
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Norbornene chaotropic salts as low molecular mass ionic organogelators (LMIOGs)†
Jordan R.
Engstrom
 a,
Aramballi J.
Savyasachi
b,
Marzieh
Parhizkar
c,
Alessandra
Sutti
c,
Chris S.
Hawes
d,
Jonathan M.
White
e,
Thorfinnur
Gunnlaugsson
*b and
Frederick M.
Pfeffer
*a
a,
Aramballi J.
Savyasachi
b,
Marzieh
Parhizkar
c,
Alessandra
Sutti
c,
Chris S.
Hawes
d,
Jonathan M.
White
e,
Thorfinnur
Gunnlaugsson
*b and
Frederick M.
Pfeffer
*a
aSchool of Life and Environmental Sciences, Deakin University, Waurn Ponds, Victoria 3216, Australia. E-mail: fred.pfeffer@deakin.edu.au
bSchool of Chemistry and Trinity Biomedical Sciences Institute (TBSI), Trinity College Dublin, The University of Dublin, Dublin 2, Ireland. E-mail: gunnlaut@tcd.ie
cInstitute for Frontier Materials, Deakin University, Waurn Ponds, Victoria 3216, Australia. E-mail: asutti@deakin.edu.au
dSchool of Chemical and Physical Sciences, Keele University, Staffordshire, ST5 5BG, UK
eBio21 Institute, School of Chemistry, University of Melbourne, Parkville, 3010, Australia
First published on 16th May 2018
Abstract
Phenylalanine functionalised norbornene (9:Na) functions as a potent, low molecular-mass (MW = 333 Da) ionic organogelator with a minimum gelating concentration of 0.5 wt% in THF, i-PrOH, 1,4-dioxane and n-BuOH. Fibrous crystals form in the gel and X-ray crystallography identified a cation mediated helical assembly process controlled by the chirality of the phenylalanine. In addition to excellent gelating properties 9:Na readily forms aqueous biphasic and triphasic systems.
Introduction
The development of efficient organogelating agents remains an important pursuit as the corresponding organogels have found use in sensing,1–3 cosmetics,4,5 water purification6,7 and drug delivery applications.8–12 Low molecular mass organogelators (LMOGs) are of particular interest as these can self-assemble using non-covalent interactions9,13–16 to give rise to novel soft materials with a range of entangled and solvent dependent morphologies.13,17,18 Recently we have developed various examples of LMOGs from simple organic ligands that can, through self-association, or via coordination to either d- or f-metal ions, give rise to functional self-assembled gels.19–25 In parallel, our ongoing interest in applications of functionalised norbornanes and related norbornylogous systems,26–33 led us to synthesise simple norbornanes modified with amino-acids and here we report the development of such functionalised norbornenes as new low molecular mass ionic organogelators (LMIOGs) (MW < 350 Da).Ionic compounds capable of gelating organic solvents are rare and the examples that do exist are typically either based on steroidal frameworks or possess long alkyl chains that give rise to MW ranging from ≈350 up to 1000 Da (e.g.1–4,Fig. 1).9,13,17,34–40 Reports of compact LMIOGs, in which large hydrophobic groups have not been deliberately incorporated are extremely rare (e.g.5 and 6, Fig. 1).40,41
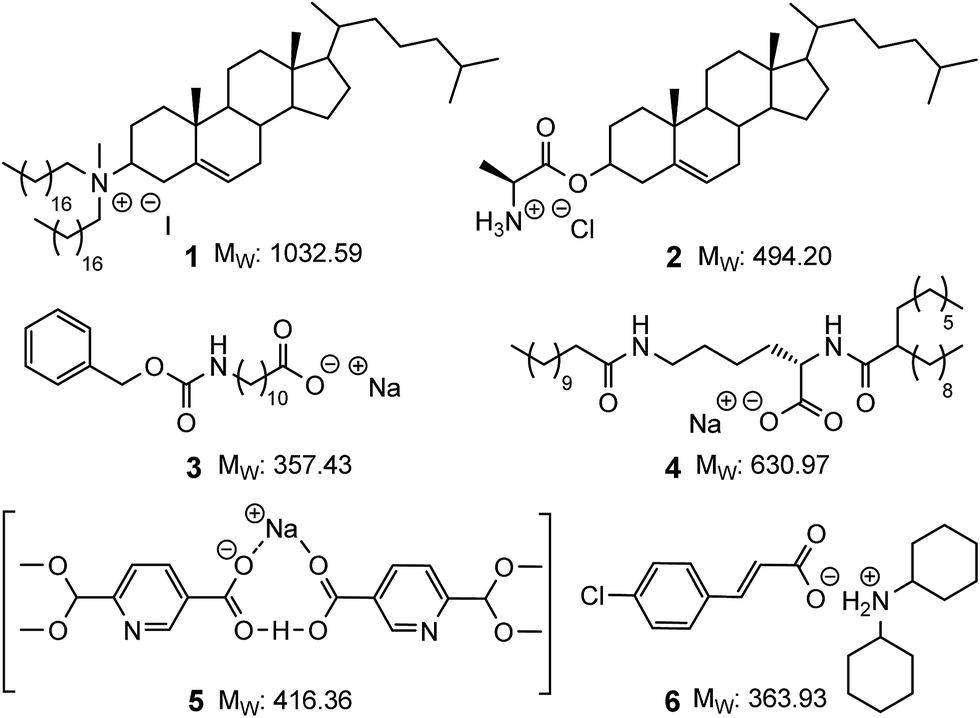 | ||
| Fig. 1 Examples of low molecular mass ionic organogelators. Compounds 1–4 with (and compounds 5 and 6 without) large hydrophobic groups. | ||
Herein, we present phenylalanine norbornene 9, a highly attractive building block for use in supramolecular self-assembly and as a LMIOG. We demonstrate that both the (R) and (S) enantiomers participate in a chirality controlled, helical assembly process that has been characterised using X-ray crystallographic analysis, and that as a LMIOG, these form gels with fast healable rheological properties. The corresponding xerogels consist of hollow tubular microcrystalline structures as identified using scanning electron microscopy (SEM).
Results and discussion
During the synthesis of (S)-phenylalanine-functionalised norbornene (S)-9 (Scheme 1) from readily available precursors, a triphasic system formed upon workup using aqueous Na2CO3. While there are a number of literature reports describing compound 9, no mention of this very interesting phenomenon has been noted.42–46 The triphasic system (Scheme 1) was characterised as EtOAc (top layer), the added Na2CO3 (bottom) and the sodium salt of 9 (9:Na, middle). | ||
| Scheme 1 Top: Synthesis of phenylalanine-functionalised norbornene (S)-9. Bottom: Triphasic system formed by mixing (S)-9 with sat. Na2CO3 and EtOAc. | ||
The corresponding aqueous biphasic system could be initiated by simply mixing a concentrated (≥1.8 M) aqueous solution of 9:Na with sat. Na2CO3 (see ESI Fig. S1.4†). Aqueous biphasic systems have demonstrated utility in delicate separations such as those involving aliphatic carboxylic acids.47–50 The biphasic systems also formed when sat. K2CO3, 5 M NaOH and 5 M KOH solutions were used. As these bases are known kosmotropic salts, the norbornene sodium salt 9:Na, was suspected of being strongly chaotropic and the two phase system was a result of high concentrations of a kosmotrope and chaotrope.50–52 To confirm this theory ammonium sulphate (a kosmotropic salt) was added to a concentrated aqueous solution of 9:Na and the rapid formation of a biphasic system validated these suspicions.
Further evidence of the chaotropic nature was obtained using 1H NMR diffusion experiments that showed an increase in the diffusion rate as the concentration of 9:Na was elevated (Fig. 2, see ESI section S2† for details). This is likely caused by a decrease in the viscosity of the D2O, which can in turn, be attributed to the known ability of chaotropic salts to “disrupt” the structure of water.53–55
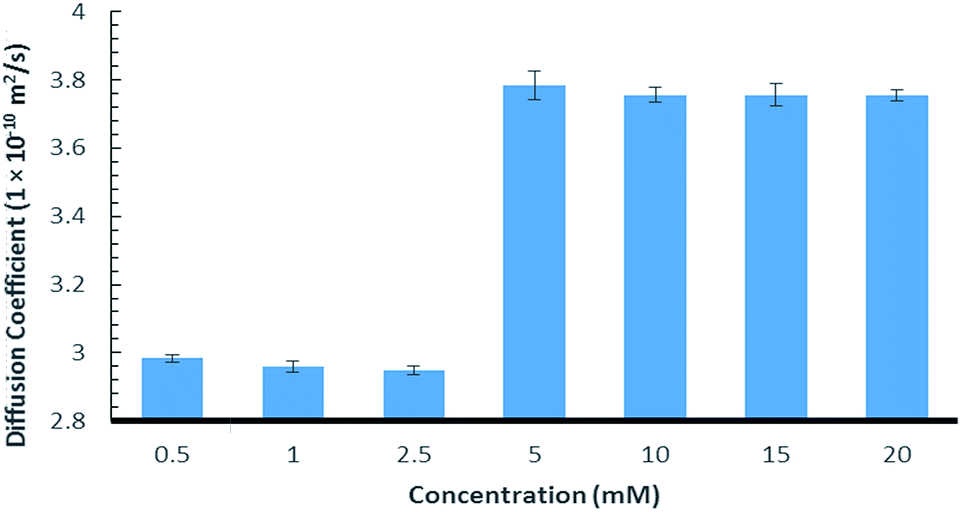 | ||
| Fig. 2 Diffusion coefficient of 9:Na in water in relation to concentration. | ||
Organogelation
It was also noted that 9:Na was exceptionally water soluble, up to 3 g mL−1 with heating. Upon cooling, this highly concentrated solution formed a hydrogel (rather than a precipitate) with gel formation likely caused by assembly of a supramolecular polymer.56,57 Other concentrated solutions (2–2.5 g mL−1), did not form gels; instead resulting in the formation of highly viscous solutions, as shown in Fig. 3, again indicating that a larger assembly was likely to be forming under these experimental conditions. | ||
| Fig. 3 Concentrated aqueous solutions of 9:Na; from left to right: 1, 2, 2.5 and 3 g mL−1. | ||
During the characterisation of the biphasic and triphasic systems, it was serendipitously discovered that addition of small amounts (10 μL) of aqueous 9:Na (≥3 M) to a variety of organic solvents (1.0 mL) resulted in the rapid formation of clear gels. As such, a range of salts of 9 were prepared, including Ca2+, K+, Na+, Li+ as well as the tetramethylammonium (TMA), tetraethylammonium (TEA) and tetrabutylammonium (TBA), and their gelating properties were investigated in various protic and aprotic solvents including EtOH, i-PrOH, n-BuOH, 1,4-dioxane, THF, CHCl3 and PhMe.
The calcium salt failed to elicit gel formation in any of the solvents trialled, demonstrating that this salt was not functioning as a LMIOG. However, when small amounts (4 mg) of the potassium salt 9:K were added to a range of these solvents (200 μL) and heated, on cooling the formation of soft-materials with strong gel-like properties (withstanding the classic inversion test) was observed in both i-PrOH and n-BuOH, while a weak gel was formed in PhMe (Table 1). These results clearly demonstrate the ability of 9:K to function as LMIOG in a broad range of solvents.
| Solvent | 9:K | 9:Na |
|---|---|---|
| a Gelator added as a powder (2 mg mL−1). b Minimum gelator concentration (MGC established using solid 9:K or 9:Na—see ESI section S3 for full details. c MGC established using 3.0 M solution of 9:Na—see ESI for full details. G = clear gel, WG = weak gel, PG = partial gel, S = suspension, D = dissolved, I = insoluble, A = aggregate, TG = turbid gel. | ||
| Methanol | D | D |
| Ethanol | D | Ga (2 wt%)b |
| Isopropanol | TG (2 wt%)b | Ga (0.5 wt%)b,c |
| n-Butanol | TG (2 wt%)b | Ga (0.5 wt%)b,c |
| Dioxane | A | Gb (0.5 wt%)c |
| Tetrahydrofuran | I | Gb (0.5 wt%)c |
| Diethyl ether | I | I |
| Acetone | A | PG |
| Ethyl acetate | A | PG |
| Dimethylformamide | D | D |
| Dimethyl sulfoxide | D | D |
| Acetonitrile | I | A |
| Chloroform | A | WG (1 wt%)b |
| Dichloromethane | A | S |
| Toluene | S | A |
| Pet. spirits (40–60 °C) | I | I |
| Heptane | I | I |
We next investigated the morphological features of the gels generated from the chaotropic salt 9:K. Imaging, using SEM, of the xerogel formed from the 9:Ki-PrOH gel identified an entangled web of fibrils with average diameter ≈ 200 nm (shown in Fig. 4a, see also ESI section S4†) and such morphology is typical for gelatinous materials. While the fibres identified from the gel formed in n-BuOH (Fig. 4b) appeared more linear and needle-like, the typical entangled morphology was maintained.
 | ||
| Fig. 4 SEM images of 9:K xerogels obtained from (a) i-PrOH gel (2 wt%), (b) n-BuOH gel (2 wt%). | ||
The sodium salt 9:Na was a potent organogelator (Table 1) and using the solid material (4 mg added to 200 μL of a solvent) robust soft-materials, that withstood inversion tests, were obtained from EtOH, i-PrOH, n-BuOH, THF and 1,4-dioxane, whereas weaker gels were observed to form in CHCl3 and CH2Cl2.
The gelation experiments were also carried out by using a 5 μL addition of a 3 M aqueous solution of 9:Na (50 μg in 50 μL of H2O, see ESI section S3† for full details) to the organic solvents (1 mL) followed by sonication of the resulting mixture. Formation of gels in i-PrOH, THF and 1,4-dioxane was again observed at low gelator concentration (0.5 wt%). The formation of gels in both THF and 1,4-dioxane occurred extremely rapidly, especially in the case of THF, which was observed to gelate immediately after addition of the 9:Na solution without the use of either heat or sonication to trigger the gelation process, again demonstrating that this salt could function as a potent LMIOG.
The SEM images of the xerogels formed from the EtOH and i-PrOH gels were largely devoid of the long fibrils observed previously, instead a fibrous matlike residue was seen (Fig. 5). The lack of fibrils was believed to be due to the extremely rapid gelation process that occurred before extended fibril formation could take place.
 | ||
| Fig. 5 SEM images of xerogel obtained from 9:Na EtOH gel (2 wt%). | ||
When the SEM imaging experiments were performed using a larger sample of the i-PrOH gel (20–30 μL) hollow hexagonal columnar structures (1–3 μm across) were observed in the resulting xerogel (Fig. 6). Recently, the group of Hardie identified a similar architecture from the crystallisation of a large bowl shaped cyclotriveratrylene (MW 723.74) with CuBr2,58 and previous work by Li and co-workers revealed that a structurally similar compound, diphenylalanine, was also capable of assembly into hollow hexagonal crystal.59 Hollow hexagonal crystals were also reported from the self-assembly of cyclic peptides in a liquid crystal by Dory et al.60 Very recently, Pasán et al. reported the use of Ostwald ripening to grow ∼300 μm sized hollow crystals, from a planar trinuclear Cu(II)–cyamelurate complex.61 The crystals identified by Pasán possessed hollow hexagonal prismatic morphology, very similar to that identified here, demonstrating the growth of larger sized (micron) hollow tubular structures from a bulk solution.61 Nevertheless, such nanometre-scale morphology is remarkably rare and to the best of our knowledge, the work presented herein, is the first instance where such topology has been observed in a xerogel formed from LMIOGs. The hollow hexagons were reproducibly formed from i-PrOH at a range of gelator concentrations (0.5–2 wt%). These organogels were found to be stable over several months, and no change was observed in their morphological features when SEM images were subsequently recorded.
 | ||
| Fig. 6 SEM images of 9:Na xerogel obtained from (a) i-PrOH gel (1 wt%) showing hollow hexagons and (b) n-BuOH gel (1 wt%) showing rods. | ||
These hollow tubes were not observed in any other solvent indicating a remarkable solvent effect on the nanoscale morphology. In n-BuOH (Fig. 6) similar intertwined rods (0.5–2 μm) formed; yet in comparison to the hollow hexagons systems, these appeared more solid, narrower, and somewhat flexible. Gels from 9:Na in hydrophilic solvents, especially i-PrOH and n-BuOH, were stable to the inversion test for periods of several weeks before fine needle-like crystals became evident (Fig. 7) eventually leading to gel collapse. Gels in volatile hydrophobic solvents, especially CH2Cl2 and CHCl3, were not as stable, lasting only a few days (unless carefully sealed to prevent solvent evaporation) before crystals formed and the gel collapsed.
 | ||
| Fig. 7 (a) A clear gel formed using 9:Na in i-PrOH (1 wt%); (b) crystals of 9:Na forming in a 1,4-dioxane gel (1 wt%) after 7 days. | ||
In contrast to these results, for the lithium salt 9:Li, no gels could be reliably obtained, although evidence of aggregation was observed in 1,4-dioxane solution at slightly higher concentration (2 wt%) and only if this hygroscopic salt was rigorously dried prior to the gelation tests, ruling out the use of this system as a LMIOG. When this aggregate was examined using SEM, unique hexagonal microcrystals (ca. 15 × 3 μm) were revealed (Fig. 8).
 | ||
| Fig. 8 SEM images of 9:Li xerogel from 1,4-dioxane (2 wt%). | ||
The structural variation in xerogel morphology clearly indicates the role of both the counterions and the solvent. Unfortunately, the tetraalkylammonium salts of 9 proved to be very hygroscopic, and consequently, none were capable of initiating a biphasic system nor the formation of a soft-material in any solvent, instead these tetraalkylammonium salts were readily soluble in most of the organic solvents tested (see ESI section S3† for details of evaluation).
Varying the gelator structure
In addition to examining the role of the counterion in gel formation, additional structural modifications using amino acids were pursued. The tryptophan (10), tyrosine (11) and alanine (12) analogues of 9 (Fig. 9) were all readily synthesised (see ESI section S1† for details). While the tryptophan functionalised norbornene 10 formed a triphasic system when washed with a saturated Na2CO3 solution, the isolated sodium salt 10:Na failed to elicit gel formation in any solvent trialled. Similarly, both 11:Na and 12:Na showed similar behaviour and were not able to function as LMIOG. | ||
| Fig. 9 Tryptophan, tyrosine and alanine functionalised norbornenes 10–12. | ||
To investigate the effects of chirality on the self-assembly and gelation process, (R)-phenylalanine (Fig. 10) was also synthesised and used to generate (R)-9:Na. As expected, this enantiomer and its behaviour was identical in all aspects to the (S) stereoisomer including the formation of the biphasic system, confirming that this enantiomer could also function as a LMIOG. Of interest, it was noted that a racemic mixture of (R)-9:Na and (S)-9:Na failed to form gel material in any of the solvent systems employed above, indicating that the supramolecular assembly process leading to solvent entanglement possibly also relied on the presence of a single enantiomer.8,62–66
 | ||
| Fig. 10 (R)-Phenylalanine functionalised norbornene (R)-9. | ||
Rheological evaluation
Having investigated the morphological features of the various xerogels, we set out to probe the mechanical properties of the gels formed by 9:K and 9:Na by carrying out dynamic rheological measurements, which included frequency and strain sweep and recovery tests. Prior to these investigations, differential scanning calorimetry (DSC) was performed on the gel samples and the results revealed that these gels had sharp gel–sol transition points 20–30 °C below the boiling point of the solvent (see ESI Table S5.1, section S5†).Frequency sweep experiments showed elastic response in the linear viscoelastic regime, wherein, the storage modulus (G′) was consistently higher than the loss modulus (G′′) across all samples, as shown for the 1% 9:Na gel formed in i-PrOH in Fig. 11 (see ESI† section S6† for all rheological results), confirming the solid like behaviour of the gels. Both the i-PrOH and n-BuOH gels of 9:K (2 wt%) possessed comparable rheological behaviour; a result that correlated well with the SEM observations where both gels had similar morphological features. Both the elasticity (G′ = 1.35 × 104 and 7.3 × 104 Pa for 2 wt% i-PrOH and n-BuOH gels respectively) and the stiffness (G′/G′′ = 6.2 and 7.3 for 6.2 for i-PrOH and n-BuOH) were similar, though the n-BuOH gel was found to be slightly stronger, as summarised in Table 2. The yield stress for both gels were also similar (σ* ∼ 10%). As 9:Na formed gels in a larger assortment of organic solvents, a broader range of rheological behaviour was observed (Table 2). Typically the 9:Na gels were stronger than their 9:K counterparts, however, gel strength did not show any particular relationship with the solvent used.
 | ||
| Fig. 11 Characterisation of the 1 wt% gel formed by 9:Na in i-PrOH (a) DSC (b) rheological strain sweep and (c) rheological recovery test. | ||
| Gelator (wt%)/solvent | G′ (Pa) | Stiffness (G′/G′′) | σ* (Pa) |
|---|---|---|---|
| a G′, G′′ and σ* values were obtained from strain sweep experiments performed at fixed frequency of 1 Hz. | |||
| 9:K (2 wt%)/i-PrOH | 1.4 × 104 | 6.2 | 9.4% |
| 9:K (2 wt%)/n-BuOH | 7.3 × 104 | 7.3 | 9.8% |
| 9:Na (2 wt%)/EtOH | 6.9 × 103 | 7.7 | 5.6% |
| 9:Na (1 wt%)/i-PrOH | 3.8 × 104 | 7.6 | 7.5% |
| 9:Na (2 wt%)/i-PrOH | 1.1 × 105 | 10.1 | 15% |
| 9:Na (2 wt%)/n-BuOH | 2 × 104 | 3.8 | 8.6% |
| 9:Na (2 wt%)/1,4-dioxane | 1.2 × 105 | 4.8 | 4.4% |
| 9:Na (2 wt%)/CHCl3 | 1.6 × 105 | 4.6 | 4.5% |
| 9:Na (2 wt%)/THF | 1.7 × 105 | 3.2 | 0.4% |
As mentioned above, the stiffness (G′/G′′) and yield strain (σ*) values correlated well to the morphological features of the xerogels that were observed using SEM (Fig. 12), and such a relationship suggests that the fibrils may be organised in similar arrangements in the corresponding gels. The lower G′ value for EtOH gel is likely to be due to the formation of smaller fibrils that lack the extended entanglement. In contrast, for the stronger 9:Nai-PrOH gel, the larger micrometre sized (1–3 μm diameter) hollow columnar structures in its gel matrix (vide supra) are likely contributors to the greater overall gel strength.
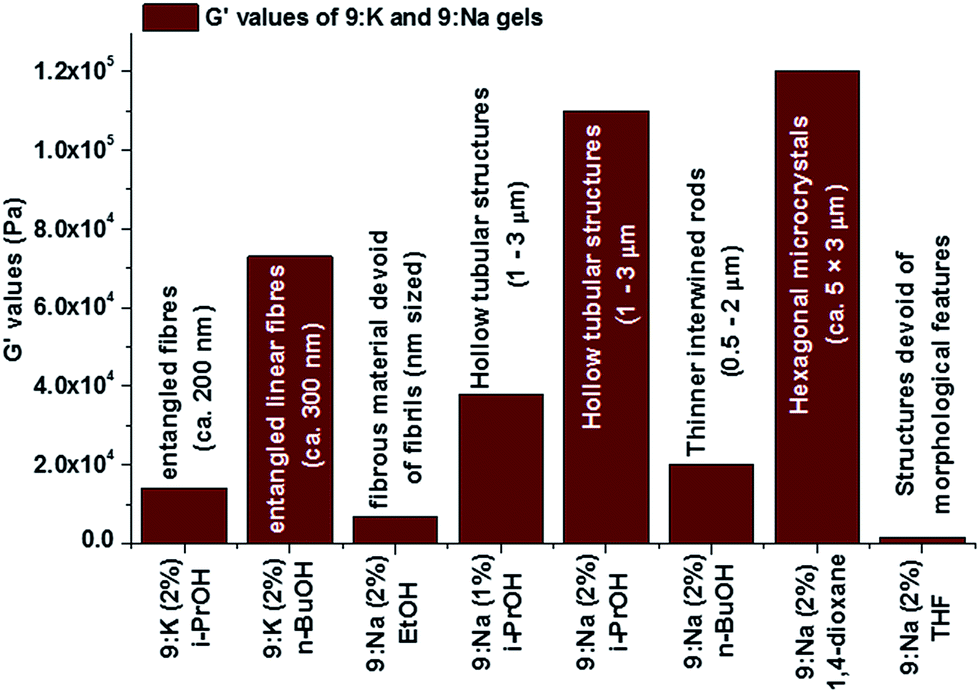 | ||
| Fig. 12 The variation of G′ values of 9:K and 9:Na gels and comparison to the morphology of the corresponding gels. | ||
Both 9:K and 9:Na gels showed remarkable recovery properties; for the 9:Na gel in i-PrOH (1 wt%) (Fig. 11c) applying a strain amplitude of 100% resulted in liquid like behaviour (G′′ > G′). However, the original gel state (G′ > G′′) instantly recovered when the strain was reduced to 0.1%. Recovery was observed over multiple cycles of measurements and all 9:K and 9:Na gels showed similar recovery behaviour within the 30–40 second interval between alternating strain regimes (see ESI section S6† for full details). These results demonstrate the thixotropic properties of the gels generated from these LMIOGs and suggest their potential as self-healing materials.
Supramolecular assembly and crystal structure
In addition to carrying out the investigation into the ability of these LMIOGs to form soft-materials, we were also able to form crystalline material from these gels. Long needle-like crystals of (S)-9:Na that were suitable for X-ray analysis were obtained upon crystallisation from its i-PrOH gel (Fig. 7) and revealed that individual units of the norbornene adopt an amphiphilic structure in the solid state, with the norbornene framework and the phenylalanine aromatic positioned in close proximity (potentially through C(sp2)–H⋯π interactions)67–69 and the carboxylate residue oriented away from the hydrophobic portions (Fig. 13). This conformational arrangement was also evident in solution as the 1H NMR spectrum of 9 (see ESI Fig. S1.1†) revealed one of the norbornene C(sp2)–H resonances significantly upfield (δ = 5.33 ppm) from the other (δ = 5.62 ppm). Such a shift is indicative of anisotropic shielding due to the proximity of the aromatic ring current from the closely positioned phenylalanine residue.70,71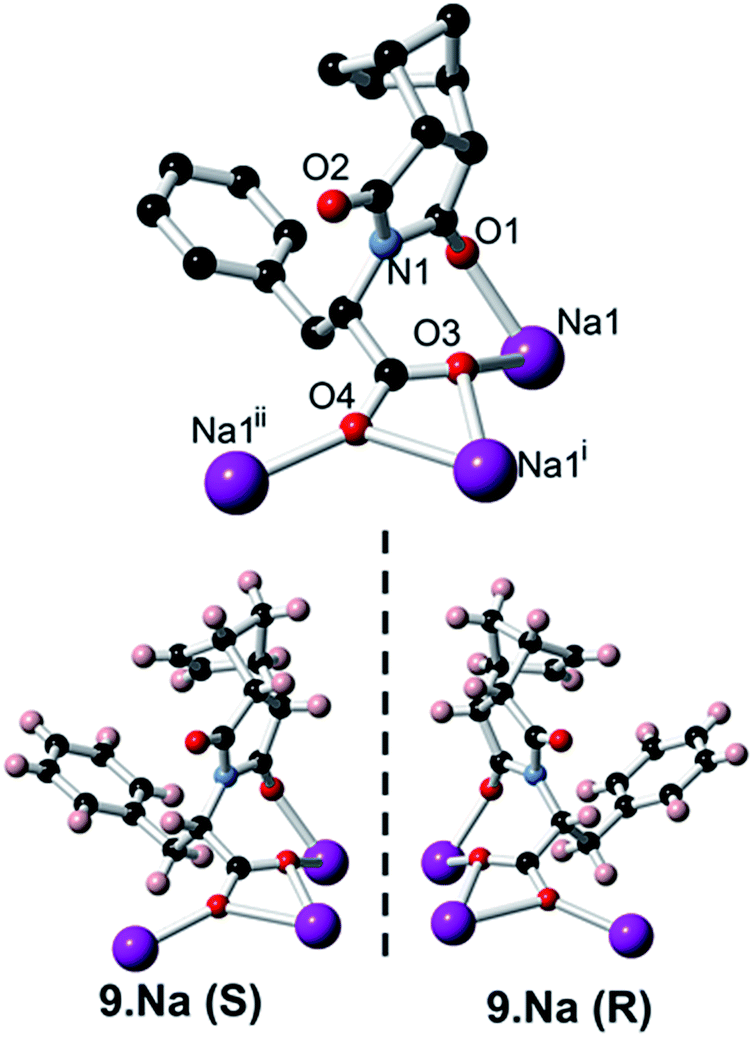 | ||
| Fig. 13 Crystal structure of both (S)-9:Na and (R)-9:Na. | ||
Using identical conditions, the structure of the (R)-9:Na enantiomer was also characterised and, as predicted, revealed the structural mirror image (Fig. 13). The racemate showed no evidence of gelation or crystal formation.
Further investigation into the long range interactions in the crystal structures revealed that the individual monomers assembled into hollow, helical columns (shown in Fig. 14a) with the helical assembly being mediated by the Na+ ions that adopt an irregular six-coordinate geometry. Four of these coordination sites are occupied by carboxylate groups with the final two sites being occupied by a water ligand and the imide oxygen syn to the chiral centre. It is this imide coordination that controls the direction of the helix, resulting in a right-handed conformation for (S)-9:Na and left-handed for (R)-9:Na (Fig. 14b).72 The helix has a pitch of 19.8 Å and a diameter of 19.6 Å, with 6 monomer residues per complete turn. Each additional monomer represents a 60° turn in the helix and a 3.2 Å translation along the helical axis.
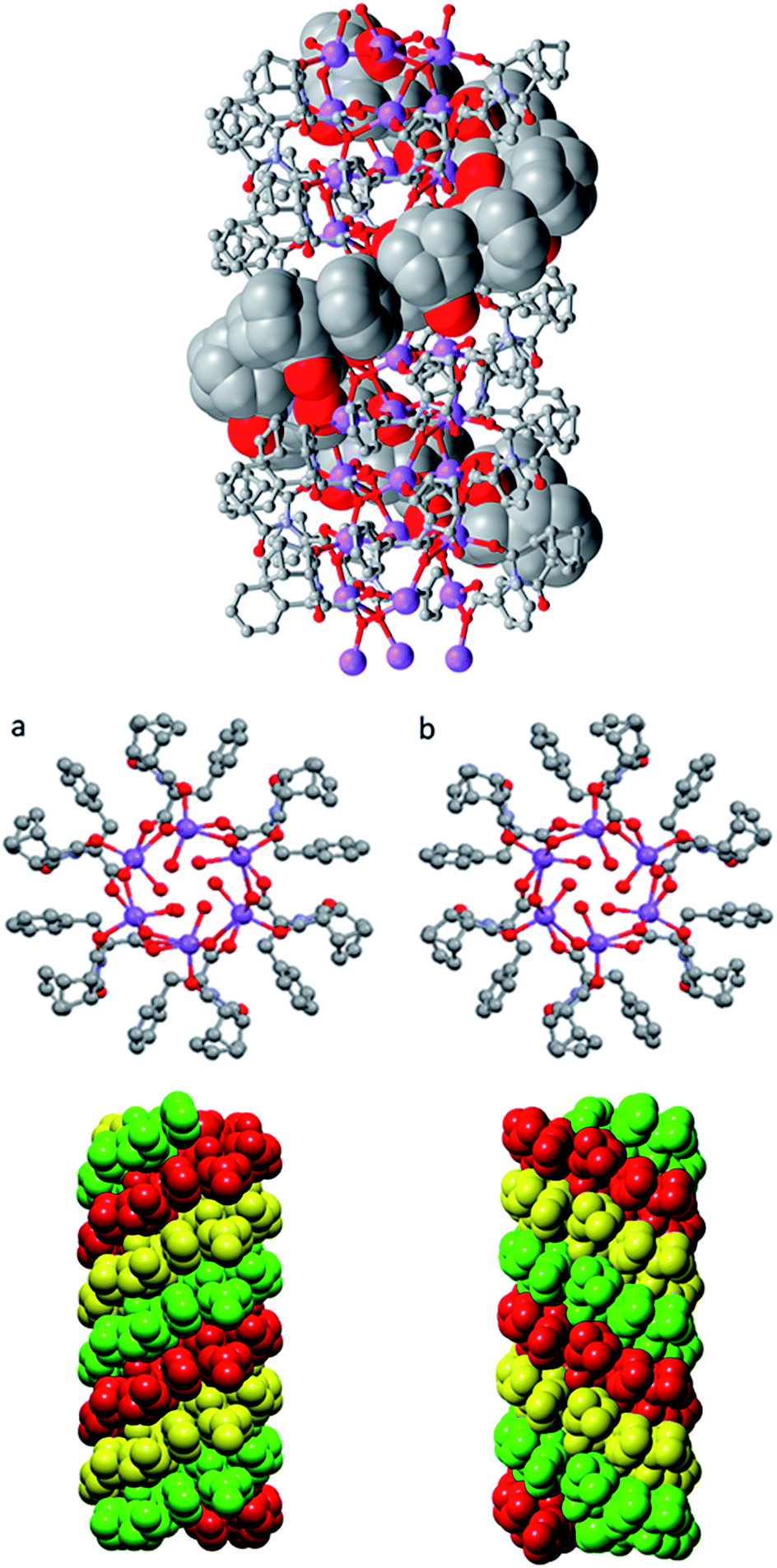 | ||
| Fig. 14 Helical columns formed by the self-assembly of (a) (S)-9:Na and (b) (R)-9:Na in i-PrOH observed in the crystal structure. | ||
Adjacent stacks in the structure of (S)-9:Na associate in a hexagonal rod packing fashion with hydrophobic interactions between the outwards facing organic periphery (Fig. 15). No significant voids were observed in the interstitial spaces, indicating efficient packing of the homochiral helices. This observation offers some justification as to the need for enantiomeric purity as given the significant undulation in the external surface of the columns and the numerous intermolecular contacts (clearly visible using Hirshfeld surface mappings, see ESI, Fig. S7.3†), co-crystallisation of helices with alternate handedness would be expected to lead to a significant decrease in packing efficiency.
 | ||
| Fig. 15 The full crystal network of (S)-9:Na showing two views of the repeating columnar helical assemblies and their interactions. | ||
As a model for assembly, the largely hydrophobic structure of 9 protects the hydrophilic carboxylate by assembling such that the carboxylate is shielded from the organic solvent. As the amphiphile is chiral, a helical assembly occurs in which all carboxylates are oriented to the interior of the helix ensuring that they are not solvent exposed.
A crystal structure of the non-gelating salt (S)-9:TEA was also obtained and revealed the formation of thin ribbons (Fig. 16). In this example the counterions did not take part in the formation of the ribbons, instead only being loosely associated with the carboxylate moieties, demonstrating the importance of the small sodium cation in the helical assembly process for 9:Na. Of additional interest, the X-ray structure of (S)-9:TEA revealed alternating cyclic tetramers of water and loops involving both water molecules and the carboxylate receptor as demonstrated in Fig. 16.73–77
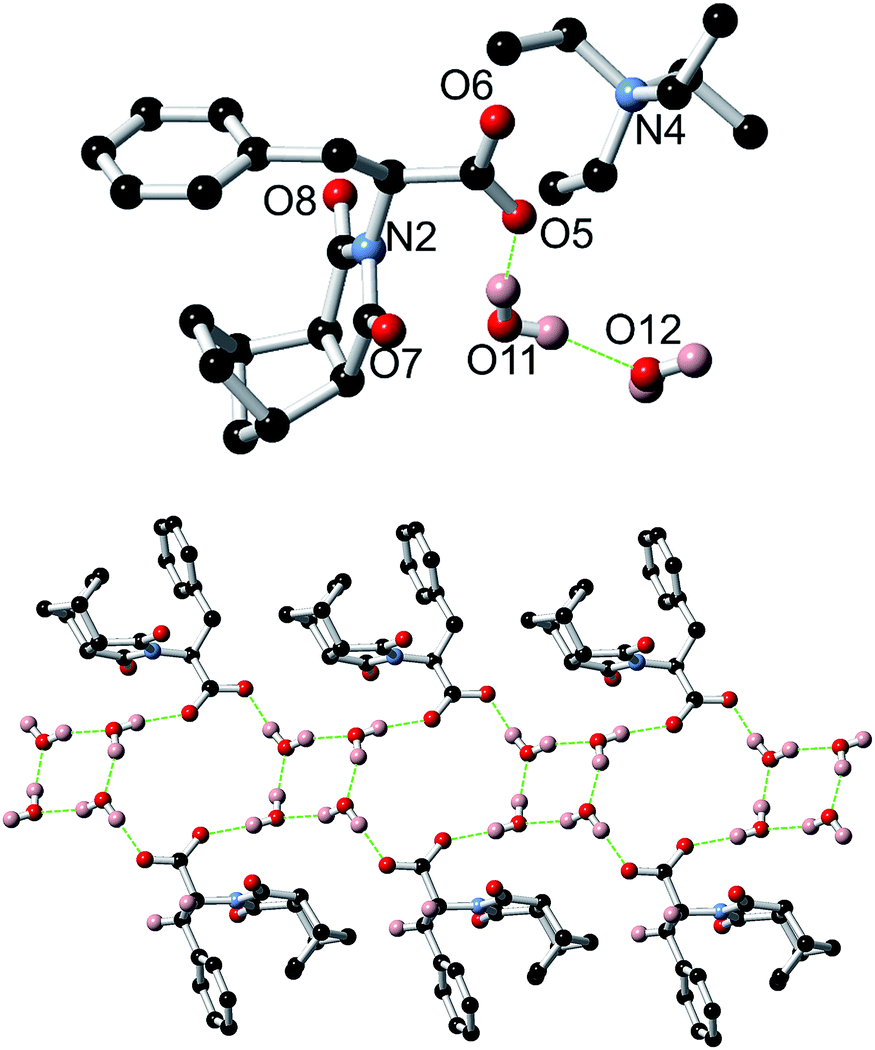 | ||
| Fig. 16 Top: Crystal structure of 9:TEA showing the loose association of the TEA counterion. Non-bonding hydrogens have been omitted for clarity. Bottom: Crystal structure of 9:TEA highlighting both the ribbon structure and the role of water in the assembly. The TEA counterions and non-bonding hydrogens have been omitted for clarity. | ||
Conclusions
The results presented here show that compound 9:Na is a highly capable small molecule . This ionic compound can readily form biphasic and triphasic systems and, in the solid state, assembles to form chiral helical columns. In addition, 9:Na acts as an efficient low molecular weight ionic organogelator (LMIOG) in a variety of organic solvents at concentrations as low as 0.5 wt%. The rheological measurements of the gels demonstrate rapid recovery and that these organogels have potential as self-healing materials. Such interesting behaviour reinforces the importance of supramolecular assembly as a means to control macroscopic behaviour and also the requirement for careful characterisation to aid in the understanding of fundamental molecular-level interactions. We are in the process of developing other LMIOGs and investigating their properties and applications.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors acknowledge Dr Surya Subianto for insightful discussions. FP and JE thank the Deakin Centre for Chemistry and Biotechnology and also the Australian Research Council (DP140100227) for financial support. This research was supported in part by the Australian Research Council World Class Future Fibre Industry Transformation Research Hub (IH140100018). TG thanks Science Foundation Ireland (SFI) for financial support (SFI PI Award 13/IA/1865). Donna Edwards is acknowledged for the photos of the triphasic system and gels (Scheme 1, Fig. 3 and 7).References
- B. K. An, D. S. Lee, J. S. Lee, Y. S. Park, H. S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232–10233 CrossRef PubMed.
- P. C. Xue, Q. X. Xu, P. Gong, C. Qian, A. M. Ren, Y. Zhang and R. Lu, Chem. Commun., 2013, 49, 5838–5840 RSC.
- S. Bhattacharya, S. Sarkar and R. Shunmugam, J. Mater. Chem. A, 2013, 1, 8398–8405 Search PubMed.
- M. E. Morales, V. Gallardo, B. Clares, M. B. Garcia and M. A. Ruiz, J. Cosmet. Sci., 2009, 60, 627–636 Search PubMed.
- P. L. Luisi, R. Scartazzini, G. Haering and P. Schurtenberger, Colloid Polym. Sci., 1990, 268, 356–374 Search PubMed.
- S. Debnath, A. Shome, S. Dutta and P. K. Das, Chem.–Eur. J., 2008, 14, 6870–6881 CrossRef PubMed.
- E. J. Cho, I. Y. Jeong, S. J. Lee, W. S. Han, J. K. Kang and J. H. Jung, Tetrahedron Lett., 2008, 49, 1076–1079 CrossRef.
- A. Vintiloiu and J.-C. Leroux, J. Controlled Release, 2008, 125, 179–192 CrossRef PubMed.
- K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644–656 CrossRef PubMed.
- J. A. Foster, K. K. Damodaran, A. Maurin, G. M. Day, H. P. Thompson, G. J. Cameron, J. C. Bernal and J. W. Steed, Chem. Sci., 2017, 8, 78–84 RSC.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef PubMed.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef PubMed.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42 CrossRef PubMed.
- A. J. Savyasachi, O. Kotova, S. Shanmugaraju, S. J. Bradberry, G. M. Ó'Máille and T. Gunnlaugsson, Chem, 2017, 3, 764–811 Search PubMed.
- D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237–1247 CrossRef.
- R. Daly, O. Kotova, M. Boese, T. Gunnlaugsson and J. J. Boland, ACS Nano, 2013, 7, 4838–4845 CrossRef PubMed.
- M. Martínez-Calvo, O. Kotova, M. E. Möbius, A. P. Bell, T. McCabe, J. J. Boland and T. Gunnlaugsson, J. Am. Chem. Soc., 2015, 137, 1983–1992 CrossRef PubMed.
- J. I. Lovitt, C. S. Hawes, A. D. Lynes, B. Haffner, M. E. Möbius and T. Gunnlaugsson, Inorg. Chem. Front., 2017, 4, 296–308 RSC.
- S. J. Bradberry, A. J. Savyasachi, R. D. Peacock and T. Gunnlaugsson, Faraday Discuss., 2015, 185, 413–431 RSC.
- C. S. Hawes, A. D. Lynes, K. Byrne, W. Schmitt, G. Ryan, M. E. Möbius and T. Gunnlaugsson, Chem. Commun., 2017, 53, 5989–5992 RSC.
- C. S. Hawes, Y. Nolvachai, C. Kulsing, G. P. Knowles, A. L. Chaffee, P. J. Marriott, S. R. Batten and D. R. Turner, Chem. Commun., 2014, 50, 3735–3737 RSC.
- E. P. McCarney, J. P. Byrne, B. Twamley, M. Martínez-Calvo, G. Ryan, M. E. Möbius and T. Gunnlaugsson, Chem. Commun., 2015, 51, 14123–14126 RSC.
- O. Kotova, R. Daly, C. l. M. dos Santos, P. E. Kruger, J. J. Boland and T. Gunnlaugsson, Inorg. Chem., 2015, 54, 7735–7741 CrossRef PubMed.
- M. D. Johnstone, E. K. Schwarze, G. H. Clever and F. M. Pfeffer, Chem.–Eur. J., 2015, 21, 3948–3955 CrossRef PubMed.
- M. D. Johnstone, M. Frank, G. H. Clever and F. M. Pfeffer, Eur. J. Org. Chem., 2013, 5848–5853 CrossRef.
- R. N. Robson and F. M. Pfeffer, Chem. Commun., 2016, 52, 8719–8721 RSC.
- A. J. Lowe, B. M. Long and F. M. Pfeffer, Chem. Commun., 2013, 49, 3376–3388 RSC.
- F. M. Pfeffer, T. Gunnlaugsson, P. Jensen and P. E. Kruger, Org. Lett., 2005, 7, 5357–5360 CrossRef PubMed.
- F. M. Pfeffer, P. E. Kruger and T. Gunnlaugsson, Org. Biomol. Chem., 2007, 5, 1894–1902 Search PubMed.
- S. M. Hickey, T. D. Ashton, S. K. Khosa, R. N. Robson, J. M. White, J. Li, R. L. Nation, Y. Y. Heidi, A. G. Elliott and M. S. Butler, Org. Biomol. Chem., 2015, 13, 6225–6241 Search PubMed.
- S. M. Hickey, T. D. Ashton, J. M. White, J. Li, R. L. Nation, Y. Y. Heidi, A. G. Elliott, M. S. Butler, J. X. Huang and M. A. Cooper, RSC Adv., 2015, 5, 28582–28596 RSC.
- X. Huang and R. G. Weiss, Tetrahedron, 2007, 63, 7375–7385 CrossRef.
- M. George, G. P. Funkhouser and R. G. Weiss, Langmuir, 2008, 24, 3537–3544 CrossRef PubMed.
- G. Mieden-Gundert, L. Klein, M. Fischer, F. Vögtle, K. Heuzé, J. L. Pozzo, M. Vallier and F. Fages, Angew. Chem., Int. Ed., 2001, 40, 3164–3166 CrossRef PubMed.
- M. Suzuki, M. Yumoto, M. Kimura, H. Shirai and K. Hanabusa, Helv. Chim. Acta, 2004, 87, 1–10 CrossRef.
- Y. Li, K. Liu, J. Liu, J. Peng, X. Feng and Y. Fang, Langmuir, 2006, 22, 7016–7020 CrossRef PubMed.
- D. J. Abdallah and R. G. Weiss, J. Braz. Chem. Soc., 2000, 11, 209–218 CrossRef.
- D. Bardelang, F. Camerel, A. C. Hotze, B. Kariuki, B. Paik, M. Schmutz, R. Ziessel and M. J. Hannon, Chemistry, 2007, 13, 9277–9285 CrossRef PubMed.
- D. R. Trivedi, A. Ballabh and P. Dastidar, Chem. Mater., 2003, 15, 3971–3973 CrossRef.
- S. K. Baloch, L. Ma, X.-L. Wang, J. Shi, Y. Zhu, F.-Y. Wu, Y.-J. Pang, G.-H. Lu, J.-L. Qi and X.-M. Wang, RSC Adv., 2015, 5, 31759–31767 RSC.
- A. Bodtke and H.-H. Otto, Pharmazie, 2005, 60, 803–813 Search PubMed.
- H. Zhou, E.-M. Schön, M. Wang, M. J. Glassman, J. Liu, M. Zhong, D. Díaz Díaz, B. D. Olsen and J. A. Johnson, J. Am. Chem. Soc., 2014, 136, 9464–9470 CrossRef PubMed.
- C. P. Hackenberger, I. Schiffers, J. Runsink and C. Bolm, J. Org. Chem., 2004, 69, 739–743 CrossRef PubMed.
- M. R. Buchmeiser, F. Sinner, M. Mupa and K. Wurst, Macromolecules, 2000, 33, 32–39 CrossRef.
- J. Rydberg, Solvent extraction principles and practice, revised and expanded, CRC Press, 2004 Search PubMed.
- M. Iqbal, Y. Tao, S. Xie, Y. Zhu, D. Chen, X. Wang, L. Huang, D. Peng, A. Sattar and M. A. B. Shabbir, Biol. Proced. Online, 2016, 18, 18 CrossRef PubMed.
- Y. Pei, J. Wang, K. Wu, X. Xuan and X. Lu, Sep. Purif. Technol., 2009, 64, 288–295 CrossRef.
- M. T. Zafarani-Moattar and S. Hamzehzadeh, J. Chem. Eng. Data, 2010, 55, 1598–1610 CrossRef.
- N. J. Bridges, K. E. Gutowski and R. D. Rogers, Green Chem., 2007, 9, 177–183 RSC.
- N. J. Bridges and R. D. Rogers, Sep. Sci. Technol., 2008, 43, 1083–1090 CrossRef.
- B. Hribar, N. T. Southall, V. Vlachy and K. A. Dill, J. Am. Chem. Soc., 2002, 124, 12302–12311 CrossRef PubMed.
- A. S. Parmar and M. Muschol, Biophys. J., 2009, 97, 590–598 CrossRef PubMed.
- R. Zangi, J. Phys. Chem. B, 2009, 114, 643–650 CrossRef PubMed.
- L. N. Neumann, M. B. Baker, C. M. Leenders, I. K. Voets, R. P. Lafleur, A. R. Palmans and E. W. Meijer, Org. Biomol. Chem., 2015, 13, 7711–7719 Search PubMed.
- B. Isare, S. Pensec, M. Raynal and L. Bouteiller, C. R. Chim., 2016, 19, 148–156 CrossRef.
- F. L. Thorp-Greenwood, A. N. Kulak and M. J. Hardie, Nat. Chem., 2015, 7, 526–531 CrossRef PubMed.
- X. Yan, J. Li and H. Möhwald, Adv. Mater., 2011, 23, 2796–2801 CrossRef PubMed.
- S. Leclair, P. Baillargeon, R. Skouta, D. Gauthier, Y. Zhao and Y. L. Dory, Angew. Chem., Int. Ed., 2004, 43, 349–353 CrossRef PubMed.
- C. Martínez-Benito, A. Bauzá, A. B. Lago, C. Ruiz-Pérez, C. A. Jiménez, M. E. Torres, A. Frontera and J. Pasán, Cryst. Growth Des., 2018, 18, 2636–2644 Search PubMed.
- A. R. Hirst, D. K. Smith, M. C. Feiters and H. P. Geurts, Chem.–Eur. J., 2004, 10, 5901–5910 CrossRef PubMed.
- K. Hanabusa, Y. Maesaka, M. Kimura and H. Shirai, Tetrahedron Lett., 1999, 40, 2385–2388 CrossRef.
- K. Hanabusa, K. Okui, K. Karaki, M. Kimura and H. Shirai, J. Colloid Interface Sci., 1997, 195, 86–93 CrossRef PubMed.
- J. H. Fuhrhop, P. Schnieder, J. Rosenberg and E. Boekema, J. Am. Chem. Soc., 1987, 109, 3387–3390 CrossRef.
- J. H. Fuhrhop and W. Helfrich, Chem. Rev., 1993, 93, 1565–1582 CrossRef.
- Z. Zhang, Y. Luo, J. Chen, S. Dong, Y. Yu, Z. Ma and F. Huang, Angew. Chem., 2011, 123, 1433–1437 CrossRef.
- J. F. Malone, C. M. Murray, M. H. Charlton, R. Docherty and A. J. Lavery, J. Chem. Soc., Faraday Trans., 1997, 93, 3429–3436 RSC.
- M. Nishio, Y. Umezawa, M. Hirota and Y. Takeuchi, Tetrahedron, 1995, 51, 8665–8701 CrossRef.
- R. M. Silverstein and G. C. Bassler, J. Chem. Educ., 1962, 39, 546 CrossRef.
- C. S. Wannere and P. v. R. Schleyer, Org. Lett., 2003, 5, 605–608 CrossRef PubMed.
- Z. Shen, T. Wang and M. Liu, Angew. Chem., Int. Ed., 2014, 53, 13424–13428 CrossRef PubMed.
- R. Ludwig, Angew. Chem., Int. Ed., 2001, 40, 1808–1827 CrossRef PubMed.
- M. Mascal, L. Infantes and J. Chisholm, Angew. Chem., Int. Ed., 2006, 45, 32–36 CrossRef PubMed.
- O. Fabelo, J. Pasán, L. Cañadillas-Delgado, F. S. Delgado, A. Labrador, F. Lloret, M. Julve and C. Ruiz-Pérez, CrystEngComm, 2008, 10, 1743–1746 RSC.
- L.-S. Long, Y.-R. Wu, R.-B. Huang and L.-S. Zheng, Inorg. Chem., 2004, 43, 3798–3800 CrossRef PubMed.
- S. Supriya, S. Manikumari, P. Raghavaiah and S. K. Das, New J. Chem., 2003, 27, 218–220 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, NMR diffusion, gelation, SEM images, DSC thermograms, rheological data and crystallographic data. CCDC 1826288–1826290. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01798k |
| This journal is © The Royal Society of Chemistry 2018 |