
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Radical-mediated direct C–H amination of arenes with secondary amines†
Sebastian C.
Cosgrove
 ,
John M. C.
Plane
and
Stephen P.
Marsden
*
,
John M. C.
Plane
and
Stephen P.
Marsden
*
School of Chemistry, University of Leeds, Leeds LS2 9JT, UK. E-mail: s.p.marsden@leeds.ac.uk
First published on 11th July 2018
Abstract
Aryl dialkyl amines, valuable subunits of a wide range of effect chemicals, are accessed by intramolecular amination of aromatic C–H bonds employing UV photolysis of N-chloroamines. The reactions show good functional group tolerance and allow access to a range of fused and bridged polycyclic structures. The homogeneous reaction conditions allow for the one-pot conversion of secondary amines to their arylated derivatives. Experimental and theoretical evidence supports the involvement of electrophilic aminium radicals which react via direct ortho-attack on the arene.
Introduction
Aromatic amines are fundamental components of a broad range of effect molecules including dyes, pharmaceuticals and agrochemicals.1 Studies within the pharmaceutical industry, for example, have shown that construction of aryl alkyl amines comprises ca. 5–10% of reactions carried out in the discovery phase2 and 5% of those undertaken in process development.3 Classically this motif is constructed by multi-step syntheses through N-alkylation of anilines or nucleophilic substitution of electron-poor haloarenes. Recent advances in metal-catalysed aminations of aromatic (pseudo)halides have been transformative,4 becoming one of the most significant reaction classes employed by industrial medicinal chemists.5 Nevertheless, the requirements for pre-functionalised aromatic (pseudo)halides and expensive precious metal catalysts and bespoke ligands are still limitations.Interest in approaches to aromatic amines by the direct amination of aryl C–H bonds has therefore grown significantly. Metal-catalysed amination of substituted benzenes has been variously reported using electrophilic aminating species such as O-acyl-6 and O-sulfonylhydroxylamines,7 dioxazolones,8N-chloroamines,9 azides10 or amine derivatives in conjunction with oxidants,11 but in nearly all cases a coordinating group is required to direct C–H activation. The chemistry of nitrogen-centred radicals has seen a renaissance in recent years, and metal-catalysed intermolecular12 and photochemical/photoredox-mediated inter-13 and intramolecular14 methods for direct N-functionalisation of (hetero)arenes using such species have been reported,15 but are predominantly limited to the introduction of non-basic nitrogen substituents such as imides,12c,13c,d,f amides,13a,c phosphonamides14a,b or sulfonamides13b,g,h,14d,e (e.g.Scheme 1, panel a). There remains a significant need for a general direct amination of aromatic C–H bonds with simple alkylamine precursors. Outstanding progress has recently been made in this regard (Scheme 1, panel b): Leonori has demonstrated the amination of a range of mono- and bicyclic aromatics with secondary aminium radicals generated by photoredox-mediated homolysis of (2,4-dinitrophenyloxy)amines,16 while Nicewicz has demonstrated the intermolecular amination of (predominantly) electron-rich arenes with aminium radicals, generated directly from primary amines using acridinium photoredox catalysis coupled with aerobic oxidation.17
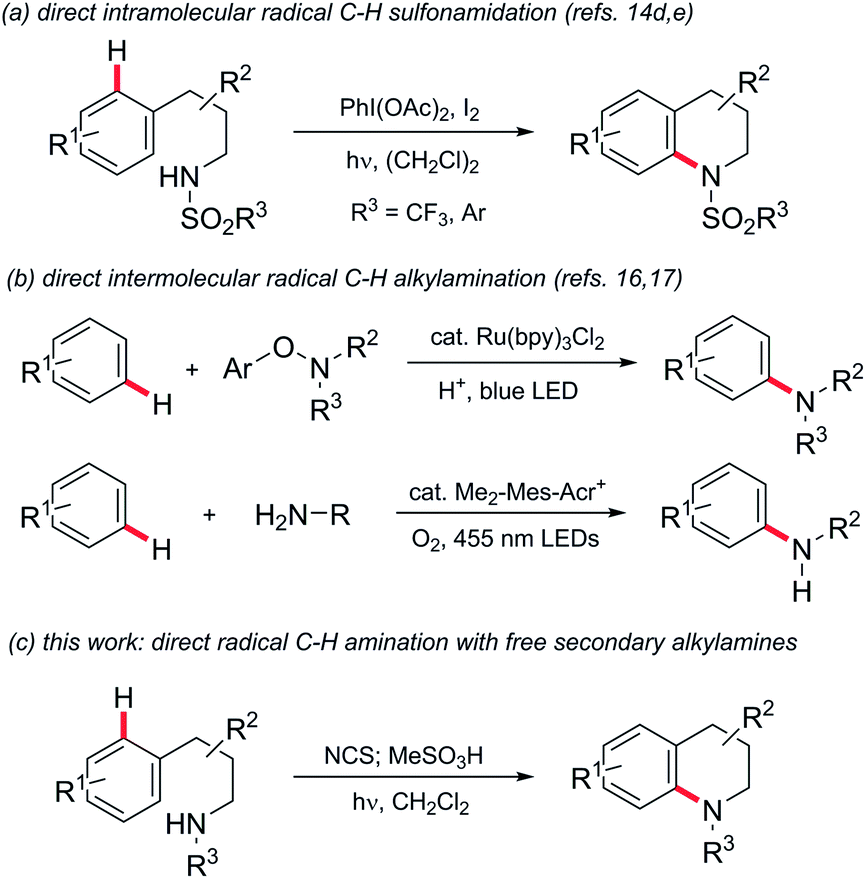 | ||
| Scheme 1 Direct radical C–H amination of arenes. | ||
Despite these groundbreaking advances, a method for the direct arylation of secondary amines is still elusive: the aryloxyamine radical precursors employed successfully by Leonori require multi-step synthesis from secondary amines, while the direct functionalization reported by Nicewicz is thus far limited to primary amines.
Minisci and Kompa reported the direct amination of aromatics using aminium radicals generated from N-chloroamines in acidic media using, respectively, iron(II) salts18 and UV photolysis.19 Despite the simplicity of these methods they have remarkably remained unexploited in the literature in the intervening 50 years.20 As noted elsewhere,17 the reasons for this are most likely due to the requirement for the use of preformed N-chloroamines which have a reputation as unstable/hazardous intermediates. Additionally, the reaction media were typically mixtures of concentrated sulfuric and acetic acid, which has limited scope as a medium for organic reactions and also precludes in situ generation of the radical precursors. Herein we describe the development of practical homogeneous conditions for N-chloroamine-mediated amination which: (a) allows us to demonstrate that the reaction has functional group tolerance, (b) allows access to a range of different polycyclic skeleta and, significantly, (c) facilitates the conversion of secondary amines to arylated derivatives in a single pot (Scheme 1, panel c). We also provide experimental and theoretical support for the involvement of electrophilic aminium radicals as intermediates.
Results and discussion
Given the significance of the tetrahydroquinoline structure in a range of biologically active molecules,21 we began by examining the cyclisation of N-chloroamine 1a, initially employing classical Hofmann–Löffler–Freytag (HLF) conditions in concentrated sulfuric acid18,19 (Table 1). Clean cyclisation to N-methyltetrahydroquinoline 2a was observed in 81% yield (comparable to the non-photolytic literature variant mediated by FeCl2 (ref. 18b)), but as expected the viscous, heterogeneous reaction mixtures were difficult to process.|
|
|||
|---|---|---|---|
| Entrya | Reaction medium | Ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3b :3b |
Yield 2ac |
|
a Reaction conditions: 1a (0.5 mmol.), 125W high pressure Hg-lamp, RT.
b Ratio of 2a:3, determined by 1H NMR analysis.
c Isolated yield.
d Non-photolytic reaction using FeCl2 (result taken from ref. 18b).
e Plus 18% of chlorinated tetrahydroquinolines.
f In absence of UV or visible light.
g Using 24W visible light.
|
|||
| 1 | c. H2SO4 | 100:0 |
81% |
| 2d | c. H2SO4, FeCl2 | n/a | 81% |
| 3 | 3 N HCl/MeOH | 0:100 |
— |
| 4 | AcOH, 5 h | 0:100 |
— |
| 5 | TFA, 5 h | 55:27e |
50% |
| 6 | MeSO3H/DCM (1:1) |
100:0 |
80% |
| 7 | MeSO 3 H (10 eq.), DCM |
100:0 |
91% |
| 8f | MeSO3H (10 eq.), DCM | 0:100 |
— |
| 9g | MeSO3H (10 eq.), DCM | 25:75 |
17% |
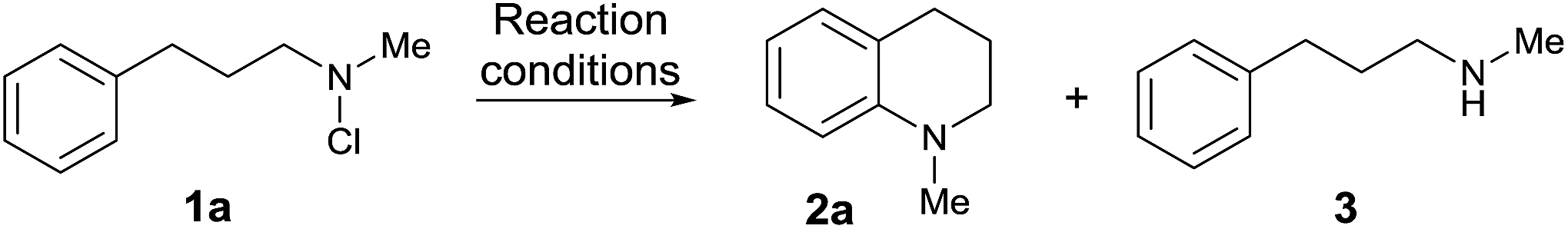
Amination was not observed in organic media such as acetic acid or methanolic HCl; in neat TFA amination occurred but ring-chlorinated products (presumably from competing electrophilic substitution pathways with the chloroamine as chlorinating agent22) were also observed. Reaction in mixtures of methanesulfonic acid and dichloromethane, however, gave clean conversion to 2a, with best results found using 10 equivalents of acid (ESI†). Control experiments (entries 7, 8) confirmed the necessity for UV light irradiation.
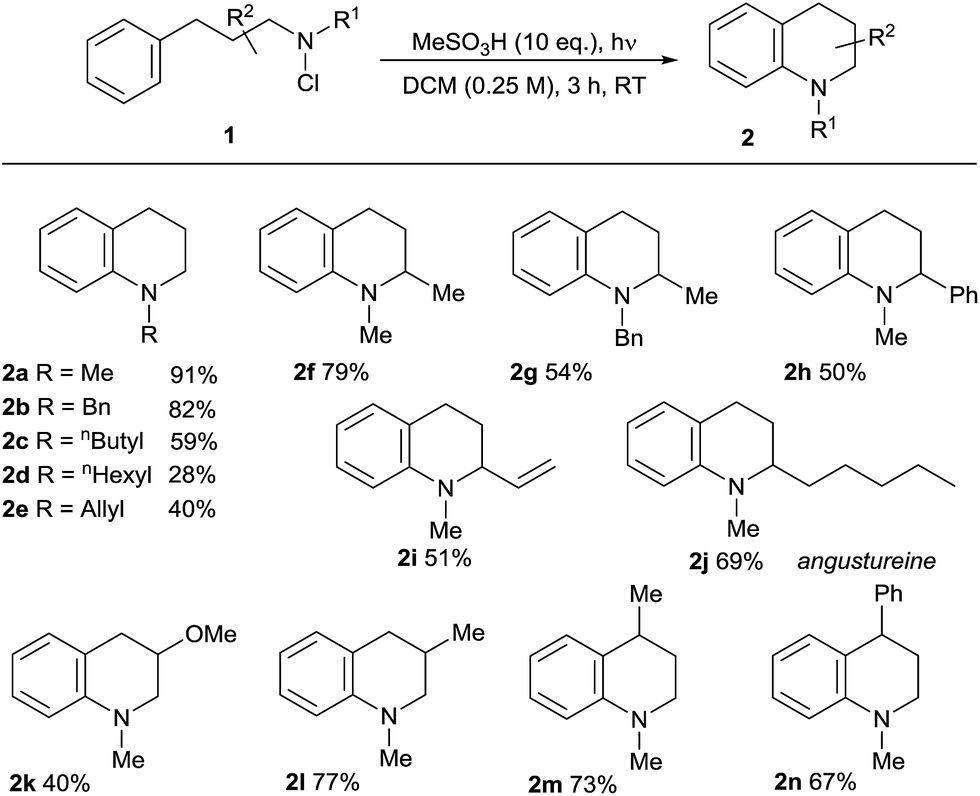
We then applied the optimised conditions in an examination of the breadth and scope of the amination process, commencing with substitution in the non-aromatic ring (Table 2). Variation of the N-alkyl substituent was possible in products 2a–e, with the tolerance of the removable N-benzyl substituent in 2b being noteworthy from a synthetic perspective. Substitution in the aliphatic backbone of substrates 1 is also tolerated, allowing variously for incorporation of alkyl, alkenyl, aryl and heteroatom functionality at C2, 3 or 4 of the tetrahydroquinoline products 2f–n. With longer alkyl chains on either the nitrogen atom or in the backbone, the potential for competing aliphatic C–H functionalisation through classical HLF chemistry exists, and indeed products of this process (<25%) accompanied the formation of the N-hexyl-substituted 2d, lowering the isolated yield. However, the presence of abstractable hydrides in the carbon backbone was less problematic, illustrated by the relatively clean formation of the natural product angustureine232j bearing a 2-pentyl substituent in 69% yield. Additionally, the potential for scission of the aminium radical intermediate where a radical-stabilising substituent is present on the beta-carbon18b means that 3-aryltetrahydroquinolines are not formed in preparatively useful yield.
|
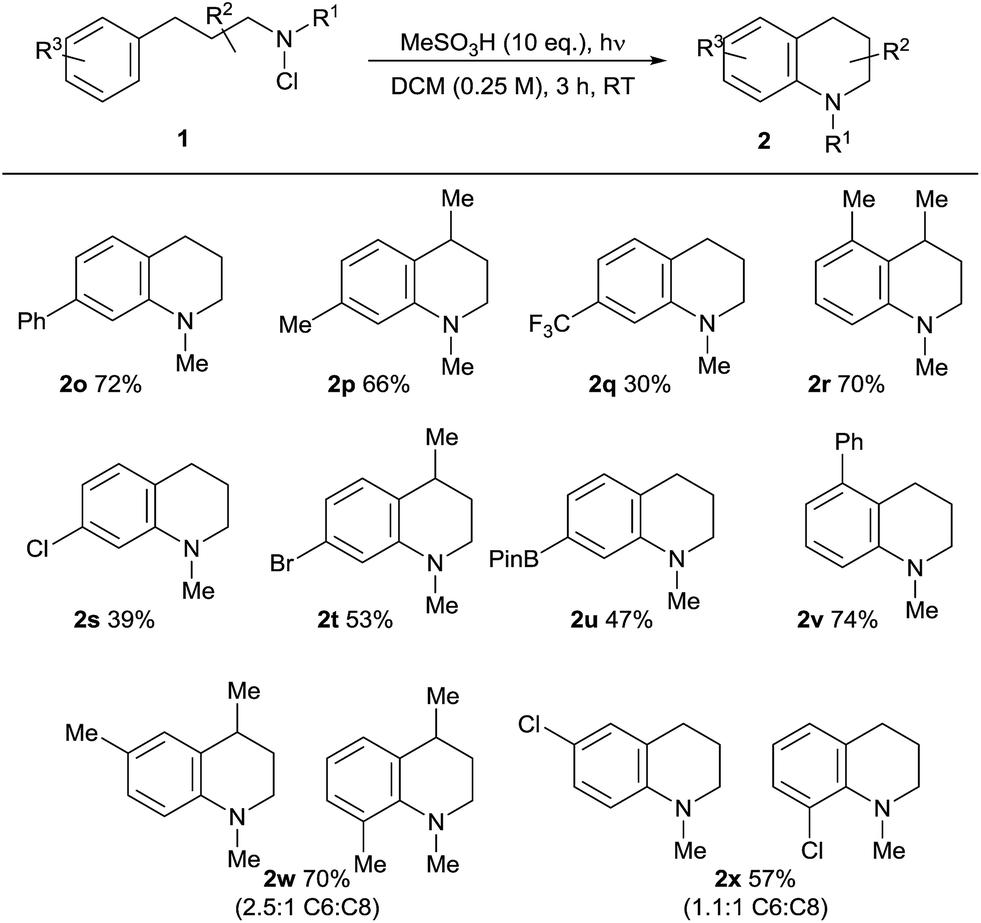
The scope of the reaction in terms of the aromatic partner was also investigated (Table 3). Both alkyl and aryl substituents are tolerated in the ortho-, meta- and para-positions of 1. The effective cyclisation of ortho-substituted substrates notably leads to the effective formation of challenging contiguously trisubstituted arene structures 2r/v. Alkyl and aromatic substituents are well tolerated, but the presence of an electron-withdrawing para-trifluoromethyl substitutent results in a lower conversion to 2q, supporting the electrophilic character of the aminating species. We found that electron-rich arene-containing substrates (e.g. methoxyphenyl) were not tolerated, resulting in complex mixtures and evidence of direct electrophilic aromatic chlorination, presumably by the chloroamine agent itself.22 As expected, the use of meta-substituted substrates 1w/x gives rise to a mixture of regioisomeric products with a moderate preference for the formation of the less-hindered C6-isomeric product. Most notably, the amination reaction demonstrates tolerance towards both halide substituents in 2s, t, x and a boronate ester function in 2u, which is synthetically significant, given the potential utility of both functionalities in downstream cross-coupling chemistries.
|
We next assessed the potential of the direct amination in the preparation of diverse polycyclic assemblies (Scheme 2). We were pleased to find that the use of N-chloroamines 4a/b derived from cyclic secondary amines lead to the highly effective synthesis of angularly-fused tricyclic amines 5a/b, while the linearly-fused tricycle 7 was accessed efficiently from 2-benzylcyclohexylamine-derived 6. Most remarkably, the N-chloroamine of the 3-phenylcyclohexylamine 8 underwent cyclisation to give bridged skeleton 9 (a framework found in complex alkaloids such as sespenine24), despite the requirement for a highly unfavourable 1,3-diaxial disposition of the reacting substituents in this conformationally-unbiased substrate.
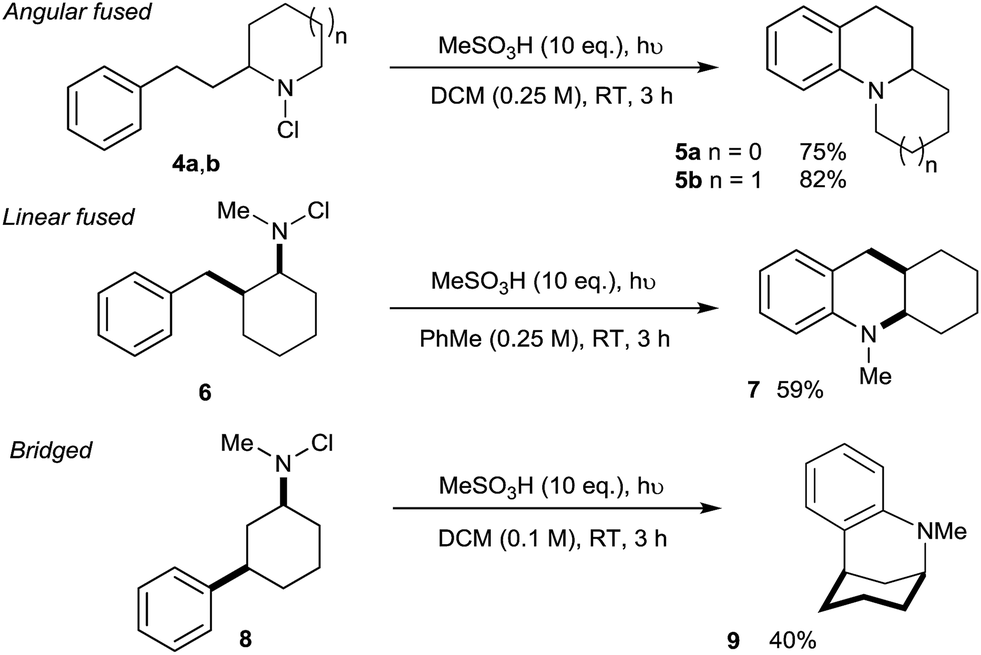 | ||
| Scheme 2 Tricyclic skeleta accessible through direct C–H amination. | ||
With the substrate scope of the protocol established, we next turned our attention to the development of a protocol for the direct amination of secondary amines in a single vessel. We were very pleased to find that this could be effected simply by treatment of the parent secondary amine with N-chlorosuccinimide (NCS) in dichloromethane followed by addition of MsOH and irradiation with UV light (Scheme 2). The presence of the succinimide by-product does not appear to adversely affect the direct amination, with isolated yields of the simple tetrahydroquinoline 2a, the alkaloid angustureine 2j, and the angularly-fused tricycle 5a comparable to the two-step process (separate N-chlorination/amination) in both cases. The formation of enantioenriched 2j proceeds with extremely high stereochemical fidelity, as predicted. This protocol constitutes the first examples of the one-pot metal-free arylation of secondary amines, and the operational simplicity of the method (obviating the need for prior functionalization of the nitrogen) should enable further synthetic applications of the method (Scheme 3).
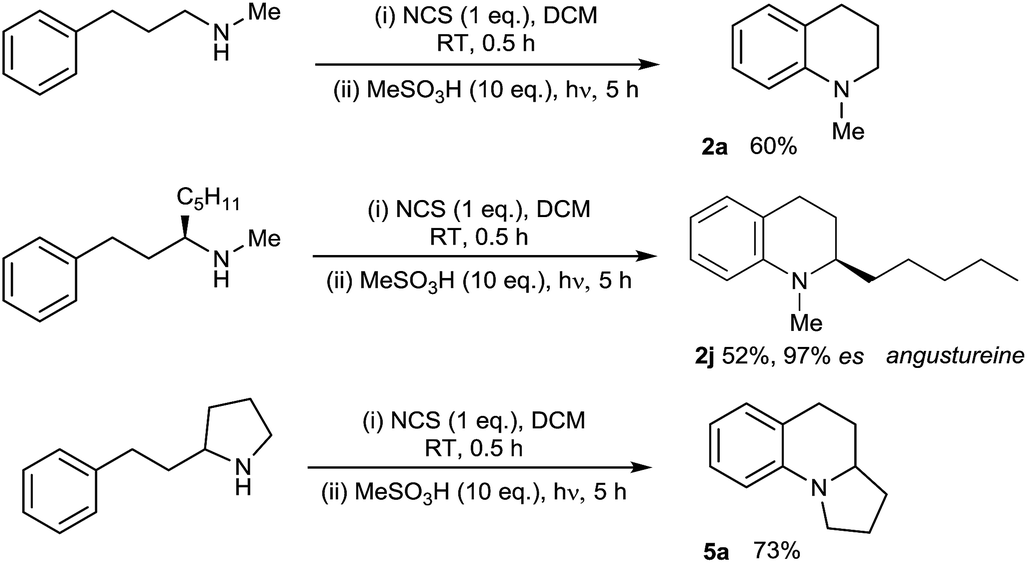 | ||
| Scheme 3 Direct one-pot arylation of secondary amines. | ||
The mechanism of amination by photolysis of N-chloroamines has been proposed to involve the intermediacy of protonated aminium radicals,12d but we sought to provide further evidence for the precise pathway to the tetrahydroquinoline products. The electrophilic nature of the aminating species was probed by internal competition experiments between differentially-substituted 3,3-diarylpropylamine substrates 10 and 11 (Scheme 4). In competition between a phenyl and a 4-methylphenyl substituent, amination of the more electron-rich ring in 10 predominates, favouring 12a by a factor of 10:1, whereas in competition with a 3-trifluoromethyl group exclusive amination of the phenyl ring of 11 is observed, giving 13a. Both results support a strongly electrophilic nature for the aminating species.
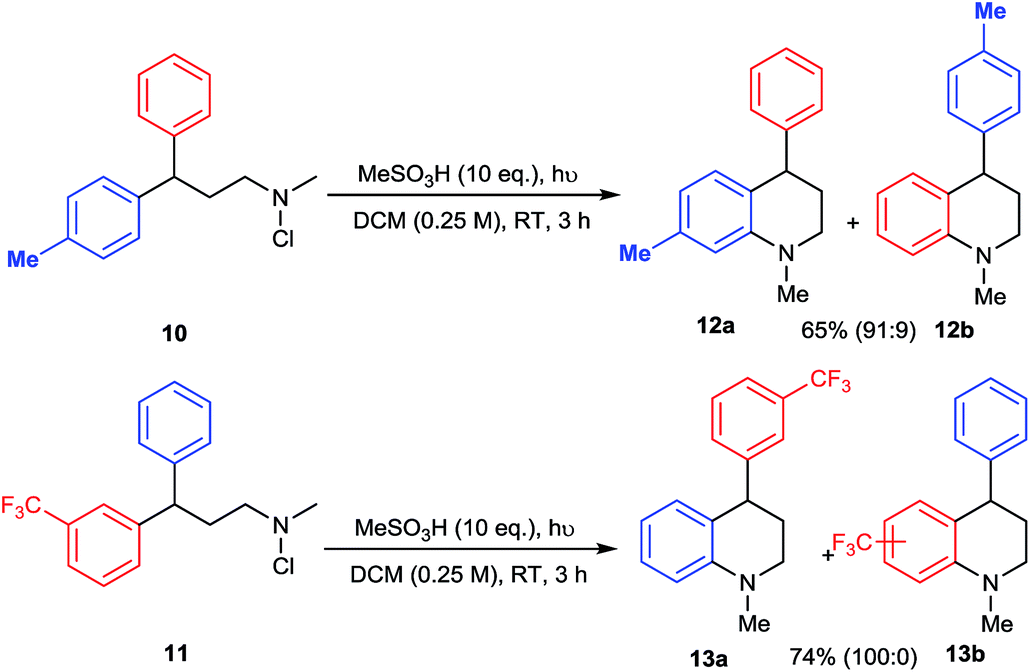 | ||
| Scheme 4 Intramolecular competition experiments. | ||
The pathway of the cyclisation reaction was further probed by DFT calculations (B3LYP method with the 6-311+G(2d,p) triple zeta basis set; solvation by dichloromethane included by the Polarisable Continuum Model; see ESI† for full details). Although the ultimate product is that of ortho-amination, the intramolecular addition of radicals to arenes can arise through 5-membered spiro-addition or direct 6-membered ortho-addition modes, and examples of each manifold are known for heteroatom-centred radicals.25 Our calculations revealed that cyclisation of the neutral N-methyl-3-phenylpropan-1-aminyl radical in both spiro- and ortho-addition modes was, as expected, highly endergonic (ΔG(298 K) = 67 and 54 kJ mol−1, respectively), and was discounted. The cyclisations of the corresponding protonated aminium radicals were energetically more reasonable, consistent with the known rate enhancement in 1,5-hydrogen atom transfer26 and cyclisation to alkenes27 for aminium versus aminyl radicals. Of the two cyclisation pathways, the transition state for the ortho-cyclisation mode was found to be 14.2 kJ mol−1 more favourable than the spiro-mode and we conclude that this is the mode of attack (Fig. 1).
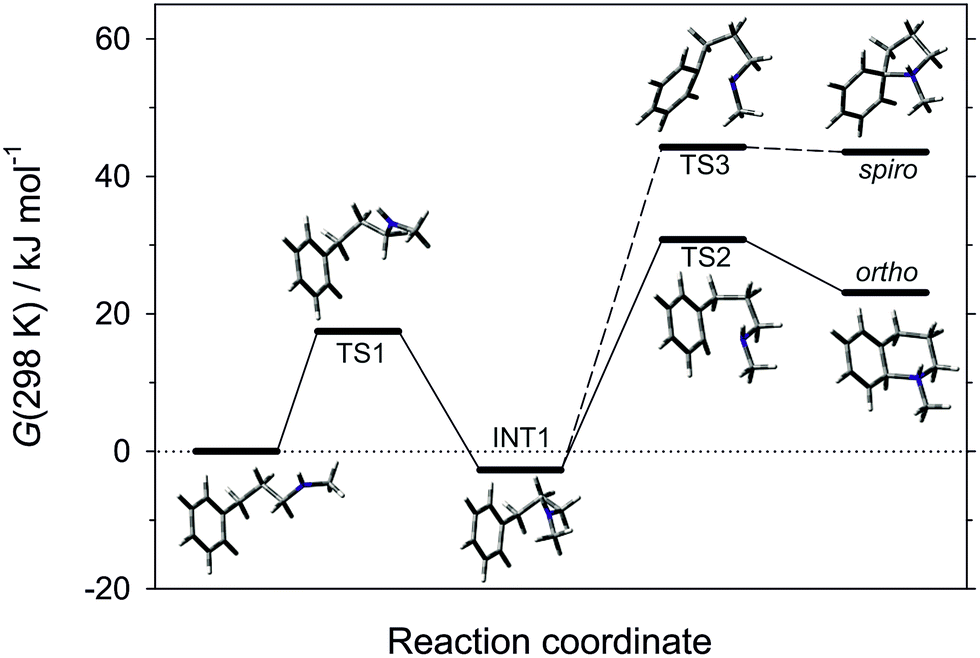 | ||
| Fig. 1 Energy plot for the cyclisation modes of aminium radical derived from 1a. | ||
In light of this, we examined the behaviour of 2,6-disubstituted substrates in which both ortho-positions are blocked. 2,6-Dichlorophenyl substrate 14 underwent formal C–Cl amination to give 15, presumably via ortho-cyclisation and ipso-radical substitution. This constitutes a metal-free equivalent to Buchwald-Hartwig and Ullman amination reactions, occurring under acidic conditions which contrast with the basic conditions used in such processes. With 2,6-dimethylphenyl substrate 16, the contiguously tetrasubstituted arene 17 was unexpectedly formed. This compound presumably arises by 1,2-methyl migration in the delocalised cyclohexadienyl radical cationic (Wheland) intermediate derived from this. Both of these reaction manifolds may find further synthetic application (Scheme 5).
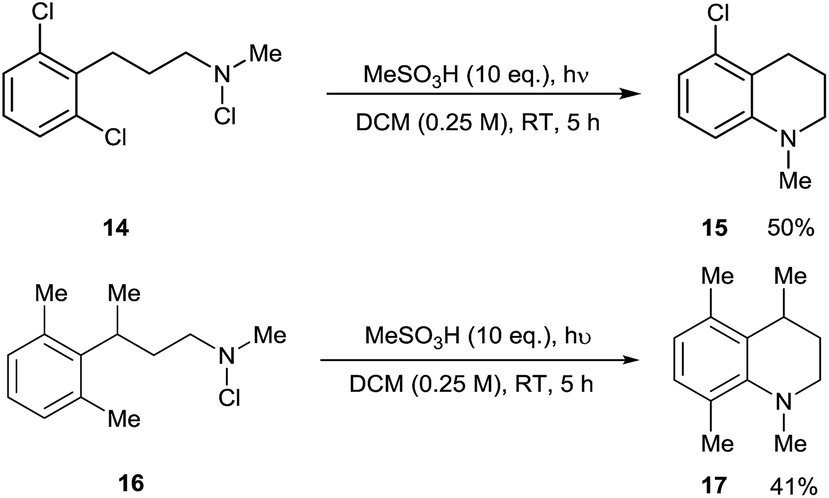 | ||
| Scheme 5 Substitution and rearrangement reactions of 2,6-disubstituted arene substrates. | ||
Conclusions
In summary, direct amination of aromatic compounds by photolytically-generated aminium radicals is revealed as an effective and functional group tolerant method for the construction of the valuable aryl dialkyl amine function. The reaction allows access to a range of polycyclic skeleta including bridged variants of relevance to complex alkaloids. The development of homogeneous reaction conditions for the amination allows for the direct one-pot conversion of secondary amines to their arylated derivatives under metal-free radical-based conditions. The mechanism has been probed experimentally and theoretically, with direct ortho-addition of electrophilic aminium radicals favoured. The functional group tolerance of the reaction coupled with the ability to access diverse polycyclic ring systems supports future applications in target synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for a DTA studentship award to SCC.Notes and references
- (a) R. Hili and A. K. Yudin, Nat. Chem. Biol., 2006, 2, 284–287 CrossRef PubMed; (b) The Chemistry of Anilines, Part 1, ed. Z. Rapoport, Wiley VCH, 2007 Search PubMed; (c) P. F. Vogt and J. J. Gerulis, Amines, Aromatic, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2005 Search PubMed; (d) Amino Group Chemistry: From Synthesis to the Life Sciences, ed. A. Ricci, Wiley VCH, 2008 Search PubMed.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef PubMed.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC.
- (a) M. Tomas-Gamasa, in Science of Synthesis: Cross Coupling and Heck-Type Reactions 2, ed. J. P. Wolfe, Thieme, Stuttgart, 2013 Search PubMed; (b) U. Scholz, W. Dong, J. Feng and W. Shi, in Science of Synthesis: Cross Coupling and Heck-Type Reactions 2, ed. J. P. Wolfe, Thieme, Stuttgart, 2013 Search PubMed.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef PubMed.
- (a) D. Zhu, G. Yang, J. He, L. Chu, G. Chen, W. Gong, K. Chen, M. D. Eastgate and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 2497–2500 CrossRef PubMed; (b) P. Patel and S. Chang, ACS Catal., 2015, 5, 853–858 CrossRef; (c) Z. Dong and G. Dong, J. Am. Chem. Soc., 2013, 135, 18350–18353 CrossRef PubMed; (d) E. J. Yoo, S. Ma, T.-S. Mei, K. S. L. Chan and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 7652–7655 CrossRef PubMed.
- (a) M. Paudyal, A. M. Adebesin, S. R. Burt, D. H. Ess, Z. Ma, L. Kurti and J. R. Falck, Science, 2016, 353, 1144–1147 CrossRef PubMed; (b) M. R. Yadav, M. Shankar, E. Ramesh, K. Ghosh and A. K. Sahoo, Org. Lett., 2015, 17, 1886–1889 CrossRef PubMed; (c) S. Yu, B. Wan and X. Li, Org. Lett., 2013, 15, 3706–3709 CrossRef PubMed; (d) K.-H. Ng, A. S. C. Chan and W.-Y. Yu, J. Am. Chem. Soc., 2010, 132, 12862–12864 CrossRef PubMed.
- J. Park and S. Chang, Angew. Chem., Int. Ed., 2015, 54, 14103–14107 CrossRef PubMed.
- (a) T. Matsubara, S. Asako, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2014, 136, 646 CrossRef PubMed; (b) C. Grohmann, H. Wang and F. Glorius, Org. Lett., 2012, 14, 656–659 CrossRef PubMed.
- (a) K. Shin, Y. Baek and S. Chang, Angew. Chem., Int. Ed., 2013, 52, 8031–8036 CrossRef PubMed; (b) Y. Lian, J. R. Hummel, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2013, 135, 12548–12551 CrossRef PubMed; (c) V. S. Thirunavukkarasu, K. Raghuvanshi and L. Ackermann, Org. Lett., 2013, 15, 3286–3289 CrossRef PubMed.
- (a) J. Roane and O. Daugulis, J. Am. Chem. Soc., 2016, 138, 4601–4607 CrossRef PubMed; (b) Q. Yan, Z. Chen, W. Yu, H. Yin, Z. Liu and Y. Zhang, Org. Lett., 2015, 17, 2482–2485 CrossRef PubMed; (c) L. Marchetti, A. Kantak, R. Davis and B. DeBoef, Org. Lett., 2015, 17, 358–361 CrossRef PubMed; (d) M. Yang, B. Su, Y. Wang, K. Chen, X. Jiang, Y.-F. Zhang, X.-S. Zhang, G. Chen, Y. Cheng, Z. Cao, Q.-Y. Guo, L. Wang and Z.-J. Shi, Nat. Commun., 2014, 5, 4707 CrossRef PubMed; (e) H. Kim, K. Shin and S. Chang, J. Am. Chem. Soc., 2014, 136, 5904–5907 CrossRef PubMed; (f) M. Shang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 3354–3357 CrossRef PubMed; (g) Q. Li, S.-Y. Zhang, G. He, Z. Ai, W. A. Nack and G. Chen, Org. Lett., 2014, 16, 1764 CrossRef PubMed; (h) L. D. Tran, J. Roane and O. Daugulis, Angew. Chem., Int. Ed., 2013, 52, 6043–6046 CrossRef PubMed; (i) R. Shrestha, P. Mukherjee, Y. Tan, Z. C. Litman and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 8480–8483 CrossRef PubMed; (j) E. T. Nadres and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 7–10 CrossRef PubMed; (k) G. He, Y. Zhao, S. Zhang, C. Lu and G. Chen, J. Am. Chem. Soc., 2012, 134, 3–6 CrossRef PubMed; (l) B. Xiao, T.-J. Gong, J. Xu, Z.-J. Liu and L. Liu, J. Am. Chem. Soc., 2011, 133, 1466–1474 CrossRef PubMed; (m) A. John and K. M. Nicholas, J. Org. Chem., 2011, 76, 4158–4162 CrossRef PubMed; (n) T.-S. Mei, X. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 10806–10807 CrossRef PubMed; (o) X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790–6791 CrossRef PubMed; (p) U. Takeshi, I. Shinya and C. Naoto, Chem. Lett., 2006, 35, 842–843 CrossRef; (q) H.-Y. Thu, W.-Y. Yiu and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 9048–9049 CrossRef PubMed.
- Intermolecular: (a) L. Legnani, G. Prina Cerai and B. Morandi, ACS Catal., 2016, 6, 8162–8165 CrossRef; (b) J. Liu, K. Wu, T. Shen, Y. Liang, M. Zou, Y. Zhu, X. Li, X. Li and N. Jiao, Chem.–Eur. J., 2017, 23, 563–567 CrossRef PubMed; (c) K. Foo, E. Sella, I. Thome, M. D. Eastgate and P. S. Baran, J. Am. Chem. Soc., 2014, 136, 5279–5282 CrossRef PubMed; (d) L. Stella, Angew. Chem., Int. Ed. Engl., 1983, 22, 337–422 CrossRef; (e) F. Minisci, Synthesis, 1973, 1–24 Search PubMed.
- Intermolecular: (a) J. Davies, T. D. Svejstrup, D. Fernandez Reina, N. S. Sheikh and D. Leonori, J. Am. Chem. Soc., 2016, 138, 8092–8095 CrossRef PubMed; (b) K. Tong, X. Liu, Y. Zhang and S. Yu, Chem.–Eur. J., 2016, 22, 15669–15673 CrossRef PubMed; (c) T. W. Greulich, C. G. Daniliuc and A. Studer, Org. Lett., 2015, 17, 254–257 CrossRef PubMed; (d) L. J. Allen, P. J. Cabrera, M. Lee and M. S. Sanford, J. Am. Chem. Soc., 2014, 136, 5607–5610 CrossRef PubMed; (e) J.-D. Wang, Y.-X. Liu, D. Xue, C. Wang and J. Xiao, Synlett, 2014, 25, 2013–2018 CrossRef; (f) H. Kim, T. Kim, D. G. Lee, S. W. Roh and C. Lee, Chem. Commun., 2014, 50, 9273–9276 RSC; (g) Q. Qin and S. Yu, Org. Lett., 2014, 16, 3504–3507 CrossRef PubMed; (h) L. Song, L. Zhang, S. Luo and J.-P. Cheng, Chem.–Eur. J., 2014, 20, 14231–14234 CrossRef PubMed.
- Intramolecular: (a) Y.-N. Ma, M.-X. Cheng and S.-D. Yang, Org. Lett., 2017, 19, 600–603 CrossRef PubMed; (b) Y.-N. Ma, X. Zhang and S.-D. Yang, Chem.–Eur. J., 2017, 23, 3007–3011 CrossRef PubMed; (c) C. Martinez, A. E. Bosnidou, S. Allmedinger and K. Muniz, Chem.–Eur. J., 2016, 22, 9929–9932 CrossRef PubMed; (d) H. Togo, Y. Harada and M. Yokoyama, J. Org. Chem., 2000, 65, 926–929 CrossRef; (e) H. Togo, Y. Hoshina, T. Muraki, H. Nakayama and M. Yokoyama, J. Org. Chem., 1998, 63, 5193–5200 CrossRef.
- For a perspective article, see: K. Murakami, G. J. P. Perry and K. Itami, Org. Biomol. Chem., 2017, 15, 6071–6075 RSC.
- T. D. Svejstrup, A. Ruffoni, F. Julia, V. M. Aubert and D. Leonori, Angew. Chem., Int. Ed., 2017, 56, 14948–14952 CrossRef PubMed.
- K. A. Margrey, A. Levens and D. A. Nicewicz, Angew. Chem., Int. Ed., 2017, 56, 15644–15648 CrossRef PubMed.
- (a) F. Minisci and R. Galli, Tetrahedron Lett., 1965, 8, 433–436 CrossRef; (b) F. Minisci and R. Galli, Tetrahedron Lett., 1966, 9, 2531–2533 CrossRef.
- (a) H. Bock and K.-L. Kompa, Angew. Chem., Int. Ed., 1965, 4, 783 CrossRef; (b) H. Bock and K.-L. Kompa, Chem. Ber., 1966, 99, 1357–1360 CrossRef.
- For an isolated and unexpected intramolecular example of this reaction, see: A. U. Dey and B. Pathak, J. Med. Chem., 1970, 13, 152–153 CrossRef ; for confirmation of this reaction, see: P. S. Anderson, G. F. Lundell, J. L. Cias and F. M. Robinson, Tetrahedron Lett., 1971, 12, 2787–2790 CrossRef.
- V. Sridharan, P. A. Suryavanshi and J. C. Menendez, Chem. Rev., 2011, 111, 7157–7259 CrossRef PubMed.
- (a) J. R. Lindsay Smith, L. C. McKeer and J. M. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, 1533–1537 RSC; (b) J. R. Lindsay Smith and L. C. McKeer, Tetrahedron Lett., 1983, 24, 3117–3120 CrossRef.
- I. Jacquemond-Collet, S. Hannedouche, N. Fabre, I. Fourasté and C. Moulis, Phytochemistry, 1999, 51, 1167–1169 CrossRef.
- L. Ding, A. Maier, H.-H. Fiebig, W.-H. Lin and C. Hertweck, Org. Biomol. Chem., 2011, 9, 4029–4031 RSC.
- See, for example: spiro-cyclisation of O-centred radicals: R. T. McBurney, A. Eisenschmidt, A. M. Z. Slawin and J. C. Walton, Chem. Sci., 2013, 4, 2028–2035 RSC ; ortho-cyclisation of iminyl radicals: R. T. McBurney and J. C. Walton, Beilstein J. Org. Chem., 2013, 9, 1083–1092 CrossRef PubMed.
- D. Sakic and H. Zipse, Adv. Synth. Catal., 2016, 358, 3983–3991 CrossRef.
- J. H. Horner, F. N. Martinez, O. M. Musa, M. Newcomb and H. E. Shahin, J. Am. Chem. Soc., 1995, 117, 11124–11133 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures, spectral characterisation and details of DFT calculations. See DOI: 10.1039/c8sc01747f |
| This journal is © The Royal Society of Chemistry 2018 |