
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiastereo- and enantioselective copper catalyzed hydroallylation of disubstituted cyclopropenes†
Heiko
Sommer
and
Ilan
Marek
 *
*
The Mallat Family Laboratory of Organic Chemistry, Schulich Faculty of Chemistry, Technion-Israel Institute of Technology, Haifa, 3200009 Israel. E-mail: chilanm@technion.ac.il
First published on 2nd July 2018
Abstract
A highly diastereo- and enantioselective protocol for the hydroallylation of 1,1- and 1,2-disubstituted cyclopropenes has been developed utilizing an in situ formed copper hydride. A variety of allyl electrophiles could be utilized yielding a diverse range of trisubstituted cyclopropanes. Finally a preliminary enantioselective variant could be established employing a recently described P-stereogenic xantphos derivative as ligand.
Introduction
The synthesis of enantioenriched cyclopropane containing scaffolds constitutes an ongoing challenge in organic synthesis despite significant advancements over the past few decades.1–4 The dominant strategies5–7 construct stereoselectively cyclopropanes from olefins and the metal-catalyzed decomposition of diazoesters,8–11 the addition of zinc carbenoid12,13 or the Michael addition followed by an 1,3-elimination reaction.14–21 An alternative approach consists in the manipulation of an existing scaffold and in this context the carbometalation of cyclopropenes has proven to be a very efficient method leading to a large variety of polysubstituted cyclopropane derivatives from a single precursor.22,23 Our group24–33 and others34–40 have contributed to the last approach by reporting several transition-metal catalyzed protocols for the addition of carbon nucleophiles to cyclopropenes with high diastereo- and enantioselectivities (Scheme 1a). However, as the incoming alkyl residues (nucleophiles) and the organometallic species are always introduced in a cis-relationship across the unsaturation, all the above discussed strategies of carbometalation provide only access to 1,2-syn functionalized cyclopropanes. The formation of 1,2-anti di-functionalized cyclopropane derivatives remained an unsolved synthetic problem (Scheme 1c). On the other hand, the asymmetric addition of nucleophiles (alkyl, vinyl, aryl, acyl, metalloids) to C2-symmetric cyclopropenyl rings led to various functionalized cyclopropanes in high diastereo- and enantiomeric ratios (Scheme 1b).41,42 Yet, the catalytic asymmetric allylation of non-functionalized cyclopropenes represent one of the missing structural scaffolds (Scheme 1c). This class of racemic structure can solely be accessed through stoichiometric addition of allylindium,43,44 allylgallium45 on 1,2-disubstituted cyclopropenes, asymmetric allylzincation of cyclopropenone ketal,46 or cyclopropenyl metal species,47 respectively. To address the issues stated above, we decided to pursue a different approach for the synthesis of 1,2-anti trisubstituted cyclopropanes. | ||
| Scheme 1 (a) Carbometalation of cyclopropenes: syn-1,2-bisalkylation reaction. (b) Asymmetric hydrofunctionalization of cyclopropenes. (c) Proposed strategy for ‘anti’-selective hydrofunctionalization and asymmetric allylation. | ||
At the outset of this study, we were interested in the development of a broadly applicable protocol that would allow us to access a variety of hydrofunctionalized cyclopropane derivatives in a concise manner. Inspired by the recent developments in copper mediated hydro- and boroallylations of olefins and alkynes,48–54 we expected that a similar strategy might be amenable to the synthesis of allylated cyclopropanes (Scheme 1c). As the initiating step involves a hydrometalation of the cyclopropene followed by an electrophilic trapping, this strategy places the two substituents in the 2,3-position in an anti-relationship. The hydroallylation of cyclopropenes would therefore provide a valuable solution for the synthesis of these desirable products.55 Based on previous observations in carbometalations of 1,2-disubstituted cyclopropenes, we expected the hydrometalation to occur similarly in a highly regio- and diastereoselective fashion. Accordingly, an in situ generated ligated copper hydride species would add across the cyclopropene double bond furnishing a cyclopropyl copper intermediate that would be trapped by an allyl electrophile. From earlier studies on copper mediated boro56- and hydrometalations,57 we anticipated that the highly reactive cyclopropyl copper intermediate might undergo a variety of undesired pathways, e.g. oxidation, dimerization or cycloaddition.
Results and discussion
Despite all potential pitfalls, we decided to investigate the above mentioned transformation and determine the most suitable reaction parameters, initially on the readily available 1,2-disubstituted cyclopropenyl ester 1a.58 Optimization of reaction conditions quickly revealed a major influence of the ligand on the diastereomeric ratio (Table 1). In the presence of a variety of bidentate phosphine ligands (L1 to L10), the hydroallylation product 3a was obtained in moderate to excellent diastereomeric ratios (Table 1, entries 1 to 10). In the presence of triphenylphoshine no conversion was observed (Table 1, entry 11) whereas in the absence of ligand (Table 1, entry 12), an equimolar mixture of the two diastereomers was formed. Stronger bases ca. NaOtBu or KOtBu only led to decomposition while weaker bases did not furnish any product (not shown in Table 1). A reduction of the amount of silane led to a pronounced decrease in the reaction rate but the quantity of LiOtBu could be reduced to 1.5 equivalents without considerable loss of reactivity. It should be noted that from the 4 possible diastereoisomers, only two were detected where the major isomer 3a, as indicated in Table 1, results from a syn-hydroallylation reaction proceeding anti to the ester group. With the optimized conditions in hand for the formation of the formally 1,2-trans-cyclopropanes with excellent diastereoselectivity (Table 1, entry 13), we surveyed a variety of different cyclopropenyl esters 1a–f with allyl electrophiles 2a–d (Scheme 2).|
|
|||
|---|---|---|---|
| Entry | Ligand | Conv. [%] | dr |
| a Cyclopropenyl ester 1a (0.2 mmol), allylOP(O)(OEt)22a, (0.4 mmol), CuI (5.0 mol%), ligand (6.0 mol%), LiOtBu (200 mol%), (MeO)2MeSiH (400 mol%), THF (0.25 M), room temperature. b On 0.5 mmol scale with LiOtBu (150 mol%). | |||
| 1 | L1 | 91 | 68![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :32:0:0 :32:0:0 |
| 2 | L2 | 100 | 86:14:0:0 |
| 3 | L3 | 100 | 89:11:0:0 |
| 4 | L4 | 100 | 85:15:0:0 |
| 5 | L5 | 100 | 87:13:0:0 |
| 6 | L6 | 100 | 93:07:0:0 |
| 7 | L7 | 100 | 93:07:0:0 |
| 8 | L8 | 100 | 91:09:0:0 |
| 9 | L9 | 100 | 91:09:0:0 |
| 10 | L10 | 74 | 82:18:0:0 |
| 11 | PPh3 | No conv. | — |
| 12 | — | 100 | 50:50:0:0 |
| 13 | L8 | 100b (62% isol.) | 95:05:0:0 |

 | ||
Scheme 2 Copper-catalyzed hydroallylation of 1,3-disubstituted cyclopropenes 1a–f. Reaction conditions: 1a–f (0.5 mmol), H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(R1)CH2OP(O)(OEt)22a–d, (1.0 mmol), CuI (5.0 mol%), L8 (6.0 mol%), LiOtBu (150 mol%), (MeO)2MeSiH (400 mol%), THF (0.25 M), room temperature. Diastereomeric ratios determined by GC. C(R1)CH2OP(O)(OEt)22a–d, (1.0 mmol), CuI (5.0 mol%), L8 (6.0 mol%), LiOtBu (150 mol%), (MeO)2MeSiH (400 mol%), THF (0.25 M), room temperature. Diastereomeric ratios determined by GC. | ||
We found that besides allyl phosphate 2a, various substitutions at the 2-position of the allylic fragment (2b–d), including sterically encumbered groups, are well tolerated (Scheme 2, 3b–d). It should be noted that substitutions at the terminal position of the allyl fragment and electron-rich arenes at the 2-position are not tolerated. On the cyclopropene moiety, longer (Scheme 2, 3e–j and 3m,n) or branched (Scheme 2, 3k,l) alkyl residues are well tolerated. Furthermore, a TIPS protected alcohol (3g,h), a terminal chlorine (3i,j) or internal olefins (3m,n) do not impede the reaction either. Finally, the reaction could also be conducted on a 10-fold scale giving access to more than 1 gram of allylated cyclopropane in similar yields (Scheme 2, 3a and 3k). All transformations proceeded with a good to excellent diastereoselectivity leading to a unique trans-relationship between the allyl unit and all the substituents on the cyclopropyl ring. The relative configuration of 3g has been determined by comparison with an authentic sample independently prepared by the reported allylindation strategy43 and by analysis of NOE experiments on 3a (see the ESI†). The configurations of all other products were assigned by analogy.
Having established an easy access to the general 1,2-anti-1,3-syn-trisubstituted cyclopropyl framework 3, we sought to expand the scope to the formation of 1,1,2-trisubstituted cyclopropanes 5.
In contrast to the disubstituted cyclopropenes 1 where we only had to develop a diastereoselective protocol, 3,3-disubstituted cyclopropenes 4 required the additional development of an enantioselective protocol. At the outset we decided first to develop a reliable diastereoselective approach which could eventually be transformed into a catalytic, asymmetric variant. As a model substrate, we investigated the readily accessible cyclopropane 4a which have been extensively studied in stereoselective carbometalations.59,60 Employing xantphos as ligand, we quickly learned that this substrate class posed considerably harder challenges as the choice of copper salt, ligand and silane exhibited a more pronounced influence on the reaction outcome (Table 2). Dimethoxymethylsilane proved again to be the silane of choice (Table 2, entries 1–7). Additionally, lowering the concentration had a positive effect on both yield and diastereoselectivity. Copper iodide provided optimal results among all copper salts screened and, after further lowering catalyst loading to 5 mol%, we could isolate the product in 80% yield as a single diastereoisomer (Table 2, entry 16). The relative configuration of 5a was determined by NOE experiment and confirmed that the allyl substituent is introduced in syn to the Me group. With the optimized reaction conditions in hand, we have subsequently evaluated the scope of cyclopropenes 4 and allyl electrophiles 2 that could be used in this transformation. Simple modifications on the arene moiety were well tolerated as well as spiro- or naphthalene substituted cyclopropenes (Scheme 3, 5a–e). Surprisingly, electron-rich arenes or longer alkyl chains than methyl resulted in no detectable product formation. Furthermore, as expected 1,1-dialkyl cyclopropene furnished the product 5f with low diastereoselectivity (as also observed in the copper-catalyzed carbozincation and carbomagnesiation reactions).32 Various substitutions at the 2-position of the allyl phosphate 2 were again well tolerated. Simple methyl and more complex linear and branched alkyl chains as well as an arene and a silane cleanly underwent this transformation (Scheme 3, 5g–n). Again, substituting the 3-position of the electrophile or embedding electron rich substituents on the olefin prohibited product formation. Finally, this reaction could be scaled to give the desired cyclopropane in good yield on a 5.0 mmol scale for 5a. Additionally, we found that the electrophile can be utilized as the limiting component provided that the diethyl phosphate is replaced by the more reactive bis(trichloroethyl) phosphate leaving group and similar yields and diastereoselectivities were obtained for three representative examples (5a, 5k and 5n).
|
|
|||||
|---|---|---|---|---|---|
| Entry | CuX | Silane | Conc. | Yielde [%] | dr |
| a 4a (0.2 mmol), allylOP(O)(OEt)22a, (0.4 mmol), CuX (10 mol%), xantphos (12 mol%), LiOtBu (200 mol%), (MeO)2MeSiH (400 mol%), THF, room temperature. b CuI (5.0 mol%), xantphos (6.0 mol%). c (MeO)2MeSiH (200 mol%). d On 5.0 mmol scale. e GC yield using tetradecane as internal standard. | |||||
| 1 | CuI | PMHS | 0.33 M | 6 | 12:1 |
| 2 | CuI | (TMSO)2MeSiH | 0.33 M | 47 | 18:1 |
| 3 | CuI | TMS3SiH | 0.33 M | 7 | 7:1 |
| 4 | CuI | Me2PhSiH | 0.33 M | 26 | 20:1 |
| 5 | CuI | PhSiH3 | 0.33 M | 59 | 10:1 |
| 6 | CuI | (MeO)2MeSiH | 0.33 M | 62 | 17:1 |
| 7 | CuI | (EtO)2MeSiH | 0.33 M | 63 | 14:1 |
| 8 | CuBr | (MeO)2MeSiH | 0.25 M | 84 | 21:1 |
| 9 | CuI | (MeO)2MeSiH | 0.25 M | 87 | 24:1 |
| 10 | Cu(OAc)2 | (MeO)2MeSiH | 0.25 M | 41 | 10:1 |
| 11 | CuOAc | (MeO)2MeSiH | 0.25 M | 74 | 12:1 |
| 12 | CuTC | (MeO)2MeSiH | 0.25 M | 58 | 13:1 |
| 13 | CuIb | (MeO)2MeSiH | 0.25 M | 83 | >50:1 |
| 14 | CuI | (MeO)2MeSiHc | 0.25 M | 74 | >50:1 |
| 15 | CuIb | (MeO)2MeSiHc | 0.25 M | 68 | 21:1 |
| 16 | CuId | (MeO)2MeSiH | 0.25 M | 80 (isol.) | >50:1 |

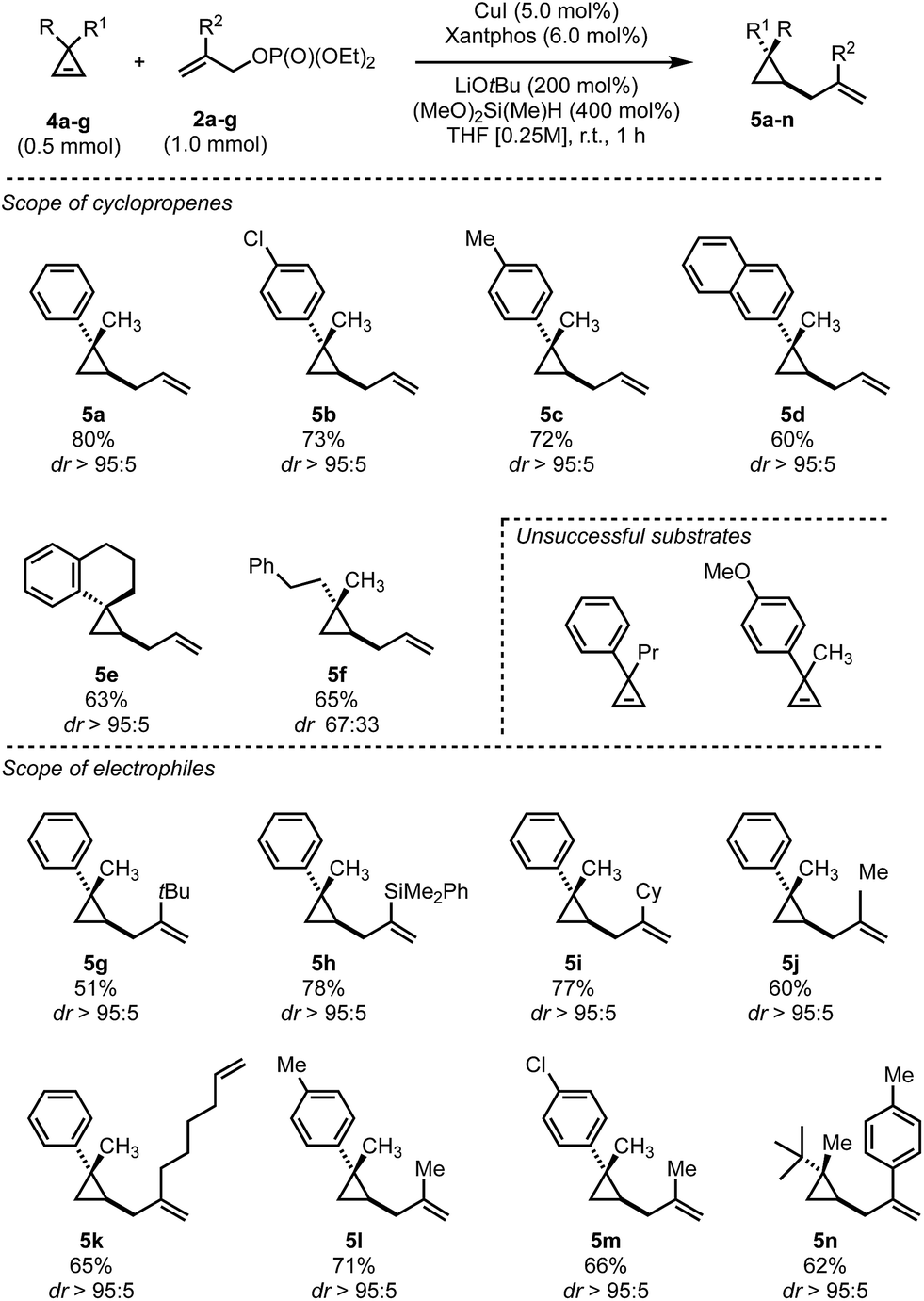 | ||
| Scheme 3 Substrate scope of copper catalyzed hydroallylation of 3,3-disubstituted cyclopropenes 4a–g. Reaction conditions: 4a–g (0.5 mmol), allylOP(O)(OEt)2, (1.0 mmol), CuI (5.0 mol%), xantphos (6.0 mol%), LiOtBu (200 mol%), (MeO)2MeSiH (400 mol%), THF (0.25 M), room temperature; 5i, 5k and 5n were obtained from the corresponding (bistrichloroethyl)phosphates, respectively. | ||
Having established this new diastereoselective protocol, we felt confident to identify conditions that would allow us to access these substrates through asymmetric catalysis. The enantioselective, copper catalyzed hydroallylation has recently received considerable attention and a variety of catalytic systems have been established.48,49 Furthermore, our groups has shown that different nucleophiles can undergo enantioselective copper-catalyzed asymmetric additions to this class of substrates.32
After extensive screening of commercially available ligands, copper salts and bases, we found that none of the combinations delivered the product in a satisfactory fashion (see vide infra). Although good yields were obtained at room temperature, only low levels of enantioinduction were observed. Lowering the reaction temperature led to considerable increase in stereoselectivity at the expense of dramatic loss of yield. This observation can be explained by competing undesired pathways of the intermediate cyclopropyl copper species. We surmised that the electrophile had to be rendered more reactive in order to outcompete potential side-reactions. After screening a variety of different allylating reagents in the presence of (S)-DTBM-SEGPHOS as ligand, we eventually found that the bis(trichloroethyl) phosphate leaving group proved to be the most effective. Nevertheless, we were not able to obtain high levels of enantioinduction with concomitant high yields.
As we obtained high yields in the diastereoselective protocol in the presence of xantphos as ligand, we decided to test the possibility to employ a P-chiral analog of this parent ligand. Recently an optimized route towards bidentate P-stereogenic ligands was disclosed.61 Under the initial reaction conditions, these ligands did not provide any improvement but we found that by decreasing the reaction temperature and utilizing the allyl electrophile as the limiting reagent, high levels of enantioinduction with good yields could be achieved (Table 3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | CuX | Ligand | T [°C] | er | Yieldf [%] |
| a 4a (0.2 mmol), allylOP(O)(OR)22a (0.4 mmol), CuX (5.0 mol%), ligand (6.0 mol%), LiOtBu (200 mol%), (MeO)2MeSiH (400 mol%), THF (0.25 M). b AllylOP(O)(OEt). c AllylOP(O)(OCH2CCl3)2. d 4a (200 mol%). e 4a (300 mol%). f GC yield using tetradecane as internal standard. | |||||
| 1b | CuI | L11 | 25 | 62:38 |
41 |
| 2b | CuI | L12 | 25 | 64:36 |
56 |
| 3b | CuI | L13 | 25 | 72:28 |
54 |
| 4c | CuI | L13 | 25 | 79:21 |
51 |
| 5c | CuTC | L13 | 25 | 86:14 |
33 |
| 6c | Cu(OAc)2 | L13 | 25 | 86:14 |
34 |
| 7c,d | Cu(OAc)2 | L15 | 25 | 94:06 |
30 |
| 8c,d | Cu(OAc)2 | L13 | 25 | 82:18 |
69 |
| 9c,e | Cu(OAc)2 | L13 | 0 | 87:13 |
72 |
| 10c,e | CuI | L14 | −20 | 94:06 |
38 |
| 11c,e | Cu(OAc)2 | L14 | −20 | 96:04 |
31 |
| 12c,e | Cu(OAc)2 | L13 | −20 | 92:08 |
70 |

To test the applicability of this preliminary finding we applied this protocol to the synthesis on two previously described examples (Scheme 4).
 | ||
| Scheme 4 Preliminary results of the enantioselective hydroallylation of 3,3-disubstituted cyclopropene 4a. | ||
Nevertheless, we could show that an enantioselective approach is feasible and can allow for the synthesis of enantioenriched 1,1,2-trisubstituted cyclopropanes.
Conclusions
In summary we described a broadly applicable copper catalyzed hydroallylation of 3,3- and 1,3-disubstituted cyclopropenes. The present method features an in situ formed copper hydride species that undergoes stereoselective hydrometalation of cyclopropenes and is trapped with a variety of electrophilic allylphosphates. This protocol allows for the diastereoselective synthesis of 1,2,3- and 1,1,2-trisubstituted cyclopropanes in good to high yields with good to excellent diastereoselectivity. The robustness of this method has been showcased in the synthesis of more than 30 examples and scaled up to >1 g of product in each class of substrates. Finally, we established preliminary conditions that give rise to enantioenriched products utilizing a P-chiral xantphos derivative.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the European Research Council under the European Community's Seventh Framework Program (ERC grant agreement no. 338912) and by the Israel Science Foundation administrated by the Israel Academy of Sciences and Humanities (140/12). H. S. acknowledges the Max-Planck-Society for a Minerva Fellowship. I. M. is holder of the Sir Michael and Lady Sobell Academic Chair.Notes and references
- H. N. C. Wong, M. Y. Hon, C. W. Tse, Y. C. Yip, J. Tanko and T. Hudlicky, Chem. Rev., 1989, 89, 165–198 CrossRef.
- A.-H. Li, L.-X. Dai and V. K. Aggarwal, Chem. Rev., 1997, 97, 2341–2372 CrossRef PubMed.
- H.-U. Reissig and R. Zimmer, Chem. Rev., 2003, 103, 1151–1196 CrossRef PubMed.
- C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651–11679 CrossRef PubMed.
- H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977–1050 CrossRef PubMed.
- A. B. Charette and A. Beauchemin, in Organic Reactions, John Wiley & Sons, Inc., 2004, vol. 58, pp. 330–415 Search PubMed.
- G. Bartoli, G. Bencivenni and R. Dalpozzo, Synthesis, 2014, 46, 979–1029 CrossRef.
- A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981–10080 CrossRef PubMed.
- H. M. L. Davies and D. Morton, Chem. Soc. Rev., 2011, 40, 1857–1869 RSC.
- D. Chao, W. Li-Jia, Z. Jun and T. Yong, Angew. Chem., Int. Ed., 2012, 51, 11620–11623 CrossRef PubMed.
- L. Jun, L. Sai-Hu, X. Hu, Z. You-Yun, S. Xiu-Li, Z. Yue, Z. Xiao-Guang and T. Yong, Angew. Chem., Int. Ed., 2012, 51, 8838–8841 CrossRef PubMed.
- A. B. Charette and J. Lemay, Angew. Chem., Int. Ed., 1997, 36, 1090–1092 CrossRef.
- L. E. Zimmer and A. B. Charette, J. Am. Chem. Soc., 2009, 131, 15624–15626 CrossRef PubMed.
- X. Xie, G. Yue, S. Tang, X. Huo, Q. Liang, X. She and X. Pan, Org. Lett., 2005, 7, 4057–4059 CrossRef PubMed.
- R. K. Kunz and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 3240–3241 CrossRef PubMed.
- S. Norsikian, I. Marek, S. Klein, J. F. Poisson and J. F. Normant, Chem.–Eur. J., 1999, 5, 2055–2068 CrossRef.
- S. L. Riches, C. Saha, N. F. Filgueira, E. Grange, E. M. McGarrigle and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 7626–7630 CrossRef PubMed.
- V. K. Aggarwal and E. Grange, Chem.–Eur. J., 2006, 12, 568–575 CrossRef PubMed.
- V. K. Aggarwal, E. Alonso, G. Fang, M. Ferrara, G. Hynd and M. Porcelloni, Angew. Chem., Int. Ed., 2001, 40, 1433–1436 CrossRef PubMed.
- V. K. Aggarwal, H. W. Smith, R. V. H. Jones and R. Fieldhouse, Chem. Commun., 1997, 1785–1786 RSC.
- V. K. Aggarwal, H. W. Smith, G. Hynd, R. V. H. Jones, R. Fieldhouse and S. E. Spey, J. Chem. Soc., Perkin Trans. 1, 2000, 3267–3276 RSC.
- D. S. Müller and I. Marek, Chem. Soc. Rev., 2016, 45, 4552–4566 RSC.
- I. Marek, S. Simaan and A. Masarwa, Angew. Chem., Int. Ed., 2007, 46, 7364–7376 CrossRef PubMed.
- P. O. Delaye, D. Didier and I. Marek, Angew. Chem., Int. Ed., 2013, 52, 5333–5337 CrossRef PubMed.
- D. Didier, P. O. Delaye, M. Simaan, B. Island, G. Eppe, H. Eijsberg, A. Kleiner, P. Knochel and I. Marek, Chem.–Eur. J., 2014, 20, 1038–1048 CrossRef PubMed.
- M. Simaan, P. O. Delaye, M. Shi and I. Marek, Angew. Chem., Int. Ed., 2015, 54, 12345–12348 CrossRef PubMed.
- F. G. Zhang, G. Eppe and I. Marek, Angew. Chem., Int. Ed., 2016, 55, 714–718 CrossRef PubMed.
- M. Preshel-Zlatsin, F.-G. Zhang, G. Eppe and I. Marek, Synthesis, 2016, 48, 3279–3286 CrossRef.
- S. R. Roy, D. Didier, A. Kleiner and I. Marek, Chem. Sci., 2016, 7, 5989–5994 RSC.
- L. Dian, D. S. Müller and I. Marek, Angew. Chem., Int. Ed., 2017, 56, 6783–6787 CrossRef PubMed.
- M. Simaan and I. Marek, Angew. Chem., Int. Ed., 2018, 57, 1543–1546 CrossRef PubMed.
- D. S. Müller and I. Marek, J. Am. Chem. Soc., 2015, 137, 15414–15417 CrossRef PubMed.
- F.-G. Zhang and I. Marek, J. Am. Chem. Soc., 2017, 139, 8364–8370 CrossRef PubMed.
- L.-a. Liao and J. M. Fox, J. Am. Chem. Soc., 2002, 124, 14322–14323 CrossRef PubMed.
- X. Liu and J. M. Fox, J. Am. Chem. Soc., 2006, 128, 5600–5601 CrossRef PubMed.
- N. Yan, X. Liu and J. M. Fox, J. Org. Chem., 2008, 73, 563–568 CrossRef PubMed.
- V. Tarwade, X. Liu, N. Yan and J. M. Fox, J. Am. Chem. Soc., 2009, 131, 5382–5383 CrossRef PubMed.
- K. Krämer, P. Leong and M. Lautens, Org. Lett., 2011, 13, 819–821 CrossRef PubMed.
- X. Xie and J. M. Fox, Synthesis, 2013, 45, 1807–1814 CrossRef.
- T. Nakano, K. Endo and Y. Ukaji, Synlett, 2015, 26, 671–675 CrossRef.
- M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2003, 125, 7198–7199 CrossRef PubMed.
- M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2004, 126, 3688–3689 CrossRef PubMed.
- S. Araki, H. Nakano, K. Subburaj, T. Hirashita, K. Shibutani, H. Yamamura, M. Kawai and Y. Butsugan, Tetrahedron Lett., 1998, 39, 6327–6330 CrossRef.
- T. Hirashita, F. Shiraki, K. Onishi, M. Ogura and S. Araki, Org. Biomol. Chem., 2007, 5, 2154–2158 RSC.
- S. Araki, T. Tanaka, T. Hirashita and J.-i. Setsune, Tetrahedron Lett., 2003, 44, 8001–8003 CrossRef.
- M. Nakamura, H. Isobe and E. Nakamura, Chem. Rev., 2003, 103, 1295–1326 CrossRef PubMed.
- A. Levin and I. Marek, Chem. Commun., 2008, 4300–4302 RSC.
- J. T. Han, W. J. Jang, N. Kim and J. Yun, J. Am. Chem. Soc., 2016, 138, 15146–15149 CrossRef PubMed.
- Y.-M. Wang and S. L. Buchwald, J. Am. Chem. Soc., 2016, 138, 5024–5027 CrossRef PubMed.
- W. J. Jang, J. T. Han and J. Yun, Synthesis, 2017, 49, 4753–4758 CrossRef.
- N. Kim, J. T. Han, D. H. Ryu and J. Yun, Org. Lett., 2017, 19, 6144–6147 CrossRef PubMed.
- J. Lee, S. Torker and A. H. Hoveyda, Angew. Chem., Int. Ed., 2017, 56, 821–826 CrossRef PubMed.
- M. Mailig, A. Hazra, M. K. Armstrong and G. Lalic, J. Am. Chem. Soc., 2017, 139, 6969–6977 CrossRef PubMed.
- G. Xu, H. Zhao, B. Fu, A. Cang, G. Zhang, Q. Zhang, T. Xiong and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 13130–13134 CrossRef PubMed.
- A. Reichelt and S. F. Martin, Acc. Chem. Res., 2006, 39, 433–442 CrossRef PubMed.
- A. Parra, L. Amenós, M. Guisán-Ceinos, A. López, J. L. García Ruano and M. Tortosa, J. Am. Chem. Soc., 2014, 136, 15833–15836 CrossRef PubMed.
- W. M. Sherrill and M. Rubin, J. Am. Chem. Soc., 2008, 130, 13804–13809 CrossRef PubMed.
- N. Petiniot, A. J. Anciaux, A. F. Noels, A. J. Hubert and P. Teyssié, Tetrahedron Lett., 1978, 19, 1239–1242 CrossRef.
- L. Dian, D. S. Müller and I. Marek, Angew. Chem., Int. Ed., 2017, 56, 6783–6787 CrossRef PubMed.
- L. Dian and I. Marek, Angew. Chem., Int. Ed., 2018, 57, 3682–3686 CrossRef PubMed.
- J. Holz, K. Rumpel, A. Spannenberg, R. Paciello, H. Jiao and A. Börner, ACS Catal., 2017, 7, 6162–6169 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02085j |
| This journal is © The Royal Society of Chemistry 2018 |