
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photocatalytic reverse polarity Povarov reaction†
Jamie A.
Leitch
 a,
Angel L.
Fuentes de Arriba
a,
Joanne
Tan
b,
Oskar
Hoff
a,
Carlos M.
Martínez
c and
Darren J.
Dixon
*a
a,
Angel L.
Fuentes de Arriba
a,
Joanne
Tan
b,
Oskar
Hoff
a,
Carlos M.
Martínez
c and
Darren J.
Dixon
*a
aDepartment of Chemistry, Chemical Research Laboratory, University of Oxford, 12 Mansfield Road, Oxford, UK. E-mail: darren.dixon@chem.ox.ac.uk
bDavenport Research Laboratories, Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, ON, Canada
cJanssen Research and Development, C/Rio Jarama, 75A, Toledo, Spain
First published on 6th July 2018
Abstract
A visible light mediated iridium photocatalysed reverse polarity Povarov reaction of aryl imines and electron deficient alkenes is described. Operating via a putative nucleophilic α-amino radical, generated by a proton coupled electron transfer process, addition to a range of conjugated electron deficient alkene substrates affords substituted tetrahydroquinoline products in high yields and with typically good to excellent diastereoselectivity in favor of the trans diastereoisomer. Sub-stoichiometric quantities of Hantzsch ester were found to be key to initiate the overall redox-neutral, free radical cyclization cascade. This new reaction complements existing two electron Lewis acid mediated variants and expands the capabilities of imine umpolung chemistry to synthetically relevant cyclisation methodology.
Introduction
Photoredox catalysis, through its ability to generate reactive radical intermediates under mild yet highly tunable reaction conditions and experimental set-ups,1 offers great potential for new reaction discovery.2 Pioneering developments have significantly expanded the synthetic toolbox3 and have been applied in the construction of building blocks4 and natural products,5 and also in the derivatization of biologically relevant molecules6 and macromolecular peptide structures.7Photocatalytic approaches to the synthesis and functionalisation of amines and their derivatives – with direct impact on medicinal chemistry programmes8 – have been particularly prevalent.9 Recent efforts have focussed on the photocatalytic generation and trapping of α-amino radicals derived from a range of starting materials.10 Noteworthy examples include single electron transfer (SET) decarboxylation of amino acid derivatives,11 direct hydrogen atom transfer (HAT) of aliphatic amines,12 or via the SET reduction of imine derivatives.13 The resulting α-amino radicals have been shown to subsequently engage in radical–radical coupling reactions,14 partake in transition metal catalysed cross coupling processes or react with a range of electrophilic species.15
Building on Knowles' seminal studies on proton coupled electron transfer (PCET) for the generation of α-heteroatom radicals,13a our group recently reported the photocatalytic reductive coupling of imines with allyl sulfone electrophiles using Eosin Y as the photocatalyst, and Hantzsch ester as the stoichiometric reductant, under green LED light irradiation.15b Contemporaneously Chen,15a and later Ngai,15c reported similar reactivity of in situ generated α-amino radicals from imines. Such a reversal of the natural imine polarity via the PCET manifold establishes a new umpolung approach for the synthesis of α-functionalised amines (Scheme 1a), and creates many opportunities for new reaction discovery.
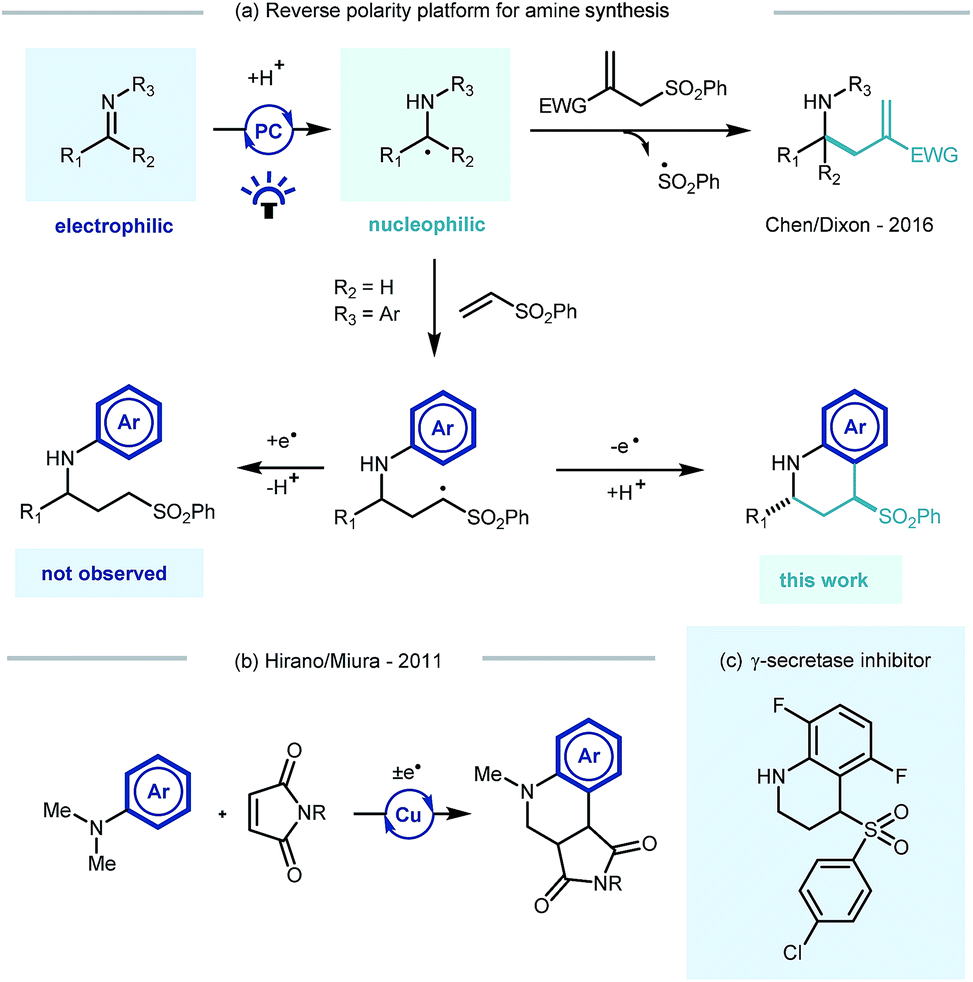 | ||
| Scheme 1 Previous reports in the context of this work. PC = photocatalyst. | ||
In a continuation of our programme, we sought to explore and expand the synthetic utility of photocatalytic imine umpolung chemistry. Our previous work demonstrated that the α-allylated amine product was the outcome of the reaction with Michael acceptors bearing a sulfone leaving group.15b With prospective coupling partners not possessing a suitable leaving group it was unclear as to what the outcome of the reaction would be. Following nucleophilic addition, the intermediate γ-amino radical could either gain a further electron and proton to form the Michael addition product,15a or potentially cyclize onto the pendant electron rich aromatic ring and subsequently rearomatize (Scheme 1a). Both pathways would be synthetically useful but the latter would allow the direct construction of the biologically relevant tetrahydroquinoline scaffold, examples of which have shown to possess biological activity in γ-secretase inhibitors (Scheme 1c)16 and androgen agonists/antagonists.17 Previous studies by Hirano and Miura on the copper catalysed redox coupling of N,N-dimethylaniline and maleimide derivatives provided some precedent for the cyclisation pathway (Scheme 1b)18 and encouraged us to investigate along these lines of enquiry. Herein, we wish to report our findings.
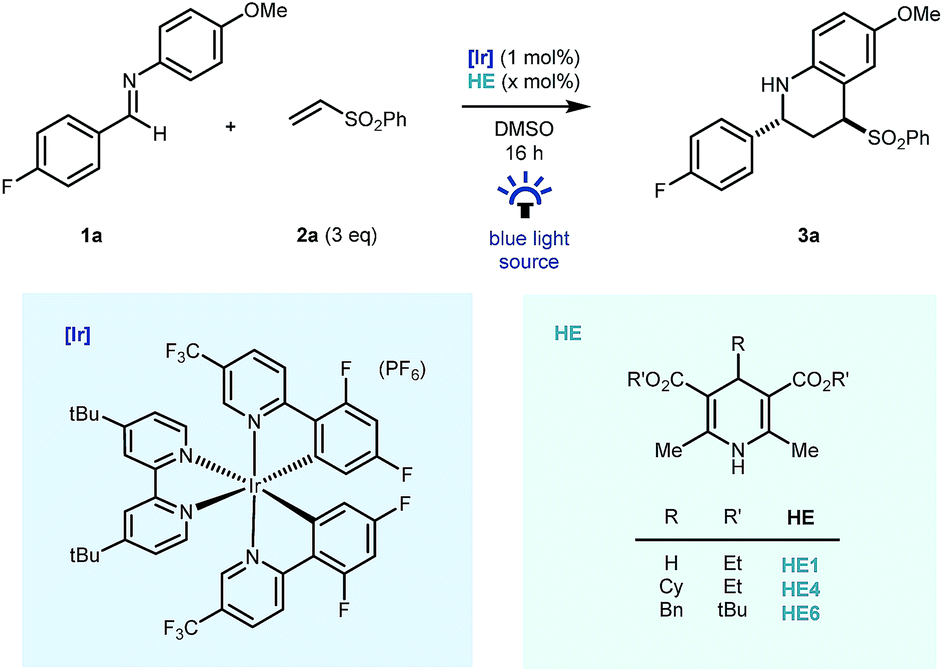
Preliminary studies were carried out using fluorine tagged aldimine (1a), phenyl vinyl sulfone (2a, 3 equiv.) as the Michael acceptor, (Ir[dF(CF3)ppy]2(dtbbpy))PF6 ([Ir], 1 mol%) as photocatalyst and the commercial Hantzsch ester (HE1, 1.2 equiv.) as a stoichiometric reductant, in DMSO, under blue LED light irradiation. Pleasingly, good reactivity was identified early and importantly cyclised product 3a – a reverse polarity Povarov product19 – was formed as the sole coupling product in the reaction mixture (Table 1, entry 1) as a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers. Despite the typical use of Hantzsch esters as a superstoichiometric reductive quencher in photoredox coupling reactions of imines,20,21 our redox-neutral process benefited from the use of 60 mol% HE1. This was largely due to suppression of over-reduction and aza-pinacol side products (entry 2) and the use of substituted Hantzsch esters (HE4 & HE6, entries 3 & 4) further suppressed their formation.16,22 Further reduction of the Hantzsch ester loading to 10 mol% led to lower reaction conversions (entry 5) using blue LED irradiation, however by switching to a commercial photoreactor, full conversion was achieved in 16 hours and the product was isolated in excellent yield (90%) as a 10:1 mixture of diastereomers. The reaction methodology was also amenable to a reduction in equivalents of vinyl sulfone coupling partner without major impact on reaction efficiency (entry 7) and reactivity was completely lost on removal of Hantzsch ester (entry 8), or iridium photocatalyst (entry 9), and in the absence of light (entry 10).
:1 mixture of diastereomers. Despite the typical use of Hantzsch esters as a superstoichiometric reductive quencher in photoredox coupling reactions of imines,20,21 our redox-neutral process benefited from the use of 60 mol% HE1. This was largely due to suppression of over-reduction and aza-pinacol side products (entry 2) and the use of substituted Hantzsch esters (HE4 & HE6, entries 3 & 4) further suppressed their formation.16,22 Further reduction of the Hantzsch ester loading to 10 mol% led to lower reaction conversions (entry 5) using blue LED irradiation, however by switching to a commercial photoreactor, full conversion was achieved in 16 hours and the product was isolated in excellent yield (90%) as a 10:1 mixture of diastereomers. The reaction methodology was also amenable to a reduction in equivalents of vinyl sulfone coupling partner without major impact on reaction efficiency (entry 7) and reactivity was completely lost on removal of Hantzsch ester (entry 8), or iridium photocatalyst (entry 9), and in the absence of light (entry 10).
|
|
||||
|---|---|---|---|---|
| Entry | HE (x mol%) | Light source | 3a | dr |
| a General reaction conditions: 1a (0.25 mmol), phenyl vinyl sulfone (1.25 mmol, 5 eq.), (Ir[dF(CF3)ppy]2(dtbbpy))PF6 (0.0025 mmol), Hantzsch ester derivatives (x mol%), DMSO (1 mL). b For further details on blue light source see ESI. c 19F NMR yield determined by direct conversion between 1a and 3a (major + minor diastereoisomer) including by-products. d Isolated yield after silica gel column chromatography. e 2 eq. phenyl vinyl sulfone used. f Without the iridium catalyst. | ||||
| 1 | HE1 (120 mol%) | LED bath | 64 | 7:1 |
| 2 | HE1 (60 mol%) | LED bath | 84 | 7:1 |
| 3 | HE4 (60 mol%) | LED bath | 100 (56)d | 10:1 |
| 4 | HE6 (60 mol%) | LED bath | 99 | 11:1 |
| 5 | HE4 (10 mol%) | LED bath | 54 | 8:1 |
| 6 | HE4 (10 mol%) | Photoreactor | 95 (90) |
10:1 |
| 7 | HE4 (10 mol%) | Photoreactor | 93 (88) |
10:1 |
| 8e | — | Photoreactor | — | — |
| 9e,f | HE4 (10 mol%) | Photoreactor | — | — |
| 10e | HE4 (10 mol%) | — | — | — |
With optimal conditions established, we looked to probe the scope of the photocatalytic reverse polarity Povarov reaction (Scheme 2a). Initially, the tolerance to variation on the aniline portion of the aldimine was investigated. Pleasingly, the reaction proceeded well with a number of substituents in the 4-position (3b–d). Good reaction efficiency and excellent diastereoselectivity towards the trans diastereoisomeric product was noted in all cases.23 This is in contrast to Brønsted or Lewis acid catalysed Povarov reactions in which cis-configured stereoisomeric products often predominate.19 When variations to the aromatic ring of the aldehyde moiety were explored, electronics were found to play a pivotal role, with electron rich and neutral arenes giving excellent yields (3e–g, 3j–n) whereas electron poor aromatics led to longer reaction times (3h) and even complete nullification of reactivity (3i). 3-Fluoro and 2-fluoro-substituted aldimines were tolerated in this chemistry however longer reaction times were required and yields diminished with proximity to the imine C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond (3p–r). A substituted pyridyl substrate was also shown to be effective in the reaction mixture leading to an excellent yield of product (3s).
N bond (3p–r). A substituted pyridyl substrate was also shown to be effective in the reaction mixture leading to an excellent yield of product (3s).
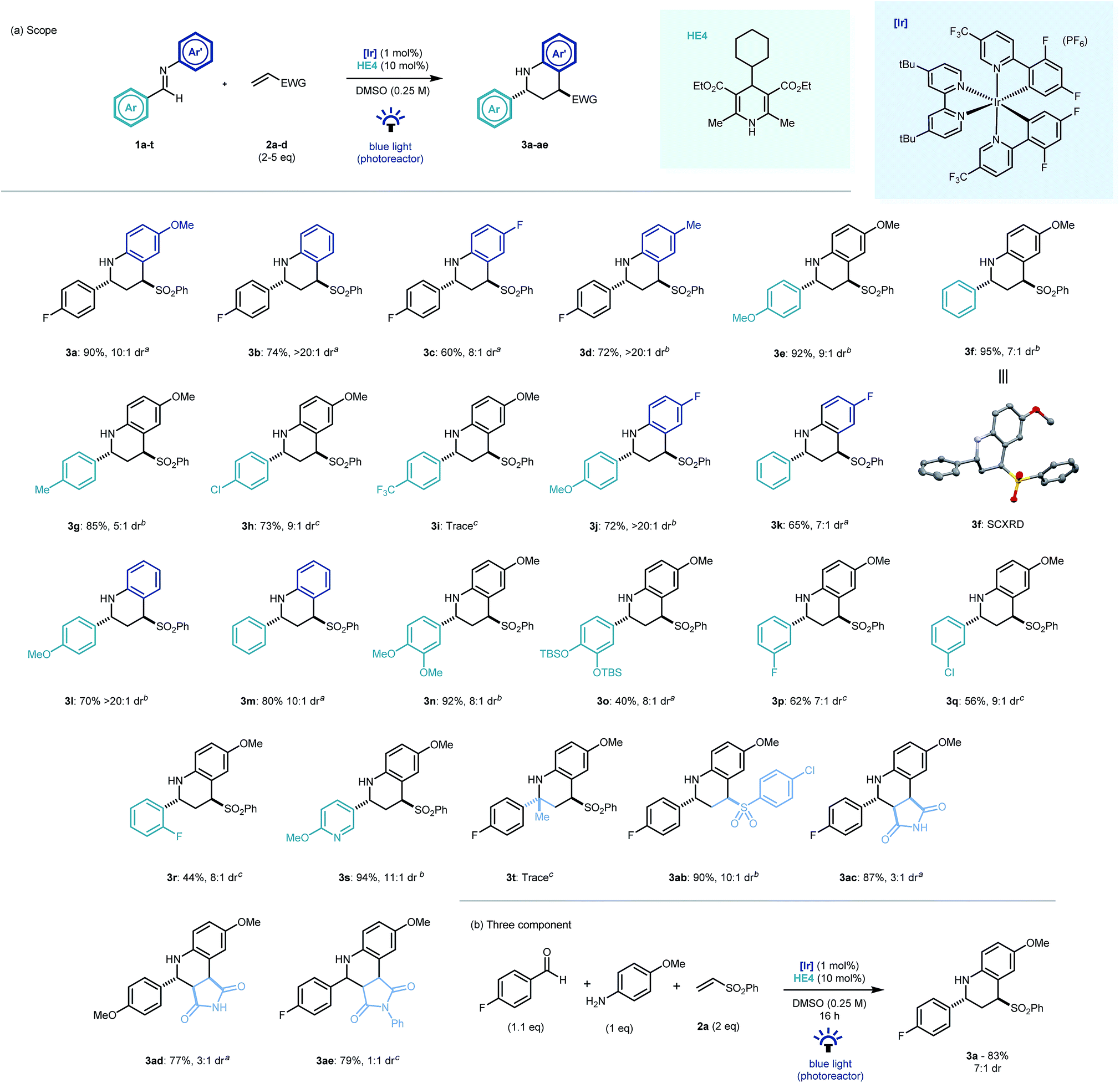 | ||
| Scheme 2 Scope of the photocatalytic reverse polarity Povarov reaction. Reaction time: a16 h, b40 h, c64 h. | ||
Disappointingly, ketimines were not found to be viable substrates for this reaction (3t) even after prolonged reaction times. 4-Chlorophenyl vinyl sulfone was found to be an excellent electrophile (3ab). Similarly, maleimide and N-phenylmaleimide electrophiles were shown to be excellent coupling partners in the new cyclisation methodology, however diastereoselectivity was respectively reduced or absent in the reaction products (3ac–ae). The chemistry was extendable to a three component one-pot process (Scheme 2b) and demonstrated the expedient construction of complex tetrahydroquinoline structures using simple building blocks, as well as its potential application to modular library synthesis.
An interesting observation made throughout the optimisation studies was that after the imine substrate 1a was consumed in the reaction vessel, product diastereomeric ratio continued to increase, suggesting that epimerization was occurring under the reaction conditions. To investigate this further, Povarov product 3a (dr 10:1) was resubmitted to the reaction conditions. Pleasingly an increase to 18:1 was observed and mass balance was maintained (Scheme 3a). As this epimerization does not take place without the iridium catalyst, or in the absence of light, a photoredox based mechanism for the formation of a planar α-amino radical is proposed.24 Monitoring the reaction over time from initiation using 19F NMR spectroscopy, demonstrated that the formation of 3a had an intrinsic kinetic dr of ∼10:1 in favor of the trans diastereoisomer. Then from the point of ∼90% conversion, the diastereoselectivity increased gradually up to 20:1 after 48 hours (Scheme 3b).
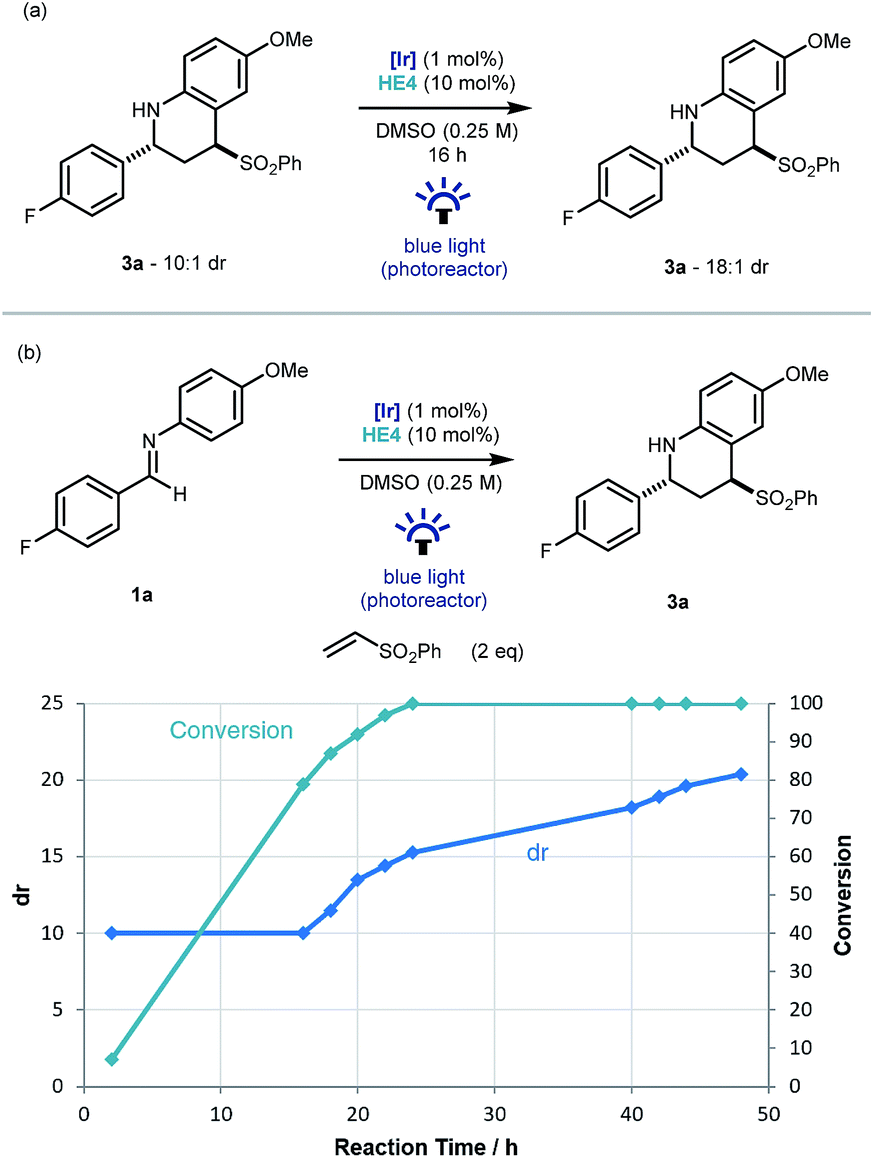 | ||
| Scheme 3 Investigation into the diastereoselectivity of the reaction. | ||
Recent investigations have shown that imines (E01/2 = −1.90 V vs. SCE in CH3CN),25 can be reduced more readily using proton-coupled electron transfer. Chen's,15a ours,15b and Ngai's15c previous investigations have demonstrated that partially oxidized Hantzsch esters are acidic enough to be used as proton donors in PCET mechanisms.26 To probe this further, the reaction was repeated using the reduced amine – which is a by-product of the reaction – as starting material (4a, Scheme 4a). Only trace amounts of product were observed, suggesting the reverse polarity Povarov reaction does not proceed via initial reduction of the imine. This reinforces the proposal of PCET construction of the key α-amino radical.
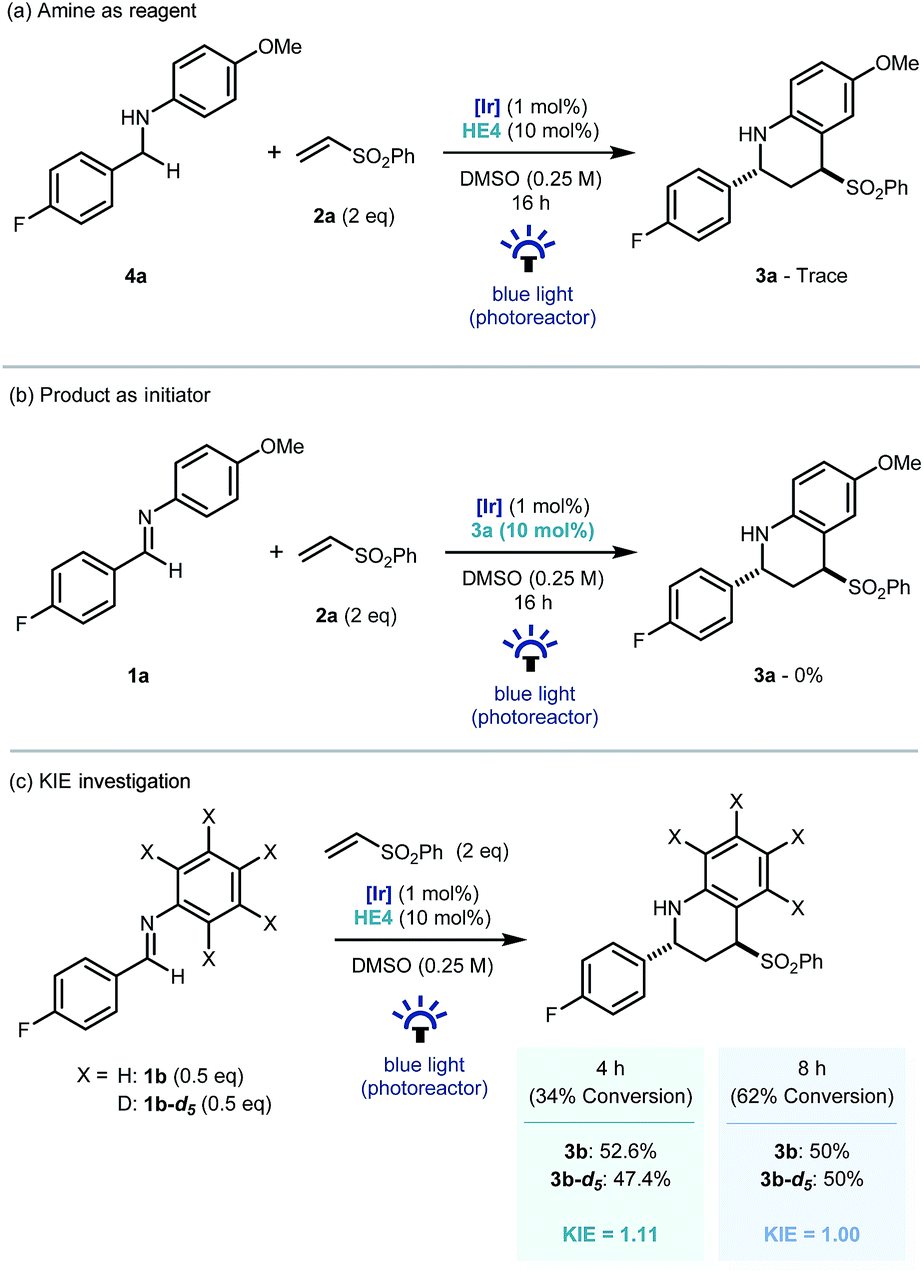 | ||
| Scheme 4 Control and mechanistic experiments. | ||
As only sub stoichiometric quantities of the Hantzsch ester are needed for full conversion, its role is likely as an initiator in a self-propagating mechanism. We were intrigued to identify whether either the cyclised product 3a or an intermediate could act as a reductive quencher to propagate further product formation. To this end, we carried out a control experiment where HE4 (10 mol%) was replaced with 3a (Scheme 4b). Importantly, no product formation was observed suggesting that an intermediate – not the product of the reaction – is capable of propagating this reaction mechanism. Kinetic isotope effect (KIE) studies demonstrated that the isotopic sensitivity of selectivity determining steps has a value of ∼1, suggesting that the substrates are kinetically committed toward product formation prior to any C–H cleavage events (Scheme 5c).27
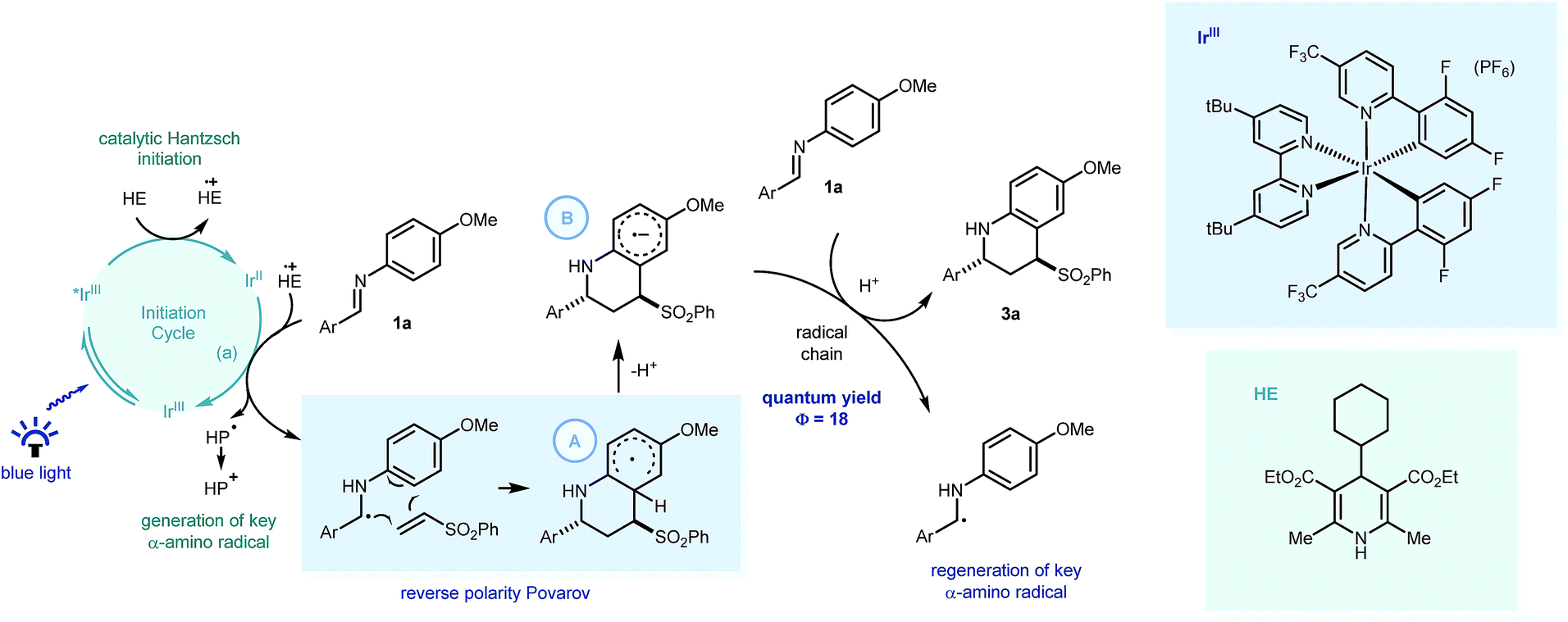 | ||
| Scheme 5 Postulated mechanism for the visible light mediated reverse polarity Povarov reaction with key PCET processes highlighted. HE = Hantzsch ester, HP = Hantzsch pyridine. | ||
From these insights, we propose that PCET would enable Ir[dF(CF3)ppy]2(dtbbpy)PF6 (E01/2 = −1.37 V vs. SCE in CH3CN)1a to reduce the imine readily to form the key α-amino radical (Scheme 5) which can react with the phenyl vinyl sulfone in a step-wise radical cyclization to give the stabilised Povarov radical intermediate A. As this methodology only requires sub-stoichiometric quantities of the substituted Hantzsch ester (HE4), we suggest this Povarov radical intermediate A can lose a proton (kinetically facile) to form radical anion intermediate B, in a base assisted homolytic aromatic substitution-type mechanism.28 Using Yoon's method, we calculated the quantum yield of the reaction between 1a and 2a, to be 18.29 This suggests that a radical chain process is in operation, with the strongly reducing radical anion intermediate able to reduce fresh imine substrate with concomitant protonation,30 thus forming the product (3a) and regenerating the key α-amino radical.
Conclusions
In conclusion we have developed a new photocatalytic reverse polarity Povarov reaction to construct decorated tetrahydroquinolines in high yield and diastereoselectivity. This polarity reversal was postulated to stem from an α-amino radical formed via the PCET of imine derivatives. 10 mol% of a Hantzsch ester was found to be optimal as a reductive initiator, a feature which has not previously been disclosed in photoredox catalysis. Further investigations are ongoing to establish further coupling partners and scaffolds for this reverse polarity platform for the synthesis of α-functionalised amines.Conflicts of interest
There are no conflicts to declare.Acknowledgements
JAL wishes to thank the Leverhulme Trust (RPG-2017-069) for a research fellowship. ALFA wishes to thank the Marie Curie Actions-MSCA-IF-EF-ST (660125) and Dr Soledad Encinas-Madrazo for fruitful discussion. JT would like to thank NSERC CGS-D and NSERC MSFSS. OH is supported by the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine, generously supported by AstraZeneca, Diamond Light Source, Defence Science and Technology Laboratory, Evotec, GlaxoSmithKline, Janssen, Novartis, Pfizer, Syngenta, Takeda, UCB, and Vertex. We also thank Heyao Shi for X-ray structure determination and Dr Amber L. Thompson and Dr Kirsten E. Christensen (Oxford Chemical Crystallography Service) for X-ray mentoring and help. We also thank Dr Hamish Hepburn for helpful discussions.Notes and references
- P. V. Pham, D. A. Nagib and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2011, 50, 6119 CrossRef PubMed.
- J. K. Matsui, S. B. Lang, D. R. Heitz and G. A. Molander, ACS Catal., 2017, 7, 2563 CrossRef PubMed.
- For selected reviews on photoredox catalysis see: (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef PubMed; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef PubMed; (c) J. Twilton, C. Lee, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem., 2017, 1, 1 CrossRef.
- Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef PubMed.
- (a) S. Inuki, K. Sato, T. Fukuyama, I. Ryu and Y. Fujimoto, J. Org. Chem., 2017, 82, 1248 CrossRef PubMed; (b) Y. Slutskyy, C. R. Jamison, P. Zhao, J. Lee, Y. H. Rhee and L. E. Overman, J. Am. Chem. Soc., 2017, 139, 7192 CrossRef PubMed.
- D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224 CrossRef PubMed.
- S. Bloom, C. Liu, D. K. Kölmel, J. X. Qiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. MacMillan, Nat. Chem., 2017, 10, 205 CrossRef PubMed.
- J. J. Douglas, M. J. Sevrin and C. R. J. Stephenson, Org. Process Res. Dev., 2016, 20, 1134 CrossRef.
- (a) T. L. Lemke, Review of Organic Functional Groups: Introduction to Medicinal Organic Chemistry, Lippincott Williams & Williams, Baltimore, 4th edn, 2003 Search PubMed; (b) J. W. Beatty and C. R. J. Stephenson, Acc. Chem. Res., 2015, 48, 1474 CrossRef PubMed.
- K. N. Lee and M.-Y. Ngai, Chem. Commun., 2017, 53, 13093 RSC.
- For key publications see: (a) Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 5257 CrossRef PubMed; (b) Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832 CrossRef PubMed; (c) A. Noble and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602 CrossRef PubMed; (d) L. Fan, J. Jia, H. Hou, Q. Lefebvre and M. Rueping, Chem.–Eur. J., 2016, 22, 16437 CrossRef PubMed; (e) R. S. Procter, H. J. Davis and R. J. Phipps, Science, 2018 DOI:10.1126/science.aar6376.
- For key publications see: (a) A. G. Condie, J. C. Gonzalez-Gomez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464 CrossRef PubMed; (b) D. A. DiRocco and T. J. Rovis, J. Am. Chem. Soc., 2012, 134, 8094 CrossRef PubMed.
- For review see: (a) E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546 CrossRef PubMed; For key publications see: (b) G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268 CrossRef PubMed; (c) K. T. Tarantino, O. Lium and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 10022 CrossRef PubMed; (d) L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 17735 CrossRef PubMed.
- (a) J. L. Jeffrey, F. R. Petronijevic and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 8404 CrossRef PubMed; (b) D. Hager and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 16986 CrossRef PubMed; (c) D. Uraguchi, N. Kinoshita, T. Kizu and T. Ooi, J. Am. Chem. Soc., 2015, 137, 13768 CrossRef PubMed; (d) M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828 CrossRef PubMed; (e) E. Fava, A. Millet, M. Nakajima, S. Loescher and M. Rueping, Angew. Chem., Int. Ed., 2016, 55, 6776 CrossRef PubMed; (f) N. R. Patel, C. B. Kelly, A. P. Siegenfeld and G. A. Molander, ACS Catal., 2017, 7, 1766 CrossRef PubMed.
- (a) L. Qi and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 13312 CrossRef PubMed; (b) A. L. Fuentes de Arriba, F. Urbitsch and D. J. Dixon, Chem. Commun., 2016, 52, 14434 RSC; (c) K. N. Lee, Z. Lei and M.-Y. Ngai, J. Am. Chem. Soc., 2017, 139, 5003 CrossRef PubMed; (d) H.-H. Zhang and S. Yu, J. Org. Chem., 2017, 82, 9995 CrossRef PubMed; (e) M. Chen, X. Zhao, C. Yang and W. Xia, Org. Lett., 2017, 19, 3807 CrossRef PubMed.
- T. Asberom, T. Bara, C. E. Bennett, D. A. Burnett, M. A. Caplen, J. W. Clader, D. J. Cole, M. S. Domalski, H. B. Josien, C. E. Knutson, H. Li, M. D. McBriar, D. A. Pissarnitski, L. Qiang, M. Rajagopalan, T. K. Sasikumar, J. Su, H. Tang, W.-L. Wu, R. Xu and Z. Zhao, PCT Int. Appl., WO 2007084595 A2 20070726, 2007.
- M. Miyakawa, S. Amano, M. Kamei, K. Hanada, K. Furuya and N. Yamamoto, PCT Int. Appl., WO 2002022585A1 20020321, 2002.
- (a) M. Nishino, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2011, 76, 6447 CrossRef PubMed; (b) S. Zhu, A. Das, L. Bui, H. Zhou, D. Curran and M. Rueping, J. Am. Chem. Soc., 2013, 135, 1823 CrossRef PubMed; (c) X. Ju, D. Li, W. Li, W. Yu and F. Bian, Adv. Synth. Catal., 2012, 354, 3561 CrossRef.
- (a) L. S. Povarov and B. M. Mikhailov, IIzv. Akad. Nauk SSSR, Ser. Khim., 1963, 953 Search PubMed; (b) L. S. Povarov, V. I. Grigos and B. M. Mikhailov, IIzv. Akad. Nauk SSSR, Ser. Khim., 1963, 2039 Search PubMed; (c) L. S. Povarov, Russ. Chem. Rev., 1967, 36, 656 CrossRef; (d) H. Liu, G. Dagousset, G. Masson, P. Retailleau and J. Zhu, J. Am. Chem. Soc., 2009, 131, 4598 CrossRef PubMed; (e) S. Kobayashi and S. Nagayama, J. Am. Chem. Soc., 1996, 118, 8977 CrossRef.
- Hantzsch ester contamination of product led to lower isolated yields. Experimentally, both HE4 and 1a were shown to be capable of quenching the photoexcited iridium species, see ESI.†.
- It must be noted that despite their use as alkyl radical precursors in photoredox catalysis, no alkyl addition products were observed, see (a) A. Gutiérrez-Bonet, J. C. Tellis, J. K. Matsui, B. A. Vara and G. A. Molander, ACS Catal., 2016, 6, 8004 CrossRef PubMed; (b) A. Gutiérrez-Bonet, C. Remeur, J. K. Matsui and G. A. Molander, J. Am. Chem. Soc., 2017, 139, 12251 CrossRef PubMed and ref. 15d.
- W. Huang, X. Cheng, W. Huang and X. Cheng, Synlett, 2017, 28, 148 Search PubMed.
- 3f was characterized further by SCXRD – CCDC 1831373.†.
- A postulated epimerization mechanism is given in the ESI.†.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714 Search PubMed.
- Previous investigations have shown that when used in conjugation with photocatalyst Eosin Y (also a pH indicator), the reduced Hantzsch leads to a colour change, which shows presence of acid in the reaction medium, see ref. 15b.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef PubMed.
- (a) S. Wertz, D. Leifert and A. Studer, Org. Lett., 2013, 15, 928 CrossRef PubMed; (b) D. Leifert, C. G. Daniliuc and A. Studer, Org. Lett., 2013, 15, 6286 CrossRef PubMed; (c) T. D. Svejstrup, A. Ruffoni, F. Julià, V. M. Aubert and D. Leonori, Angew. Chem., Int. Ed., 2017, 56, 14948 CrossRef PubMed. For similar example on aniline structures. (d) J. A. Leitch, C. L. McMullin, A. J. Paterson, M. F. Mahon, Y. Bhonoah and C. G. Frost, Angew. Chem., Int. Ed., 2017, 56, 15131 CrossRef PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC.
- It is not known at this time whether the proton shuttle between B and the propagation PCET reduction is pseudo-concerted or whether it transfers via the Hantzsch pyridine formed in situ.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1831373. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01704b |
| This journal is © The Royal Society of Chemistry 2018 |