DOI:
10.1039/C8SC02215A
(Edge Article)
Chem. Sci., 2018,
9, 6053-6061
Single-ribonucleotide repair-mediated ligation-dependent cycling signal amplification for sensitive and specific detection of DNA methyltransferase†
Received
18th May 2018
, Accepted 17th June 2018
First published on 18th June 2018
Abstract
DNA methylation is a predominant epigenetic modification that plays crucial roles in various cellular processes. DNA methyltransferase (MTase) is responsible for DNA methylation, and its dysregulation may induce aberrant methylation patterns that are closely related to cancers. Conventional methods for DNA MTase assay are usually cumbersome and laborious with poor sensitivity. Alternatively, some signal amplification strategies are employed to improve the sensitivity, but they suffer from poor specificity and consequently limited sensitivity due to the nonspecific amplification. Herein, we develop for the first time a new fluorescence method to specifically and sensitively detect DNA MTase activity on the basis of single-ribonucleotide repair-mediated ligation-dependent cycling signal amplification. In the presence of DNA MTase, the hairpin substrate is methylated and cleaved by endonuclease Dpn I, releasing a 24-nt cleavage product. The 24-nt cleavage product may function as a primer and adjacently hybridize with the ligation probes (LP1 and LP2) to form the template (LP1–LP2) for strand displacement amplification (SDA), initiating the single-ribonucleotide repair-mediated cyclic ligation-dependent SDA to produce a large number of reporter probes. The reporter probe can subsequently hybridize with the signal probe that is modified with FAM and BHQ1 to form a stable double-stranded DNA (dsDNA) duplex with a ribonucleotide mismatch. Ribonuclease HII (RNase HII) can excise the single ribonucleotide, resulting in the cyclic cleavage of signal probes and the generation of an enhanced fluorescence signal. Taking advantage of the high specificity of RNase HII-catalyzed single-ribonucleotide excision and the high amplification efficiency of cyclic ligation-dependent SDA, this assay exhibits the highest sensitivity reported so far with a detection limit of 4.8 × 10−6 U mL−1 and a large dynamic range of 5 orders of magnitude. Moreover, this method can be used for the discrimination of Dam MTase from other DNA MTases, the accurate quantification of Dam MTase activity in E. coli cells, and the screening of Dam MTase inhibitors, providing a new paradigm for biomedical research and clinical diagnosis.
Introduction
Genomic DNA methylation frequently occurs at the carbon 5/nitrogen 4 positions of cytosine (C) and the nitrogen 6 position of adenine (A),1 and it is the most important epigenetic modification in genomic DNA,2 playing critical roles in the regulation of gene transcription, chromatin structure, embryonic development, and cellular senescence.3 DNA methyltransferase (MTase) is responsible for the genomic DNA methylation modification, and it catalyzes the transfer of the methyl group to the adenine/cytosine residues in the specific genomic DNA sequences with S-adenosyl-L-methionine (SAM) as the methyl donor to maintain cellular methylation patterns.4,5 The aberrant DNA MTase activity may destroy the normal DNA methylation patterns, inducing a variety of cancers including lung, breast, prostate, colon, liver, kidney, cervix, thyroid, retinoblastoma, and hematologic cancers.6–10 Importantly, the inhibition of DNA MTase activity by specific inhibitors may provide useful information for drug discovery.11 Therefore, DNA MTase may function as both a potential biomarker for early clinical diagnosis and a pharmacological target for anticancer therapy, and the development of accurate and sensitive methods for DNA MTase assay is essential to the understanding of the carcinogenesis mechanism and the discovery of anticancer drugs.
So far, a variety of approaches have been developed for DNA MTase assay including radioactive labeling-based gel electrophoresis,12 enzyme-linked immunoassay,13 and high-performance liquid chromatography (HPLC).14 However, they usually suffer from hazardous radiation,12 poor sensitivity,12,13 sophisticated instruments,14 and labor- and time-consuming experimental procedures,12,13 greatly limiting their practical applications. Alternatively, some new approaches including colorimetric,15 electrochemical,16–19 fluorescence,20–24 and chemiluminescence25 methods have been developed for the detection of DNA MTase activity. The colorimetric assay may realize the visualized detection of DNA MTase activity through the oxidation of ABTS2− by hydrogen peroxide (H2O2) for the generation of colored ABTS˙.15 Electrochemical assays combine a series of nanomaterials (e.g. graphene oxide (GO),16 silver nanoparticles (AgNPs)17 and gold nanoparticles (AuNPs)18,19) with the methylation-sensitive restriction endonuclease to monitor the DNA MTase activity. Fluorescence assays use GO-20 and quantum dot (QD)21-based fluorescence resonance energy transfer (FRET) to quantify DNA MTase activity. However, the sensitivities of these methods are not significantly enhanced15–21 with the involvement of sophisticated manipulation,21 complicated synthesis of nanomaterials,16–20 and tedious surface modification of electrodes.16–19 To improve the detection sensitivity, some signal amplification strategies have been introduced for DNA MTase assay. Typical examples include exonuclease III-aided target recycling22 and nicking endonuclease (e.g. Nt.BbvCI and Nt.Alwl)-assisted cyclic signal probe cleavage23,24-based fluorescence assays, and rolling circle amplification (RCA)-based chemiluminescence assay.25 Despite the improved sensitivity, these assays involve the careful design of molecular beacons22–24 and the complicated preparation of circular templates,25 and usually suffer from a high background signal resulting from either nonspecific digestion22–24 or nonspecific amplification.25 Notably, conventional nucleic acid amplification approaches (e.g. polymerase chain reaction (PCR),26 strand displacement amplification (SDA),27,28 rolling circle amplification (RCA),25 and exponential isothermal amplification reaction (EXPAR)29) are usually based on either DNA polymerase or the combination of nickase and DNA polymerase to produce large amounts of DNA fragments for the achievement of signal amplification, but they inevitably suffer from a high background signal caused by nonspecific amplification, because (1) some DNA polymerases have no proofreading exonuclease activity to repair the mismatched deoxyribonucleotides,30,31 which leads to the generation of nonspecific fragments; (2) the DNA polymerase mediates ab initio synthesis32 and the elongation of DNA duplexes in which the recognition site of nickase will be randomly incorporated,33 which results in exponential amplification of nonspecific DNA.34 To eliminate the high background signal, uracil-DNA glycosylase and endonuclease IV are introduced to coordinate with DNA polymerase to initiate uracil repair-mediated nucleic acid amplification,34,35 but this enzymatic repair-based amplification (ERA) requires the coexistence of two repair enzymes (i.e., uracil-DNA glycosylase and endonuclease IV) and involves multi-step cleaving reactions (i.e. uracil-DNA glycosylase-mediated uracil base excision and endonuclease IV-mediated apurinic/apyrimidinic (AP) site cleavage) and the careful design of the DNA template with a uracil mismatch. To simplify the experimental design and improve the detection specificity and sensitivity, we introduce ribonuclease HII (RNase HII) that can specifically excise any single ribonucleotide misincorporated in a one-step hydrolysis reaction of the phosphodiester bond. RNase HII is an endoribonuclease widely distributed in living organisms, and it plays an essential role in the repair of ribonucleotides existing in the genomic DNA.36,37 RNase HII can specifically recognize the single ribonucleotide misincorporated within the 5′-DNA-RNA-DNA-3′/3′-DNA-5′duplexes and then efficiently hydrolyze the phosphodiester bonds 5′ to the ribonucleotide at the DNA-RNA junction, leaving a single nucleotide gap with the 5′ phosphate and 3′ hydroxyl ends.36,38 In this research, we utilize the unique feature of RNase HII to develop a new fluorescence method for specific and sensitive detection of DNA MTase activity on the basis of single-ribonucleotide repair-mediated ligation-dependent cycling signal amplification. Taking advantage of the high specificity of RNase HII-catalyzed single-ribonucleotide excision, the high amplification efficiency of cyclic ligation-dependent SDA, and the low background signal resulting from the inhibition of nonspecific amplification by single-ribonucleotide excision and specific ligation-dependent strand displacement polymerase extension, this method can detect Dam MTase activity with a detection limit of as low as 4.8 × 10−6 U mL−1 (this is the lowest detection limit for DNA MTase assay reported so far) and exhibits a large dynamic range of 5 orders of magnitude from 1 × 10−4 to 40 U mL−1. This method can be further applied for the discrimination of Dam MTase from other DNA MTases, the screening of Dam MTase inhibitors, and the quantification of Dam MTase activity in E. coli cells, holding great potential in biomedical research and clinical diagnosis.
Results and discussion
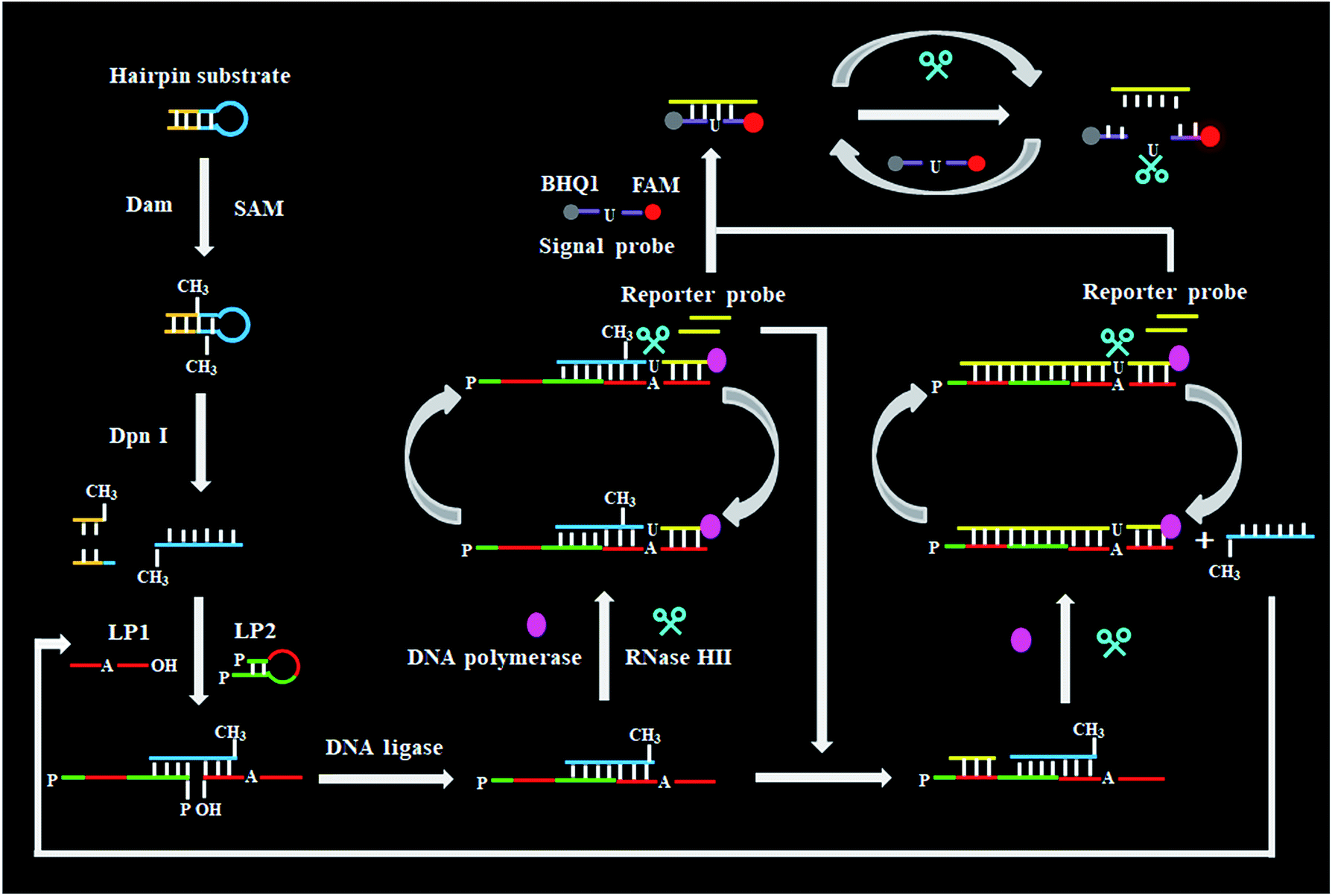
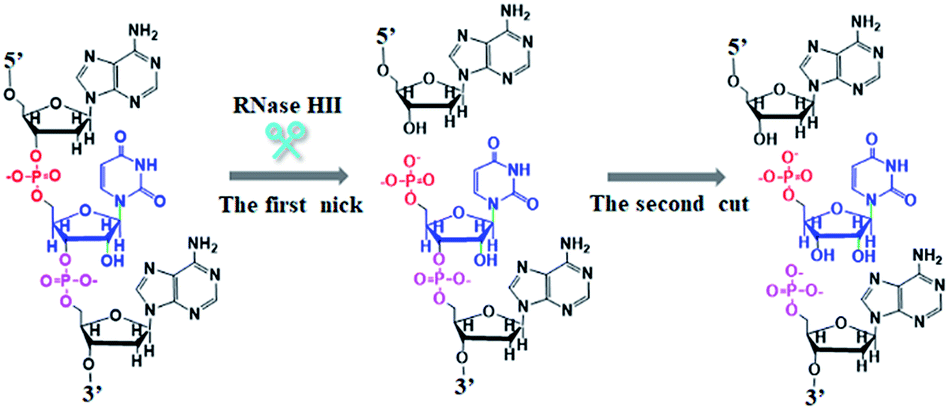
As a proof of concept, we used Dam MTase as a model target. Dam MTase is distributed in prokaryotic organisms, and it can methylate the N6 position of the adenine residue in the palindromic sequence of 5′-G-A-T-C-3′,39 playing a crucial role in regulating various cellular processes (e.g. DNA transcription, DNA replication, and RNA repair).40–42 The principle of single-ribonucleotide repair-mediated ligation-dependent cycling signal amplification for DNA MTase assay is illustrated in Scheme 1. We designed a hairpin substrate with a stem and a loop. The stem contains the palindromic sequence of 5′-G-A-T-C-3′, which functions as the DNA substrate of Dam MTase. The loop (Scheme 1, blue color) is partly complementary to the ligation probes LP1 and LP2, respectively, whose ligation reaction may form LP1–LP2 as the template of SDA. The linear LP1 (Scheme 1, red color) with a hydroxyl (OH) group modification at the 3′ end is partly complementary to the blue sequence of the hairpin substrate (Scheme 1, blue color). In addition, LP1 contains a single adenine (A) base which is matched with the uracil (U) base and may actuate the RNase HII-catalyzed single uracil nucleotide repair-mediated SDA. The LP2 with a hairpin structure (Scheme 1, green color) consists of a stem and a loop. The stem contains two complementary strands (the sense strand and the antisense strand). In the upper strand, the 3′ end is modified with a phosphate group (PO4) to prevent the occurrence of the polymerization reaction. In the lower strand, the 5′ end is modified with a PO4 group for the ligation reaction. In the loop, a sequence (Scheme 1, LP2, red color) the same as the part sequence of LP1 is designed to be complementary to the reaction product (i.e. reporter probe) (Scheme 1, yellow color), and it may trigger single uracil nucleotide repair-mediated cyclic ligation-dependent SDA. The signal probe (Scheme 1, purple color) containing a uracil ribonucleotide is a 15-nt sequence flanked by nucleotides modified with a fluorophore (FAM) and a quencher (BHQ1). The signal probe is complementary to the reporter probe and can be cleaved by RNase HII to generate a distinct fluorescence signal. This assay consists of three reaction steps: (1) Dam MTase-directed cleavage of hairpin substrates, (2) single-ribonucleotide repair-mediated cyclic ligation-dependent SDA, and (3) RNase HII-catalyzed cyclic cleavage of signal probes for the generation of a fluorescence signal. In the first step, Dam MTase will transfer the methyl group from SAM to the adenine residue in the palindromic sequence of 5′-G-A-T-C-3′, enabling the methylation (i.e. 5′-G-mA-T-C-3′) of hairpin substrates. Subsequently, the methylated hairpin substrate is cleaved at the site of methylated adenine by endonuclease Dpn I,43 generating a 24-nt cleavage product (Scheme 1, blue color). In the second step, LP1 and LP2 hybridize adjacently with the 24-nt cleavage product and are then ligated to form the template (LP1–LP2) of SDA. In the presence of DNA polymerase and four nucleotides (i.e., dATP, dGTP, dCTP and dUTP), the 24-nt cleavage product will function as a primer to initiate the polymerization extension, generating a stable double-stranded DNA (dsDNA) duplex with the incorporation of a uracil (U) ribonucleotide. The uracil ribonucleotide can be specifically excised by RNase HII (Fig. 1), generating a single nucleotide gap with phosphate (PO4) and hydroxyl (OH) groups at the 5′ and 3′ ends, respectively. The site with a 3′-OH end will become a new replication site for DNA polymerase to initiate SDA, producing abundant reporter probes (Scheme 1, yellow color). The released reporter probe may act as a primer to hybridize with the LP1–LP2 template, initiating a new polymerization extension to form a new dsDNA duplex with the incorporation of a uracil nucleotide. The subsequent RNase HII-catalyzed uracil nucleotide excision induces the release of a 30-nt cleavage product (Scheme 1, blue color). The released 30-nt cleavage product will hybridize with the free LP1 and LP2 to form a new LP1–LP2 template, initiating a new cycle of single-ribonucleotide repair-mediated ligation-dependent SDA. Through cycles of ligation, polymerization, cleavage and release, multiple SDA reactions will be initiated to generate large numbers of reporter probes. In the third step, the signal probe (Scheme 1, purple color) with a uracil (U) ribonucleotide can hybridize with the resultant reporter probe, forming a dsDNA duplex with a uracil ribonucleotide mismatch. RNase HII may specifically excise the uracil ribonucleotide, leading to the breaking of the signal probe and consequently the generation of the fluorescence signal and the release of the reporter probe. The released reporter probe can bind with another signal probe to initiate cycles of hybridization, cleavage and release, eventually resulting in significant fluorescence enhancement. In comparison with the reported nucleic acid amplification approaches,25–29 this single-ribonucleotide repair-mediated ligation-dependent cycling signal amplification has significant advantages: (1) ligation-dependent strand displacement polymerization extension and RNase HII-catalyzed single ribonucleotide excision can prevent the nonspecific amplification, greatly reducing the background signal; (2) cyclic ligation-dependent SDA can achieve high amplification efficiency, improving the detection sensitivity; (3) both the ligation reaction and RNase HII-catalyzed single-ribonucleotide excision can be implemented in a homogeneous format without the involvement of any thermal cycling, washing and separation steps, greatly simplifying the experimental procedures.
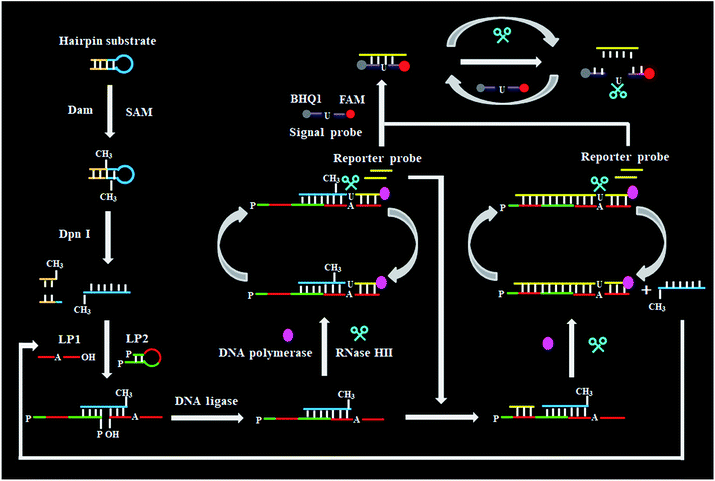 |
| | Scheme 1 Schematic illustration of DNA MTase assay based on single-ribonucleotide repair-mediated ligation-dependent cycling signal amplification. This assay involves three reaction steps: (1) Dam MTase-directed cleavage of hairpin substrates, (2) single-ribonucleotide repair-mediated cyclic ligation-dependent SDA, and (3) RNase HII-catalyzed cyclic cleavage of signal probes for the generation of a distinct fluorescence signal. | |
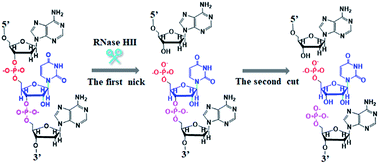 |
| | Fig. 1 Mechanism of RNase HII-catalyzed single-ribonucleotide repairing. The single ribonucleotide misincorporated within the 5′-DNA-RNA-DNA-3′/3′-DNA-5′duplexes can be specifically recognized and excised by RNase HII through hydrolyzing the phosphodiester bonds 5′ to the ribonucleotide at the DNA–RNA junction, leaving a single nucleotide gap with 5′ PO4 and 3′ OH ends. | |
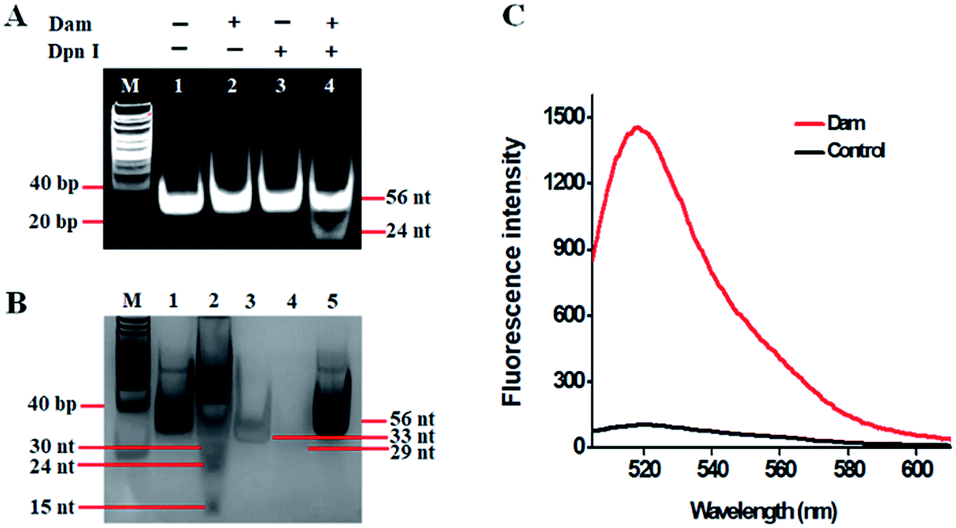
This assay is dependent on the successful cleavage of hairpin substrates to initiate single-ribonucleotide repair-mediated ligation-dependent cycling signal amplification. To verify whether Dam MTase can direct the cleavage of hairpin substrates in the presence of Dpn I, we used nondenaturing gel electrophoresis to analyze the reaction products with SYBR gold as the fluorescent indicator (Fig. 2A). No matter whether in the absence of Dam MTase and Dpn I (Fig. 2A, lane 1) or in the presence of either Dam MTase (Fig. 2A, lane 2) or Dpn I (Fig. 2A, lane 3), only a 56-nt band is observed, the same as the size of the synthesized hairpin substrates (56 nt), suggesting no occurrence of methylation and cleavage reactions, while in the presence of Dam MTase and Dpn I, a new band of 24 nt is observed (Fig. 2A, lane 4), indicating that Dam MTase may induce the methylation of the DNA substrate which can subsequently be cleaved by Dpn I at the sites of methylated adenines. To verify the single-ribonucleotide repair-mediated cyclic ligation-dependent SDA, we used nondenaturing gel electrophoresis to analyze the reaction products with a silver staining kit as the indicator (Fig. 2B). In the absence of Dam MTase, only the bands of original LP1 (33 nt, Fig. 2B, lane 4), LP2 (29 nt, Fig. 2B, lane 3) and the hairpin substrate (56 nt, Fig. 2B, lane 5) are observed (Fig. 2B lane 1), indicating no occurrence of the amplification reaction, while in the presence of Dam MTase, the characteristic bands of 30, 24 and 15 nt are observed (Fig. 2B, lane 2), corresponding to the sizes of the 30-nt cleavage product, 24-nt cleavage product and 15-nt reporter probe, respectively, suggesting that Dam MTase can direct the cleavage of hairpin substrates in the presence of Dpn I to release the 24-nt cleavage product which subsequently acts as an initiator to induce the single-ribonucleotide repair-mediated cyclic ligation-dependent SDA for the generation of the 30-nt cleavage product and 15-nt reporter probe (Fig. 2B, lane 2). We further monitor the fluorescence signal under different reaction conditions (Fig. 2C). No significant fluorescence signal is detected in the control group without Dam MTase (Fig. 2C, black curve), indicating that no amplification reaction is initiated in the absence of Dam MTase, while in the presence of Dam MTase, a significant fluorescence signal is observed (Fig. 2C, red curve), suggesting that Dam MTase can induce the single-ribonucleotide repair-mediated ligation-dependent cycling signal amplification for the generation of a large number of reporter probes and an enhanced fluorescence signal. Both the gel electrophoresis (Fig. 2A and B) and fluorescence experiments (Fig. 2C) clearly demonstrate that Dam MTase can direct the cleavage of hairpin substrates to initiate the single-ribonucleotide repair-mediated ligation-dependent cycling signal amplification. Notably, no other bands are observed in the absence of Dam MTase (Fig. 2B, lane 1) and in the presence of Dam MTase (Fig. 2B, lane 2), demonstrating the high specificity of single-ribonucleotide repair-mediated cyclic ligation-dependent SDA without the production of nonspecific DNA fragments. The high specificity may be ascribed to (1) the specific ligation-dependent strand displacement polymerization extension which can prevent the nonspecific amplification in the absence of target Dam MTase and (2) RNase HII-catalyzed single-ribonucleotide excision which can efficiently inhibit the nonspecific amplification caused by template- and primer-independent DNA synthesis.
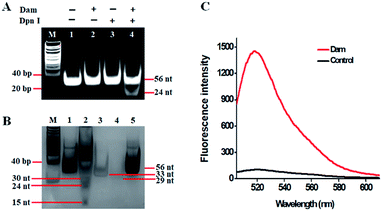 |
| | Fig. 2 (A) Nondenaturing PAGE analysis of the products of the Dam MTase-directed cleavage reaction under different experimental conditions. Lane M, the DNA ladder marker; lane 1, in the absence of Dam MTase and Dpn I; lane 2, in the absence of Dpn I; lane 3, in the absence of Dam MTase; lane 4, in the presence of Dam MTase and Dpn I. SYBR gold is used as the fluorescent indicator. (B) Nondenaturing PAGE analysis of the products of the single-ribonucleotide repair-mediated cyclic ligation-dependent SDA reaction under different experimental conditions. Lane M, the DNA ladder marker; lane 1, in the absence of Dam MTase; lane 2, in the presence of Dam MTase; lane 3, the synthesized LP1; lane 4, the synthesized LP2; lane 5, the synthesized hairpin substrate. A silver staining kit is used to stain the gel. (C) Measurement of the fluorescence signal in the presence of Dam MTase (red curve) and in the absence of Dam MTase (control, black curve). The concentration of Dam MTase is 40 U mL−1. | |
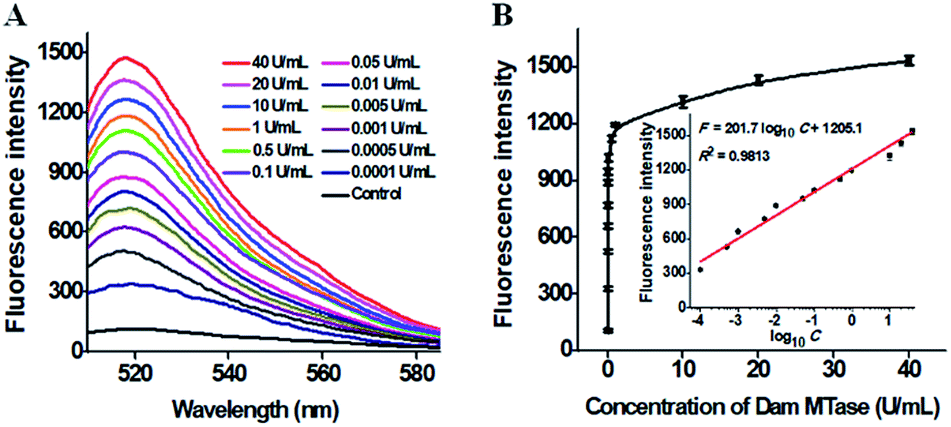
The sensitivity of the proposed method is dependent on the amount of products (i.e. the reporter probes) of the single-ribonucleotide repair-mediated cyclic ligation-dependent SDA reaction, which is mainly influenced by the amounts of Taq DNA ligase and RNase HII, the concentration of signal probes, and the reaction time of single-ribonucleotide repair-mediated ligation-dependent cycling signal amplification. In order to achieve the best performance, we optimized the above experimental conditions (see ESI, Fig. S1–S4†). To investigate the sensitivity of the proposed method, we monitored the fluorescence intensity in response to different concentrations of Dam MTase under the optimal conditions (Fig. 3). As shown in Fig. 3, the fluorescence intensity is enhanced significantly with the Dam MTase concentration from 1 × 10−4 to 10 U mL−1 and reaches a plateau at a concentration of 40 U mL−1. Notably, a good linear correlation is obtained between the fluorescence intensity and the logarithm of Dam MTase concentration from 1 × 10−4 to 10 U mL−1 (inset of Fig. 3B). The corresponding equation is F = 201.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log10C + 1205.1 with a correlation coefficient of 0.9813, where F is the fluorescence intensity and C is the concentration of Dam MTase. The detection limit is determined to be 4.8 × 10−6 U mL−1 on the basis of evaluation of the average response of the negative control plus three times the standard deviation. To the best of our knowledge, this is the most sensitive method for DNA MTase assay reported so far. In comparison with the reported DNA MTase assays,15,18,21–23,25 the sensitivity of the proposed method has been improved by 5 orders of magnitude as compared with that of methylation-responsive DNAzyme-based colorimetric assay (0.25 U mL−1),15 2 orders of magnitude as compared with that of rolling circle amplification (RCA)- and G-quadruplex DNAzyme-based chemiluminescence assay (1.29 × 10−4 U mL−1),25 5 orders of magnitude as compared with AuNP–DNA and Ru(III)-mediated dual signal amplification-based electrochemical assay (0.3 U mL−1),18 4 orders of magnitude as compared with that of nicking enzyme-assisted signal amplification-based fluorescence assay (0.06 U mL−1),23 4 orders of magnitude as compared with that of exonuclease III-mediated target recycling-based fluorescence assay (0.01 U mL−1),22 and 3 orders of magnitude as compared with that of QD-mediated FRET assay (0.002 U mL−1).21 The enhanced sensitivity can be ascribed to (1) the high specificity of RNase HII-catalyzed single-ribonucleotide excision, (2) the high amplification efficiency of cyclic ligation-dependent SDA, and (3) the low background signal resulting from the inhibition of nonspecific amplification by single-ribonucleotide excision and specific ligation-dependent strand displacement polymerase extension.
log10C + 1205.1 with a correlation coefficient of 0.9813, where F is the fluorescence intensity and C is the concentration of Dam MTase. The detection limit is determined to be 4.8 × 10−6 U mL−1 on the basis of evaluation of the average response of the negative control plus three times the standard deviation. To the best of our knowledge, this is the most sensitive method for DNA MTase assay reported so far. In comparison with the reported DNA MTase assays,15,18,21–23,25 the sensitivity of the proposed method has been improved by 5 orders of magnitude as compared with that of methylation-responsive DNAzyme-based colorimetric assay (0.25 U mL−1),15 2 orders of magnitude as compared with that of rolling circle amplification (RCA)- and G-quadruplex DNAzyme-based chemiluminescence assay (1.29 × 10−4 U mL−1),25 5 orders of magnitude as compared with AuNP–DNA and Ru(III)-mediated dual signal amplification-based electrochemical assay (0.3 U mL−1),18 4 orders of magnitude as compared with that of nicking enzyme-assisted signal amplification-based fluorescence assay (0.06 U mL−1),23 4 orders of magnitude as compared with that of exonuclease III-mediated target recycling-based fluorescence assay (0.01 U mL−1),22 and 3 orders of magnitude as compared with that of QD-mediated FRET assay (0.002 U mL−1).21 The enhanced sensitivity can be ascribed to (1) the high specificity of RNase HII-catalyzed single-ribonucleotide excision, (2) the high amplification efficiency of cyclic ligation-dependent SDA, and (3) the low background signal resulting from the inhibition of nonspecific amplification by single-ribonucleotide excision and specific ligation-dependent strand displacement polymerase extension.
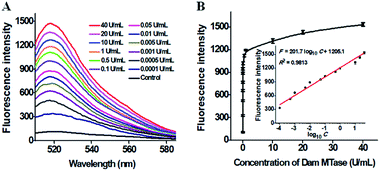 |
| | Fig. 3 (A) Fluorescence spectra in response to different-concentration Dam MTase. (B) Variance of fluorescence intensity with the concentration of Dam MTase from 1 × 10−4 to 40 U mL−1. The inset shows the linear relationship between the fluorescence intensity and the logarithm of Dam MTase concentration from 1 × 10−4 to 10 U mL−1. Error bars show the standard deviations of three experiments. | |
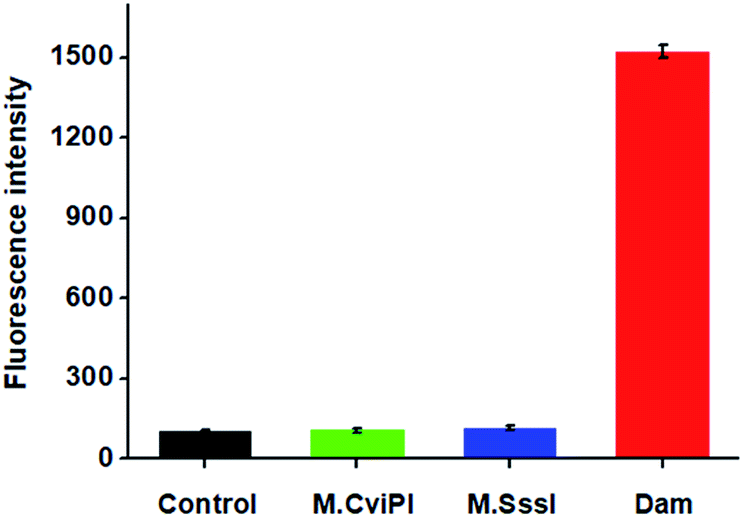
DNA MTase is a superfamily encompassing a large group of members. To investigate the specificity of the proposed method, we used M.CviPI MTase and M.SssI MTase as the negative controls. As shown in Fig. 4, under identical conditions, a high fluorescence signal is detected in the presence of Dam MTase (Fig. 4, red column). In contrast, no distinct fluorescence signal is observed in the presence of either M.CviPI MTase (Fig. 4, green column) or M.SssI MTase (Fig. 4, blue column), with a similar signal to the control group with only reaction buffer (Fig. 4, black column). Even though both M.CviPI MTase and M.SssI MTase belong to the DNA MTase superfamily, they can only recognize and methylate the cytosine residues in the palindromic sequences of 5′-G-C-3′44 and 5′-C-G-3′,45 respectively, instead of the adenine residues in the palindromic sequence of 5′-G-A-T-C-3′. As a result, they can neither cleave the hairpin substrates nor initiate single-ribonucleotide repair-mediated ligation-dependent cycling signal amplification. These results (Fig. 4) clearly demonstrate that the proposed method can detect Dam MTase activity with high specificity.
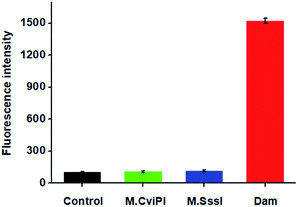 |
| | Fig. 4 Measurement of fluorescence intensity in response to the reaction buffer (control, black column), 40 U mL−1 M.CviPI (green column), 40 U mL−1 M.SssI (blue column), and 40 U mL−1 Dam MTase (red column), respectively. Error bars show the standard deviation of three independent experiments. | |
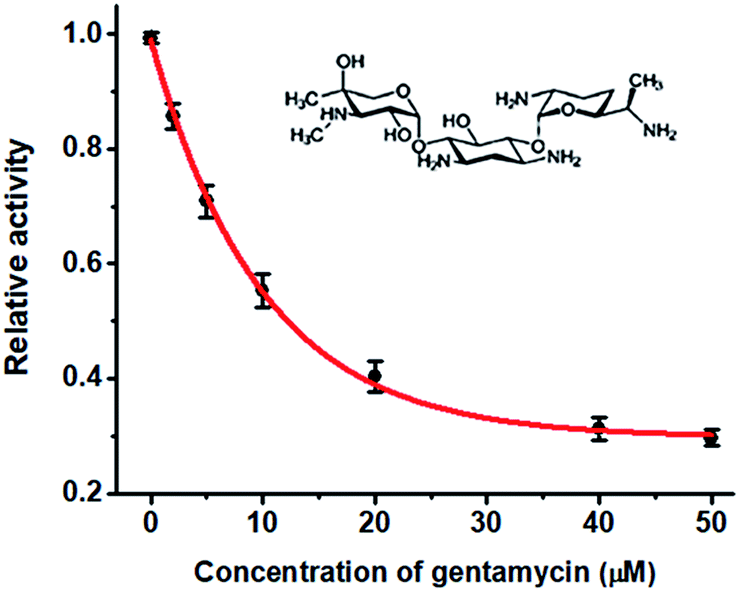
DNA MTase is recognized as both an important biomarker and a critical pharmacological target,21 and the efficient screening of its inhibitors is crucial to the clinical diagnosis and therapy. To verify the feasibility of the proposed method for DNA MTase inhibition assay, we used gentamycin as a model inhibitor. Gentamycin is a well-known broad-spectrum antibiotic and is widely used as an inhibitor of methyl transferase.46 As shown in Fig. 5, when the concentration of Dam MTase is fixed at 40 U mL−1, the relative activity of Dam MTase decreases with increasing concentration of gentamycin from 0 to 50 μM. The half maximal inhibition (IC50) is defined as the concentration of the inhibitor required to inhibit the enzyme activity by 50%. The IC50 value is calculated to be 12.21 μM according to the fitted calibration curve (Fig. 5), consistent with the value (10.0 μM) obtained by the exonuclease III-mediated target recycling-based fluorescence assay22 and the value (11.25 μM) obtained by the QD-mediated FRET assay.21 This result demonstrates that the proposed method can be used to screen DNA MTase inhibitors, holding great potential in anticancer drug discovery.
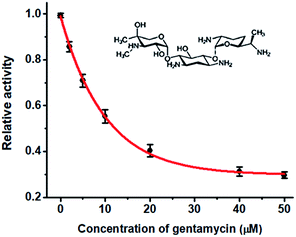 |
| | Fig. 5 Variance of the relative activity of Dam MTase in response to different-concentration gentamycin. The inset shows the chemical structure of gentamicin. The concentration of Dam MTase is 40 U mL−1. Error bars show the standard deviation of three experiments. | |
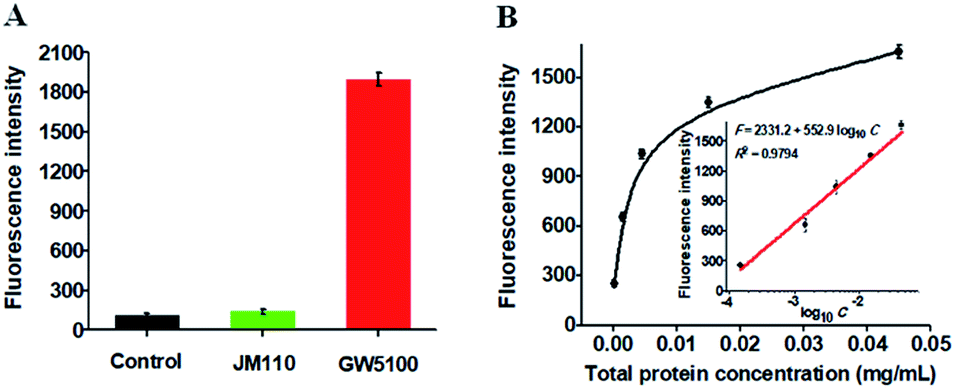
To investigate the feasibility of the proposed method for real sample analysis, we evaluated the recovery of Dam MTase by spiking different concentrations of Dam MTase (1–40 U mL−1) to the normal human serum. As shown in Table 1, the recovery ratio is calculated to be 97.5–112.8% with a relative standard deviation (RSD) of 0.16–2.16%, consistent with the values (recovery ratio of 98.6–102.8% and RSD of 1.04–2.62%) obtained by the QD-mediated FRET assay.21 These results demonstrate that the proposed method can accurately detect Dam MTase activity in human serum samples. We further measured Dam MTase activity in E. coli cells with GW5100 and JM110 E. coli cells as Dam MTase-positive and Dam MTase-negative samples, respectively.47 As shown in Fig. 6A, a high fluorescence signal is detected in GW5100 E. coli cells (Fig. 6A, red column). In contrast, only a low fluorescence signal is observed in JM110 E. coli cells (Fig. 6A, green column), the same as the value detected in the control group with only lysis buffer (Fig. 6A, black column). These results indicate that the fluorescence signal in GW5100 E. coli cells is only derived from Dam MTase instead of other enzymes existing in the total proteins. Moreover, the fluorescence intensity is enhanced with increasing amount of total proteins from GW5100 E. coli cells (Fig. 6B), with a good linear correlation between the fluorescence intensity and the logarithm of the total protein concentration in the range from 1.5 × 10−4 to 4.5 × 10−2 mg mL−1 (inset of Fig. 6B). The corresponding equation is F = 2331.2 + 552.9log10C with a correlation coefficient of 0.9794, where F is the measured fluorescence intensity and C is the concentration of total proteins from GW5100 E. coli cells (mg mL−1), respectively. The detection limit is calculated to be 3.14 × 10−5 mg mL−1 based on the evaluation of the average response of the control plus 3 times the standard deviation. The sensitivity of the proposed method is much higher than that (4.5 × 10−5 mg mL−1) obtained by exonuclease III-mediated target recycling-based fluorescence assay.22 In addition, we measured the recovery of Dam MTase in the spiked cancer cell extracts (see ESI, Table S1†), which is consistent with the result obtained by using the spiked human serum samples (Table 1). These results clearly demonstrate that the proposed method can be used for accurate quantification of Dam MTase activity in complex real samples, holding great potential in practical applications.
Table 1 Recovery studies in the spiked human serum samples
| Sample |
Added (U mL−1) |
Measured (U mL−1) |
Recovery (%) |
RSD (%) |
| 1 |
1 |
1.04 |
103.5 |
0.16 |
| 2 |
5 |
4.98 |
99.5 |
0.37 |
| 3 |
10 |
9.69 |
109.5 |
1.82 |
| 4 |
20 |
21.02 |
97.5 |
2.16 |
| 5 |
40 |
43.62 |
112.8 |
2.12 |
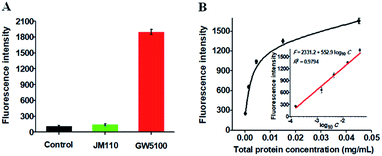 |
| | Fig. 6 (A) Measurement of Dam MTase in lysis buffer (control, black column), JM110 (green column) and GW5100 (red column) E. coli cells, respectively. The total protein concentration is 0.015 mg mL−1 for GW5100 and 0.015 mg mL−1 for JM110 E. coli cells, respectively. (B) Variance of the fluorescence intensity with different-concentration total protein extracted from GW5100 E. coli cells. The inset shows the linear relationship between the fluorescence intensity and the logarithm of the total protein concentration of GW5100 in the range from 1.5 × 10−4 to 4.5 × 10−2 mg mL−1. Error bars represent the standard deviation of three independent experiments. | |
Conclusions
In summary, we have developed for the first time a new fluorescence method to specifically and sensitively detect DNA MTase activity on the basis of single-ribonucleotide repair-mediated ligation-dependent cycling signal amplification. In comparison with conventional nucleic acid amplification approaches (e.g., PCR,26 SDA,27,28 RCA,25 EXPAR29 and ERA34,35), this assay utilizes RNase HII-catalyzed single-ribonucleotide repair and ligation-dependent strand displacement polymerase extension to achieve excellent amplification specificity and high amplification efficiency, effectively inhibiting the nonspecific amplification independent of the target enzyme and templates/primers. This method is extremely simple, easily manipulated and highly sensitive, and it can be performed in a homogeneous format without the involvement of any thermal cycling, washing and separation steps. Taking advantage of the high specificity of RNase HII-catalyzed single-ribonucleotide excision, the high amplification efficiency of cyclic ligation-dependent SDA, and the low background signal resulting from the inhibition of nonspecific amplification by single-ribonucleotide excision and specific ligation-dependent strand displacement polymerase extension, this method exhibits the highest sensitivity reported so far with a detection limit of as low as 4.8 × 10−6 U mL−1 and a large dynamic range of 5 orders of magnitude, which is more sensitive than the reported colorimetric (0.25 U mL−1),15 chemiluminescence (1.29 × 10−4 U mL−1),25 electrochemical (0.3 U mL−1)18 and fluorescence assays (0.01, 0.06 and 0.002 U mL−1).21–23 Moreover, this method can be applied for the discrimination of Dam MTase from other DNA MTases, screening of potential Dam MTase inhibitors, and accurate quantification of Dam MTase activity in real complex samples. Importantly, this method may provide a new paradigm for efficient nucleic acid amplification and may find wide applications in biomedical research and clinical diagnosis.
Experimental section
Chemicals and materials
All oligonucleotides (Table 2) were synthesized and purified by HPLC at Shanghai Sangon Biological Engineering Technology & Services Co. Ltd. (Shanghai, China). DNA adenine methyltransferase (Dam MTase), 10× Dam MTase reaction buffer (500 mM trizma hydrochloride (Tris–HCl), 50 mM 2-mercaptoethanol (β-ME), 100 mM ethylenediaminetetraacetic acid (EDTA), pH 7.5), CpG methyltransferase (M.SssI), GpC methyltransferase (M.CviPI), Dpn I, 10× CutSmart buffer (500 mM potassium acetate (KAc), 200 mM tris-acetate, 100 mM magnesium acetate (Mg(Ac)2), 1 mg mL−1 bovine serum albumin (BSA), pH 7.9), S-adenosylmethionine (SAM), Taq DNA ligase, 10× Taq DNA ligase reaction buffer (200 mM Tris–HCl, 250 mM KAc, 100 mM Mg(Ac)2, 100 mM DL-dithiothreitol (DTT), 10 mM nicotinamide adenine dinucleotide (NAD), 1% triton X-100, pH 7.6), Bst DNA polymerase (large fragment), 10× ThermoPol reaction buffer (200 mM Tris–HCl, 100 mM potassium chloride (KCl), 100 mM ammonium sulfate (NH4)2SO4), 20 mM magnesium sulfate (MgSO4), 1% triton X-100, pH 8.8), ribonuclease HII (RNase HII), deoxyadenosine triphosphate (dATP), deoxyuridine triphosphate (dUTP), deoxyguanosine triphosphate (dGTP) and deoxycytidine triphosphate (dCTP) were purchased from New England Biolabs (Ipswich, MA, USA). SYBR Gold was purchased from Life Technologies (Carlsbad, CA, USA). Gentamycin, bovine serum albumin (BSA), fetal bovine serum (FBS) and other chemicals of analytical grade were bought from Sigma-Aldrich Co (St. Louis, MO, USA). The E. coli strains (GW5100 and JM110) were provided by Wuhan University (Wuhan, China). Ultrapure water used in all experiments was obtained from a Millipore filtration system (Millipore, Milford, MA, USA).
Table 2 Sequences of the oligonucleotidesa
| Note |
Sequences (5′–3′) |
|
In the hairpin substrate, the underlined bases “A” indicate the recognition sites of Dam MTase. In ligation probe 2, the two “P”s indicate the phosphate (PO4) groups. In the signal probe, the underlined bases “T” indicate the modification of a fluorescein (FAM) and a black hole quencher 1 (BHQ1), respectively, and the italicized base “U” indicates the uracil ribonucleotide.
|
| Hairpin substrate |
ACT TAT CAG CTT AAG ![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) TC CAC GAC AAA AAA GCA AGC AGG TC TTA AGC TGA TAA GT TC CAC GAC AAA AAA GCA AGC AGG TC TTA AGC TGA TAA GT |
| Ligation probe 1 |
CCT GCT CTT CTC TTC ACT CTC GTC CTG CTT GCT |
| Ligation probe 2 |
P-TTT TTG TCG TGG ACC TGC TCT CCA CGA CA-P |
| Signal probe |
(FAM) ![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) CC TGC TCU TCT CT (BHQ1) CC TGC TCU TCT CT (BHQ1) |
| Reporter probe |
GAA GAG AAG AGC AGG |
Single-ribonucleotide repair-mediated ligation-dependent cycling signal amplification
The DNA MTase assay involves two consecutive reaction steps. First, all oligonucleotides were diluted with 1× Tris–EDTA buffer (10 mM Tris, 1 mM EDTA, pH 8.0) for the preparation of stock solutions. Then the hairpin substrates and ligation probe 2 (LP2) were diluted to 10 μM in a buffer (1.5 mM MgCl2, 10 mM Tris–HCl, pH 8.0) and incubated at 95 °C for 5 min, followed by slow cooling to room temperature for perfect folding of a hairpin structure. Subsequently, 1 μL of hairpin substrates (10 μM) was added into 20 μL of the reaction system containing Dam MTase, 160 μM SAM, 6 U of Dpn I, 2 μL of 10× Dam MTase reaction buffer and 2 μL of 10× CutSmart buffer, and incubated at 37 °C for 2 h, followed by inactivation at 80 °C for 20 min. Second, 5 μL of the above reaction products were added into 20 μL of the reaction system containing 500 nM LP1, 500 nM LP2, 500 nM dATPs, 500 nM dUTPs, 500 nM dGTPs, 500 nM dCTPs, 900 nM signal probes, 15 U of Taq DNA ligase, 1 U of Bst DNA polymerase, 2 U of RNase HII, 2 μL of 10× Taq DNA ligase reaction buffer and 2 μL of 10× ThermoPol reaction buffer, followed by incubation at 45 °C for 60 min.
Steady-state fluorescence measurements and electrophoresis analysis
20 μL of reaction products were diluted to 60 μL with ultrapure water. Then the fluorescence spectra were measured using a Hitachi F-7000 fluorescence spectrophotometer (Tokyo, Japan) at an excitation wavelength of 492 nm, and the fluorescence intensity at an emission wavelength of 518 nm was used for data analysis. To analyze the products of the cleavage reaction, 12% nondenaturing polyacrylamide gel electrophoresis (PAGE) was performed in 1 × TBE buffer (9 mM Tris–HCl, 9 mM boric acid, 0.2 mM EDTA, pH 7.9) at 110 V constant voltage for 50 min at room temperature, and SYBR gold was used as the fluorescent indicator. To analyze the products of the single-ribonucleotide repair-mediated cyclic ligation-dependent SDA reaction, 12% nondenaturing PAGE was performed in 1× TBE buffer. After electrophoresis at 110 V for 45 min, the gel was stained with a silver staining kit (81104-1000, Tiandz Inc., Beijing, China). The above stained gels were visualized with a ChemiDoc MP Imaging system (Hercules, California, U.S.A.).
Inhibition of Dam MTase activity
To investigate the inhibition effect of gentamicin on the activity of Dam MTase, different concentrations of gentamycin were preincubated with the reaction mixture containing 500 nM hairpin substrates and 10× Dam MTase reaction buffer at 37 °C for 15 min. Then 40 U mL−1 Dam MTase, 6 U of Dpn I, 160 μM SAM and 2 μL of 10× CutSmart buffer were added into the reaction mixture and incubated at 37 °C for 2 h. The reaction was terminated by inactivation at 80 °C for 20 min. The Dam MTase activity was measured using the procedure described above, and its relative activity (RA) was calculated based on eqn (1):| | | RA = (Fi − Fo)/(Ft − Fo) | (1) |
where Fo, Ft, and Fi represent the fluorescence intensity in the absence of Dam MTase, in the presence of 40 U mL−1 Dam MTase, and in the presence of 40 U mL−1 Dam MTase and different concentrations of gentamicin, respectively.
Cell culture and preparation of cell extracts
The colonies of GW5100 and JM110 E. coli cells were incubated in 3 mL of liquid medium (5 g L−1 yeast extracts, 10 g L−1 trypton, 10 g L−1 NaCl) at 37 °C in a rotary shaker at 250 rpm for 12 h. Then 100 μL of the above cell suspension was added into 3 mL of liquid medium and incubated under the same conditions for 2.5 h. At the exponential phase of growth, the E. coli cells were harvested and centrifuged at 8000 rpm for 10 min to obtain the cell pellet. After washing three times with 10× phosphate buffer saline (PBS, 1.6 M NaCl, 10.6 mM potassium phosphate monobasic (KH2PO4), 29.7 mM sodium phosphate dibasic (Na2HPO4·7H2O), pH 7.4), the cells were lysed with radio-immunoprecipitation assay (RIPA) lysis buffer (P0013J, Beyotime Biotech, Shanghai, China), and the concentration of the extracted proteins was quantified using a BCA protein assay kit (P0009, Beyotime Biotech, Shanghai, China). The lung adenocarcinoma cell line (A549 cells) was cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco, USA) with 10% fetal bovine serum (FBS, Gibco, USA) and 1% penicillin–streptomycin (Gibco, USA) in a 5% CO2 incubator at 37 °C. At the exponential phase of growth, cells (1 × 106) were collected and washed twice with ice-cold PBS (pH 7.4, Gibco, USA). The cell extracts were prepared by using a nuclear extract kit (Active Motif, Carlsbad, CA, USA) according to the manufacturer's protocol. The obtained supernatant was used to investigate the feasibility of the proposed method in a complex reaction system.
Detection of Dam MTase activity in human serum and cell extracts
A total volume of 20 μL of sample containing 5% human serum spiked with various concentrations of Dam MTase (1–40 U mL−1), 500 nM hairpin substrates, 160 μM SAM, 6 U of DpnI, 2 μL of 10× Dam MTase reaction buffer and 2 μL of 10× CutSmart buffer was incubated at 37 °C for 2 h, followed by inactivation at 80 °C for 20 min. The Dam MTase activity was measured using the same procedures described above. For the detection of Dam MTase activity in cancer cell extracts, 5 μL of A549 cell extracts were added into the above solution without human serum and incubated at 37 °C for 2 h, followed by inactivation at 80 °C for 20 min. Subsequently, the Dam MTase activity was measured using the same procedures described above and calculated according to the fitting equation (inset of Fig. 3B). The recovery ratio (R) is calculated based on eqn (2):| | | R (%) = (Cm/C0) × 100% | (2) |
where Cm and C0 represent the Dam MTase concentration in the presence and absence of A549 cell extracts, respectively.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 21325523, 21527811, 21735003, and 21705097) and the Award for Team Leader Program of Taishan Scholars of Shandong Province, China.
Notes and references
- R. J. Wood, J. C. McKelvie, M. D. Maynard-Smith and P. L. Roach, Nucleic Acids Res., 2010, 38, e107 CrossRef PubMed.
- D. Subramaniam, R. Thombre, A. Dhar and S. Anant, Frontiers in Oncology, 2014, 4, 80–93 CrossRef PubMed.
- R. Z. Jurkowska, T. P. Jurkowski and A. Jeltsch, ChemBioChem, 2011, 12, 206–222 CrossRef PubMed.
- G. E. Lee, J. H. Kim, M. Taylor and M. T. Muller, J. Biol. Chem., 2010, 285, 37630–37640 CrossRef PubMed.
- Z. D. Smith and A. Meissner, Nat. Rev. Genet., 2013, 14, 204 CrossRef PubMed.
- P. W. Laird, Nat. Rev. Cancer, 2003, 3, 253–266 CrossRef PubMed.
- M. Esteller, P. G. Corn, S. B. Baylin and J. G. Herman, Cancer Res., 2001, 61, 3225–3229 Search PubMed.
- D. K. Vanaja, M. Ehrich, D. Van den Boom, J. C. Cheville, R. J. Karnes, D. J. Tindall, C. R. Cantor and C. Y. F. Young, Canc. Invest., 2009, 27, 549–560 CrossRef PubMed.
- C. Boltze, S. Zack, C. Quednow, S. Bettge, A. Roessner and R. Schneider-Stock, Pathol. Res. Pract., 2003, 199, 399–404 CrossRef PubMed.
- K. Miyamoto, T. Fukutomi, S. Akashi-Tanaka, T. Hasegawa, T. Asahara, T. Sugimura and T. Ushijima, Int. J. Cancer, 2005, 116, 407–414 CrossRef PubMed.
- B. Brueckner, D. Kuck and F. Lyko, Cancer J., 2007, 13, 17–22 CrossRef PubMed.
- S. Pradhan, A. Bacolla, R. D. Wells and R. J. Roberts, J. Biol. Chem., 1999, 274, 33002–33010 CrossRef PubMed.
- J. Pröll, M. Födermayr, C. Wechselberger, P. Pammer, M. Sonnleitner, O. Zach and D. Lutz, DNA Res., 2006, 13, 37–42 CrossRef PubMed.
- L. Lennard and H. J. Singleton, J. Chromatogr. B: Biomed. Sci. Appl., 1994, 661, 25–33 CrossRef PubMed.
- W. Li, Z. Liu, H. Lin, Z. Nie, J. Chen, X. Xu and S. Yao, Anal. Chem., 2010, 82, 1935–1941 CrossRef PubMed.
- W. Li, P. Wu, H. Zhang and C. Cai, Anal. Chem., 2012, 84, 7583–7590 CrossRef PubMed.
- H.-F. Zhao, R.-P. Liang, J.-W. Wang and J.-D. Qiu, Biosens. Bioelectron., 2015, 63, 458–464 CrossRef PubMed.
- H. Zhang, H. Dong, G. Yang, H. Chen and C. Cai, Anal. Chem., 2016, 88, 11108–11114 CrossRef PubMed.
- X. Jing, X. Cao, L. Wang, T. Lan, Y. Li and G. Xie, Biosens. Bioelectron., 2014, 58, 40–47 CrossRef PubMed.
- J. Lee, Y.-K. Kim and D.-H. Min, Anal. Chem., 2011, 83, 8906–8912 CrossRef PubMed.
- F. Ma, W.-j. Liu, B. Tang and C.-y. Zhang, Chem. Commun., 2017, 53, 6868–6871 RSC.
- X.-W. Xing, F. Tang, J. Wu, J.-M. Chu, Y.-Q. Feng, X. Zhou and B.-F. Yuan, Anal. Chem., 2014, 86, 11269–11274 CrossRef PubMed.
- Y. Zhao, F. Chen, Y. Wu, Y. Dong and C. Fan, Biosens. Bioelectron., 2013, 42, 56–61 CrossRef PubMed.
- F. Chen and Y. Zhao, Analyst, 2013, 138, 284–289 RSC.
- Y.-p. Zeng, J. Hu, Y. Long and C.-y. Zhang, Anal. Chem., 2013, 85, 6143–6150 CrossRef PubMed.
- C.-Y. Yu, B.-C. Yin, S. Wang, Z. Xu and B.-C. Ye, Anal. Chem., 2014, 86, 7214–7218 CrossRef PubMed.
- L.-j. Wang, M. Ren, Q. Zhang, B. Tang and C.-y. Zhang, Anal. Chem., 2017, 89, 4488–4494 CrossRef PubMed.
- L.-j. Wang, Y. Zhang and C.-y. Zhang, Chem. Commun., 2014, 50, 15393–15396 RSC.
- F. Ma, Y. Yang and C.-y. Zhang, Anal. Chem., 2014, 86, 6006–6011 CrossRef PubMed.
- H. Kong, R. B. Kucera and W. E. Jack, J. Biol. Chem., 1993, 268, 1965–1975 Search PubMed.
- Y. Tang, H. Chen and Y. Diao, Sci. Rep., 2016, 6, 27605–27617 CrossRef PubMed.
- N. Ogata and T. Miura, Nucleic Acids Res., 1998, 26, 4652–4656 CrossRef PubMed.
- E. Tan, B. Erwin, S. Dames, T. Ferguson, M. Buechel, B. Irvine, K. Voelkerding and A. Niemz, Biochemistry, 2008, 47, 9987–9999 CrossRef PubMed.
- D.-M. Zhou, W.-F. Du, Q. Xi, J. Ge and J.-H. Jiang, Anal. Chem., 2014, 86, 6763–6767 CrossRef PubMed.
- L.-j. Wang, Z.-Y. Wang, Q. Zhang, B. Tang and C.-y. Zhang, Chem. Commun., 2017, 53, 3878–3881 RSC.
- J. A. Bastock, M. Webb and J. A. Grasby, J. Mol. Biol., 2007, 368, 421–433 CrossRef PubMed.
- B. Rydberg and J. Game, Proc. Natl. Acad. Sci. U.S.A., 2002, 99, 16654–16659 CrossRef PubMed.
- Y. V. Gerasimova, S. Peck and D. M. Kolpashchikov, Chem. Commun., 2010, 46, 8761–8763 RSC.
- J. R. Horton, K. Liebert, M. Bekes, A. Jeltsch and X. Cheng, J. Mol. Biol., 2006, 358, 559–570 CrossRef PubMed.
- S. R. Coffin and N. O. Reich, Biochemistry, 2009, 48, 7399–7410 CrossRef PubMed.
- M. G. Marinus and J. Casadesus, FEMS Microbiol. Rev., 2009, 33, 488–503 CrossRef PubMed.
- D. A. Low, N. J. Weyand and M. J. Mahan, Infect. Immun., 2001, 69, 7197–7204 CrossRef PubMed.
- G. E. Geier and P. Modrich, J. Biol. Chem., 1979, 254, 1408–1413 Search PubMed.
- M. Xu, M. P. Kladde, R. T. Simpson and J. L. Van Etten, Nucleic Acids Res., 1998, 26, 3961–3966 CrossRef PubMed.
- K. Matsuo, J. Silke, K. Gramatikoff and W. Schaffner, Nucleic Acids Res., 1994, 22, 5354–5359 CrossRef PubMed.
- E. A. Elsinghorst, Meth. Enzymol., 1994, 236, 405–420 CrossRef PubMed.
- A. Løbner-Olesen, O. Skovgaard and M. G. Marinus, Curr. Opin. Microbiol., 2005, 8, 154–160 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02215a |
| ‡ These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*