
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrophile-promoted Fe-to-N2 hydride migration in highly reduced Fe(N2)(H) complexes†
Meaghan M.
Deegan
and
Jonas C.
Peters
 *
*
Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, CA 91125, USA. E-mail: jpeters@caltech.edu
First published on 29th June 2018
Abstract
One of the emerging challenges associated with developing robust synthetic nitrogen fixation catalysts is the competitive formation of hydride species that can play a role in catalyst deactivation or lead to undesired hydrogen evolution reactivity (HER). It is hence desirable to devise synthetic systems where metal hydrides can migrate directly to coordinated N2 in reductive N–H bond-forming steps, thereby enabling productive incorporation into desired reduced N2-products. Relevant examples of this type of reactivity in synthetic model systems are limited. In this manuscript we describe the migration of an iron hydride (Fe-H) to Nα of a disilylhydrazido(2-) ligand (Fe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NNR2) derived from N2via double-silylation in a preceding step. This is an uncommon reactivity pattern in general; well-characterized examples of hydride/alkyl migrations to metal heteroatom bonds (e.g., (R)MNR′ → M–N(R)R′) are very rare. Mechanistic data establish the Fe-to-Nα hydride migration to be intramolecular. The resulting disilylhydrazido(1-) intermediate can be isolated by trapping with CNtBu, and the disilylhydrazine product can then be liberated upon treatment with an additional acid equivalent, demonstrating the net incorporation of an Fe-H equivalent into an N-fixed product.
NNR2) derived from N2via double-silylation in a preceding step. This is an uncommon reactivity pattern in general; well-characterized examples of hydride/alkyl migrations to metal heteroatom bonds (e.g., (R)MNR′ → M–N(R)R′) are very rare. Mechanistic data establish the Fe-to-Nα hydride migration to be intramolecular. The resulting disilylhydrazido(1-) intermediate can be isolated by trapping with CNtBu, and the disilylhydrazine product can then be liberated upon treatment with an additional acid equivalent, demonstrating the net incorporation of an Fe-H equivalent into an N-fixed product.
Introduction
The Haber–Bosch process has been used since early in the last century for the industrial conversion of nitrogen (N2) and hydrogen (H2) to the bio-available N-atom source ammonia (NH3). This energy intensive process accounts for some 1–2% of annual global energy consumption, with high associated CO2 emissions.1 Attractive alternatives to this process that operate under mild temperature and pressure conditions have long been sought, motivated by biological nitrogenase enzymes that facilitate the proton-coupled reduction of N2 under ambient temperature and pressure conditions.2,3Despite extensive study of the enzymatic system, the detailed mechanism of biological N2 fixation is not reliably known. Based on kinetic and pulsed EPR studies, it has been hypothesized that the build-up of a reduced hydride cluster state, generated by alternate addition of protons and electrons, precedes N2 binding and fixation.4 Given inherent challenges associated with direct study of the biological system, molecular model chemistry provides a means to develop and constrain mechanistic possibilities concerning reactivity patterns germane to N2-to-NH3 conversion.5
In recent years, Fe-based synthetic systems have garnered significant attention, including the development of Fe-based molecular catalysts that facilitate proton-coupled N2 reduction to ammonia.6 As has been proposed for the biological cofactor, hydride species appear to play a significant role during catalysis for a number of these iron systems (Fig. 1). In initial reports, the generation of Fe-H species under turnover relevant conditions was hypothesized to play a role in catalyst deactivation;6a,b more recent studies show that Fe-H species can be observed as off-path resting states and, as such, can serve as competent pre-catalysts for N2 fixation.7,6d,f Of particular relevance are in situ spectroscopic studies on the PB3Fe (PB3 = B(o-iPr2PC6H4)3) system from our lab, which allowed for the observation of a hydride resting state, PB3(μ-H)Fe(H)(N2) 1, under catalytic conditions for a specific acid/reductant combination.8 In examples where hydride species have been shown to be amenable to productive N2 fixation, it has generally been suggested that unproductive H2 evolution pathways are accessible, and precede the generation of a catalytically active, hydride-free species.9
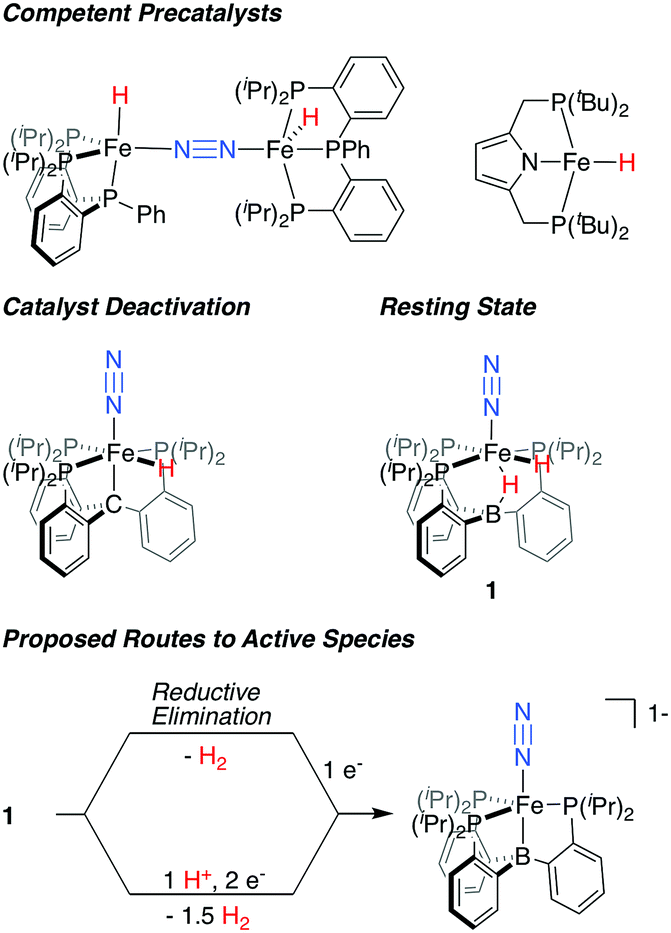 | ||
| Fig. 1 Fe-H species relevant to catalytic N2 fixation by molecular Fe complexes and proposed routes for the generation of an on path Fe–N2 intermediate from dihydride resting state 1.6b,d,f,8 | ||
An alternative possibility can be considered where Fe-H intermediates are able to undergo Fe-to-N migration steps, and therefore be incorporated into productive overall schemes for N2 fixation. While such a scenario has been suggested to occur within the iron-molybdenum cofactor during catalysis,2c relevant N2 model chemistry demonstrating such a reactivity pattern is limited to a fascinating set of TinHm clusters, which have been shown to promote N2-cleavage with N–H bond formation (Scheme 1A).10 Conceptually related reactivity has also been reported for a number of multimetallic systems, where bridged N2 ligands are reactive towards a variety of E-H substrates (Scheme 1B).11–13
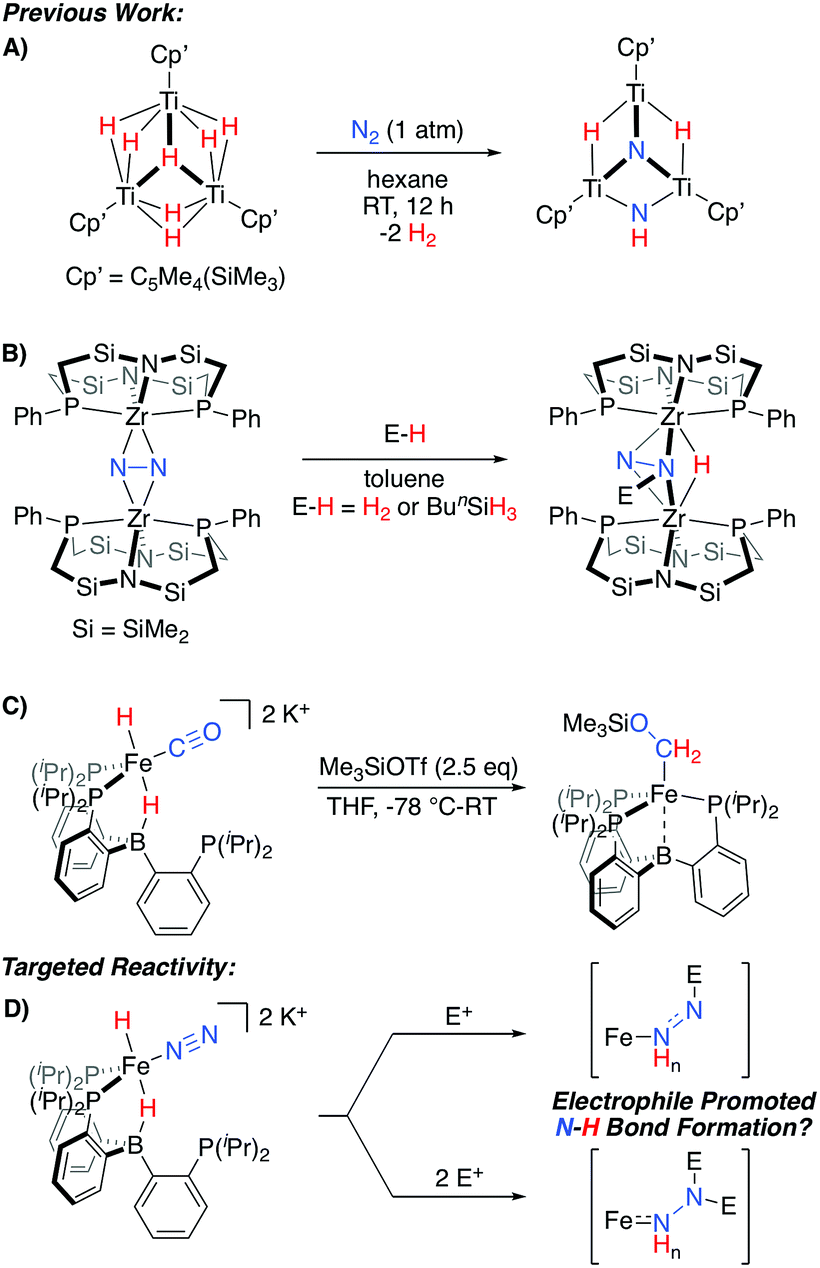 | ||
| Scheme 1 Representative stoichiometric reactions exhibiting relevant reactivity patterns for N2 (A and B)10,11a or CO (C)14 functionalization, and the electrophile-promoted Fe-H to N2 migration targeted herein (D). | ||
Recently, we demonstrated that a PB3Fe system supports a series of highly reduced hydride complexes with CO as a co-ligand.14 These species are able to undergo facile O-functionalization that promotes the rapid intramolecular formation of two new C–H bonds from an Fe(H)2 precursor (Scheme 1C). Thus, we wondered whether related N2 complexes might also be supported by this platform. If so, they would be of interest as targets for exploring electrophile-promoted Fe-H migration, to generate new N–H bond(s), as a new reactivity pattern (Scheme 1D).
Herein we describe the synthesis and characterization of a series of [PB3Fe(N2)(H)n]m− complexes. One of these species, in a highly reduced state, is reactive toward β-functionalization with silyl electrophiles; intramolecular Fe-to-Nα hydride migration is observed upon difunctionalization. The N–H bond-forming step proceeds slowly in solution at low temperature and is thereby amenable to a detailed kinetic analysis with the determination of associated activation parameters. Trapping of the product with an exogenous CNtBu ligand allows for its isolation as a hydrazido(1-) species, with N2-derived disilylhydrazine products liberated upon protonolysis of this complex. Overall, this reactivity demonstrates a plausible series of steps through which an Fe-H equivalent can be incorporated as part of a productive N2 fixing scheme, in contrast to unproductive H2 evolution. The utility of silyl, as opposed to proton, electrophiles is highlighted by the ability to kinetically stabilize and characterize extremely reactive species along the reaction pathway.
Results and discussion
Synthesis of Fe(N2)(H)n complexes
We initially targeted [Fe(N2)(H)n]m− complexes analogous to the previously reported [Fe(CO)(H)n]m− species supported by the PB3 ligand platform.14 Thus, reduction of PB3(μ-H)Fe(H)(N2) 1,8,15 with an excess of KC8 at −78 °C led to the formation of the dianionic complex [PB3(μ-H)Fe(H)(N2)][K2(S)n] 2 (Scheme 2). The 31P NMR spectrum of this species shows one of the phosphine arms dechelated from the Fe center (δ = 92.6 (2P), −1.5 (1P)), with both of the hydride resonances apparent in the 1H NMR spectrum at −18.19 (Fe-H–B) and −20.78 (Fe-H) ppm. The IR spectrum of dianionic 2 exhibits a highly activated N2 ligand (νNN = 1746 cm−1), with a 325 cm−1 increase in N2 activation compared to the neutral precursor 1 (νNN = 2071 cm−1). Crystallographic characterization of this species confirmed its assignment, with hydride ligands occupying axial sites of an approximate trigonal bipyramid (Fig. 2). Strong π-backbonding to the phosphine and N2 ligands is apparent, with quite short Fe–P (2.15 Å, average) and Fe–N (1.743(2) Å) distances, and an elongated N–N distance (1.169(3) Å) when compared to 1 (1.128(4) Å). Taken together, the spectroscopic and structural data suggest an N–N bond order that can be described as lying between a double and triple bond. The extreme sensitivity of the dianionic complex 2 precluded prolonged handling and storage of this species, so its spectroscopic characterization and further reactivity studies were carried out on samples that were generated in situ.16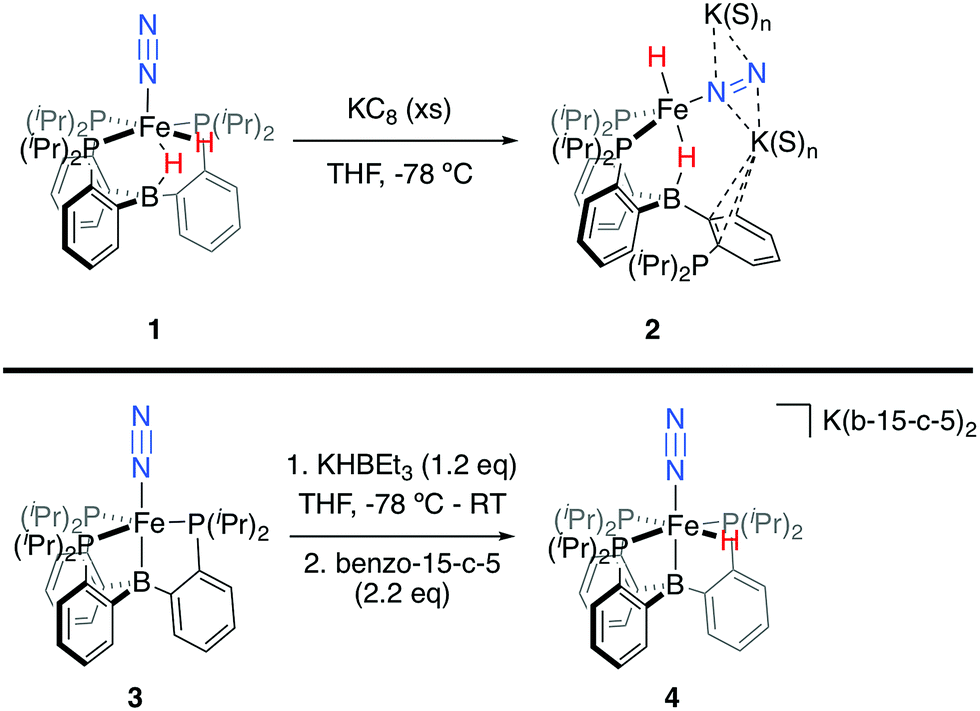 | ||
| Scheme 2 Synthesis of [Fe(N2)(H)n]m− complexes 2 and 4. | ||
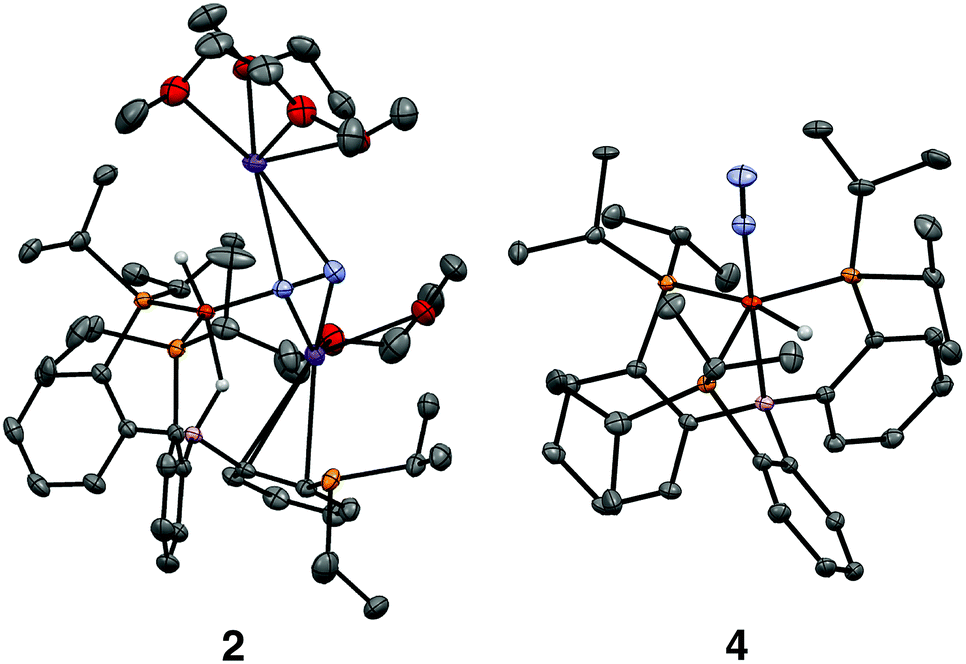 | ||
| Fig. 2 Crystal structures of the dianionic complex 2 (left) and the anionic complex 4 (right). Displacement ellipsoids shown at 50% probability; C-bound H atoms, disorder of iPr groups, and the crown encapsulated potassium countercation in 4 have been omitted for clarity. | ||
We also found that the monoanionic hydride complex [PB3Fe(H)(N2)][K(benzo-15-crown-5)2] 4 could be generated by the addition of a solution of KHBEt3 to a THF solution of PB3Fe(N2) 3 at −78 °C.17 The product was readily isolated as a crystalline orange material following workup as its benzo-15-crown-5 encapsulated K salt (Scheme 2). This species exhibits a sharp N2 stretch (1956 cm−1) that is 51 cm−1 higher than the hydride-free anion [PB3Fe(N2)][Na(12-crown-4)2] (1905 cm−1).17 The hydride of 4 could be identified by 1H NMR spectroscopy via a quartet resonance at −12.59 ppm, with equal coupling to each of the three coordinated phosphine ligands. This hydride was crystallographically located as a terminal ligand in the plane of the three Fe-ligated phosphines (Fig. 2). In solution, 4 exhibits three-fold symmetry at room temperature, reflected by a single 31P NMR resonance (δ = 77.1); the asymmetry observed in the solid state is borne out in solution upon cooling of 4 to lower temperatures (at −80 °C: δ = 95.5, 79.3, 59.7; see ESI†). Similar solution state behavior has been observed for the analogous CO complex 4-CO and we presume that in both cases this may be attributed to rapid equilibration of the hydride position through a borohydride intermediate.14,18,19
Bond metrics for a series of structurally related PE3-supported (E = B, Si) Fe–N2 complexes are shown in Table 1 for direct comparison. For complex 4, relevant metrics compare quite well to the related, formally Fe(0)–N2 complex [PSi3Fe(N2)][Na(12-crown-4)2] (Table 1).20 Direct comparison of bond metrics and the degree of N2 activation with dianionic 4 is complicated by structural differences and interactions with K countercations, where 4, like its analogous CO complex, was found to be unstable in the presence of encapsulating reagents (e.g., benzo-15-crown-5). As might be expected, there appears to be a substantially greater degree of N2 activation and associated π-backdonation in 4 than for any of the other crystallographically characterized N2 complexes supported by these ligand frameworks.
| Complex | ν NN (cm−1) | FOS | S | N–N (Å) | Fe–N (Å) | Fe–P (ave, Å) | Fe–E (Å) | Reference |
|---|---|---|---|---|---|---|---|---|
| PB3(μ-H)Fe(H)(N2) 1 | 2071 | +2 | 0 | 1.128(4) | 1.801(3) | 2.283 | 2.602(3) | 15 |
| [PSi3Fe(N2)][BArF4] | 2143 | +2 | 1 | 1.091(3) | 1.914(2) | 2.390 | 2.298(7) | 20 |
| PSi3Fe(N2) | 2003 | +1 | 1/2 | 1.1245(2) | 1.8191(1) | 2.285 | 2.2713(2) | 20 |
| [PB3(μ-H)Fe(H)(N2)][K2(S)n] 2 | 1746 | 0 | 0 | 1.169(3) | 1.743(2) | 2.148 | 2.749(3) | This work |
| [PB3Fe(H)(N2)][K(b-15-c-5)2] 4 | 1956 | 0 | 0 | 1.132(2) | 1.800(1) | 2.205 | 2.289(1) | This work |
| [PSi3Fe(N2)][Na(12-c-4)2] | 1920 | 0 | 0 | 1.132(2) | 1.795(3) | 2.196 | 2.236(1) | 20 |
| [PB3Fe(N2)][Na(12-c-4)2] | 1905 | −1 | 1/2 | 1.144(3) | 1.781(2) | 2.251 | 2.293(3) | 17 |
Functionalization with silyl electrophiles
Tractable functionalization reactivity was accessible for the dianionic complex 2 at low temperature, where treatment with TiPSCl generated the thermally stable, anionic silyl diazenido complex, [PB3(μ-H)Fe(H)(NNSiiPr3)][K(THF)] 5 (Scheme 3).21 In the transformation from dianionic 2 to monoanionic 5, the phosphine asymmetry is retained, with one of the phosphines dechelated, as observed in the 31P NMR spectrum (90.6 (br, 2P), −1.0 (1P)). Further, the hydride and borohydride ligands are readily observed in the 1H NMR spectrum at −9.71 (t) and −11.10 ppm respectively, along with activated N–N stretches in the IR spectrum (1541, 1588 cm−1), consistent with the formation of an anionic silyl diazenido complex. This was confirmed by XRD analysis (Fig. 3). Complex 5 is structurally comparable to the previously reported anionic silyl diazenido complex [PB3Fe(NNSiMe3)][Na(THF)] 6, in which one of the phosphine ligands is similarly dechelated from the Fe center (Fig. 4A).22 Both 5 and 6 have tightly interacting countercations bridging the terminal silyl diazenido ligand and the phenylene linker of the dechelated phosphine arm. In 5 the hydride and borohydride ligands occupy the axial sites of an approximate trigonal bipyramidal structure, whereas the Fe center in 6 is ligated by an additional η2 interaction with the phenylene linker. Complex 5 has a similar overall geometry to the dianionic precursor 2, featuring hydride and borohydride ligands that are unperturbed upon N2-silylation. Nevertheless, the relevant bond metrics for the Fe-coordinated ligands in the anionic silyldiazenido complexes 5 and 6 compare favorably (Table 2).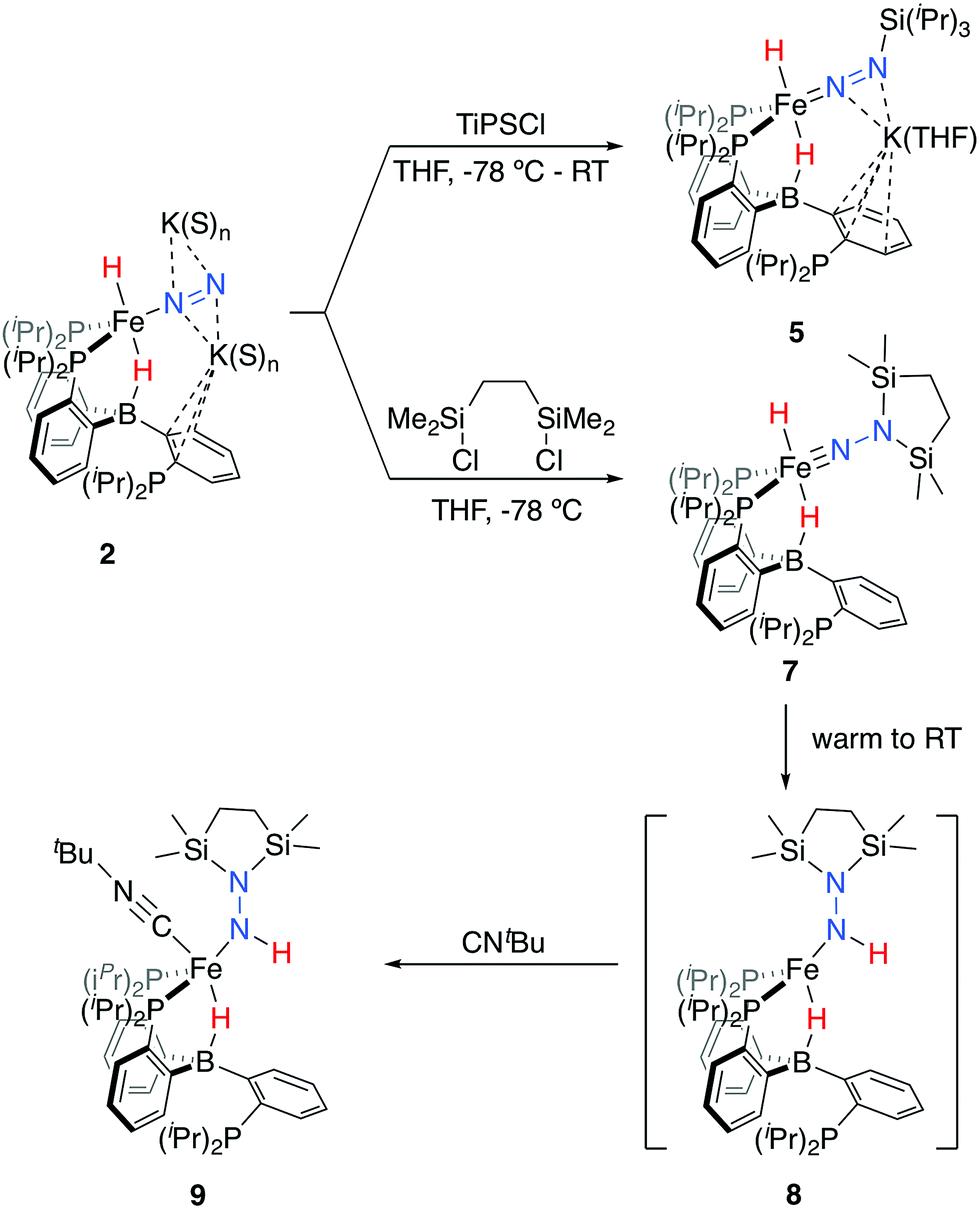 | ||
| Scheme 3 Functionalization of dianionic 2 with silyl electrophiles and subsequent hydride migration to generate intermediate 8, trapped as isolable 9. | ||
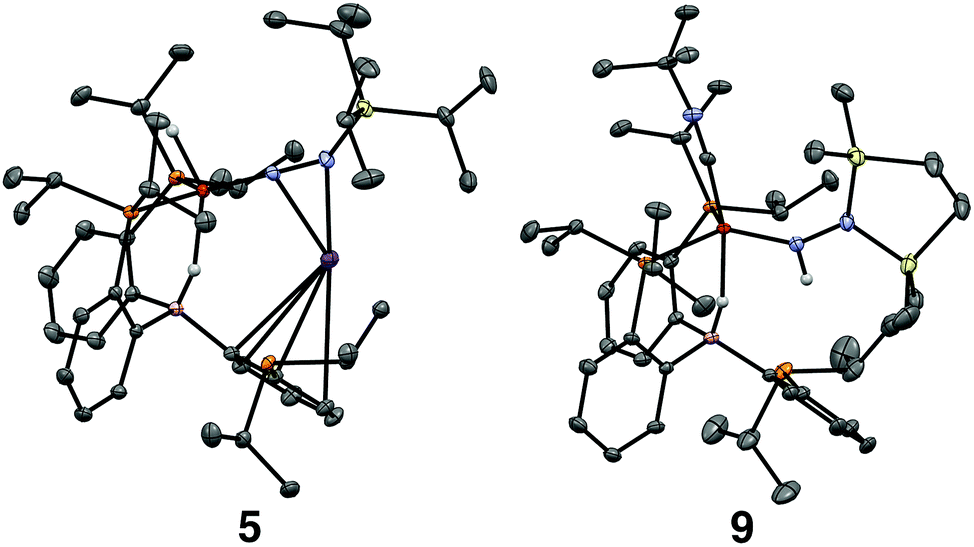 | ||
| Fig. 3 Crystal structures of the silyl diazenido complex 5 (left) and the disilylhydrazido(1-) complex 9 (right). Displacement ellipsoids shown at 50% probability; C-bound H atoms, solvent molecules and disorder of iPr groups in 5 have been omitted for clarity. | ||
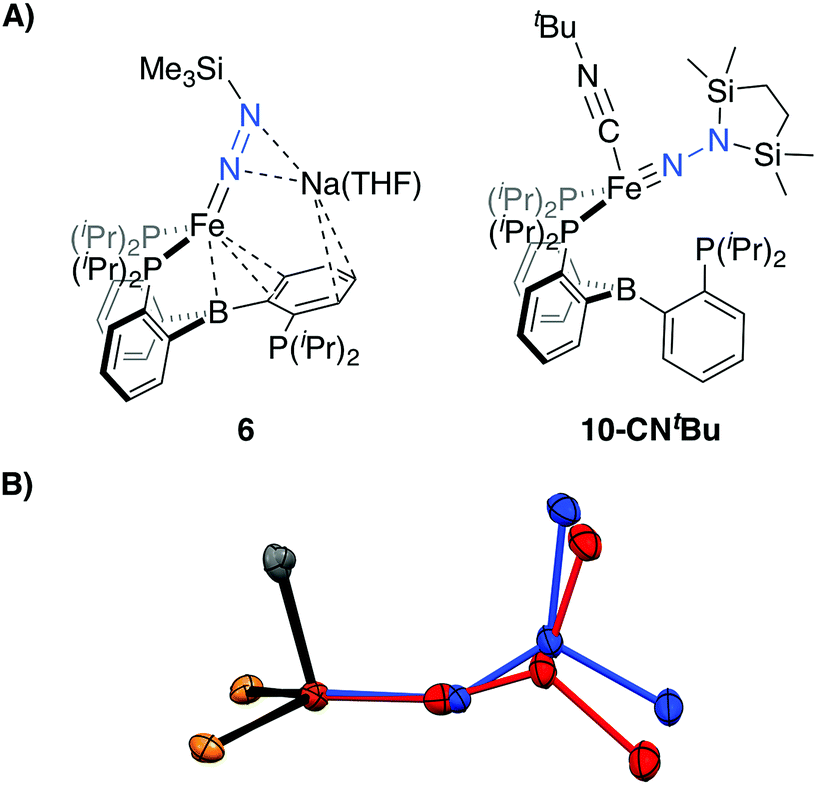 | ||
| Fig. 4 (A) Depictions of previously reported silyl functionalized N2 complexes 6 and 10-CNtBu. (B) Overlay of the core structures of the hydrazido(1-) complex 9 (blue) and the previously reported hydrazido(2-) complex 10-CNtBu (red).22 | ||
| Complex | N–N (Å) | Fe–N (Å) | Fe–P (ave, Å) | Fe–B (Å) | Fe–N–N (°) | Reference |
|---|---|---|---|---|---|---|
| [PB3(μ-H)Fe(H)(NNSiiPr3)][K(THF)] 5 | 1.249(2) | 1.672(1) | 2.219 | 2.791(1) | 169.31(9) | This work |
| [PB3Fe(NNSiMe3)][Na(THF)] 6 | 1.259(4) | 1.673(3) | 2.229 | 2.319(4) | 169.8(2) | 22 |
| PB3(μ-H)Fe(NHNSi2)(CNtBu) 9 | 1.404(2) | 1.824(2) | 2.231 | 2.885(2) | 145.8(1) | This work |
| PB3Fe(NNSi2)(CNtBu) 10-CNtBu | 1.351(3) | 1.640(2) | 2.247 | 2.863(3) | 162.7(2) | 22 |
Treatment of 2 with the disilylating reagent 1,2-bis(chlorodimethylsilyl)ethane (Si2) at −78 °C resulted in the formation of a new species. This reaction could be monitored at low temperatures (<−20 °C), allowing for the observation of a diamagnetic intermediate, with both hydride resonances retained in the 1H NMR spectrum at −16.74 (br) and −17.51 (t) ppm. The asymmetry in this species is also evident by 31P NMR spectroscopy (−80 °C; δ = 106.6, 95.1, −10.2), consistent with its tentative assignment as the five-coordinate disilylhydrazido(2-) species 7 (Scheme 3). Upon warming the reaction mixture further, decay of this intermediate was observed concurrent with the appearance of a new set of paramagnetically shifted 1H NMR resonances. A 1H NMR spectrum reflecting a mixture of paramagnetic species was obtained after several minutes at room temperature.
We hypothesized the initially generated paramagnetic intermediate to be the product of N–H bond formation via alpha-hydride migration to generate the hydrazido(1-) complex 8 (Scheme 3). Such a formulation would be consistent with IR spectra of the reaction mixture; no diagnostic N–N multiple bond stretch is observed, whereas an N–H vibration is located at 3263 cm−1; the latter is absent in the spectrum of the deuterium labeled analog 8-D2.23
To generate an isolable analog of the disilylhydrazido(1-) complex, we explored the addition of coordinating exogenous ligands to 8 (e.g., CO, PMe3) to trap it. We found that addition of an excess of CNtBu resulted in the formation of a comparatively stable, diamagnetic complex PB3(μ-H)Fe(NHNSi2)(CNtBu) 9 (Scheme 3). This species was assigned initially through the use of multinuclear NMR spectroscopy, with the hydride resonating at −12.59 ppm and the N–H at 7.84 ppm; the latter exhibits weak coupling to Si in the 29Si-HMBC spectrum (see ESI†); these assignments could be confirmed by comparison to the D2-labeled analog 9-D2. By IR spectroscopy, an intense vibration associated with the isocyanide C–N stretch is observed at 1997 cm−1, with the N–H and B–H–Fe vibrations assigned at 3197 and 2070 (br) cm−1, respectively, shifting to 2377 and 1515 (br) cm−1 in the labeled analog 9-D2. The 31P NMR spectrum reflects asymmetry with one phosphine ligand dechelated from the Fe center (δ = 88.9, 82.7, −2.1).
Confirmation of the proposed assignment for 9 was enabled by XRD analysis, with the disilylhydrazido(1-) ligand located in the plane of the coordinated phosphine ligands and the CNtBu ligand trans to the coordinated borohydride moiety (Fig. 3). The overall geometry of this complex is similar to a previously reported, CNtBu-trapped disilylhydrazido(2-) complex, 10-CNtBu (Fig. 4A).21 Examination of an overlay of these structures (Fig. 4B) highlights the key differences distinguishing the hydrazido(1-) and hydrazido(2-) ligands in 9 and 10-CNtBu, respectively; they otherwise, have remarkably similar structures. Upon formation of the new Nα–H bond in 9, multiple bond character of both the Fe–N and N–N bonds is predicted to decrease with associated bending of the Fe–N–N angle. This is readily observed in the crystallographic bond metrics for these species; hydrazido(1-) 9 exhibits substantial elongation of the Fe–N distance (1.824(2) vs. 1.640(2)) and the N–N distance (1.404(2) vs. 1.351(3)) as well as increased bending in the Fe–N–N angle (145.8(1) vs. 162.7(2)) (Table 2) when compared with the hydrazido(2-) complex 10-CNtBu.
N–H bond formation
Given the unusual nature of the N–H bond-forming step from 7 to 8 (and 9), we were interested in further understanding its mechanism. As discussed above, selective D-incorporation into the N–D bond for the labeled complex 9-D2 confirmed that the newly formed N–H(D) bond is derived from one of the original hydride/deuteride ligands (Fig. 5A). Further, this transformation does not appear to proceed through the loss of free H2 prior to the N–H bond-forming step. Consistent with this supposition, the complexes 10 and 10-CNtBu do not undergo productive reactivity with free H2,24 and the transformation from 7 to 9 can be carried out under an atmosphere of D2 without the generation of the D-labeled Fe product 9-D2 (see ESI†). The kinetics of this transformation can conveniently be monitored by UV-visible spectroscopy; the process is first order in Fe, with a kinetic isotope effect of 1.39 (measured at 10 °C; Fig. 5B).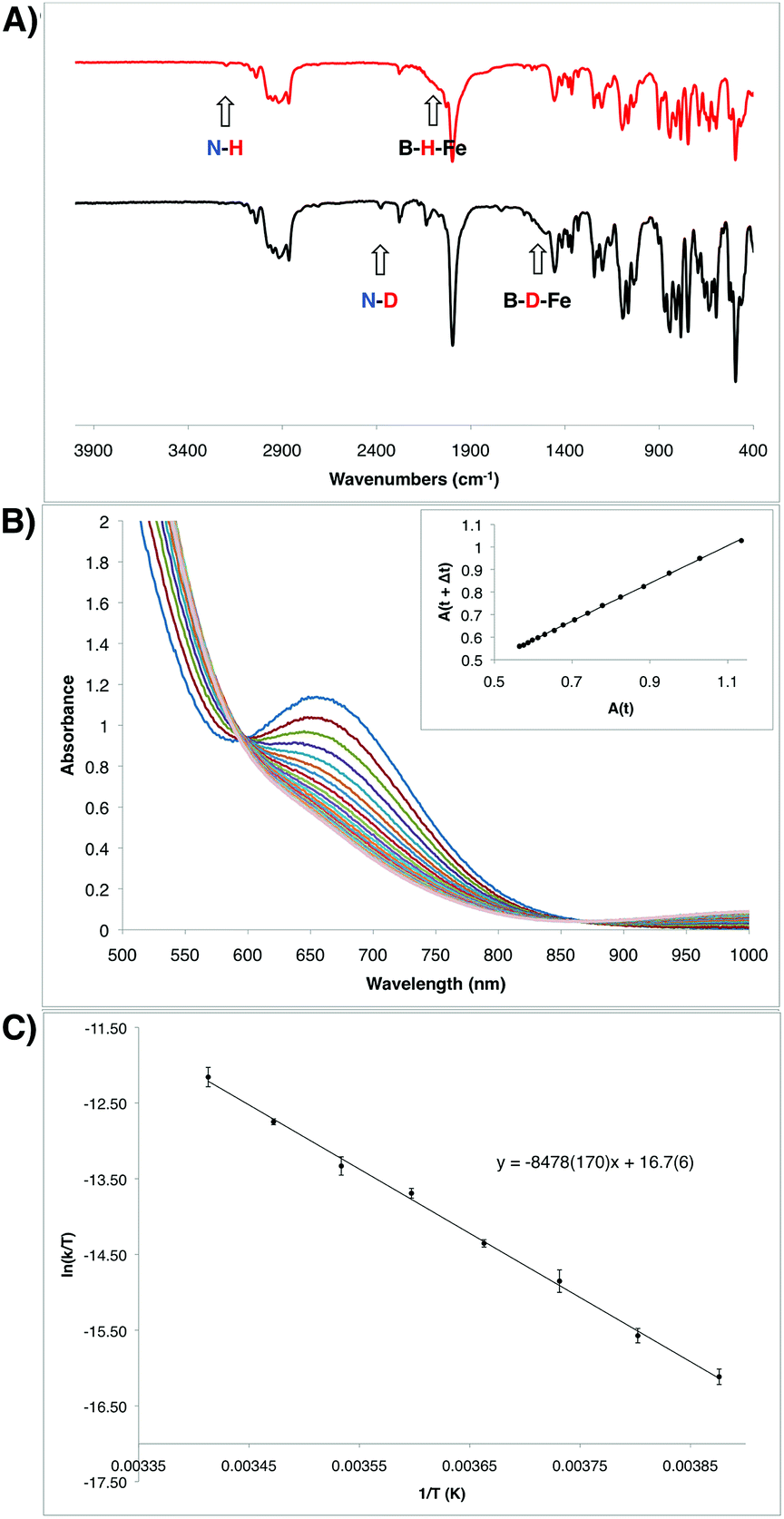 | ||
| Fig. 5 (A) IR spectra of the hydrazido(1-) complexes 9 (top) and 9-D2 (bottom). (B) Transformation of the intermediate hydrazido(2-) complex 8 to the hydrazido(1-) complex 9 observed by UV-Vis spectroscopy in 6 minute intervals at 10 °C. Inset shows the corresponding linear plot of A(t) vs. A(t + Δt), which is consistent with a first order decay process (R2 = 0.99961). Rate constant: 4.98 × 10−4 s−1 (see ESI†). (C) Eyring plot for the decay from 8 to 9 measured in 5 degree increments from −15 °C to 20 °C. Measurements were made in duplicate at each temperature. Eyring parameters: ΔH‡ = 16.8(3) kcal mol−1; ΔS‡ = −14(1) e.u.; ΔG‡ = 20.9 kcal mol−1 (at 20 °C). | ||
Combined, these observations are consistent with an intramolecular hydride migration step as the operative mechanism for N–H bond formation. This type of reaction is highly unusual, even when expanded to include the migration of any R group (R = alkyl, aryl, or hydride) to any multiply bonded heteroatom (M = E; E = O, NR, etc.). Well-studied examples of this reactivity pattern are scarce, with early detailed studies from the Mayer group reporting the migration of a variety of R groups (R = alkyl, aryl, H) to electrophilic Re(O).25 Perhaps more closely related to the present system, Wolczanski, Cundari, and coworkers very recently reported a four-coordinate Fe(IV) imido complex that formally inserts into an Fe–R bond (R = neoPe, 1-nor) to generate a new, three-coordinate Fe(II) amido complex (Scheme 4).26
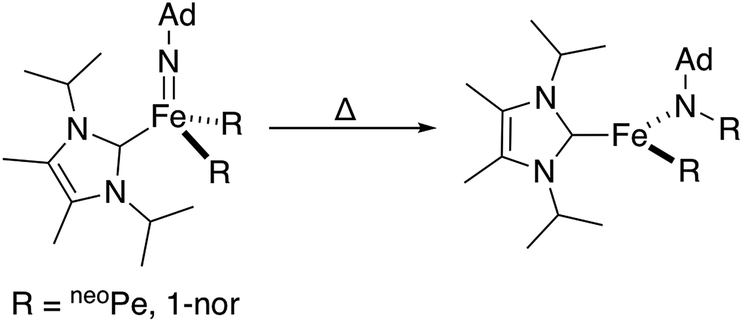 | ||
| Scheme 4 Reported Fe-imido to Fe-amido transformation proceeding through the migration of a bulky alkyl group.26 | ||
The observed transformation of 7 to 8 that leads to generation of an N–H bond could be monitored over a 35 °C temperature range27 (−15 °C up to 20 °C), enabling an Eyring analysis to obtain the following pertinent transition state parameters: ΔH‡ = 16.8(3) kcal mol−1; ΔS‡ = −14(1) e.u.; ΔG‡ = 20.9 kcal mol−1 (at 20 °C) (Fig. 5C). The negative entropy of activation observed is unexpected for an intramolecular process that is first order in Fe when compared with previous observations. In the related Fe study by Wolczanski and coworkers (Scheme 4), small, positive activation entropies (ΔS‡) were determined, with values of 6.5(1) e.u. (R = neoPe) and 1.3(1) e.u. (R = 1-nor); these values were close to those predicted computationally.26,28 We suspect that, for our present system, some sort of ordering of the flexible phosphine–borane ligand may be present in the transition state, leading to a negative entropy of activation for example via the interaction of the otherwise free phosphine arm.
Release of N-fixed products
With the isolable hydrazido(1-) complex 9-CNtBu in hand, we explored conditions for the release of reduced, N2-derived products. Initially, we explored possible conditions for the reductive elimination of the final N–H bond to liberate disilylhydrazine. In these attempts, which included both thermal and photochemical reaction conditions, N-fixed products could be detected in low yields in certain cases, but invariably these reactions yielded mixtures of ill-defined Fe-containing products (see ESI† for further details). Direct treatment of 9-CNtBu instead with a stoichiometric equivalent of [H(OEt2)2][BArF4] (BArF4 = B(C8H3F6)4) at low temperature, followed by warming the solution, proved more tractable (Scheme 5); liberated disilylhydrazine products could be detected (62%) in the absence of detectable NH3. The major Fe-containing product, cationic [PB3(μ-H)Fe(CNtBu)][B(C8H3F6)4], could be isolated in similar yields (see ESI† for details).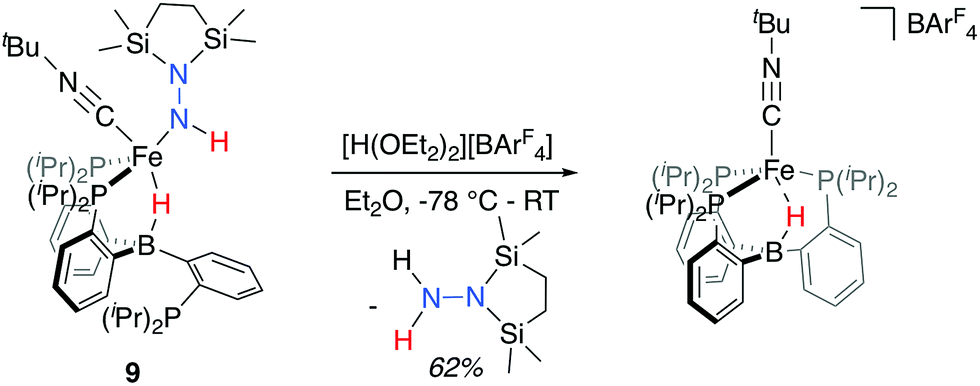 | ||
| Scheme 5 Release of disilylhydrazine upon treatment with stoichiometric acid (BArF4 = B(C8H3F6)4). | ||
Conclusions
We have described the synthesis and characterization of a series of unusual Fe(N2)(H)n complexes, one of which undergoes β-N-functionalization with silyl electrophiles to generate silyldiazenido and disilylhydrazido(2-) species featuring hydride and borohydride ligands. Of most interest, the disilylhydrazido(2-) complex is observed to undergo a highly unusual Fe-to-Nα hydride migration step to generate a dislylhydrazido(1-) complex that is isolable upon trapping with an exogenous CNtBu ligand. The hydride migration step is shown to be intramolecular and proceeds over a temperature range that enables activation parameters to be measured. The ability to generate an N–H bond at Nα of an Fe(H)(N2) species, triggered by electrophilic derivatization of the Nβ position, opens up interesting opportunities to generate new N2-derived products from Fe-mediated processes. That disilylhydrazine can be liberated in the present system provides a promising starting point in this context.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NIH (GM 070757). The authors are grateful to Larry Henling and Mike Takase for crystallographic assistance. We thank Trixia Buscagan for preliminary data on complex 4.Notes and references
- V. Smil, Enriching the Earth, MIT Press, Boston, 2001 Search PubMed.
- Reviews: (a) J. B. Howard and D. C. Rees, Chem. Rev., 1996, 96, 2965 CrossRef PubMed; (b) B. K. Burgess and D. J. Lowe, Chem. Rev., 1996, 96, 2983 CrossRef PubMed; (c) B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041 CrossRef PubMed.
- Representative experimental studies: (a) S. J. Yoo, H. C. Angove, V. Papaefthymiou, B. K. Burgess and E. Münck, J. Am. Chem. Soc., 2000, 122, 4926 CrossRef; (b) O. Einsle, F. A. Tezcan, S. L. A. Andrade, B. Schmid, M. Yoshida, J. B. Howard and D. C. Rees, Science, 2002, 297, 1696 CrossRef PubMed; (c) T. Spatzal, M. Aksoyoglu, L. Zhang, S. L. A. Andrade, E. Schleicher, S. Weber, D. C. Rees and O. Einsle, Science, 2011, 334, 940 CrossRef PubMed; (d) B. M. Hoffman, D. Lukoyanov, D. R. Dean and L. C. Seefeldt, Acc. Chem. Res., 2013, 46, 587 CrossRef PubMed; (e) J. Kowalska and S. DeBeer, Biochim. Biophys. Acta, Mol. Cell Res., 2015, 1853, 1406 CrossRef PubMed.
- (a) D. J. Lowe and R. N. F. Thornley, Biochem. J., 1984, 224, 877 CrossRef PubMed; (b) D. Lukoyanov, N. Khadka, Z.-Y. Yang, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, J. Am. Chem. Soc., 2016, 138, 10674 CrossRef PubMed; (c) D. Lukoyanov, N. Khadka, Z.-Y. Yang, D. R. Dean, L. C. Seefeldt and B. M. Hoffman, J. Am. Chem. Soc., 2016, 138, 1320 CrossRef PubMed; (d) C. N. Morrison, T. Spatzal and D. C. Rees, J. Am. Chem. Soc., 2017, 139, 10856 CrossRef PubMed.
- For representative reviews, see: (a) J. Chatt, J. R. Dilworth and R. L. Richards, Chem. Rev., 1978, 78, 589 CrossRef; (b) R. R. Schrock, Acc. Chem. Res., 2005, 38, 955 CrossRef PubMed; (c) Y. Nishibayashi, Inorg. Chem., 2015, 54, 9234 CrossRef PubMed; (d) K. C. MacLeod and P. L. Holland, Nat. Chem., 2013, 5, 559 CrossRef PubMed; (e) N. Stucke, B. M. Flöser, T. Weyrich and F. Tuczek, Eur. J. Inorg. Chem., 2018, 1337 CrossRef; (f) Y. Roux, C. Duboc and M. Gennari, ChemPhysChem, 2017, 18, 2606 CrossRef PubMed.
- (a) J. S. Anderson, J. Rittle and J. C. Peters, Nature, 2013, 501, 84 CrossRef PubMed; (b) S. E. Creutz and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 1105 CrossRef PubMed; (c) G. Ung and J. C. Peters, Angew. Chem., Int. Ed., 2015, 54, 532 Search PubMed; (d) K. Shogo, K. Arashiba, K. Nakajima, Y. Matsuo, H. Tanaka, K. Ishii, K. Yoshizawa and Y. Nishibayashi, Nat. Commun., 2016, 7, 12181 CrossRef PubMed; (e) P. J. Hill, L. R. Doyle, A. D. Crawford, W. K. Myers and A. E. Ashley, J. Am. Chem. Soc., 2016, 138, 13521 CrossRef PubMed; (f) T. M. Buscagan, P. H. Oyala and J. C. Peters, Angew. Chem., Int. Ed., 2017, 56, 6921 CrossRef PubMed.
- For a review of H2 reductive elimination preceding N2 binding and activation: J. Ballmann, R. F. Munhá and M. D. Fryzuk, Chem. Commun., 2010, 46, 1013 RSC.
- T. J. Del Castillo, N. B. Thompson and J. C. Peters, J. Am. Chem. Soc., 2016, 138, 5341 CrossRef PubMed.
- For a recent review see: T. Shima and Z. Hou, Top. Organomet. Chem., 2017, 60, 23 CrossRef.
- T. Shima, S. Hu, G. Luo, X. Kang and Z. Hou, Science, 2013, 340, 1549 CrossRef PubMed.
- Select examples: (a) M. D. Fryzuk, J. B. Love, S. J. Rettig and V. G. Young, Science, 1997, 275, 1445 CrossRef; (b) J. A. Pool, E. Lobovsky and P. J. Chirik, Nature, 2004, 427, 527 CrossRef PubMed; (c) M. D. Fryzuk, Acc. Chem. Res., 2009, 42, 127 CrossRef PubMed; (d) B. Wang, G. Luo, M. Nishiura, S. Hu, T. Shima, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2017, 139, 1818 CrossRef PubMed.
- Similar reactivity with E-H substrates has been demonstrated for pre-functionalized, or products of, N2 cleavage at monometallic centers: (a) D. L. M. Suess and J. C. Peters, J. Am. Chem. Soc., 2013, 135, 4938 CrossRef PubMed; (b) Q. Liao, A. Cavaillé, N. Saffon-Merceron and N. Mézailles, Angew. Chem., Int. Ed., 2016, 55, 11212 CrossRef PubMed.
- The reactivity of Fe-H for the cleavage of NN bonds has also been explored:
(a) J. M. Smith, R. J. Lachicotte and P. L. Holland, J. Am. Chem. Soc., 2003, 125, 15752 CrossRef PubMed;
(b) A. R. Sadique, E. A. Gregory, W. W. Brennessel and P. L. Holland, J. Am. Chem. Soc., 2007, 129, 8112 CrossRef PubMed.
- M. M. Deegan and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 2561 CrossRef PubMed.
- H. Fong, M.-E. Moret, Y. Lee and J. C. Peters, Organometallics, 2013, 32, 3053 CrossRef PubMed.
- A solution of the species that was generated in situ from the precursor 1 was observed to completely decompose under an inert atmosphere in a sealed J. Young NMR tube over the course of ∼2 hours at room temperature.
- M.-E. Moret and J. C. Peters, Angew. Chem., Int. Ed., 2011, 50, 2063 CrossRef PubMed.
- Solution symmetry has been observed in isoelectronic Rh and Ir hydride complexes at room temperature: (a) H. Kameo, Y. Hashimoto and H. Nakazawa, Organometallics, 2012, 31, 3155 CrossRef; (b) H. Kameo, Y. Hashimoto and H. Nakazawa, Organometallics, 2012, 31, 4251 CrossRef.
- Previously reported complexes supported by this ligand scaffold demonstrate that this ligand system can accommodate approximately three-fold symmetric B–H–Fe motifs. See ref. 14 and 15.
- Y. Lee, N. P. Mankad and J. C. Peters, Nat. Chem., 2010, 2, 558 CrossRef PubMed.
- For a brief discussion and data pertaining to the oxidation and functionalization reactivity of the monoanionic hydride complex 4, see the ESI.†.
- M.-E. Moret and J. C. Peters, J. Am. Chem. Soc., 2011, 133, 18118 CrossRef PubMed.
- Attempts to identify the isotopically shifted N-D stretch were unsuccessful, complicated by the very weak intensity of these vibrations and our inability to isolate 8 as an analytically pure compound.
- This observation is noted in (ref. 12a) for 10 and has been reproduced for 10-CNtBu (see ESI†).
- (a) S. N. Brown and J. M. Mayer, J. Am. Chem. Soc., 1994, 116, 2219 CrossRef; (b) S. N. Brown and J. M. Mayer, J. Am. Chem. Soc., 1996, 118, 12119 CrossRef; (c) Y. Matano, S. N. Brown, T. O. Northcutt and J. M. Mayer, Organometallics, 1998, 17, 2939 CrossRef; (d) D. S. Williams and R. R. Schrock, Organometallics, 1993, 12, 1148 CrossRef.
- (a) B. P. Jacobs, P. T. Wolczanski, Q. Jiang, T. R. Cundari and S. N. MacMillan, J. Am. Chem. Soc., 2017, 139, 12145 CrossRef PubMed; (b) T. R. Cundari, B. P. Jacobs, S. N. MacMillan and P. T. Wolczanski, Dalton Trans., 2018, 47, 6025 RSC.
- At higher temperatures, the thermal decay of the product becomes competitive and the transformation of 8 to 9 no longer exhibits isosbestic behavior.
- Direct comparison to the reported values for the Re(O) complexes reported by Mayer and coworkers is complicated by the coordination of an additional exogenous ligand; however, in the detailed examination of phenyl-to-oxo migration for those complexes, an even larger, negative entropy of activation was found (ΔS‡ = −20.5(25); see ref. 25b).
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic data, reactivity studies, crystallographic information. CCDC 1846405–1846409. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02380h |
| This journal is © The Royal Society of Chemistry 2018 |