonnenbichler and T. Kovács, Eur. J. Biochem., 1997, 246, 45–49 CrossRef PubMed;
(b) T. Kovács and J. Sonnenbichler, Liebigs Ann./Recl., 1997, 1, 211–212 CrossRef;
(c) K. Nozawa, M. Yamada, Y. Tsuda, K. Kawai and S. Nakajima, Chem. Pharm. Bull., 1981, 29, 2689–2691 CrossRef PubMed.
-
(a) S. Nakajima, K. Kawai and S. Yamada, Phytochemistry, 1976, 15, 327 CrossRef;
(b) T. Kovács, I. Sonnenbichler and J. Sonnenbichler, Liebigs Ann./Recl., 1997, 4, 773–777 CrossRef.
-
(a) Y. Nishimoto, H. Ueda, M. Yasuda and A. Baba, Chem.–Eur. J., 2011, 17, 11135–11138 CrossRef PubMed;
(b) Y. Nishimoto, R. Moritoh, M. Yasuda and A. Baba, Angew. Chem., Int. Ed., 2009, 48, 4577–4580 CrossRef PubMed.
- For selective examples:
(a) H. Wang, Y. Kuang and J. Wu, Asian J. Org. Chem., 2012, 1, 302–312 CrossRef;
(b) E. Tomás-Mendivil, F. C. Heinrich, J.-C. Ortuno, J. Starck and V. Michelet, ACS Catal., 2017, 7, 380–387 CrossRef.
- Benzopyrylium intermediate in oxymetalation was detected by NMR studies or MS sepctra:
(a) Y. Zhu and B. Yu, Chem.–Eur. J., 2015, 21, 8771–8780 CrossRef PubMed;
(b) B. N. Nguyen, L. A. Adrin, E. M. Barreiro, J. B. Brazier, P. Haycock, K. K. Hii, M. Nachtegaal, M. A. Newton and J. Szlachetko, Organometallics, 2012, 31, 2395–2402 CrossRefOther types of zwitterion intermediate have been characterized by X-ray crystallographic analysis:
(c) O. A. Egorova, H. Seo, Y. Kim, D. Moon, Y. M. Rhee and K. H. Ahn, Angew. Chem., Int. Ed., 2011, 50, 11446–11450 CrossRef PubMed;
(d) Y. Yu, G. Chen, L. Zhu, Y. Liao, Y. Wu and X. Huang, J. Org. Chem., 2016, 81, 8142–8154 CrossRef PubMed.
- I. Braun, A. M. Asiri and A. S. K. Hashmi, ACS Catal., 2013, 3, 1902–1907 CrossRef.
- T. M. Baber, G. Qadeer, G. S. Khan, N. H. Rama and W.-Y. Wong, Acta Crystallogr., 2006, 62, o5612–o5613 Search PubMed.
- J. D. Forrester, A. Zalkin and D. H. Templeton, Inorg. Chem., 1964, 3, 63–67 CrossRef.
- SN2-type elimination of MeI agrees with the inhibition of the elimination step by bulky O-alkyl group in ester moiety (see Table S1† in ESI).
- The values of the activation energy of the cyclization step shown in Fig. 3 (6-endo: 39.4 kcal mol−1, 5-exo: 31.0 kcal mol−1) are equivalent to two molecules. The activation energy, which is equivalent to one molecule, is a half of the values (6-endo: 19.7 kcal mol−1, 5-exo: 15.5 kcal mol−1).
-
(a) A. R. Katritzky, Chem. Rev., 2001, 101, 1421–1449 CrossRef PubMed;
(b) P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef PubMed.
-
(a) A. Issaian, D. J. Faizi, J. O. Bailey, P. Mayer, G. Berionni, D. A. Singleton and S. A. Blum, J. Org. Chem., 2017, 82, 8165–8178 CrossRef PubMed;
(b) J. Jiang, Z. Zhang and Y. Fu, Asian J. Org. Chem., 2017, 6, 282–289 CrossRef.
- X. Shi, N. El Hassan, A. Ikni, W. Li, N. Guiblin, A. Spasojević de-Biré and N. E. Ghermani, CrystEngComm, 2016, 18, 3289–3299 RSC.
-
P. Schwerdtfeger, Atomic Static Dipole Polarizabilities, in Atoms, Molecules and Clusters in Electric Fields, ed. G. Maroulis, Imperial College Press, London, 2006, pp. 1–32 Search PubMed.
- Silyl anion is stable than carboanion because of the larger atomic radius and higher dipole polarizability of silicon. A. C. Hopkinson and M. H. Lien, Tetrahedron, 1981, 37, 1105–1112 CrossRef.
- K. Zhao, L. Shen, Z.-L. Shen and T.-P. Loh, Chem. Soc. Rev., 2017, 46, 586–602 RSC.
- T.-H. Nguyen, A.-S. Castanet and J. Mortier, Org. Lett., 2006, 8, 765–768 CrossRef PubMed.
- M. Uemura and T. Sakan, J. Chem. Soc. D, 1971, 16, 921 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data, characterization, calculation data and experimental details. CCDC 1576342–1576344 and 1579824. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01537f |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Tetsuji
Yata
a,
Tetsuji
Yata