
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantioselective synthesis of 2-oxazolidinones by ruthenium(II)–NHC-catalysed asymmetric hydrogenation of 2-oxazolones†
Wei
Li
 ,
Marco
Wollenburg
and
Frank
Glorius
*
,
Marco
Wollenburg
and
Frank
Glorius
*
Organisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 40, 48149 Münster, Germany. E-mail: glorius@uni-muenster.de
First published on 28th June 2018
Abstract
An efficient synthesis of optically active 4-substituted 2-oxazolidinones by the ruthenium(II)–NHC-catalysed asymmetric hydrogenation of 2-oxazolones was developed. Excellent enantioselectivities (up to 96% ee) and yields (up to 99%) were obtained for a variety of substrates, bearing a range of functional groups and useful motifs. The hydrogenation reaction was successfully scaled up to gram scale using low catalyst loading. Moreover, the utility of this methodology was demonstrated by the transformation of the enantioenriched product into the corresponding chiral β-amino alcohol, a bisoxazoline ligand, and the formal synthesis of (−)-aurantioclavine.
Introduction
Chiral 2-oxazolidinones, widely used as Evans' chiral auxiliaries (Scheme 1a, left), play a prominent role in modern organic synthesis.1 Based on chiral 2-oxazolidinone auxiliaries, a wide range of asymmetric transformations has been developed to construct new chiral building blocks, which are frequently used in both natural product synthesis and drug discovery.2 Furthermore, the chiral 2-oxazolidinone motif itself is common in pharmaceutically relevant molecules (Scheme 1a, right).3 Thus, the synthesis of chiral 2-oxazolidinones has already attracted considerable attention. Conventionally, enantioenriched 2-oxazolidinones are synthesized by cyclization of the corresponding optically pure β-amino alcohols with C1-building blocks like phosgene and its derivatives (Scheme 1b). These methods often require toxic reagents and the pre-construction of the key stereocenter of the β-amino alcohols arises synthetic problems if they cannot be formed from natural enantiopure amino acids or related precursors.2 Considering this, the exploration of orthogonal, efficient and divergent catalytic strategies for the construction of diverse 2-oxazolidinone derivatives is highly important for organic synthesis and drug discovery.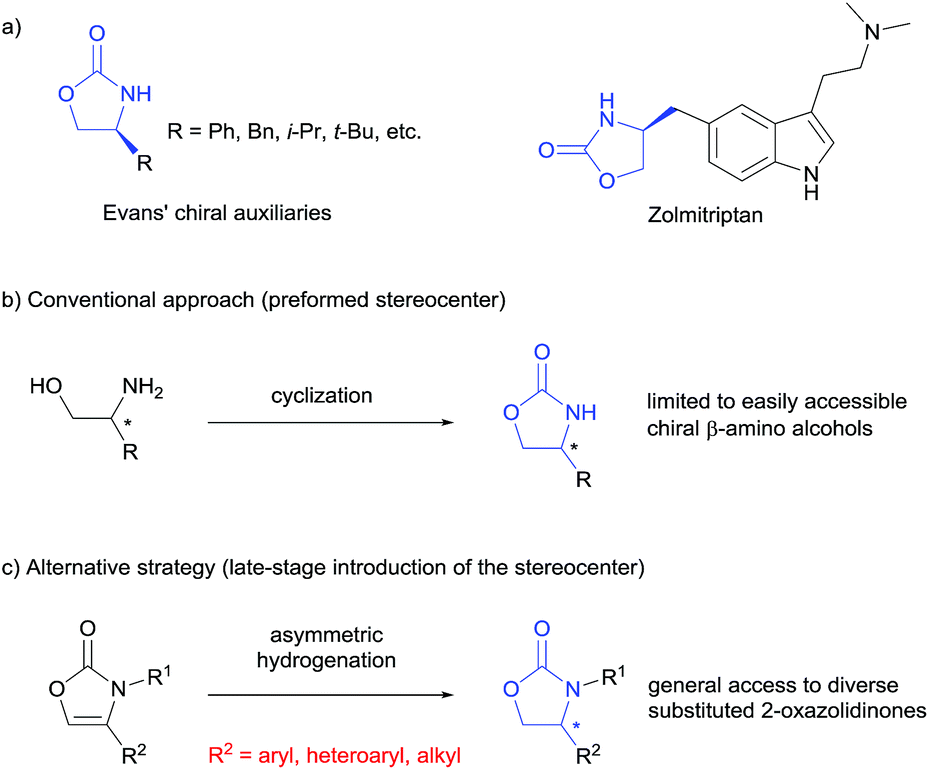 | ||
| Scheme 1 Applications and synthesis of chiral 2-oxazolidinones. | ||
In the last decade, the asymmetric hydrogenation of unsaturated heterocycles has emerged as a conceptually powerful method to produce optically active cyclic compounds and has received significant attention.4 In this regard, a synthetic method utilizing the enantioselective hydrogenation of 2-oxazolones for the late-stage construction of the key stereocenter can be envisioned as a powerful alternative to prepare diverse optically active 2-oxazolidinones (Scheme 1c). Moreover, this strategy would also provide a general way to produce optically active β-amino alcohols since the transformation from 2-oxazolidinones to free β-amino alcohols is very convenient.2 Recently, Zhang and co-workers reported the rhodium-catalysed asymmetric hydrogenation of 2-oxazolones, which afforded 4-aryl substituted 2-oxazolidinones with moderate enantioselectivities.5 To the best of our knowledge, this is the only precedent of an enantioselective synthesis of chiral 2-oxazolidinones by asymmetric hydrogenation of unsaturated heterocycles. As a continuous effort in the field of arene and heterocycle hydrogenation,6 we herein describe a highly enantioselective and practical hydrogenation of a broad scope of 2-oxazolones to access diverse enantioenriched 2-oxazolidinones catalysed by a ruthenium(II)–N-heterocyclic carbene (NHC) complex.
Results and discussion
Initially, the hydrogenation of 4-phenyloxazol-2(3H)-one (1a) was attempted under 50 bar H2 in n-hexane at room temperature in the presence of our previously-developed ruthenium(II)–NHC catalyst, which is prepared in situ from [Ru(2-methylallyl)2(COD)], the NHC precursor (R,R)-SINpEt·HBF4, and NaOt-Bu.7,8 However, the desired product was not observed, presumably due to catalyst deactivation by coordination of the metal with free N–H (Table 1, entry 1). To circumvent catalyst deactivation, a variety of protecting groups were investigated, of which the 4-methoxybenzyl (PMB) protected carbamate 1d was found to be suitable for hydrogenation, providing the desired 4-phenyloxazolidin-2-one with 85% ee and in 95% isolated yield (entry 2–4). To further improve the reaction conditions, a solvent screen was conducted (entries 4–8). No reaction occurred in dichloromethane presumably due to catalyst decomposition (entry 5), and cyclohexane (entry 8) was found to be slightly superior to other solvents (n-hexane, toluene and THF) for enantioselectivity. Decreasing the reaction temperature to 0 °C further improved enantiocontrol, providing the desired product with 95% ee and in 93% yield (entry 9). Finally, a solvent mixture of cyclohexane/THF = 20/1 was used to improve the solubility of the substrate, to afford the chiral 2-oxazolidinone 2d in 99% yield and 95% ee (entry 10).|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | Solvent | T (°C) | Yieldb (%) | ee (%) |
| a General conditions: [Ru(2-methylallyl)2(COD)] (0.10 mmol), (R,R)-SINpEt·HBF4 (0.20 mmol) and NaOt-Bu (0.24 mmol) were stirred at 70 °C in n-hexane (5.0 mL) for 16 h to perform the catalyst (0.02 M), after which 0.1 mL of the catalyst suspension were added to substrates 1a–d (0.10 mmol) in the indicated solvent (1.0 mL), and the hydrogenation was performed at 50 bar H2 for 24 h. b Yields of isolated product after column chromatography are reported. c Determined by HPLC analysis using a chiral stationary phase. d Using a solvent mixture of cyclohexane/THF = 20/1. Boc = tert-butyloxycarbonyl, Ac = acetyl, PMB = 4-methoxybenzyl. N. D. = not determined. | |||||
| 1 | H (1a) | n-Hexane | 25 | 0 | — |
| 2 | Boc (1b) | n-Hexane | 25 | Traces | N. D. |
| 3 | Ac (1c) | n-Hexane | 25 | 0 | — |
| 4 | PMB (1d) | n-Hexane | 25 | 95 | 85 |
| 5 | PMB (1d) | CH2Cl2 | 25 | 0 | — |
| 6 | PMB (1d) | Toluene | 25 | 99 | 85 |
| 7 | PMB (1d) | THF | 25 | 99 | 87 |
| 8 | PMB (1d) | Cyclohexane | 25 | 98 | 88 |
| 9 | PMB (1d) | Cyclohexane | 0 | 93 | 95 |
| 10d | PMB (1d) | Cyclohexane | 0 | 99 | 95 |
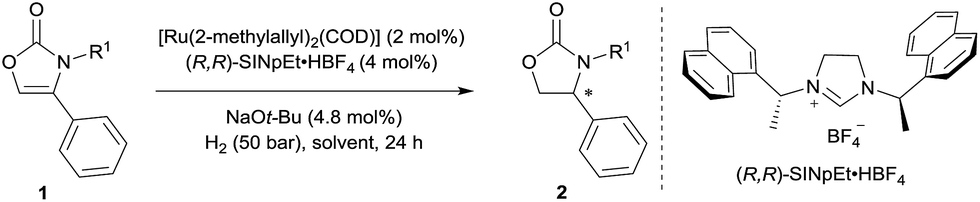
With the optimised reaction conditions in hand (Table 1, entry 10), the substrate scope of the reaction was explored (Schemes 2 and 3). First, the variation of the protecting group from N-PMB to N-methyl afforded the products with similar results (Scheme 2, 2d and 2e). Next, the positional influence of substituents on the phenyl ring was investigated. Methyl groups in the 2-, 3- and 4-positions were well-tolerated, providing the corresponding 2-oxazolidinones with excellent enantioselectivities and in high yields (2f–h). The electronic effect of the substituents was also examined. Both, electron-rich and electron-deficient substrates (1i and 1j respectively) underwent hydrogenation to smoothly afford products 2i and 2j. Halogenated substrates 1k–m were also used to provide the desired products 2k–m with excellent enantioselectivities, in very high yields and without the formation of dehalogenated byproducts. Notably, the catalytic system showcased a robust reactivity, tolerating various functional groups and useful motifs (such as SMe, 1,3-benzodioxole, morpholine, CO2Me and SO2Me) to provide the corresponding products 2o–s with 91–96% ee and in 76–99% yield. These functional groups and motifs (2i–s) provide an excellent opportunity for further applications of the 2-oxazolidinone products. In addition, the absolute configuration of 2s was determined to be (R) by X-ray crystallographic analysis.9 The absolute configuration of all other products was assigned by analogy. Additionally, substrates with condensed-ring and heteroaromatic moieties were also tested. Both 1- and 2-naphthyl-substituted substrates were tolerated under the standard conditions (2t and 2u). Remarkably, thiophene and pyridine containing substrates did not poison the Ru–NHC catalyst, producing the corresponding products (2v and 2w) with 93% ee and 94% ee respectively.
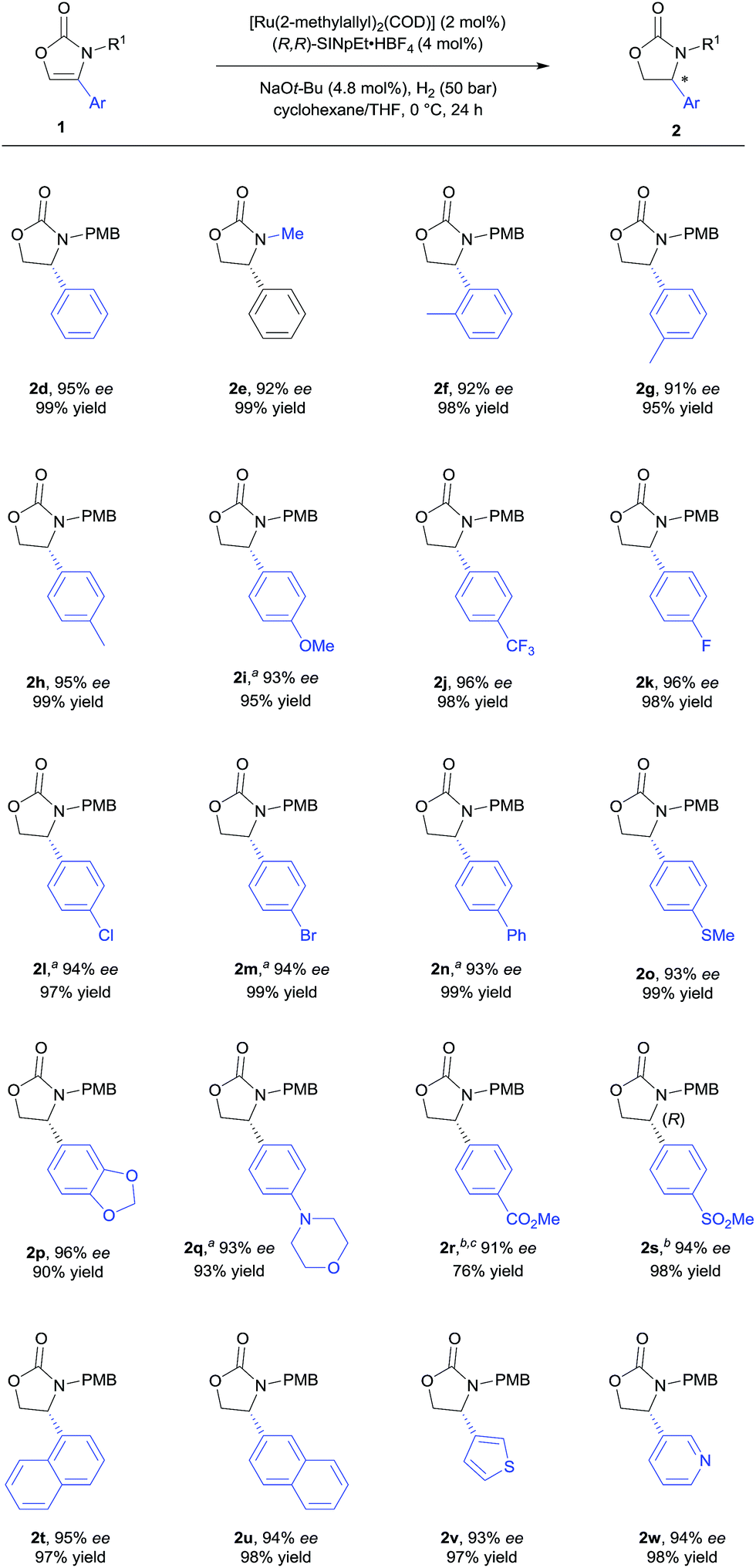 | ||
| Scheme 2 Substrate scope of 4-aryl or 4-heteroaryl substituted 2-oxazolones. General conditions: [Ru(2-methylallyl)2(COD)] (0.10 mmol), (R,R)-SINpEt·HBF4 (0.20 mmol) and NaOt-Bu (0.24 mmol) were stirred at 70 °C in n-hexane (5.0 mL) for 16 h to perform the catalyst (0.02 M), after which 0.2 mL of the catalyst suspension were added to substrates 1d–s (0.20 mmol) in cyclohexane/THF (20/1), and the hydrogenation was performed under 50 bar H2 at 0 °C for 24 h. Yields of isolated products after column chromatography are reported. % ee values were determined by HPLC analysis using a chiral stationary phase. aUsing a solvent mixture of cyclohexane/THF (1/1). bUsing THF (2.0 mL). cAt −10 °C. | ||
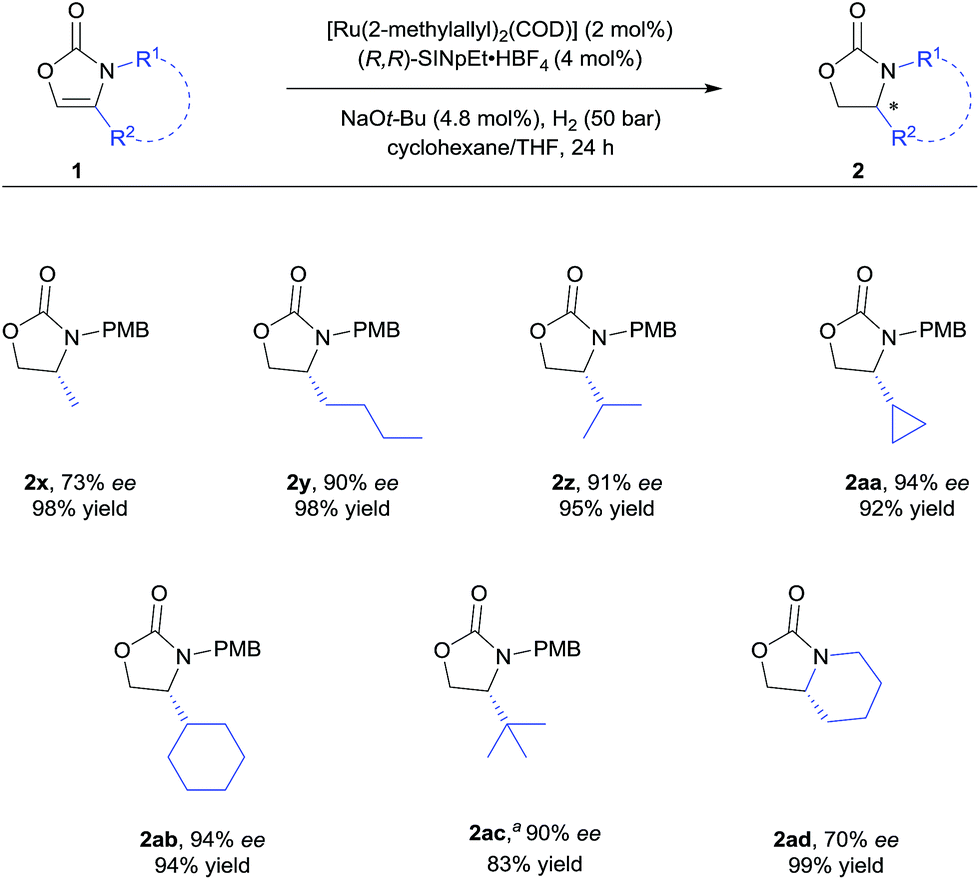 | ||
| Scheme 3 Substrate scope of 4-alkyl substituted 2-oxazolones. For detailed conditions, see ESI.† Yields of isolated products after column chromatography are reported. % ee values were determined by HPLC analysis using a chiral stationary phase. aUsing 5 mol% of the catalyst. | ||
We further explored the substrate scope with 4-alkyl substituents (Scheme 3). Alkyl substituted substrates with different steric demand were systematically tested. Simple 4-methyloxazolidin-2-one 2x was obtained with moderate enantioselectivity. Better control of the stereoselectivity was observed when introducing n-butyl substituent (2y). Isopropyl, cyclopropyl, and cyclohexyl substituents were successfully employed, affording the corresponding products with 91–94% ee and in 92–95% yield (2z–ab). Finally, tert-butyl substituted 2-oxazolone 1ac was tolerated to give the product 2ac with 90% ee and in 83% yield. These results indicate that bulky alkyl groups are beneficial for the enantioinduction. In addition, bicyclic 2-oxazolidinone 2ad was also obtained by the developed method, albeit with moderate ee.
We then proceeded to demonstrate applications of this methodology (Scheme 4). Scale-up of the hydrogenation of 4-(1-naphthyl) substituted substrate 1t to gram-scale provided the corresponding 2-oxazolidinone 2t with 95% ee and in 99% yield (Scheme 4a). Remarkably, the catalyst loading was successfully reduced to 0.2 mol%, which greatly increases the synthetic viability of this protocol. The deprotection of the PMB group was conveniently completed to afford 3 in 99% yield and without loss of enantiomeric excess.6d Hydrolysis of 2t using NaOH liberated the N-PMB β-amino alcohol 4 in 92% yield. The enantiopurity of product 3 was readily increased to >99% ee after recrystallization from ethyl acetate. Optically pure β-amino alcohol 5 was then prepared in quantitative yield by cleavage of the 2-oxazolidinone 3 using diethylenetriamine (Scheme 4b).10 The absolute configuration of β-amino alcohol 5 was reaffirmed to be (R) by comparing the optical rotation to the literature data.11 The β-amino alcohol 5 was further transformed into 1-naphthyl-substituted bisoxazoline ligand 6 in 74% yield by reaction with dimethylmalononitrile and Zn(OTf)2.11a,12 Furthermore, aryl iodide and different N-alkyl substituted substrate 1ae was well tolerated under the established reaction conditions (Scheme 4c). Hydrogenation of 1ae using (S,S)-SINpEt·HBF4 as the carbene ligand precursor, furnished the oxazolidinone 2ae (92% ee and 96% yield), a key synthetic intermediate employed in the synthesis of the alkaloid (−)-aurantioclavine.13
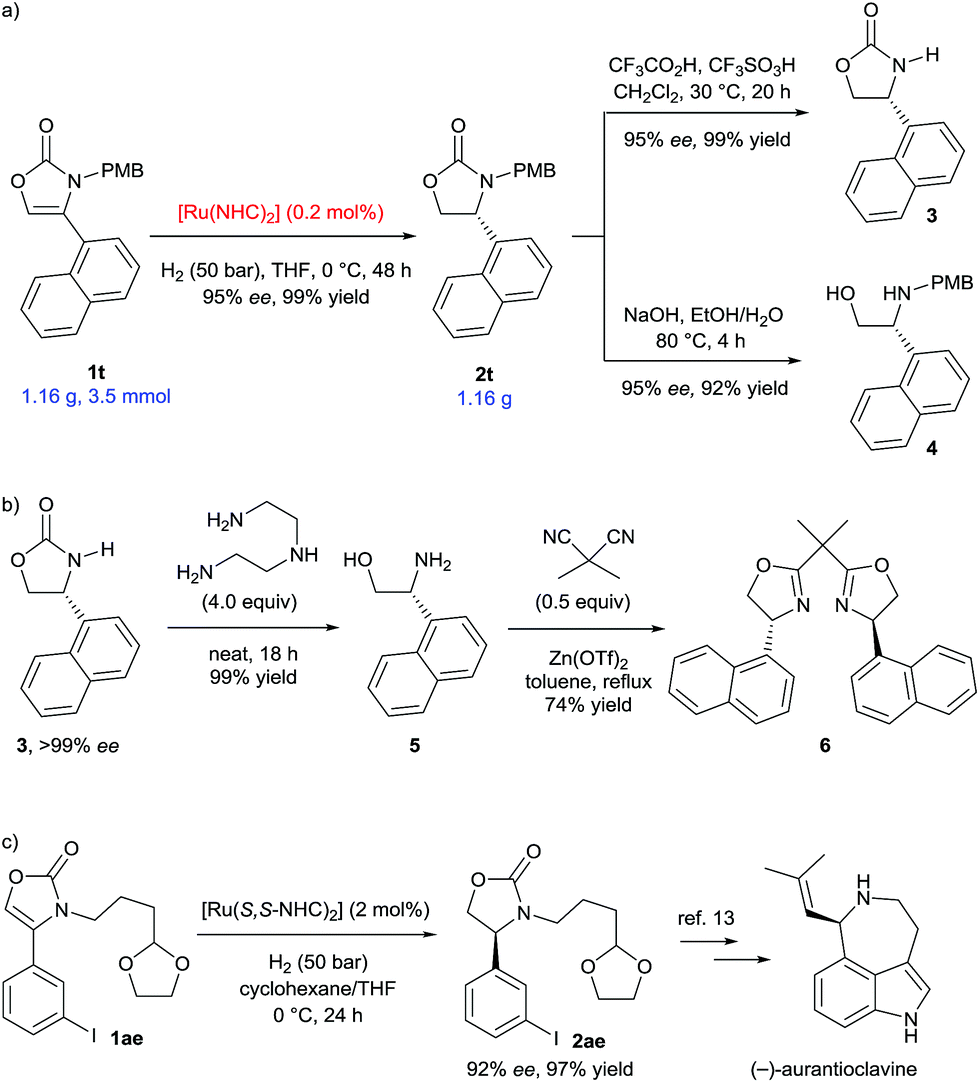 | ||
| Scheme 4 Scaled-up hydrogenation and transformations of the products. | ||
Conclusions
In summary, we have developed a protocol for the catalytic enantioselective hydrogenation of 2-oxazolones to obtain optically active 2-oxazolidinone derivatives. The ruthenium(II)–NHC catalyst system enabled a broad range of substrates to be successfully hydrogenated with excellent enantioselectivities (up to 96% ee) and in yields (up to 99%), thus to make the approach practical for the first time. Various functional groups and synthetically useful motifs were well-tolerated. The synthetic utility of this protocol was further demonstrated by performing a reaction on a gram-scale with a reduced catalyst loading; the obtained enantioenriched product was readily converted into an optically pure β-amino alcohol and subsequently a bisoxazoline ligand. The formal synthesis of (−)-aurantioclavine was enabled by the functional group tolerance towards an iodine substituent at the aryl ring and a varied N-alkyl chain.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Alexander von Humboldt Foundation (W. L.) and the Deutsche Forschungsgemeinschaft (IRTG 2027, Leibniz Award) for generous support. We thank Mario P. Wiesenfeldt, Christoph Schlepphorst and Dr Michael J. James for many helpful discussions. We thank Dr Constantin G. Daniliuc and Birgit Wibbeling for the X-ray crystallographic analysis.Notes and references
- (a) D. A. Evans, J. Bartroli and T. L. Shih, J. Am. Chem. Soc., 1981, 103, 2127 CrossRef; (b) D. A. Evans, Aldrichimica Acta, 1982, 15, 23 Search PubMed.
- For selected reviews, see: (a) J. Seyden-Penne, Chiral Auxiliaries and Ligands in Asymmetric Synthesis, Wiley, New York, 1995 Search PubMed; (b) D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835 CrossRef PubMed; (c) D. J. Ager, I. Prakash and D. R. Schaad, Aldrichimica Acta, 1997, 30, 3 Search PubMed; (d) Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley, New York, 2000 Search PubMed.
- (a) M. E. Dyen and D. Swern, Chem. Rev., 1967, 67, 197 CrossRef PubMed; (b) E. A. MacGregor, Drugs Today, 1998, 34, 1027 CrossRef PubMed; (c) A. M. Rapoport, M. E. Bigal, S. J. Tepper and F. D. Sheftell, Expert Rev. Neurother., 2004, 4, 33 CrossRef PubMed.
- For selected reviews, see: (a) W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef PubMed; (b) Y.-G. Zhou, Acc. Chem. Res., 2007, 40, 1357 CrossRef PubMed; (c) R. Kuwano, Heterocycles, 2008, 76, 909 CrossRef; (d) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713 CrossRef PubMed; (e) D.-S. Wang, Q.-A. Chen, S.-M. Lu and Y.-G. Zhou, Chem. Rev., 2012, 112, 2557 CrossRef PubMed; (f) Z. Yu, W. Jin and Q. Jiang, Angew. Chem., Int. Ed., 2012, 51, 6060 CrossRef PubMed; (g) Y.-M. He, F.-T. Song and Q.-H. Fan, Top. Curr. Chem., 2014, 343, 145 CrossRef PubMed; (h) Z. Zhang, N. A. Buttand and W. Zhang, Chem. Rev., 2016, 116, 14769 CrossRef PubMed; (i) Z.-P. Chen and Y.-G. Zhou, Synthesis, 2016, 48, 1769 CrossRef.
- Q. Wang, X. Tan, Z. Zhu, X.-Q. Dong and X. Zhang, Tetrahedron Lett., 2016, 57, 658 CrossRef.
- (a) S. Urban, N. Ortega and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 3803 CrossRef PubMed; (b) S. Urban, B. Beiring, N. Ortega, D. Paul and F. Glorius, J. Am. Chem. Soc., 2012, 134, 15241 CrossRef PubMed; (c) N. Ortega, S. Urban, B. Beiring and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 1710 CrossRef PubMed; (d) W. Li, C. Schlepphorst, C. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 3300 CrossRef PubMed; (e) W. Li, M. P. Wiesenfeldt and F. Glorius, J. Am. Chem. Soc., 2017, 139, 2585 CrossRef PubMed; (f) M. P. Wiesenfeldt, Z. Nairoukh, W. Li and F. Glorius, Science, 2017, 357, 908 CrossRef PubMed.
- Selected reviews for the use of NHCs as ligands in transition-metal catalysis, see: (a) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef; (b) S. P. Nolan, N-Heterocyclic Carbenes in Synthesis, Wiley-VCH, Weinheim, Germany, 2006 CrossRef; (c) F. Glorius, N-Heterocyclic Carbenes in Transition Metal Catalysis, Springer, Berlin, 2007 CrossRef; (d) V. Dragutan, I. Dragutan, L. Delaude and A. Demonceau, Coord. Chem. Rev., 2007, 251, 765 CrossRef; (e) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef PubMed; (f) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed; (g) S. Gaillard, C. S. J. Cazin and S. P. Nolan, Acc. Chem. Res., 2012, 45, 778 CrossRef PubMed; (h) C. Valente, S. Çalimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314 CrossRef PubMed; (i) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef PubMed.
- Selected reviews for the use of NHCs as ligands in asymmetric catalysis, see: (a) M. C. Perry and K. Burgess, Tetrahedron: Asymmetry, 2003, 14, 951 CrossRef; (b) V. César, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619 RSC; (c) L. H. Gade and S. Bellemin-Laponnaz, Coord. Chem. Rev., 2007, 251, 718 CrossRef; (d) F. Wang, L.-J. Liu, W. Wang, S. Li and M. Shi, Coord. Chem. Rev., 2012, 256, 804 CrossRef; (e) D. Zhao, L. Candish, D. Paul and F. Glorius, ACS Catal., 2016, 6, 5978 CrossRef.
- CCDC 1584965 contains the supplementary crystallographic data for this paper.†.
- M. Noshita, Y. Shimizu, H. Morimoto and T. Ohshima, Org. Lett., 2016, 18, 6062 CrossRef PubMed.
- (a) H. L. van Lingen, J. K. W. van de Mortel, K. F. W. Hekking, F. L. van Delft, T. Sonke and F. P. J. T. Rutjes, Eur. J. Org. Chem., 2003, 317 CrossRef; (b) J. M. Takacs, M. R. Jaber and A. S. Vellekoop, J. Org. Chem., 1998, 63, 2742 CrossRef PubMed.
- (a) A. Cornejo, J. M. Fraile, J. I. García, M. J. Gil, V. Martínez-Merino, J. A. Mayoral, E. Pires and I. Villalba, Synlett, 2005, 2321 Search PubMed; (b) Z. Zhou and M. B. Andrus, Tetrahedron Lett., 2012, 53, 4518 CrossRef.
- J. Park, D.-H. Kim, T. Das and C.-G. Cho, Org. Lett., 2016, 18, 5098 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1584965. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01869c |
| This journal is © The Royal Society of Chemistry 2018 |