
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilyl-mediated photoredox-catalyzed Giese reaction: addition of non-activated alkyl bromides†
Abdellatif
ElMarrouni‡
 *a,
Casey B.
Ritts
b and
Jaume
Balsells‡
*b
*a,
Casey B.
Ritts
b and
Jaume
Balsells‡
*b
aDepartment of Discovery Chemistry, MRL, Merck & Co., Inc., 770 Sumneytown Pike, West Point, Pennsylvania 19486, USA. E-mail: abdellatif.el.marrouni@merck.com
bDepartment of Process Research & Development, MRL, Merck & Co., Inc., 770 Sumneytown Pike, West Point, Pennsylvania 19486, USA. E-mail: jaume.balsells@merck.com
First published on 11th July 2018
Abstract
The emergence of photoredox catalysis has enabled the discovery of mild and efficient conditions for the generation of a variety of radical reaction platforms. Herein is disclosed the development of a conjugate addition reaction of non-activated alkyl bromides to Michael acceptors under visible-light photoredox catalysis. Optimization of the reaction was achieved using high-throughput experimentation (HTE) tools to enable the identification of mild, general and practical reaction conditions. A diverse set of alkyl bromides was successfully added to cyclic or acyclic α,β-unsaturated esters and amides. The features of this transformation allowed also access to a key intermediate of Vorinostat®, an HDAC inhibitor used to fight cancer and HIV.
Introduction
The development of general and modular synthetic methods requiring minimal activating groups for reactivity has the potential to dramatically expand the Medicinal Chemistry toolbox and accelerate the discovery of new small molecule therapeutics.1 As part of this strategy, the formation of C(sp3)–C(sp3) bonds remains one of the most challenging synthetic transformations2 with many bond disconnections still lacking robust methodology, a wide substrate scope and functional group tolerance. The conjugate addition reaction exemplifies this vacancy,3 suffering from limiting reaction conditions, mostly related to the use of strong bases, that obstruct its use in parallel or library synthesis for Medicinal Chemistry applications. Recent advances in photoredox catalysis have demonstrated the generation of carbon-centered radicals under mild conditions,4 which has substantially filled the existing methodology gaps on conjugate addition reactions to electron-deficient olefins, also known as the Giese reaction.5 Several research groups have recently reported on the generation of carbon-centered radicals from a diverse set of functional handles such as alkyl trifluoroborate salts,6 carboxylic acids,7 secondary and tertiary alcohols8 or organosilicates9 and demonstrated their utility in addition reactions to different alkenes (Scheme 1). While these synthetic methods allow access to a broad diversity of substrates, there is still an opportunity to expand the scope of this synthetic transformation to additional families of organic compounds that are widely available as building blocks in Medicinal Chemistry such as haloalkanes.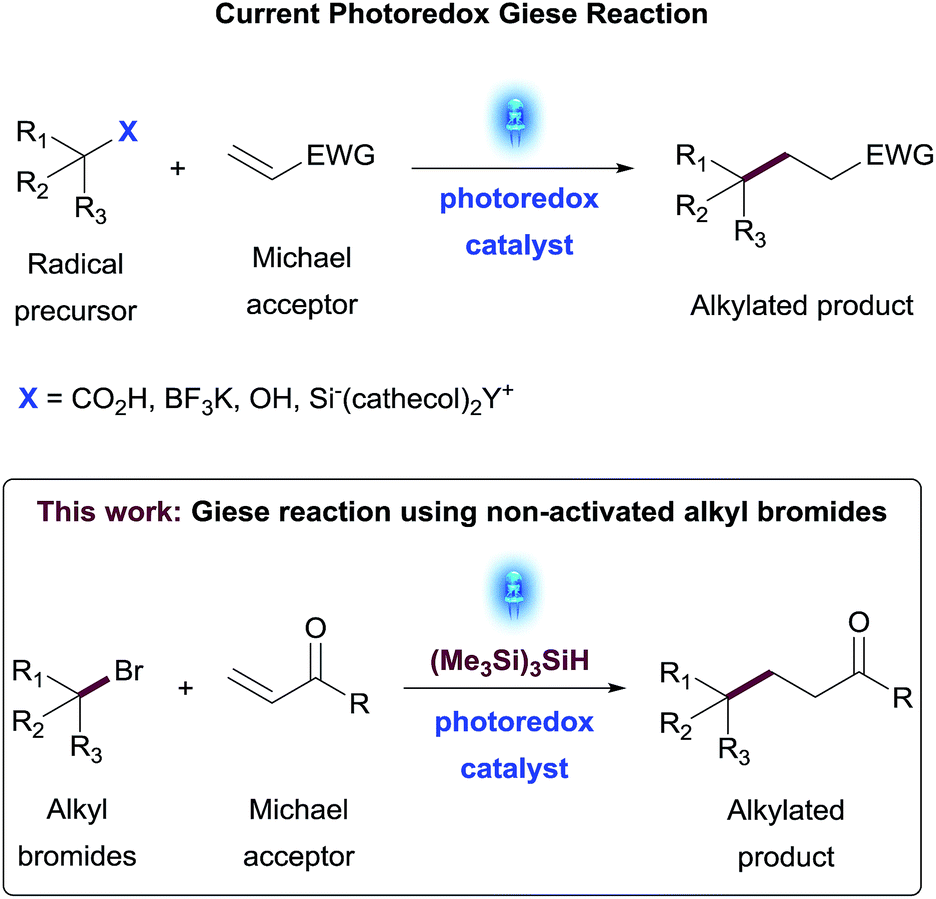 | ||
| Scheme 1 Synthesis of C–C bonds through photocatalytic Giese reaction. | ||
The thermal generation of primary and other radicals from alkyl bromides mediated by tributyltin hydride or tris(trimethylsilyl)silane as radical initiators have been widely studied.10 The radical reactions initiated using triethylborane and oxygen have been also described.11 Reactions mediated by metals such as indium, nickel, samarium, chromium, and zinc additives to promote the reduction of halogen-containing derivatives have been also reported.12
More recently, seminal work by MacMillan and co-workers13 demonstrated the generation of alkyl radicals from alkyl bromides through halogen-atom abstraction via a photocatalytically generated tris(trimethylsilyl)silyl radical species. With these considerations in mind, we became interested in developing a method for the generation of non-activated alkyl radicals in a practical and efficient manner to use them as nucleophiles in a photoredox Giese-type transformation. This reaction would allow the construction of new C(sp3)–C(sp3) bonds and provide access to a wide range of valuable compounds including clinical candidates and analogues thereof.14 Herein, we describe the development, optimization and scope of a photoredox promoted Giese addition of alkylbromides to α,β-unsaturated amides and esters.
Reaction optimization
To evaluate this transformation, we first turned our attention to the reaction of phenylacrylamide 1 and 4-bromopiperidine derivative 2 as a model system. Initially, these reactants were subjected to the silyl mediated photoredox catalysis reaction conditions, using 1 mol% Ir[dF(CF3)ppy]2(dtbbpy)PF6 as photocatalyst, equimolar amounts of (Me3Si)3SiH and Na2CO3 in MeCN as solvent. Under these initial set of conditions, the addition of alkylbromide 2 to amide 1 furnished the desired product 3 in 35% yield amongst various side products. This result reinforced our photoredox mechanistic hypothesis. However, to make sure the reaction occurred under photoredox catalysis, we performed a series of control reactions wherein we independently set up reactions in the absence of light, photocatalyst and/or silane (entries 2–4, Table 1). In all cases, no reaction was observed and starting materials were recovered. Interestingly, a test reaction without base resulted in the formation of the coupled product 3 in 30% yield together with a significant increase in side-products (entry 5, Table 1).|
|
||||
|---|---|---|---|---|
| Entry | Conditions | Yield 1b | Yield 2b | Yield 3b |
| a N-Phenylmetacrylamide (1) (1.0 equiv.), (4-bromopiperidin-1-yl)(phenyl)methanone (2) (1.0 equiv.), Na2CO3 (2.0 equiv.), (Me3Si)3SiH (1.0 equiv.) and Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1 mol%) in MeCN, Aldrich® Micro Photochemical Reactor (ring) for 16 h. b Assay yields for product and remaining starting materials were determined using HPLC techniques. | ||||
| 1 | As shown | 0% | 0% | 35% |
| 2 | No light | 100% | 100% | 0% |
| 3 | No silane | 100% | 100% | 0% |
| 4 | No [Ir] cat. | 100% | 100% | 0% |
| 5 | No base | 8% | 21% | 30% |
Ensuing optimization of the main reaction parameters aimed at improving the ratio of desired product to side-products while reaching complete conversion. For this optimization study, we applied High-Throughput Experimentation (HTE)15 techniques using the Aldrich® Micro Photochemical Reactor which allows for up to 16 reactions to be run in parallel under uniform lighting conditions. Both the amounts of desired product as well as unreacted starting materials were measured in these experiments as this provided a better understanding of mass balance and overall reaction performance.
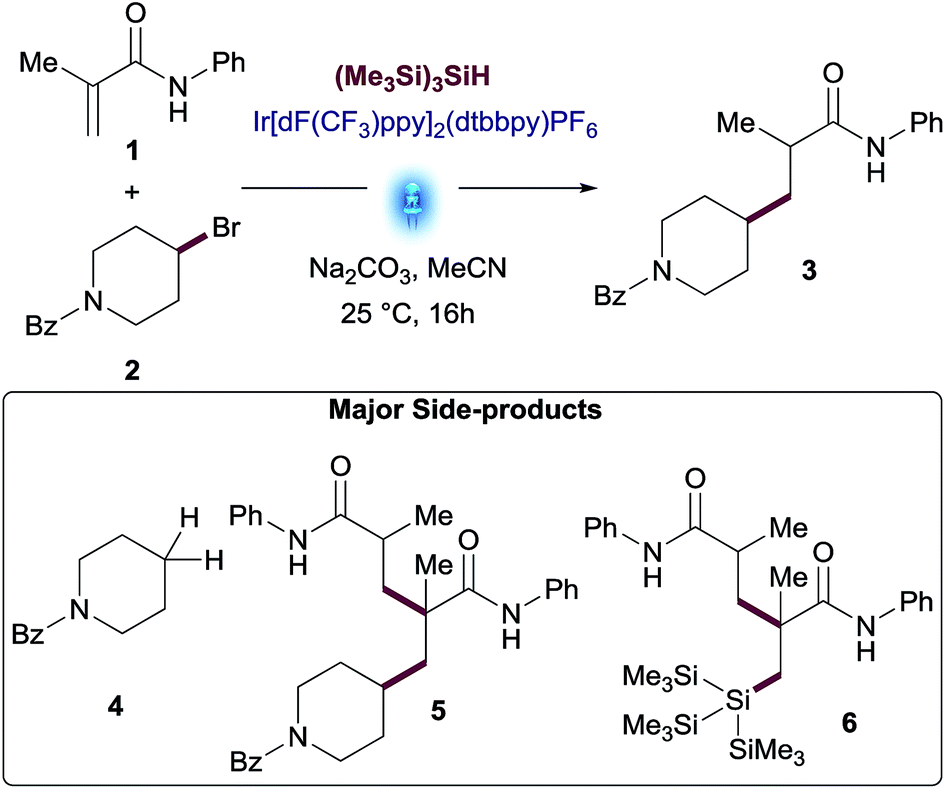
An initial screen of photocatalysts across different solvents (Fig. 1) revealed that Ir[dF(CF3)ppy]2(dtbbpy)PF6 was a superior catalyst in the solvents tested. This screening also exposed MeOH (35% yield) and MeCN (38% yield) as the two ideal solvents. Although reactions in THF or 2-propanol (IPA) performed similarly to MeCN (36% and 38% yield respectively), starting materials were completely consumed in these reactions and there was a significant increase of the amount of side-products.
 | ||
| Fig. 1 Photocatalyst and solvent screening: N-phenylmetacrylamide (1), (4-bromopiperidin-1-yl)(phenyl)methanone (2), Na2CO3 (2.0 equiv.), (Me3Si)3SiH, photocatalyst (1 mol%), Aldrich® Micro Photochemical Reactor (ring) for 16 h. [a] Assay yields for desired product was determined using standard HPLC techniques. | ||
Having unveiled an optimized photocatalyst and two preferred solvents, we decided to study the effect of bases in the reaction (see Table S2 in the ESI†). While the reaction proceeded in the absence of base, higher yields were obtained when 2 equiv. of inorganic bases such as Na2CO3 (49% yield), Li2CO3 (51% yield) or KF (52% yield) were used. We selected Na2CO3 since it is more commonly available in chemistry laboratories. Organic bases (2,6-lutidine, DIPEA, Et3N, Barton's base16) were significantly less efficient and led to an increase of side-products (see Table S2 in the ESI†). In all cases, MeOH was superior to MeCN and thus we pursued the rest of the optimization study in MeOH.
A further round of optimization focused on the impact of variable amounts of (Me3Si)3SiH (see Table S3 in the ESI†). While the reaction did not occur without silane, an excess of this reagent proved detrimental for this transformation, leading to a significant increase of dehalogenated side-product 4 and conjugate addition of silane to the Michael acceptor 6.
The best results were observed using between 0.5 equiv. to 1.0 equiv. of (Me3Si)3SiH. Yields of desired product declined when using more than 1.0 equiv. of silane, as larger amounts of side-products were observed. This non-obvious behavior prompted us to explore the performance of the reaction using variable amounts of the two key reactants, hypothesizing that excess amounts of one of the reactants might favor the desired reaction pathway over side-reactions, thus maximizing the yield.
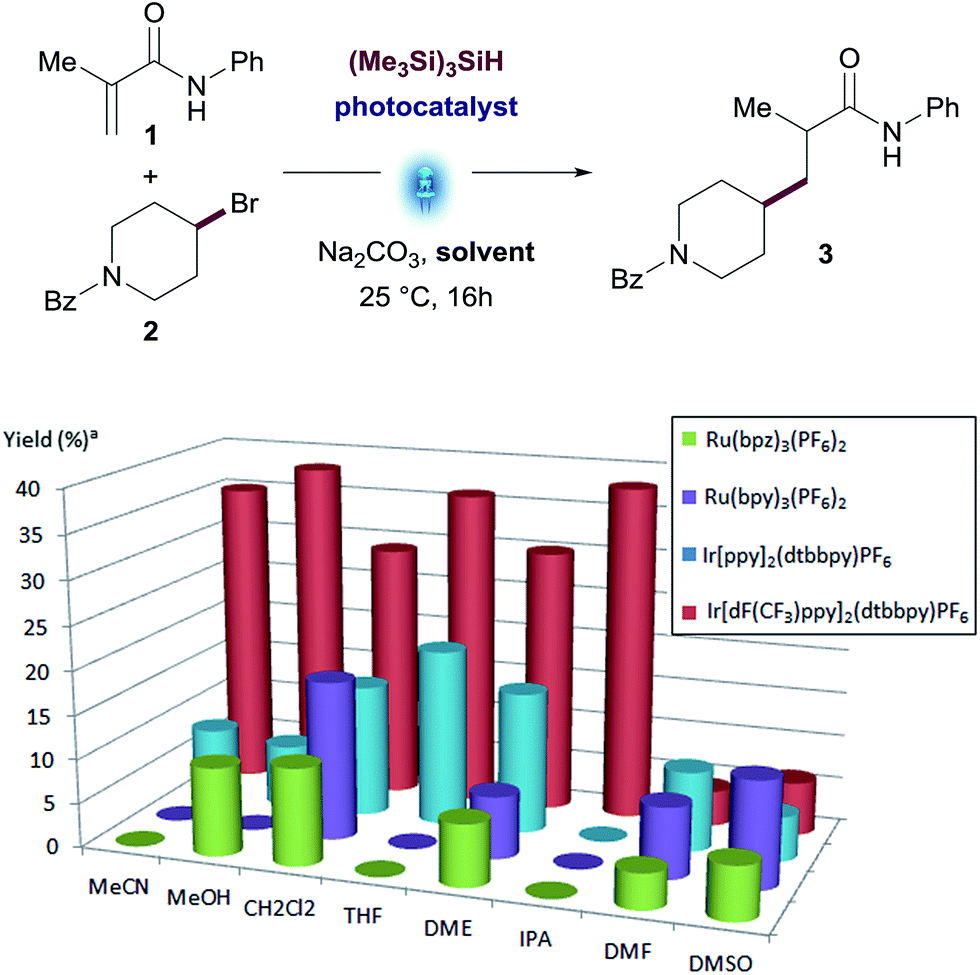
We therefore examined the ratio of Michael acceptor 1 and alkyl bromide 2 in presence of different amounts of silane (see Fig. 2 and Table S4 in the ESI†). This screening revealed that a slight excess of alkyl bromide 2 (1.5 to 3 equiv.) led to the best yields with little impact of the amount of silane. An excess of Michael acceptor 1 was deleterious in all cases. The use of 0.78 equiv. of (Me3Si)3SiH showed full consumption of the limiting reagent and a cleaner reaction profile (74% yield with 3 equiv. of bromide 2).
 | ||
| Fig. 2 Reactant ratio screening: N-phenylmetacrylamide (1), (4-bromopiperidin-1-yl)(phenyl)methanone (2), Na2CO3 (2.0 equiv.), (Me3Si)3SiH, Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1 mol%) in MeOH, Aldrich® Micro Photochemical Reactor (ring) for 16 h. [a] Assay yields for desired product was determined using standard HPLC techniques. | ||
Before engaging in the study of the scope of this transformation, we decided to evaluate the performance of this reaction using different visible-light sources. This is an important factor to ensure reproducibility of this methodology since light source setups differ from one research laboratory to another. We used three different devices differing in light intensity: (i) the Aldrich® Micro Photochemical Reactor; (ii) the previously reported setup based on Kessil® lamps with fans; and (iii) the integrated photoreactor recently developed by Merck scientists in collaboration with MacMillan's group.17 Reactions were run under the optimized conditions and monitored over 24 h. While all reactions reached similar yields on the three devices tested, drastic differences were observed in the reaction rates. Remarkably, the reaction was complete after 30 minutes in the integrated photoreactor, compared to the 24 h required for the Aldrich® Micro Photochemical Reactor (Fig. 3). Changes in the reaction rate are attributed to the different intensity of the light sources, which has been optimized in the integrated photoreactor.
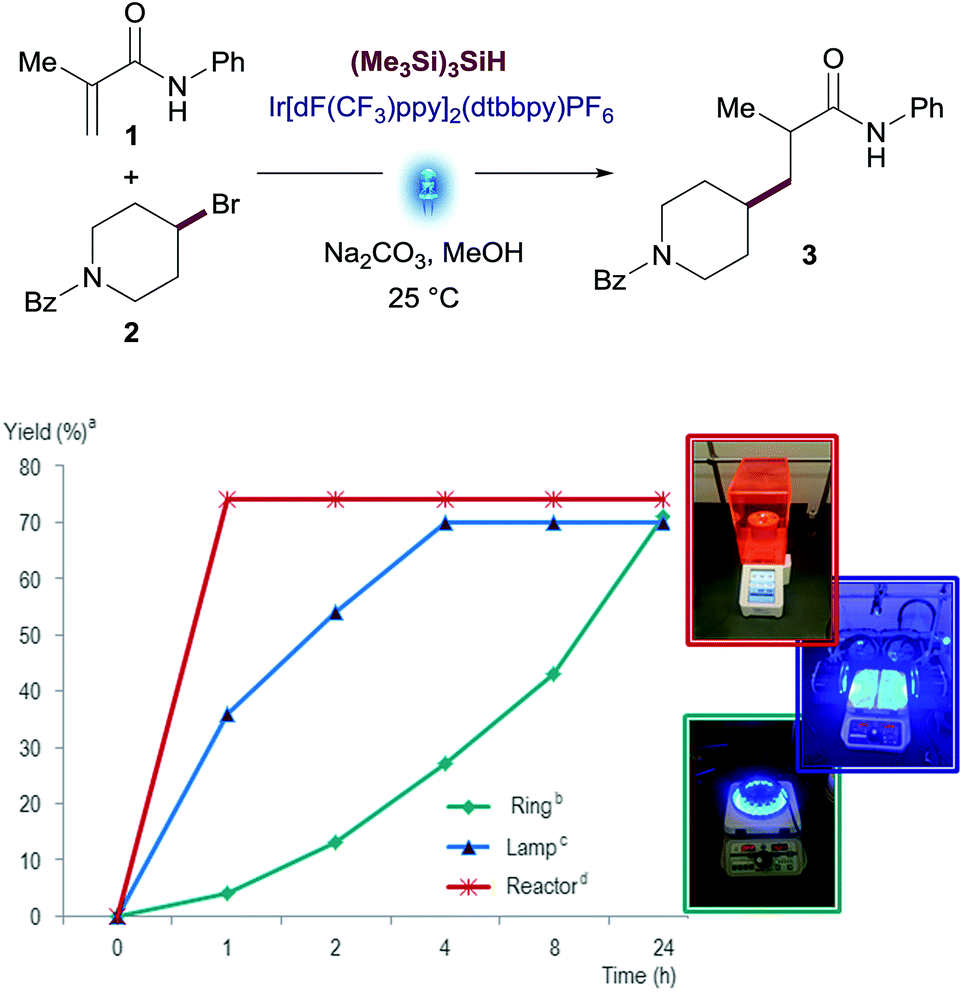 | ||
| Fig. 3 Visible-light source comparison: N-phenylmetacrylamide (1) (1.0 equiv.), (4-bromopiperidin-1-yl)(phenyl)methanone (2) (1.5 equiv.), Na2CO3 (2.0 equiv.), (Me3Si)3SiH, Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1 mol%) in MeOH. [a] Assay yields for desired product was determined using standard HPLC techniques. [b] Ring = Aldrich® Micro Photochemical Reactor. [c] Lamp = Kessil® A160WE Tuna Blue Light. [d] Reactor = 13.2W Merck Photoreactor. | ||
Reaction scope
Using the optimized conditions and performing the reactions in the integrated photoreactor, we began to explore the scope of the reaction focusing on a variety of primary, secondary and tertiary alkyl bromides (Table 2). We were pleased to find that primary alkyl bromides provided the desired products 7–15 in synthetically useful yields. The reaction conditions successfully tolerated a variety of substituents and functional groups including cyclopropyl, ester, primary Boc amine and phosphonate. Although most products were isolated in moderate yields, this transformation overcomes one of the main synthetic gaps of the existing photoredox conjugate addition methods. In addition, this method allowed access to biologically relevant phosphonates 13 and 14 with antimalarial properties in a single step.18 Unactivated secondary alkyl bromides generate more stable radicals than primary ones which led to higher isolated yields for compounds 16–22 (Table 2). Among these products, several unfunctionalized cyclic and linear alkanes were installed successfully in addition to small cyclic ethers such as tetrahydropyran and oxetane groups.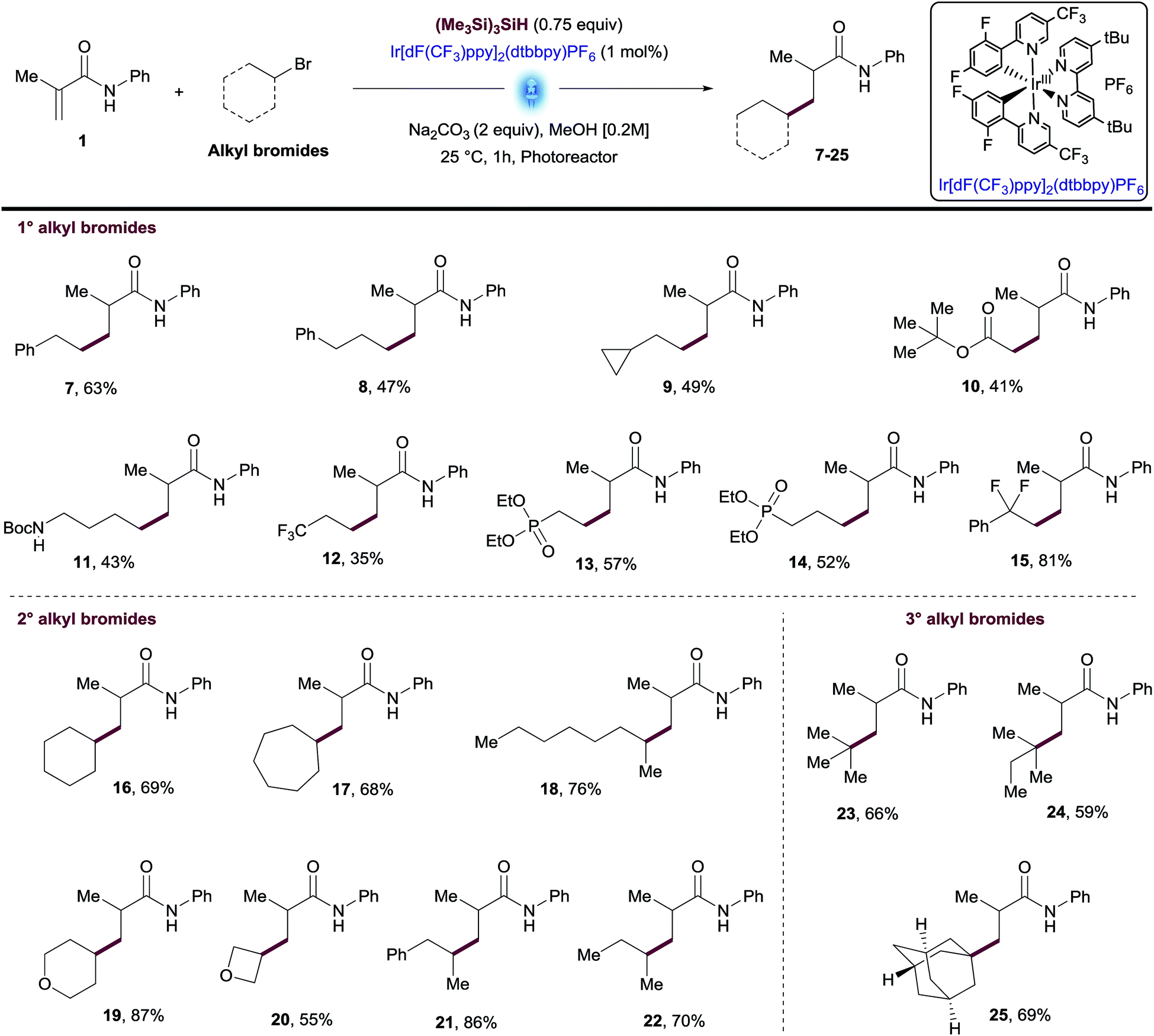
The use of tertiary alkyl bromides enabled the synthesis of quaternary centers from small alkyl groups such as tBu or 2-methylbutane (23-24, Table 2). Likewise, this methodology allowed access to the adamantyl derivative 25, an exotic moiety used often by Medicinal Chemists to increase drug-like qualities of a lead compound, without increasing toxicity.19
The reaction scope was further explored by testing a variety of Michael acceptors including esters and amides. Bromide 2 successfully reacted with diverse acceptors including fumarate, malonate, mono- and di-substituted acrylates, furanone and maleimide (26–34, Table 3). We were also pleased to find that primary, secondary and tertiary alkyl bromides such as tetrahydropyran, adamantyl and ethylbenzene reacted with different α,β-unsaturated esters such as acrylates, fumarates and malonates to provide the desired products 35–43 in good yields (Table 3). The examples shown on Tables 2 and 3 demonstrate the robustness of this transformation with a variety of alkyl bromides as well as Michael acceptors.

Tolerability of functional groups on both coupling partners, such as unprotected alcohols, esters or Boc-protected amines also surpasses that of traditional methods using organometallic species. Finally, short reaction times using the Merck integrated photoreactor render this methodology highly practical for synthetic chemists.
Mechanistic studies
A plausible mechanism for the visible light-mediated Giese addition is proposed on Scheme 2. The generation of an initial carbon-based radical A from an alkyl bromide under photoredox conditions has been explained previously by MacMillan13 in the context of their cross-electrophile coupling. Oxidation of bromide by the photocatalyst generates an electrophilic bromine radical13 which is capable of abstracting hydrogen from (Me3Si)3SiH,20 generating the silyl radical species [(Me3Si)3Si˙].10 Subsequent halogen abstraction from alkyl bromide 2 would provide the corresponding nucleophilic radical species A and the bromosilane byproduct. Giese reaction of A with an acceptor to generate radical B is also well established. The presence of stoichiometric (Me3Si)3SiH in the reaction presents two potential reaction pathways for this radical species to be reduced to the final product of the reaction, 3: (i) single-electron transfer (SET) from reductant species [IrII] to provide anion C, followed by protonation resulting in the desired product 3; (ii) radical propagation mechanism via hydrogen atom abstraction from (Me3Si)3SiH directly generating product 3 and, consequently, regenerating the stabilized silyl radical species [(Me3Si)3Si˙].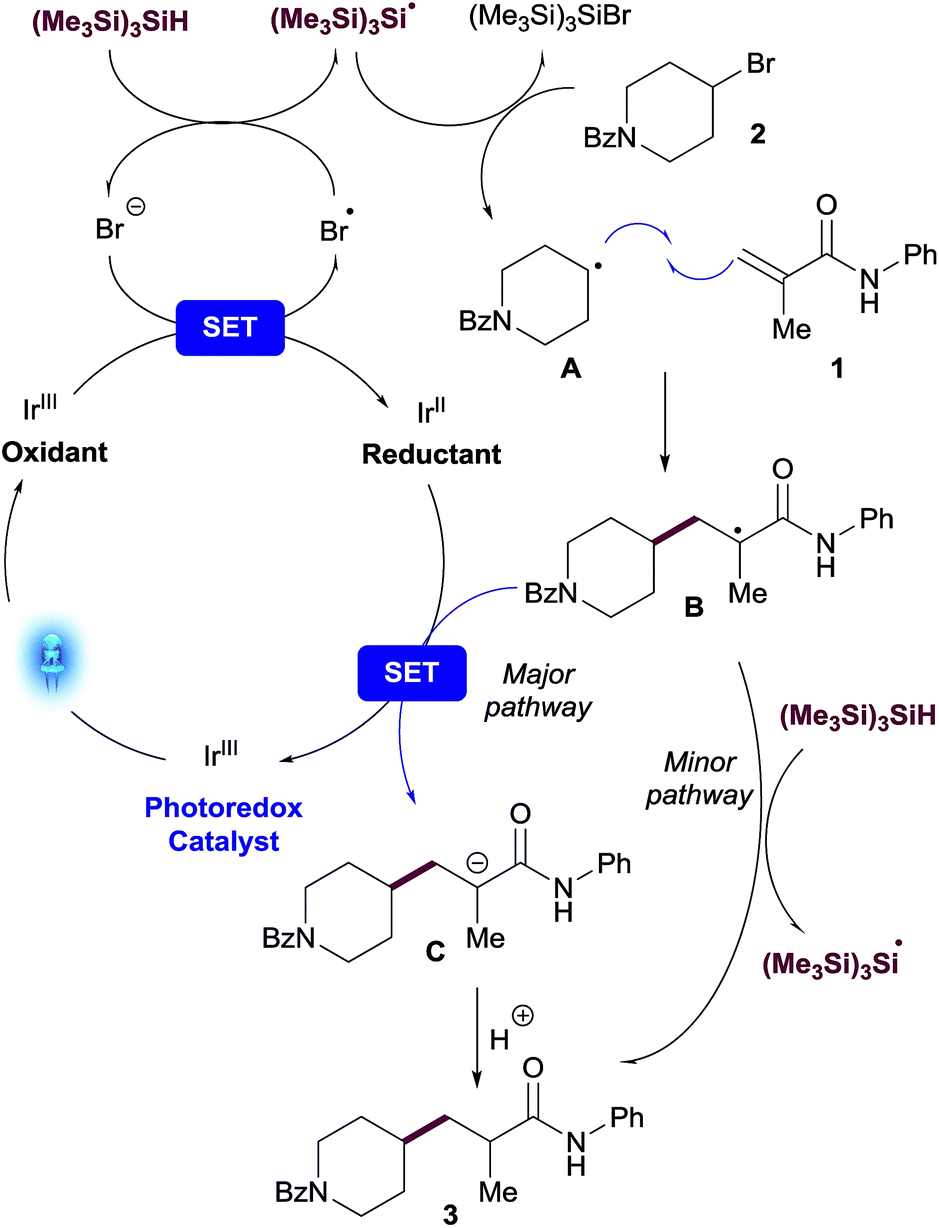 | ||
| Scheme 2 Plausible mechanism. | ||
Better understanding of this part of the mechanism was important since this is the stereodetermining step in the reaction and we envisioned that a rapid way to discern between these two pathways could be the utilization of deuterated reagents in the reaction. Therefore, we performed separate experiments using deuterated solvent (CD3OD and CH3OD) and (Me3Si)3SiD (Scheme 3).
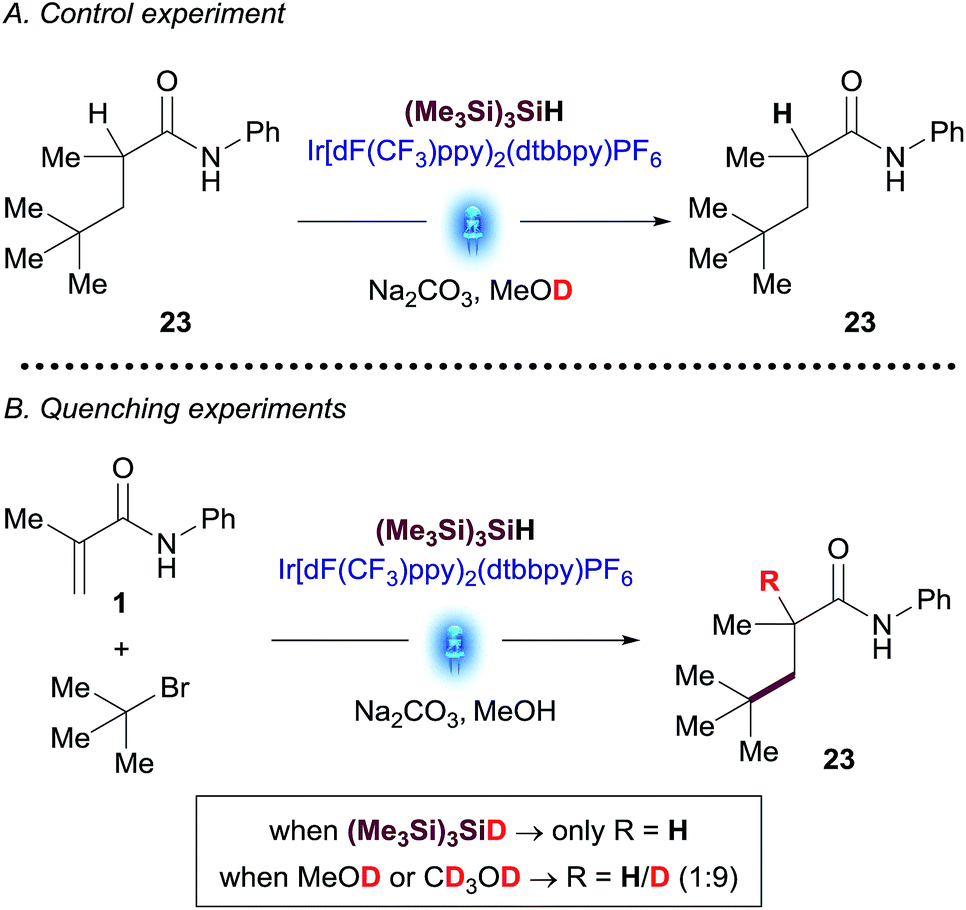 | ||
| Scheme 3 Deuterated NMR experiments: (A) control experiment; (B) reaction of amide 1 with t-butyl bromide using deuterated reagents. | ||
tButyl bromide was chosen as a substrate for these experiments due to the unambiguous assignment of the 1H-NMR spectra (see ESI†). While the reaction with (Me3Si)3SiD led to insignificant incorporation of deuterium into the desired product, the experiments in CH3OD resulted predominantly in the formation of deuterated coupled product. A control experiment subjecting purified product 23 to the reaction conditions using deuterated methanol (CD3OD) showed no deuterium incorporation, thus excluding the possibility of a base-catalyzed H/D scrambling after the product initially formed (Scheme 3). These results suggest that the major pathway to convert radical species B to desired product 3 is likely to occur through anion Cvia SET reduction by the [IrII] photocatalyst. Determination of quantum yield is also informative in characterizing chain processes in visible light photoredox catalysis.21 Using recently developed LED-NMR methodology,22 and under slightly modified reaction conditions, (see ESI†) we determined the quantum yield for the reaction is ϕ = 0.45. This result does not support the existence of an efficient radical chain and is in line with the observations of the deuteration experiments.
Medicinal Chemistry application
To demonstrate the potential value of this methodology on a Medicinal Chemistry application, we utilized this transformation to access Vorinostat® (44),23 an oncology drug approved for the treatment of cutaneous T-cell lymphoma (CTCL), and currently being studied as part of a possible strategy to cure HIV infection.24 From a Medicinal Chemistry perspective, derivatization of suberic acid 45 using conventional amide bond formation strategies easily enabled SAR exploration of both terminal carboxylic acids on the compound.23a However, derivatization or substitution on most positions of the methylene chain would be much more challenging synthetically, slowing down SAR efforts. The present chemistry enables a non-obvious bond disconnection via Giese addition and, using the standard reaction conditions described, enabled the synthesis of the Vorinostat® precursor 46 in a single step from commercially available bromovalerate 47 and acrylamide 48. The photoredox product 46 was obtained in 48% yield after 1 h using the integrated photoreactor. Within the Vorinostat® chemical space, this disconnection could be used by Medicinal Chemists to quickly explore additional sites of chemical diversity such as substitution on the α or β positions of the acrylamide or additional chain lengths, to name a few, exemplifying the value of this methodology on biologically relevant chemical motifs (Scheme 4).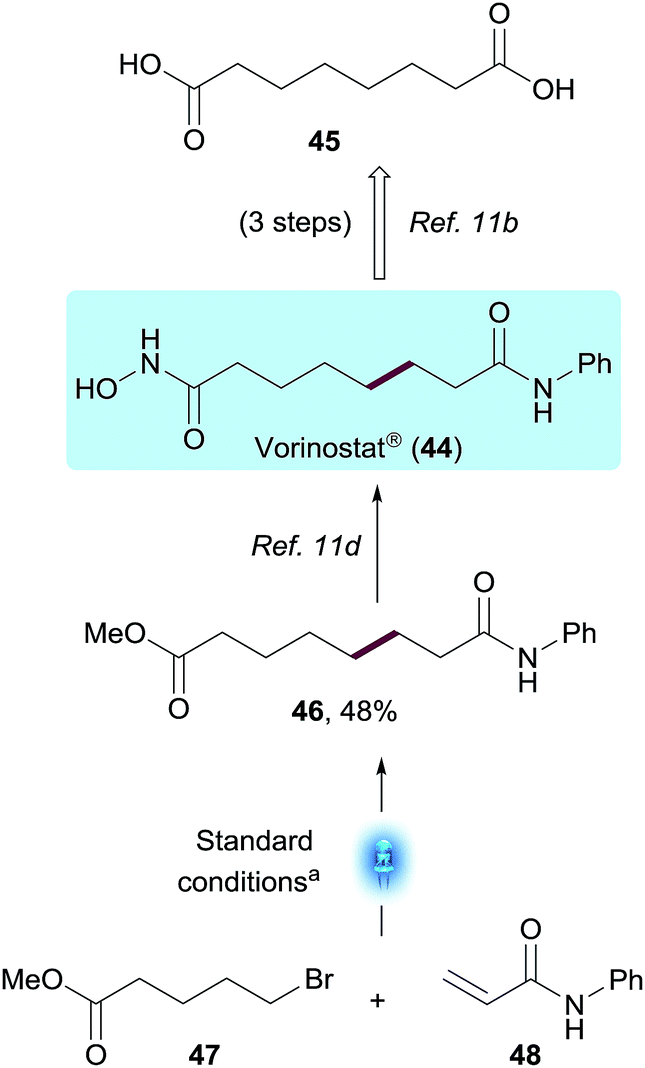 | ||
| Scheme 4 Versatile approach to Vorinostat® through photocatalytic conjugate addition. [a] 48 (1.0 equiv.), 47 (3.0 equiv.), Na2CO3 (2.0 equiv.), (Me3Si)3SiH (0.75 equiv.) and Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1 mol%) in MeOH, integrated photoreactor (reactor), 1 h. | ||
Conclusions
In conclusion, we have developed a Giese addition of unactivated alkyl bromides to α,β-unsaturated esters and amides mediated by photoredox catalysis. We have also demonstrated that this transformation proceeds under mild conditions to provide numerous primary, secondary and tertiary products, including, biologically relevant compounds such as Vorinostat®.Experimental
General procedure
In a vial were added N-phenylmetacrylamide (1) (100 mg, 0.62 mmol, 1.0 equiv.), (4-bromopiperidin-1-yl)(phenyl)methanone (2) (250 mg, 0.93 mmol, 1.5 equiv.), Na2CO3 (131 mg, 1.24 mmol, 2.0 equiv.) and Ir[dF(CF3)ppy]2(dtbbpy)PF6 (7 mg, 0.006 mmol, 0.01 equiv.). MeOH (2 mL) was added followed by (Me3Si)3SiH (145 μL, 0.46 mmol, 0.75 equiv.). The vial was purged with nitrogen and then sealed. The vial was placed in the integrated photoreactor (100% intensity, 6000 rpm fan, 1000 rpm stirring) until the starting acceptor was completely consumed (1 h). After the reaction was completed, volatiles were removed at reduced pressure. The resulting residue was dissolved in DMSO (1 mL), filtered and purified by HPLC: eluted with a 15 minute gradient of 90% water/MeCN (0.1% TFA) to 5% water/MeCN (both 0.1% TFA) with a flow rate of 25 mL min−1. The fractions were concentrated to provide pure products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank L. C. Campeau and Prof. D. W. C. MacMillan for helpful discussions. We thank Janine Brouillette and James Small for support with NMR characterization. We thank Yining Ji and Mikhail Reibarhk for their help with quantum yield experiments. We thank James J. Perkins, Antonella Converso, Mark E. Layton and Michael VanHeyst for reviewing the paper.Notes and references
-
(a) T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546 RSC
; (b) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef PubMed
-
(a) See review and references therein: E. Geist, A. Kirschning and T. Schmidt, Nat. Prod. Rep., 2014, 31, 441 RSC
- For selected reviews on 1,4-conjugate additions, see:
(a)
P. Perlmutter, Conjugate Addition Reactions in Organic Synthesis, Pergamon, Oxford, 1992 Search PubMed
- For selected reviews:
(a) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 941 CrossRef PubMed
-
(a) B. Giese and J. Dupuis, Angew. Chem., Int. Ed. Engl., 1983, 22, 622 (
Angew. Chem.
, 1983
, 95
, 633
) CrossRef
-
(a) Y. Li, K. Miyazawa, T. Koike and M. Akita, Org. Chem. Front., 2015, 2, 319 RSC
-
(a) L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886 CrossRef PubMed
-
(a) C. Nawrat, C. R. Jamison, Y. Slutskyy, D. W. C. MacMillan and L. E. Overman, J. Am. Chem. Soc., 2015, 137, 11270 CrossRef PubMed
-
(a) V. Corcé, L.-M. Chamoreau, E. Derat, J.-P. Goddard, C. Ollivier and L. Fensterbank, Angew. Chem., Int. Ed. Engl., 2015, 54, 11414 CrossRef PubMed
-
(a) C. Chatgilialoglu and J. Lalevée, Molecules, 2012, 17, 527 CrossRef PubMed
- For reviews on alkylborane, see
(a) H. Yorimitsu, H. Shinokubo and K. Oshima, Synlett, 2002, 674 CrossRef
-
(a) H. I. Tashtoush and R. Sustmann, Chem. Ber., 1992, 125, 287 CrossRef
- P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084 CrossRef PubMed
-
(a) C. Le, Y. Liang, R. W. Evans, X. Li and D. W. C. MacMillan, Nature, 2017, 547, 79 CrossRef PubMed
- M. Shevlin, ACS Med. Chem. Lett., 2017, 8, 601 CrossRef PubMed
- BTMG = 2-tert-butyl-1,1,3,3-tetramethylguanidine: D. H. R. Barton, M. Chen, J. C. Jaszberenyi and D. K. Taylor, Org. Synth., 1996, 74, 101 Search PubMed
- C. C. Le, M. K. Wismer, Z.-C. Shi, R. Zhang, D. V. Conway, G. Li, P. Vachal, I. W. Davies and D. W. C. MacMillan, ACS Cent. Sci., 2017, 3, 647 CrossRef PubMed
- T. Bodill, A. C. Conibear, M. K. M. Mutorwa, J. L. Goble, G. L. Blatch, K. A. Lobb, R. Klein and P. T. Kaye, Bioorg. Med. Chem., 2013, 21, 4332 CrossRef PubMed
-
(a) J. Liu, D. Obando, V. Liao, T. Lifa and R. Codd, Eur. J. Med. Chem., 2011, 46, 1949 CrossRef PubMed
- Oxygen has also been postulated as an initiator capable of abstracting hydrogen from (Me3Si)3SiH, see: J. J. Devery III, J. D. Nguyen, C. Dai and C. R. J. Stephenson, ACS Catal., 2016, 6, 5962 CrossRef
- M. Cismea and T. P. Yoon, Chem. Sci., 2015, 6, 5426 RSC
- Y. Ji, D. A. DiRocco, C. Hong, M. K. Wismer and M. Reibarkh, Org. Lett., 2018, 20, 2156 CrossRef PubMed
-
(a) C. Salmi-Smail, A. Fabre, F. Dequiedt, A. Restouin, R. Castellano, S. Garbit, P. Roche, X. Morelli, J. M. Brunel and Y. Collette, J. Med. Chem., 2010, 53, 3038 CrossRef PubMed
-
(a) M. A. Glozak and E. Seto, Oncogene, 2007, 26, 5420 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02253d |
| ‡ Corresponding authors and contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |