
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDegradable polymer prodrugs with adjustable activity from drug-initiated radical ring-opening copolymerization†
Elise
Guégain
 ,
Johanna
Tran
,
Quentin
Deguettes
and
Julien
Nicolas
*
,
Johanna
Tran
,
Quentin
Deguettes
and
Julien
Nicolas
*
Institut Galien Paris-Sud, CNRS UMR 8612, Univ Paris-Sud, Faculté de Pharmacie, 5 rue Jean-Baptiste Clément, 92290 Châtenay-Malabry, France. E-mail: julien.nicolas@u-psud.fr; Tel: +33 1 46 83 58 53 Web: http://www.twitter.com/julnicolas
First published on 13th September 2018
Abstract
Degradable polymer prodrugs based on gemcitabine (Gem) as an anticancer drug were synthesized by ‘drug-initiated’ nitroxide-mediated radical ring-opening copolymerization (NMrROP) of methacrylic esters and 2-methylene-4-phenyl-1,3-dioxolane (MPDL). Different structural parameters were varied to determine the best biological performances: the nature of the monomer [i.e., oligo(ethylene glycol) methacrylate (OEGMA) or methyl methacrylate (MMA)], the nature of the Gem-polymer linker (i.e., amide or amide and diglycolate) and the MPDL content in the copolymer. Depending on the nature of the methacrylate monomer, two small libraries of water-soluble copolymer prodrugs and nanoparticles were obtained (Mn ∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Đ = 1.1–1.5), which exhibited tunable hydrolytic degradation under accelerated conditions governed by the MPDL content. Drug-release profiles in human serum and in vitro anticancer activity on different cell lines enabled preliminary structure–activity relationships to be established. The cytotoxicity was independently governed by: (i) the MPDL content – the lower the MPDL content, the greater the cytotoxicity; (ii) the nature of the linker – the presence of a labile diglycolate linker enabled a greater Gem release compared to a simple amide bond and (iii) the hydrophilicity of the methacrylate monomer–OEGMA enabled a greater anticancer activity to be obtained compared to MMA-based polymer prodrugs. Remarkably, the optimal structural parameters enabled reaching the cytotoxic activity of the parent (free) drug.
000 g mol−1, Đ = 1.1–1.5), which exhibited tunable hydrolytic degradation under accelerated conditions governed by the MPDL content. Drug-release profiles in human serum and in vitro anticancer activity on different cell lines enabled preliminary structure–activity relationships to be established. The cytotoxicity was independently governed by: (i) the MPDL content – the lower the MPDL content, the greater the cytotoxicity; (ii) the nature of the linker – the presence of a labile diglycolate linker enabled a greater Gem release compared to a simple amide bond and (iii) the hydrophilicity of the methacrylate monomer–OEGMA enabled a greater anticancer activity to be obtained compared to MMA-based polymer prodrugs. Remarkably, the optimal structural parameters enabled reaching the cytotoxic activity of the parent (free) drug.
Introduction
In the field of nanomedicine, use of drug-loaded polymer nanocarriers is considered as a promising strategy to improve the efficacy of drugs such as chemotherapeutics.1,2 Traditionally, drugs are physically encapsulated during the nanocarrier formulation and thus simply entrapped into the polymer matrix. These drug delivery systems lead to protection of the drug from rapid metabolization, to longer circulation time, to lower toxicity toward healthy cells/tissues and open the door to active targeting by their surface-functionalization using biologically active ligands. Despite major advances and encouraging results, important limitations remain that may explain the small number of marketed nanomedicines and recent clinical trial disappointments: (i) the “burst-release”, that is, the quick and uncontrolled release of a significant fraction of the drug post-injection; (ii) the poor drug-loadings, usually only a few percent and (iii) the crystallization of some drugs into the polymer matrix. These three different events can lead to prohibitive toxicity and/or colloidal instability of the nanocarriers.The prodrug strategy, which consists in coupling the drug to the nanocarrier, can be used to circumvent, or at least alleviate, the above-mentioned issues.3 Among the different synthetic pathways to produce polymer prodrug nanocarriers, the most used are certainly the “grafting to” and “grafting from” approaches that consist in functionalization of the preformed polymer or monomer, respectively. The emerging “grafting from” strategy (also called “drug-initiated”), that relies on the controlled growth of a short polymer chain from a drug, used as an initiator, has appealing benefits:4 (i) the synthesis and purification are simple because only a few synthetic steps are necessary; (ii) the resulting materials have a simple, well-defined structure (one drug attached at the extremity of each polymer chain); (iii) high drug loadings can be easily reached by targeting short polymer chains; (iv) this approach can be applied to different pathologies simply by changing the nature of the drug and (v) the properties of the resulting polymer prodrug can be finely tuned by changing the nature of the growing polymer.
The robustness of the drug-initiated method has been illustrated by its application to the synthesis of a variety of different polymer prodrugs constructed by either ring-opening polymerization (ROP)5–9 or reversible-deactivation radical polymerization (RDRP),10–18 including nitroxide-mediated radical polymerization (NMP)19 or reversible addition-fragmentation chain transfer (RAFT) polymerization.20 Whereas ROP generated degradable polyester prodrug nanocarriers, they exhibited poor colloidal stability and required post-stabilization by means of macromolecular surfactants, which is a major drawback. Also, no in vivo anticancer activity has been reported from these systems. On the other hand, RDRP-constructed polymer prodrugs gave promising anticancer efficacy in vivo, relied on simpler polymerization methods (e.g., no stringent conditions, commercially available controlling agents) and offered much more versatility regarding the nature of the polymer used. However, they are not degradable because of the carbon–carbon backbone of the vinyl polymer chains. This represents an important drawback because non-degradable materials may accumulate in the body, leading to prohibitive toxicity in case chronic/repeated administration is envisioned. A global strategy combining both the advantages of ROP and RDRP for the design of efficient polymer prodrugs by the “drug-initiated” approach is thus highly desirable.
Conferring degradability to vinyl materials is currently the focus of intensive work.21 This research topic is crucial given the numerous systems based on vinyl polymers devoted to biomedical applications regularly being reported in the literature. Among the different synthetic strategies, radical ring-opening polymerization (rROP) appears to be the method of choice for incorporating labile groups into the polymer backbone and enabling significant degradation.22,23 Thanks to its radical ring-opening mechanism, rROP possesses both the versatility and simplicity of radical polymerization, together with the ability to introduce functional groups into the polymer backbone. Among the different classes of monomers that have been polymerized by rROP, cyclic ketene acetals (CKAs) are the most-studied family. Although their homopolymerization has been extensively studied in the 80s,24–27 they aroused renewed interest over the past decade as comonomers to confer degradability to vinyl polymers via insertion of ester groups from either free-radical copolymerization28–34 or RDRP.32,35–45 Other cyclic monomers deriving from cyclic allylic sulfides46–48 have also been used to incorporate cleavable ester, thioester, and disulfide functionalities into the polymer backbone through RAFT copolymerization with traditional vinyl monomers.41 Despite several applications of rROP-designed materials for biomedical applications,31,49–51 their use in the field of prodrug nanocarriers has only been reported from preformed functional copolymers via the “grafting to” approach.52,53
Herein, we report for the first time on a general approach that combines the best of two worlds, that is, the drug-initiated synthesis of degradable polymer prodrugs by rROP. We demonstrated that well-defined, degradable vinyl copolymers can be synthesized from an anticancer drug-bearing RDRP initiator by rROP, leading to nanocarriers, either water-soluble conjugates or nanoparticles (Fig. 1), with adjustable anticancer activity depending on the nature of both the drug-polymer linkage and the copolymer. Not only this new class of polymer prodrugs overcame a significant obstacle in the field, but it also disclosed important insights into the relevant parameters that govern the drug release kinetics and eventually the anticancer activity.
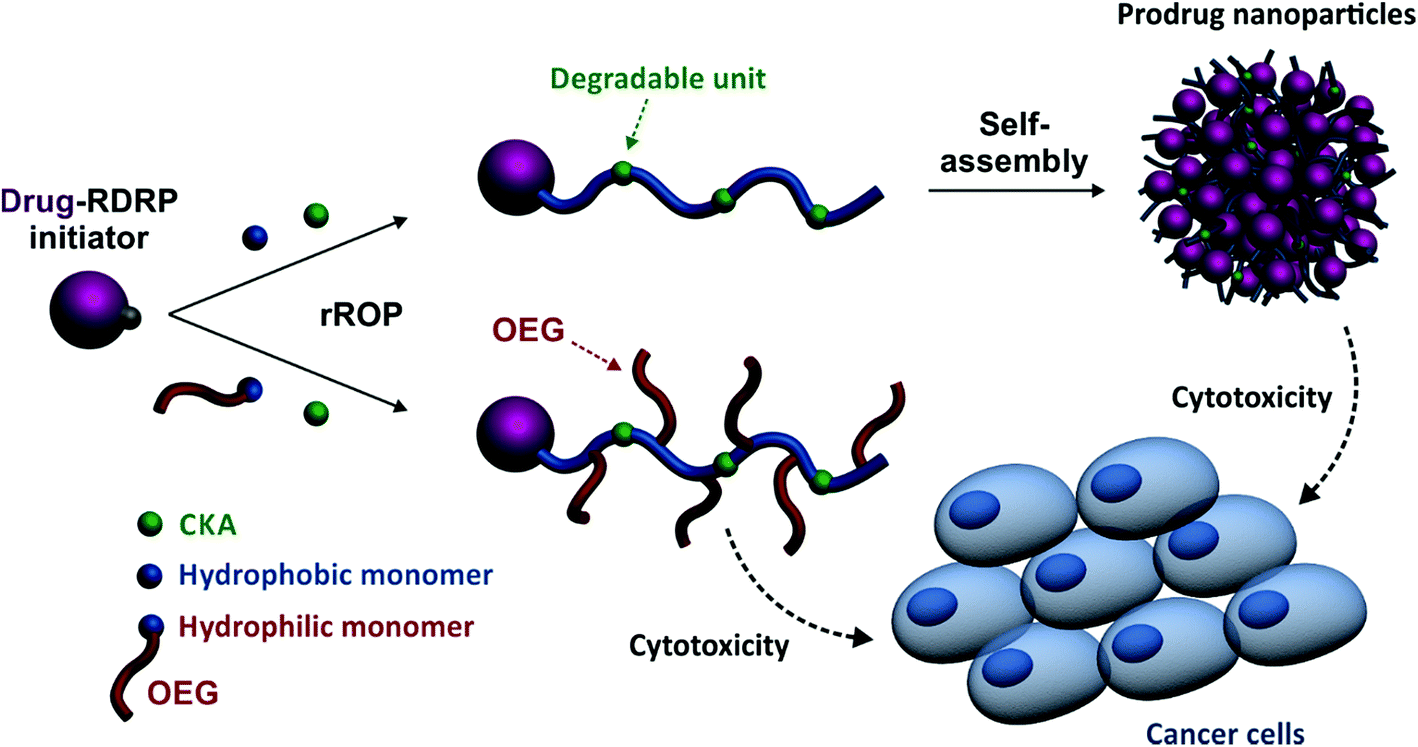 | ||
| Fig. 1 Synthetic strategy for the design of degradable Gemcitabine-based polymer prodrugs by “drug-initiated” nitroxide-mediated radical ring-opening copolymerization (NMrROP). | ||
Experimental section
Materials
Gemcitabine (>98%) was purchased from Carbosynth Limited (UK). Oligo(ethylene glycol) methyl ether methacrylate (MeOEGMA, Mn = 300 g mol−1), styrene (S, 99%), methyl methacrylate (MMA, 99%) and toluene (anhydrous, 99.8%) were purchased from Sigma-Aldrich (France) and used as received (except for MMA which was distilled under reduced pressure). 2-Methylene-4-phenyl-1,3-dioxolane (MPDL),54 4-amino-1-[4-(tert-butyl-dimethylsilanyloxy)-5-(tert-butyl-dimethyl-silanyloxymethyl)-3,3-difluorotetrahydro-furan-2-yl]-1H-pyrimidin-2-one (TBDMS-Gem),15 alkoxyamines Gem-AMA-SG1 (ref. 10) and AMA-digly11 were prepared as reported elsewhere. Tetrabutylammonium fluoride (TBAF) was purchased from Alfa Aesar (a Johnson Matthey Co., France). Perfluoro-15-Crown-5-Ether (PFCE) was obtained from FluoroChem (UK). All other reactants were purchased from Sigma-Aldrich at the highest available purity and used as received. Deuterated chloroform (CDCl3) and dimethyl sulfoxide (DMSO-d6) were obtained from Eurisotop. All other solvents were purchased from Carlo-Erba. Dulbecco's modified eagle's medium (DMEM) and fetal bovine serum (FBS) were purchased from Dulbecco (Invitrogen, France). Penicillin and streptomycin were obtained from Lonza (Verviers, Belgium). 2-Methyl-2-[N-tert-butyl-N-(1-diethoxyphosphoryl-2,2-dimethylpropyl) aminoxy]propionic acid alkoxyamine (BlocBuilder MA, 99%) and N-tert-butyl-N-(1-diethylphosphono-2,2-dimethylpropyl) nitroxide (SG1, 85%) were kindly supplied by Arkema.Analytical method
000 g mol−1) and a differential refractive index detector (Spectrasystem RI-150 from Thermo Electron Corp.), using chloroform (CHCl3) as the eluent at a flow rate of 1 mL min−1. Toluene was used as a flow-rate marker. The conventional calibration curve was based on poly(methyl methacrylate) (PMMA) standards (peak molar masses, Mp = 625–625500 g mol−1) from Polymer Laboratories. This technique allowed Mn (number-average molar mass), Mw (weight-average molar mass), and Mw/Mn (dispersity, Đ) to be determined.
Synthetic procedures
000 g mol−1 were prepared by following a similar procedure to that of expt 1 but with a polymerization time of 8 h. Experimental conditions were as follows: P1 (fMPDL,0 = 0.2) [OEGMA (1.7618 g, 5.87 mmol), MPDL (0.2382 g, 1.47 mmol), Gem-AMA-SG1 (200.0 mg, 3.27 × 10−1 mmol), SG1 (12 mg, 3.51 × 10−2 mmol) and anhydrous toluene (2.0 g, 2.31 mL)], P2 (fMPDL,0 = 0.4) [OEGMA (1.4704 g, 4.90 mmol), MPDL (0.5296 g, 3.27 mmol), Gem-AMA-SG1 (205.0 mg, 3.35 × 10−1 mmol), SG1 (12 mg, 3.51 × 10−2 mmol) and anhydrous toluene (2.0 g, 2.31 mL)] and P3 (fMPDL,0 = 0.7) [OEGMA (0.8862 g, 2.95 mmol), MPDL (1.1138 g, 6.88 mmol), Gem-AMA-SG1 (65.0 mg, 1.06 × 10−1 mmol), SG1 (3.6 mg, 1.05 × 10−2 mmol) and anhydrous toluene (2.0 g, 2.31 mL)]. The final composition of the prodrug was determined by comparing the methoxy protons of OEG from OEGMA (at 3.4 ppm) to the aromatic protons of MPDL (at 7.2 ppm). The presence of Gem was quantitatively confirmed by 19F NMR by comparing the integration of the fluorine atoms of the internal standard PFCE (δ = −88 ppm) and of Gem (δ = −117 ppm). Copolymers with lower targeted Mn were prepared (Table S1†) by following a similar procedure to that of P3: P3′ (targeted Mn = 9000 g mol−1, fMPDL,0 = 0.7) [OEGMA (0.8955 g, 2.99 mmol), MPDL (1.113 g, 6.87 mmol), Gem-AMA-SG1 (65.9 mg, 0.11 mmol), SG1 (4.1 mg, 1.20 × 10−2 mmol) and anhydrous toluene (2.0 g, 2.31 mL)] and P3′′ (targeted Mn = 3500 g mol−1, fMPDL,0 = 0.7) [OEGMA (0.9328 g, 3.11 mmol), MPDL (1.205 g, 7.44 mmol), Gem-AMA-SG1 (187.8 mg, 0.31 mmol), SG1 (9.7 mg, 2.84 × 10−2 mmol) and anhydrous toluene (2.0 g, 2.31 mL)].
000 g mol−1 were prepared by following a similar procedure to that of expt 1 but with a polymerization time of 8 h. Experimental conditions were as follows: P1d (fMPDL,0 = 0.2) [OEGMA (0.8805 g, 2.93 mmol), MPDL (0.1195 g, 0.74 mmol), Gem-digly-AMA-SG1 (266.6 mg, 2.66 × 10−1 mmol), SG1 (9 mg, 2.63 × 10−2 mmol) and toluene (1.0 g, 1.15 mL)], for P2d (fMPDL,0 = 0.4) [OEGMA (0.7375 g, 2.46 mmol), MPDL (0.2660 g, 1.64 mmol), Gem-digly-AMA-SG1 (93.3 mg, 9.32 × 10−2 mmol), SG1 (3.2 mg, 9.36 × 10−3 mmol) and toluene (1.0 g, 1.15 mL)] and for P3d (fMPDL,0 = 0.7) [OEGMA (0.4420 g, 1.47 mmol), MPDL (0.5580 g, 3.44 mmol), Gem-digly-AMA-SG1 (53.3 mg, 5.33 × 10−2 mmol), SG1 (1.7 mg, 4.89 × 10−3 mmol) and toluene (1.0 g, 1.15 mL)]. The final composition of the prodrug was determined by comparing the methoxy protons of OEG from OEGMA (at 3.4 ppm) with the aromatic protons of MPDL (at 7.2 ppm). The presence of Gem was quantitatively confirmed by 19F NMR by comparing the integration of the fluorine atoms of the internal standard PFCE (δ = −88 ppm) and of Gem (δ = −117 ppm).
000 g mol−1 were prepared by following a similar procedure to that of expt 1 but with a polymerization time of 8 h. Experimental conditions were as follows: OEGMA (1.4436 g, 4.81 mmol), S (0.0563 g, 5.41 × 10−1 mmol), Gem-AMA-SG1 (7.0 mg, 1.14 × 10−1 mmol), and anhydrous toluene (1.5 g, 1.73 mL). SEC: Mn = 10300 g mol−1, Mw/Mn = 1.29.
000 g mol−1 and fMPDL,0 = 0.4 were prepared by following a similar procedure to that of expt 1 but with a polymerization time of 8 h. Experimental conditions were as follows: OEGMA (1.1028 g, 3.68 mmol), MPDL (0.3972 g, 2.45 mmol), BlocBuilder MA (15.0 mg, 3.94 × 10−2 mmol), and anhydrous toluene (1.5 g, 1.73 mL). The final composition of the copolymer was determined by comparing the terminal methoxy protons of pendant OEG for OEGMA (at 3.4 ppm) with the aromatic protons of MPDL (at 7.2 ppm). SEC: Mn = 12100 g mol−1, Mw/Mn = 1.30. 1H NMR: FMPDL = 0.13.
000 g mol−1 were prepared by following a similar procedure to that of expt 4 but with a polymerization time of 5 h. Experimental conditions were as follows: P4 (fMPDL,0 = 0.2) [MMA (1.0673 g, 10.67 mmol), MPDL (0.4327 g, 2.67 mmol), Gem-AMA-SG1 (340.0 mg, 5.56 × 10−1 mmol), SG1 (18.1 mg, 5.29 × 10−2 mmol) and anhydrous toluene (1.5 g, 1.73 mL)], for P5 (fMPDL,0 = 0.4) [MMA (0.7215 g, 7.22 mmol), MPDL (0.7785 g, 4.81 mmol), Gem-AMA-SG1 (230 mg, 3.76 × 10−1 mmol), SG1 (12.3 mg, 3.60 × 10−2 mmol) and anhydrous toluene (1.5 g, 1.73 mL)] and for P6 (fMPDL,0 = 0.7) [MMA (0.3135 g, 3.14 mmol), MPDL (1.1865 g, 7.32 mmol), Gem-AMA-SG1 (30.0 mg, 4.90 × 10−2 mmol), SG1 (1.6 mg, 4.69 × 10−3 mmol) and anhydrous toluene (1.5 g, 1.73 mL)]. The final composition of the prodrug was determined by comparing the methyl protons in the α-position to the ester group of MMA (at 3.7 ppm) with the aromatic protons of MPDL (at 7.2 ppm). The presence of Gem was quantitatively confirmed by 19F NMR by comparing the integration of the fluorine atoms of the internal standard PFCE (δ = −88 ppm) and of Gem (δ = −117 ppm).
000 g mol−1 were prepared by following a similar procedure to that of expt 4 but with a polymerization time of 5 h. Experimental conditions were as follows: P4d (fMPDL,0 = 0.2) [MMA (1.0673 g, 10.67 mmol), MPDL (0.4327 g, 2.67 mmol), Gem-digly-AMA-SG1 (152.0 mg, 1.52 × 10−1 mmol), SG1 (5.2 mg, 1.52 × 10−2 mmol) and anhydrous toluene (1.5 g, 1.73 mL)] for P5d (fMPDL,0 = 0.4) [MMA (0.7215 g, 7.22 mmol), MPDL (0.7785 g, 4.81 mmol), Gem-digly-AMA-SG1 (100.0 mg, 9.99 × 10−2 mmol), SG1 (3.4 mg, 9.95 × 10−3 mmol) and anhydrous toluene (1.5 g, 1.73 mL)] and for P6d (fMPDL,0 = 0.7) [MMA (0.3135 g, 3.14 mmol), MPDL (1.1865 g, 7.32 mmol), Gem-digly-AMA-SG1 (25 mg, 2.50 × 10−2 mmol), SG1 (0.9 mg, 2.63 × 10−3 mmol) and anhydrous toluene (1.5 g, 1.73 mL)]. The final composition of the prodrug was determined by comparing the methyl protons in the α-position to the ester group of MMA (at 3.7 ppm) to the aromatic protons of MPDL (at 7.2 ppm). The presence of Gem was quantitatively confirmed by 19F NMR by comparing the integration of the fluorine atoms of the internal standard PFCE (δ = −88 ppm) and of Gem (δ = −117 ppm).
000 g mol−1 were prepared by following a similar procedure to that of expt 4 but with a polymerization time of 5 h. Experimental conditions were as follows: MMA (1.3393 g, 13.39 mmol), S (0.1607 g, 15.45 mmol), Gem-AMA-SG1 (2.81 mg, 4.59 × 10−2 mmol), and anhydrous toluene (1.5 g, 1.73 mL). SEC: Mn = 9751 g mol−1, Mw/Mn = 1.33.
000 g mol−1 and fMPDL,0 = 0.7 were prepared by following a similar procedure to that of expt 4 but with a polymerization time of 5 h. Experimental conditions were as follows: MMA (0.3604 g, 3.60 mmol), MPDL (0.4010 g, 2.47 mmol), BlocBuilder MA (16.0 mg, 4.20 × 10−2 mmol), and anhydrous toluene (0.77 g, 0.89 mL). The final composition of the copolymer was determined by comparing the methyl protons in the α-position to the ester group of MMA (at 3.7 ppm) to the aromatic protons of MPDL (at 7.2 ppm). SEC: Mn = 9100 g mol−1, Mw/Mn = 1.26. 1H NMR: FMPDL = 0.17.
Deprotection of the copolymers
Hydrolytic degradation
Nanoparticle preparation
Nanoparticles were prepared by the nanoprecipitation technique.55 For Gem-P(MMA-co-MPDL) and Gem-digly-P(MMA-co-MPDL), 2 mg of copolymer was dissolved in 2 mL of THF, and added dropwise to 4 mL MilliQ water under stirring. For Gem-P(OEGMA-co-MPDL) and Gem-digly-P(OEGMA-co-MPDL), 2.5 mg of copolymer was dissolved in 0.5 mL of THF, and added dropwise to 1 mL MilliQ water under stirring. In all cases, THF was evaporated at ambient temperature using a Rotavapor. Average diameter (Dz) and zeta potential (ζ) measurements were carried out in triplicate. For stability studies, the different samples were either dropped in water or PBS, or diluted in complete cell culture medium to reach a final concentration of 0.25 mg mL−1. Samples were kept at 4 °C and allowed to warm to room temperature before each measurement which was performed in triplicate at 25 °C.Nanoprecipitation yield
The amount of Gem-P(OEGMA-co-MPDL) nanoparticles formed by nanoprecipitation was determined as follows. A minimal amount of 4 mL of nanoparticle suspension (corresponding at least to 10 mg of copolymer) was ultracentrifuged (40000 rpm, 4 h, 4 °C). The supernatant and the pellet were separated and freeze-dried. The weight fraction of nanoparticles formed after nanoprecipitation was calculated according to: wnanoparticles = mpellet/(mpellet + msupernantant).
Drug release kinetics
To determine the release kinetics of Gem, 1.5 mL of Gem-P(OEGMA-co-MPDL) P1–P3 and P1d–P3d (0.5 mg mL−1) or Gem-P(MMA-co-MPDL) P4–P6 and P4d–P6d (0.5 mg mL−1) nanoparticles were added to 1.5 mL of human serum solution supplemented with 200 μg mL−1 tetrahydrouridine (THU).56,57 The mixture was incubated at 37 °C and aliquots (600 μL) of incubation medium were withdrawn at different time points (1, 4, 8 and 24 h), spiked with 60 μL of 10 μM theophylline (Internal Standard, IS) before addition of 1 mL of a mixture of acetonitrile/methanol (90/10, v/v) and ultracentrifuged (15000 g, 20 min, 4 °C). The supernatant was then evaporated to dryness under a nitrogen flow at 30 °C. The released native drug was quantified by reverse-phase HPLC (Waters, Milford, MA 01757, USA) with a C18 column. To ensure that only native Gem was quantified, the calibration curve was carried out using native Gem (elution time = 10.6 min). For drug-release experiments, only this peak was integrated to determine native Gem content. Briefly, the chromatographic system consisted of a Waters 1525 Binary HPLC pump, a Waters 2707 Autosampler, a C18 Uptisphere column (3 μm, 150 × 4.6 mm; Interchim), HPLC column temperature controllers (model 7950 column heater and chiller; Jones Chromatography, Lakewood, CO), and a Waters 2998 programmable photodiode-array detector. The HPLC column was maintained at 30 °C and detection was monitored at 270 nm. The HPLC mobile phase consisted of a mixture of methanol and water with 0.05 M sodium acetate (pH 5.0, eluent A: 5/95, v/v; eluent B 97/3, v/v). The residues were dissolved in 100 μL of eluent A. Elution was performed at a flow rate of 0.8 mL min−1 isocratically for 8 min with eluent A followed by a linear gradient (1 min) to 100% eluent B. This was followed by a 15 min hold at eluent B and a 1 min linear gradient back to 100% eluent A. The system was held for 6 min for equilibration back to initial conditions.
Biological evaluation
Results and discussion
Synthetic strategy
To illustrate our approach, gemcitabine (Gem, 2′-deoxy-2′,2′-difluorocytidine) was selected as an anticancer drug. Gem is a nucleoside analog approved for the treatment of various solid tumors including lung, pancreatic, breast, or ovarian cancers.58 However, severe limitations restrict its clinical use and drastically reduce its efficacy: (i) short plasma half-life and rapid renal excretion due to rapid deamination by deoxycitidine deaminase, (ii) induction of resistances owing to inhibition of transmembrane transporter nucleoside, and (iii) severe side effects as a result of frequent administration schedule. Therefore, new prodrug strategies applied to Gem are of high importance in the field of nanomedicine.This new class of polymer prodrugs was synthesized by nitroxide-mediated radical ring-opening copolymerization (NMrROP) between a methacrylic ester and 2-methylene-4-phenyl-1,3-dioxolane (MPDL) as a CKA, from a Gem-functionalized alkoxyamine initiator (Fig. 1). This synthetic pathway was built upon our previous findings showing that MPDL, a 5-membered ring CKA, is a very attractive monomer that can be easily obtained and efficiently incorporated into a polymethacrylate backbone by NMrROP, resulting in well-defined copolymers with controllable level of ester group insertion and up to nearly complete degradation upon hydrolysis.54,59–61
To establish structure–activity relationships and obtain polymer prodrugs with the highest activity against cancer cells, various structural parameters were varied such as the nature of the methacrylic ester, the composition of the copolymer and the nature of the drug-copolymer linker (Fig. 2). More specifically, two different methacrylic ester monomers were copolymerized with MPDL: either oligo(ethylene glycol) methyl ether methacrylate (OEGMA) as a hydrophilic monomer or methyl methacrylate (MMA) as a hydrophobic one (Fig. 2a and c). Copolymerizations were initiated by two different alkoxyamines based on the SG1 nitroxide, which only differed in the nature of the linker between Gem and the alkoxyamine moiety: an amide bond (Gem-AMA-SG1) or an amide bond connected to a labile diglycolate linker (Gem-digly-AMA-SG1) (Fig. 2b). Variable initial amounts of MPDL were also investigated to confer the resulting polymer prodrugs with distinct levels of degradability.
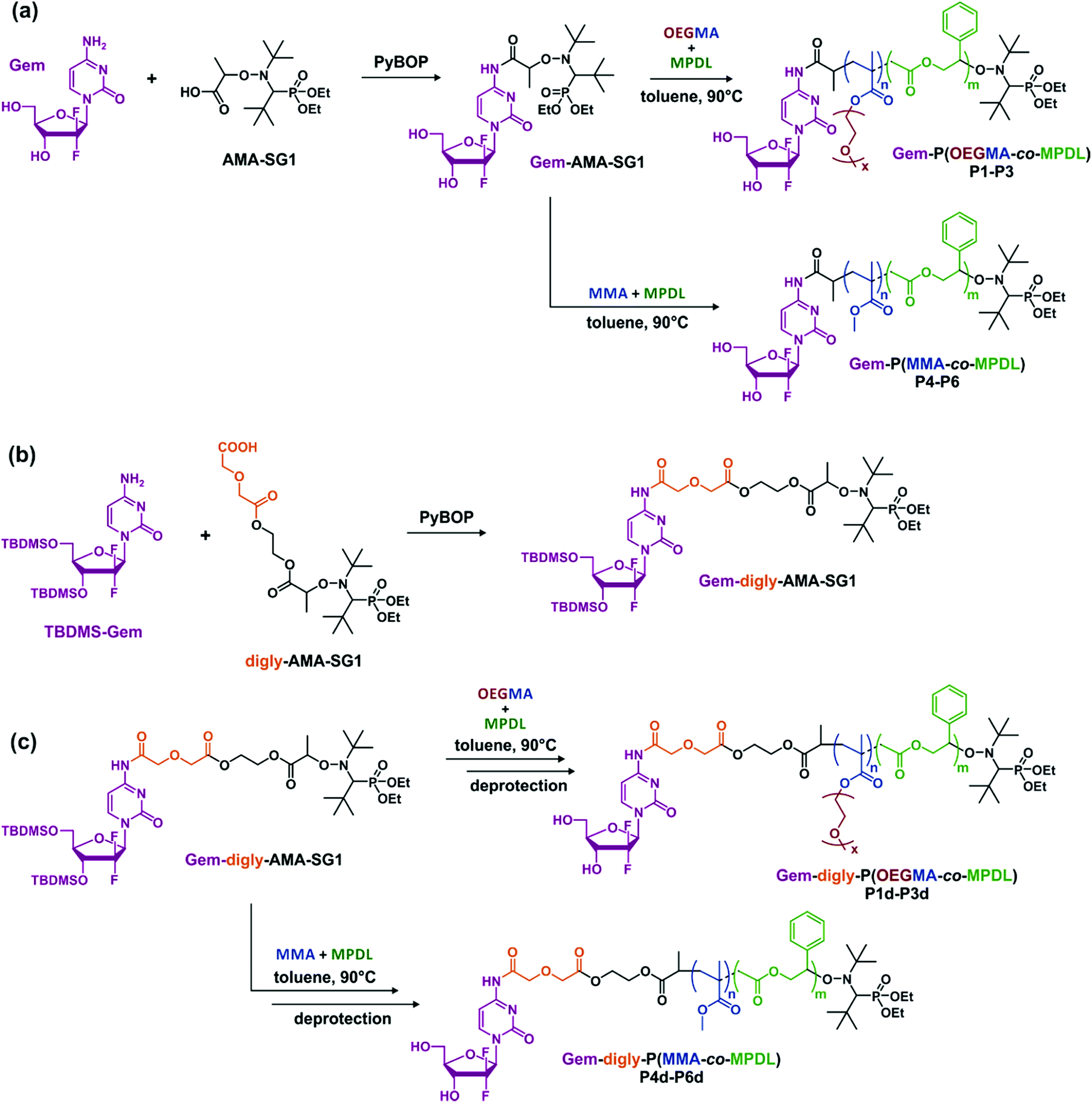 | ||
| Fig. 2 Synthesis of (a) Gem-P(OEGMA-co-MPDL) P1–P3 and Gem-P(MMA-co-MPDL) P4–P6 by NMrROP in toluene at 90 °C, (b) Gem-digly-AMA-SG1 by PyBOP-assisted coupling between TBDMS-Gem and digly-AMA-SG1 and (c) Gem-digly-P(OEGMA-co-MPDL) P1d–P3d and Gem-digly-P(MMA-co-MPDL) P4d–P6d by NMrROP in toluene at 90 °C followed by TBDMS group removal. | ||
Synthesis of Gem-based alkoxyamine initiators
Given its susceptibility to deamination,62 Gem was derivatized through its C-4 amino group. Gem-AMA-SG1 was synthesized by direct coupling between unprotected Gem and AMA-SG1 using PyBOP as a coupling agent. For Gem-digly-AMA-SG1, the best conditions were obtained via protection of the two hydroxyl groups with TBDMSCl, followed by PyBOP-assisted coupling of the resulting TBDMSGem with AMA-digly-SG1 (Fig. 2b). The expected product (Fig. S1†) was obtained with a coupling yield of 63%.Copolymerization kinetics
A comprehensive kinetic study was first performed to investigate the influence of the different parameters on the control of the copolymerization. Gem-AMA-SG1 or Gem-digly-AMA-SG1 alkoxyamines were used to initiate the NMrROP of OEGMA (expts 1–3 and 1d–3d, Fig. 3) or MMA (expts 4–6 and 4d–6d, Fig. 4) in the presence of variable initial fractions of MPDL (fMPDL,0 = 0.2–0.7) at 90 °C in 50 wt% toluene. In all cases, the higher the initial fraction of MPDL, the better the control of the copolymerization. For fMPDL,0 = 0.2, regardless of the methacrylic ester used (expts 1, 1d, 4 and 4d), the copolymerizations did not exhibit a first order kinetics while Mn values hardly increased with conversion and were much higher than the theoretical ones with rather high dispersities after 50% conversion, thus indicating a partial control. Such a low initial amount of MPDL was therefore not sufficient for efficient insertion of MPDL in the copolymer (according to the reactivity ratios) to prevent irreversible termination reactions. For fMPDL,0 = 0.4, (expts 2, 2d, 5 and 5d), the control over the polymerization was significantly improved, leading to nearly first order kinetics, linear increase of Mn with conversion, exhibiting values closer to the theoretical ones, and lower dispersities even at high conversion for expts 4–6 and 4d–6d. Control was further enhanced for fMPDL,0 = 0.7 (expt 3, 3d, 6 and 6d).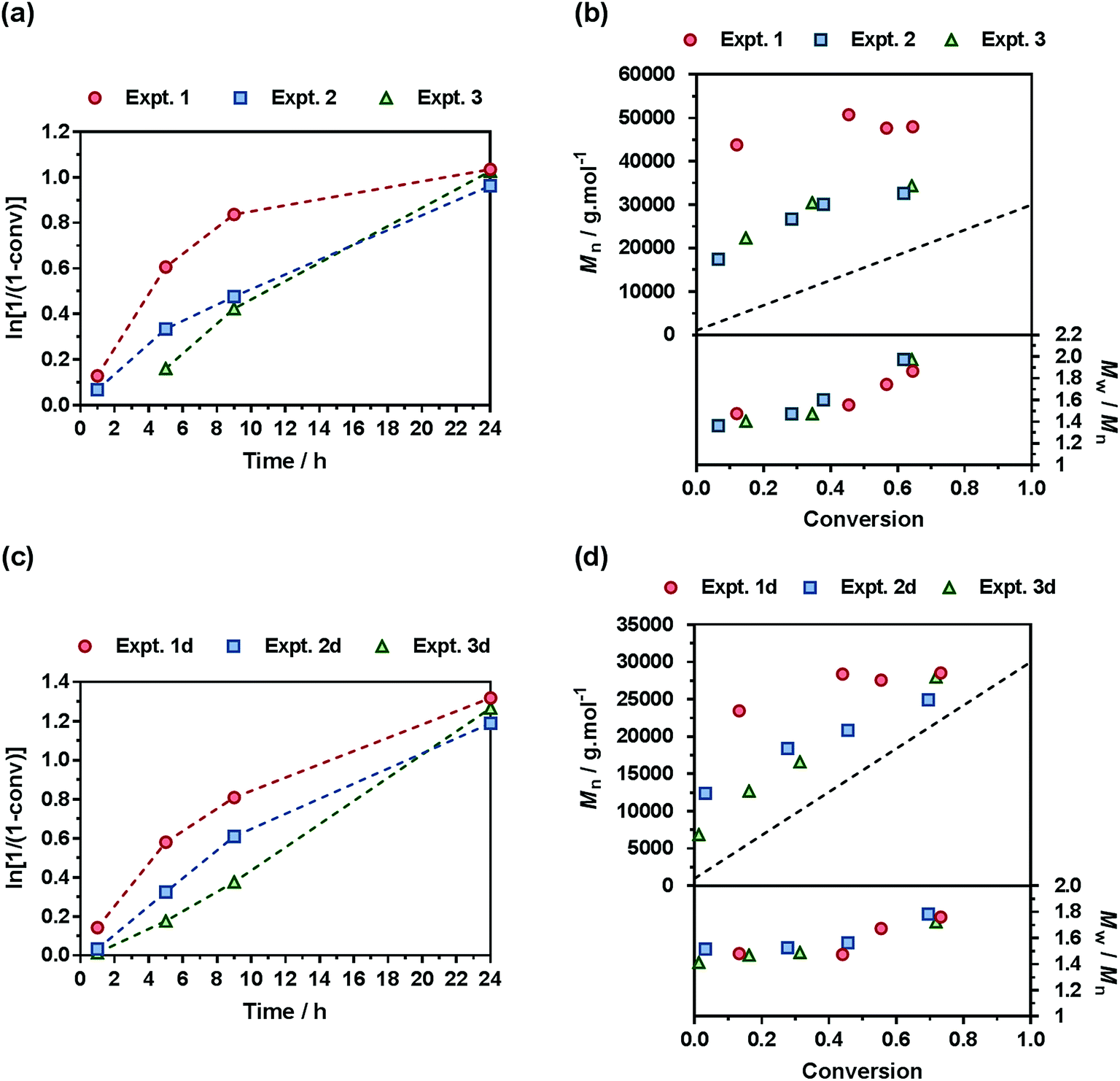 | ||
| Fig. 3 NMrROP of OEGMA and MPDL in 50 wt% toluene at 90 °C as a function of the nature of the alkoxyamine initiator [(a) and (b) Gem-AMA-SG1, (c) and (d) Gem-digly-AMA-SG1] and the initial fraction of MPDL: ● expts 1 and 1d (fMPDL,0 = 0.2), ■ expts 2 and 2d (fMPDL,0 = 0.4), ▲ expts 3 and 3d (fMPDL,0 = 0.7). (a) and (c) ln[1/(1 − conv)] vs. time (conv = OEGMA conversion). Dashed lines connecting data points are guides for the eye only. (b) and (d) Number-average molar mass, Mn and dispersity, Mw/Mn, vs. conversion. The dashed black line represents the theoretical Mn. | ||
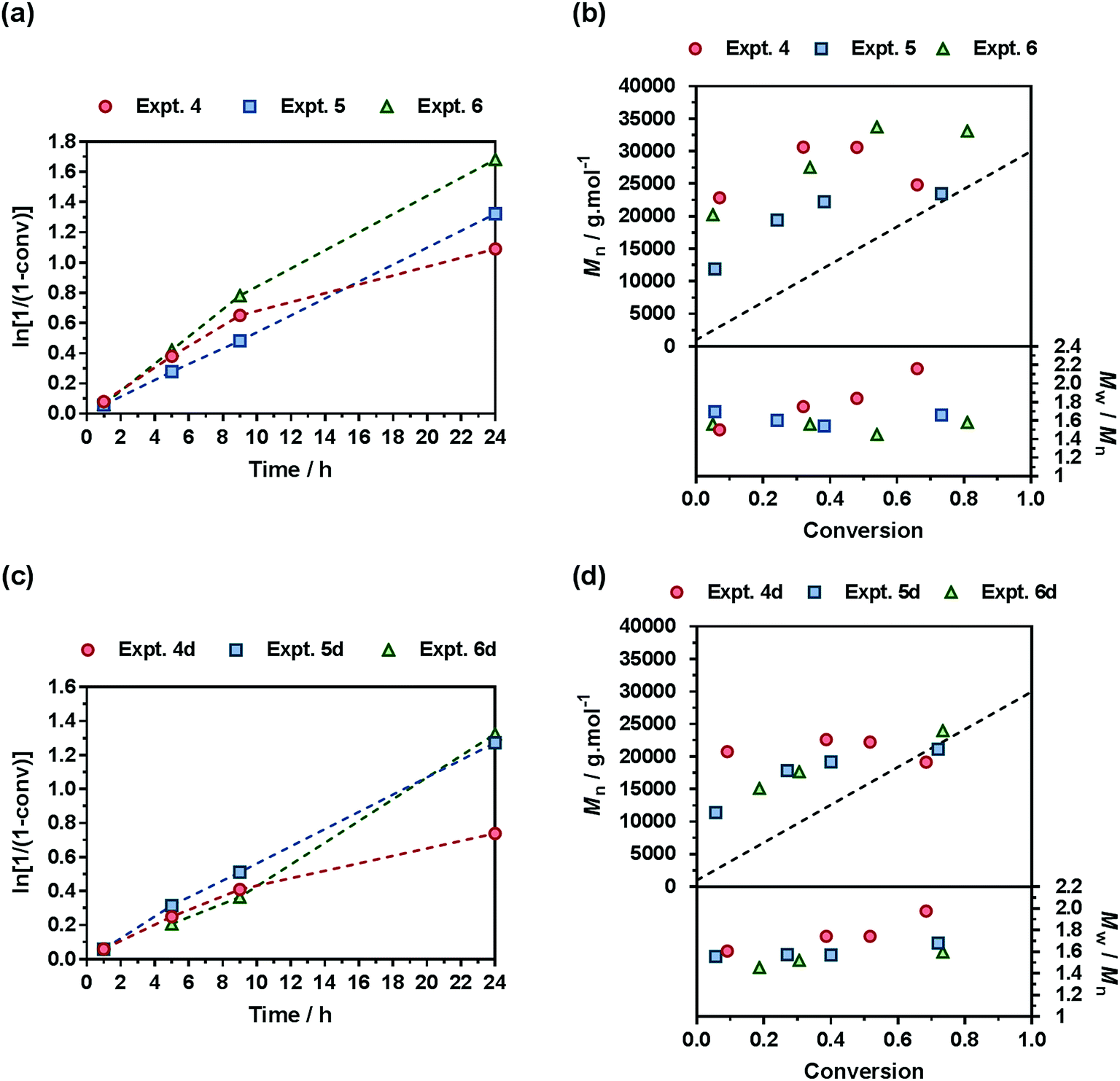 | ||
| Fig. 4 NMrROP of MMA and MPDL in 50 wt% toluene at 90 °C as a function of the nature of the alkoxyamine initiator [(a) and (b) Gem-AMA-SG1, (c) and (d) Gem-digly-AMA-SG1] and the initial fraction of MPDL: ● expts 4 and 4d (fMPDL,0 = 0.2), ■ expts 5 and 5d (fMPDL,0 = 0.4), ▲ expts 6 and 6d (fMPDL,0 = 0.7). (a) and (c) ln[1/(1 − conv)] vs. time (conv = MMA conversion). Dashed lines connecting data points are guides for the eye only. (b) and (d) Number-average molar mass, Mn and dispersity, Mw/Mn, vs. conversion. The dashed black line represents the theoretical Mn. | ||
Interestingly, the control was generally better when the copolymerization was initiated by Gem-digly-AMA-SG1 compared to those initiated by Gem-AMA-SG1. Mn values and dispersities at high conversions were indeed systematically lower with Gem-digly-AMA-SG1. This trend can be explained by the structure of the alkoxyamines and its influence on its dissociation rate constant and subsequently on the control of the polymerization. Conversely to Gem-digly-AMA-SG1, Gem-AMA-SG1 is prone to intramolecular hydrogen-bonding (IHB) between the hydrogen of the amide from the propagating radical and the nitroxide fragment.63 It resulted in slower dissociation kinetics and thus a less efficient control because of the lower amount of released nitroxide.
Synthesis of low molar mass polymer prodrugs for biological evaluations
One of the main benefits of the drug-initiated method is the facile tuning of the drug loading simply by varying the Mn of the polymer, which is of great importance for further biological evaluation. Herein, we targeted lower molar mass polymer prodrugs (Mn ∼10000 g mol−1) to obtain a drug loading of ∼2.5 wt% by adapting the reaction conditions (e.g., lower reaction times and/or lower targeted Mn, see the Experimental part). Note that, if needed, higher drug loadings can be successfully obtained (e.g., 3.6 and 9.0 wt%) by further decreasing the Mn (P3′ and P3′′, Table S1†). Four libraries of well-defined copolymer prodrugs from each series were prepared (Table 1 and Fig. S4†): Gem-P(OEGMA-co-MPDL) (P1–P3), Gem-digly-P(OEGMA-co-MPDL) (P1d–P3d), Gem-P(MMA-co-MPDL) (P4–P6) and Gem-digly-P(MMA-co-MPDL) (P4d–P6d). A lower reaction time also enabled improving the control by avoiding high monomer conversion and thus extensive occurrence of irreversible termination reactions. Overall, dispersities of the resulting copolymers ranged from 1.1 to 1.4, except for P1 whose control was difficult to achieve given the very low amount of initial MPDL59 and the use of the less efficient alkoxyamine (as detailed in the previous section). 1H NMR spectroscopy of the purified copolymers (and deprotected for P1d–P6d) in CDCl3 (Fig. S5–S8†) and in DMSO-d6 (Fig. S9–S12†) showed all signals expected for each structure. 19F NMR spectroscopy confirmed the quantitative presence of Gem at the extremity of the copolymers.
| Prodrug | Alkoxyamine | Methacrylic ester | f MPDL,0 | Conv.a (%)/temps (h) | M n (g mol−1) | Total DPn | Đ | F MPDL |
|---|---|---|---|---|---|---|---|---|
| a Methacrylic ester conversion determined by 1H NMR. b Determined by SEC after precipitation. c Determined by 1H NMR. | ||||||||
| P1 | Gem-AMA-SG1 | OEGMA | 0.2 | 40/8 | 15500 |
51 | 1.54 | 0.06 |
| P2 | Gem-AMA-SG1 | OEGMA | 0.4 | 28/8 | 10200 |
34 | 1.39 | 0.12 |
| P3 | Gem-AMA-SG1 | OEGMA | 0.7 | 18/8 | 10000 |
35 | 1.37 | 0.25 |
| P1d | Gem-digly-AMA-SG1 | OEGMA | 0.2 | 61/8 | 11500 |
36 | 1.24 | 0.07 |
| P2d | Gem-digly-AMA-SG1 | OEGMA | 0.4 | 36/8 | 13200 |
43 | 1.24 | 0.11 |
| P3d | Gem-digly-AMA-SG1 | OEGMA | 0.7 | 22/8 | 11200 |
38 | 1.13 | 0.22 |
| P4 | Gem-AMA-SG1 | MMA | 0.2 | 35/5 | 13200 |
119 | 1.27 | 0.10 |
| P5 | Gem-AMA-SG1 | MMA | 0.4 | 34/5 | 9900 | 83 | 1.28 | 0.19 |
| P6 | Gem-AMA-SG1 | MMA | 0.7 | 23/5 | 10300 |
82 | 1.21 | 0.29 |
| P4d | Gem-digly-AMA-SG1 | MMA | 0.2 | 56/8 | 15400 |
138 | 1.34 | 0.07 |
| P5d | Gem-digly-AMA-SG1 | MMA | 0.4 | 55/8 | 12400 |
106 | 1.29 | 0.12 |
| P6d | Gem-digly-AMA-SG1 | MMA | 0.7 | 33/8 | 10900 |
84 | 1.20 | 0.29 |
Despite unfavorable reactivity ratios of the different monomer pairs (rMPDL = 0 and rOEGMA = 6.95,59 and rMPDL = 0.01 and rMMA = 4.0 (ref. 54)), the molar fraction of MPDL in the copolymer (FMPDL) was finely tuned by varying the initial molar fraction of MPDL in the comonomer feed (Table 1) to induce different levels of degradability. On average, FMPDL was ∼0.07 for fMPDL,0 = 0.2, ∼0.13 for fMPDL,0 = 0.4 and ∼0.26 for fMPDL,0 = 0.7.
Hydrolytic degradation of the prodrugs
The degradation of the different copolymer prodrugs was then evaluated under accelerated conditions to probe the presence of the ester group in the polymer backbone, that is at room temperature in 5% KOH, either in water for OEGMA-based copolymers (P1–P3) or in a THF/MeOH (50:50, v/v) mixture for MMA-based copolymers (P4–P6). As expected, control copolymers without MPDL (FMPDL = 0), Gem-P(OEGMA-co-S) and Gem-P(MMA-co-S), did not undergo any degradation as shown by their constant Mn over time. Conversely, MPDL-containing copolymer prodrugs led to adjustable degradation in direct relationship with their MPDL content, as shown by the shifts of the SEC chromatograms towards lower Mn values (Fig. S13 and S14†). Whatever the nature of the methacrylic ester, the higher the MPDL content, the greater the degradation. The Mn decrease (Fig. 5) spanned from 10 to 30% for copolymers with the lowest MPDL contents (P1 and P4) to ∼70% for those with the highest amounts of MPDL (P3 and P6). These results confirmed the significant insertion of open MPDL units in the main chain of the copolymers and the possibility to fine-tune their degradation by adjusting the initial comonomer stoichiometry.
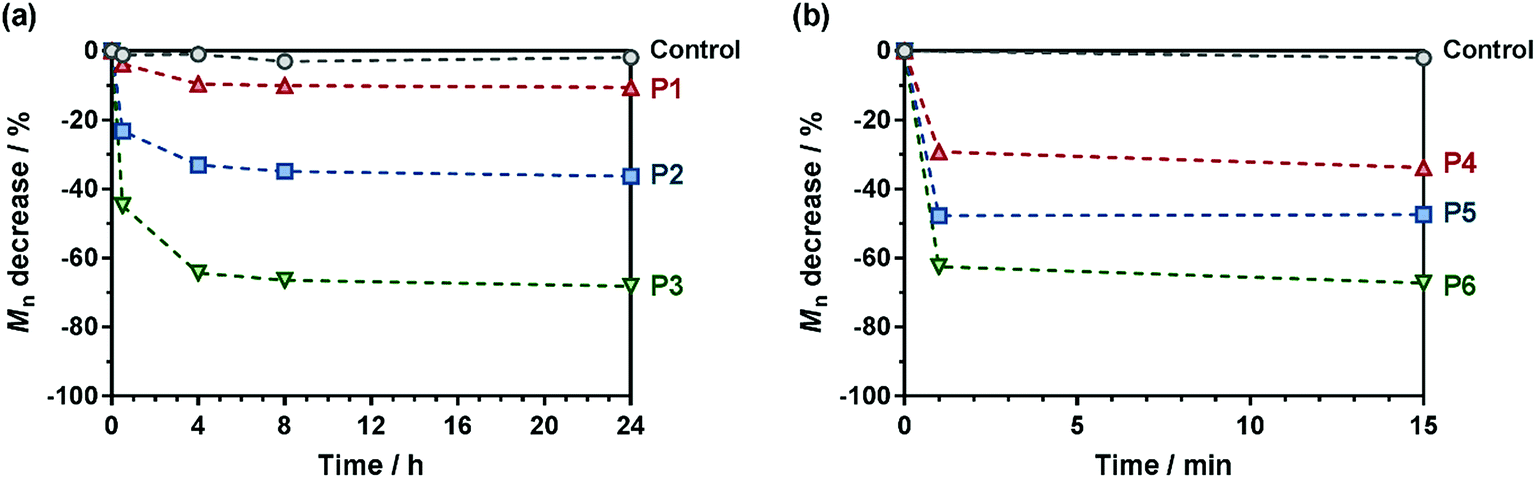 | ||
| Fig. 5 Hydrolytic degradation under accelerated conditions (5% KOH) of the different degradable polymer prodrugs as a function of the MPDL fraction and the nature of the methacrylic ester monomer: (a) Gem-P(OEGMA-co-MPDL); ●, control (FMPDL = 0); ▲, P1 (FMPDL = 0.06); ■, P2 (FMPDL = 0.12); ▼, P3 (FMPDL = 0.25). (b) Gem-P(MMA-co-MPDL); ●, control (FMPDL = 0); ▲, P4 (FMPDL = 0.10); ■, P5 (FMPDL = 0.19); ▼, P6 (FMPDL = 0.29). Dashed lines are guides for the eye only. | ||
Note that some discrepancy between theoretical Mn after degradation (calculated according to: 1/FMPDL − 1) and experimental ones may appear because of unfavorable reactivity ratios, as commonly observed with CKA monomers.59 Also, molar mass distributions stayed rather low (Đ ∼1.5–1.8) after degradation, which is in agreement with a theoretical investigation64 showing that stopping at low conversion for the synthesis of the copolymers is key to maintaining a certain homogeneity of the degraded products.
Physicochemical properties
Given their hydrophobic backbone and the water-solubility of Gem, Gem-P(MMA-co-MPDL) (P4–P6) and Gem-digly-P(MMA-co-MPDL) (P4d–P6d) prodrugs were formulated into nanoparticles in water. They displayed an average diameter in the 109–196 nm range, along with narrow particle size distributions (Table 2) and great colloidal stability in the long run either in water or in cell culture medium (Fig. S15†), whereas poor colloidal stability was observed in PBS.Interestingly, nanoparticles prepared from P4d–P6d were significantly smaller than those prepared from P4–P6, possibly because of the additional hydrophilicity provided by the diglycolate linker, promoting Gem positioning at the surface of the nanoparticles and thus inducing a more efficient stabilization. This hypothesis is supported by the increase of the predicted HLB number for a model Gem-digly-PMMA prodrug compared to Gem-PMMA using both the Davies and the Griffin methods (Table S2†). Representative Cryo-TEM images showed spherical morphologies in good agreement with DLS data (Fig. 6a and b and Fig. S16 and S17†). All nanoparticles exhibited great colloidal stability over time as shown by their constant size and size distributions for at least 25 days after nanoprecipitation (Fig. 6c). Such an efficient colloidal stability is likely the result of an efficient electrostatic stabilization, as shown by the significantly negative zeta potential measurements (Table 2).
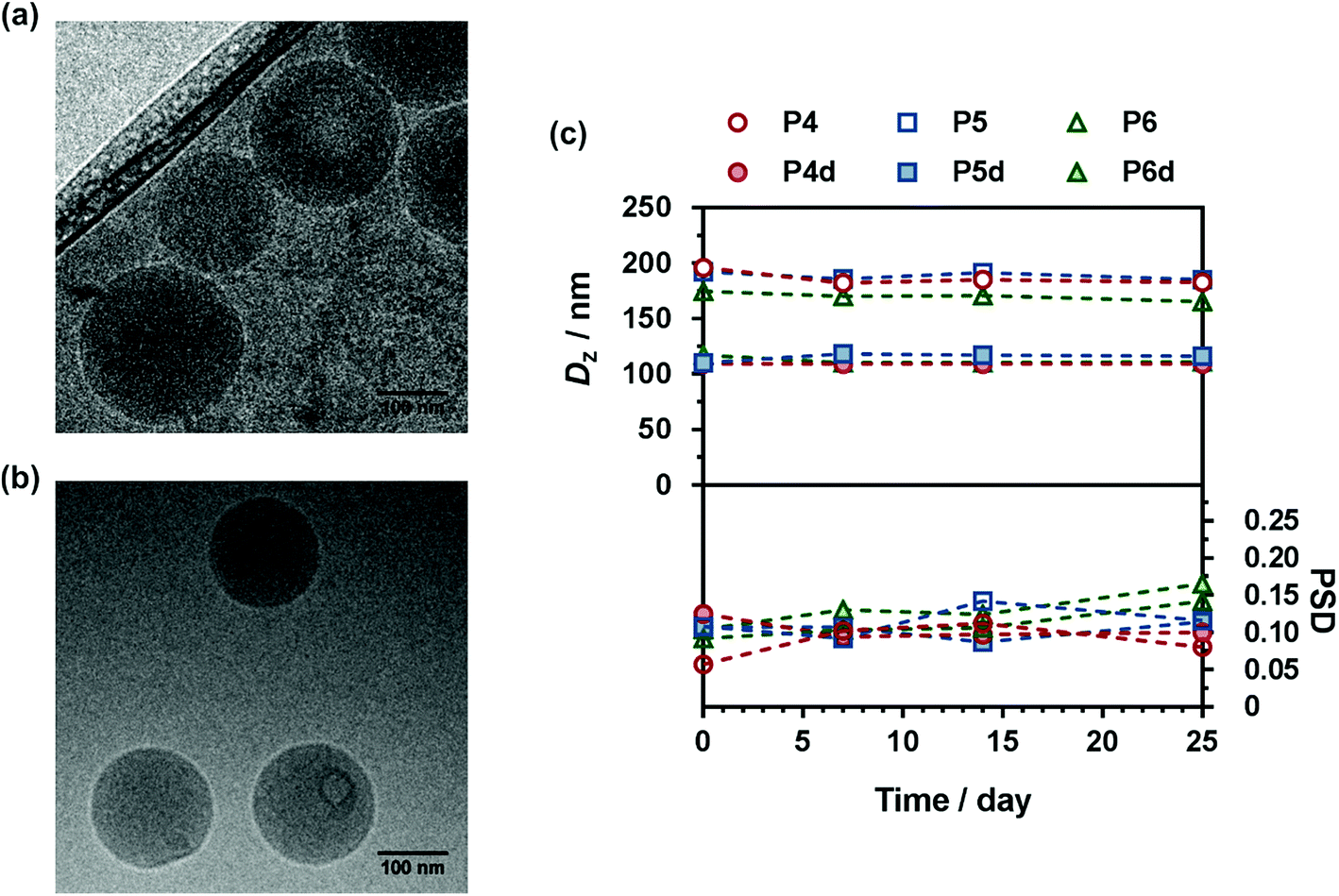 | ||
| Fig. 6 Representative Cryo-TEM images of (a) Gem-P(MMA-co-MPDL) P6 and (b) Gem-digly-P(MMA-co-MPDL) P6d nanoparticles. (c) Evolution with time of the average diameter and the particle size distribution (PSD) of Gem-P(MMA-co-MPDL) (P4–P6) and Gem-digly-P(MMA-co-MPDL) (P4d–P6d) nanoparticles in water determined by DLS. | ||
Given the relatively moderate FMPDL values for P(OEGMA-co-MPDL) copolymers, the water-solubility of OEGMA was expected to dominate over the hydrophobicity from MPDL units, and thus preferentially lead to fully water-soluble polymer prodrugs. To validate this hypothesis, Gem-P(OEGMA-co-MPDL) prodrugs with increasing contents of MPDL (P1–P3) were nanoprecipitated in water and the amount of nanoparticles, likely formed by nanoscale aggregation of amphiphilic P(OEGMA-co-MPDL) copolymers, was quantified by measurement of the dry content after ultracentrifugation (see the Experimental part and Table S3†). The weight fraction of nanoparticles was estimated to be 16 wt% maximum for the copolymer containing the highest amount of MPDL (FMPDL = 0.25) to less than 1 wt% for the one with the lowest amount (FMPDL = 0.06).
Drug release in human serum
The Gem release kinetics from the two different classes of polymer prodrugs was evaluated in human serum to better mimic the biological environment of the human body compared to accelerated degradation conditions (Fig. 7). In all cases, the maximum drug release was reached after a period of ∼1 h which is rather fast but commonly seen with water-soluble amide prodrugs.65 It is also believed that having a hydrophilic drug like Gem promotes fast drug release compared to hydrophobic ones, for instance like paclitaxel, as already observed.12 Interestingly, after the maximum drug release is reached, a plateau is observed which likely corresponds to the fraction of drug that is not easily accessible and that cannot be readily cleaved from the copolymers within the time frame of the drug release experiment. We can hypothesize for P(MMA-co-MPDL) nanoparticles that only the surface fraction of Gem is released whereas the fraction which is buried into the nanoparticle's core will be accessible and released only when the nanoparticles are degraded.66 As for soluble P(OEGMA-co-MPDL) prodrugs, which are essentially molecularly dissolved even for the highest amounts of MPDL (Fig. S18†), they will form a protective P(OEGMA-co-MPDL) shroud wrapped around the drug, efficiently protecting the drug-linker moiety from enzymes, similarly to what is observed with PEGylated peptides/proteins. Therefore, only the fraction of Gem located at the periphery of such a PEG-based random coil will be readily cleaved whereas the remaining amount of drug will be accessible after the copolymer gets degraded.66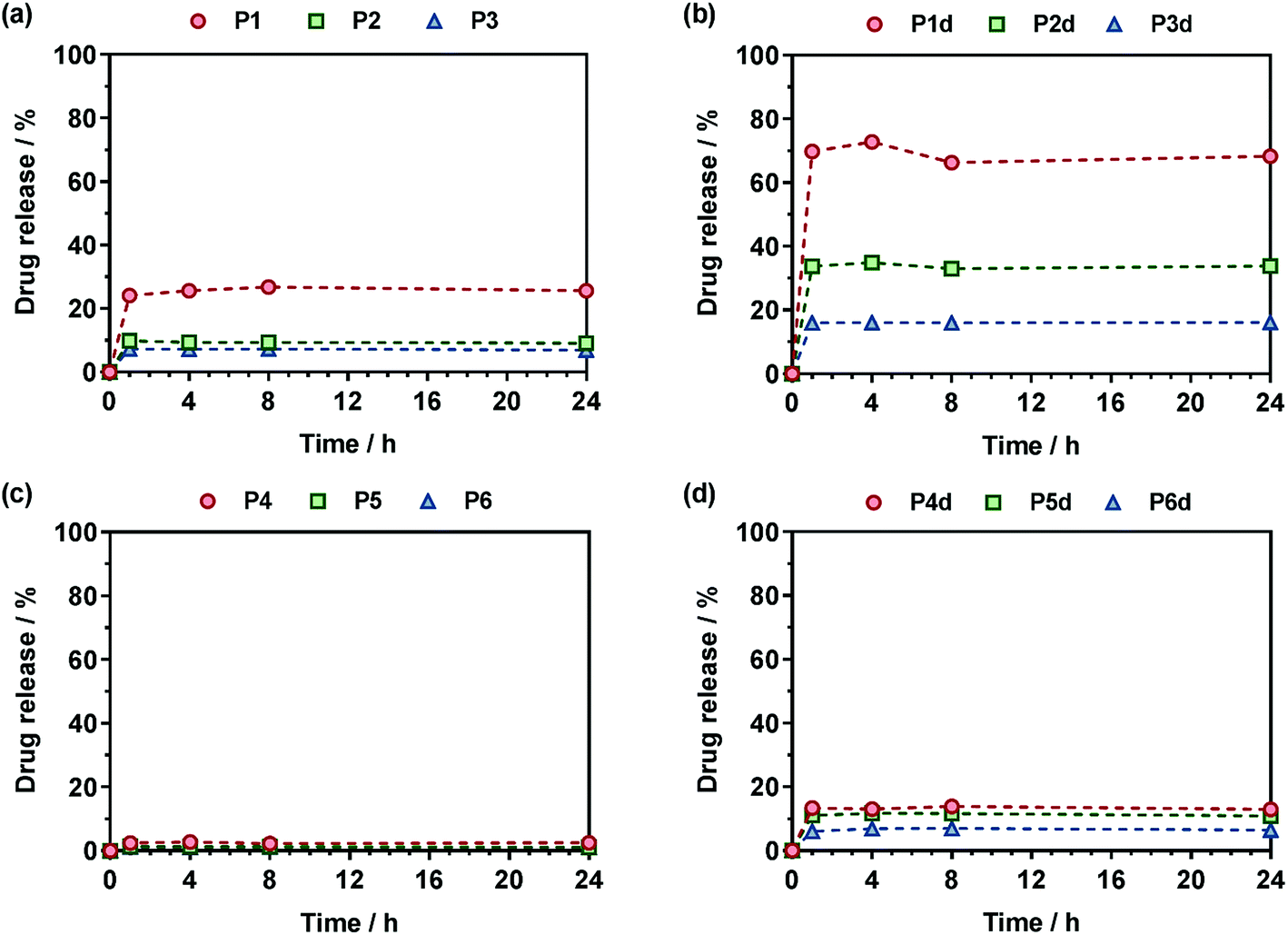 | ||
| Fig. 7 Gem release profiles at 37 °C in human serum from (a) Gem-P(OEGMA-co-MPDL) (P1–P3), (b) Gem-digly-P(OEGMA-co-MPDL) (P1d–P3d), (c) Gem-P(MMA-co-MPDL) (P4–P6) and (d) Gem-digly-P(MMA-co-MPDL) (P4d–P6d). | ||
For Gem-P(OEGMA-co-MPDL) P1–P3, the total Gem release gradually increased from ∼7 to ∼25% when decreasing FMPDL from 0.25 to 0.06 (Fig. 7a). This may be explained by the strong hydrophobic nature of MPDL that prevents enzyme access because of poor solvation of the drug-polymer linker and/or π–π stacking interactions between MPDL units. Analogous copolymers with the diglycolate linker (P1d–P3d) led to the same trend but with a significantly higher Gem release; from ∼33% for FMPDL = 0.22 to ∼70% for FMPDL = 0.07 (Fig. 7b), likely because of the increased hydrophilicity and decreased steric hindrance in the vicinity of the amide bond. Replacing OEGMA by MMA in the polymer prodrug structures (P4–P6 and P4d–P6d) led to the exact same trend but with significantly lower Gem release contents. Gem-P(MMA-co-MPDL) nanoparticles P4–P6 led to nearly no Gem release (<2%) whereas Gem-digly-P(MMA-co-MPDL) nanoparticles (P4d–P6d) allowed final Gem release contents to reach 7–13% (Fig. 7c and d). This may be explained not only by the detrimental effect of hydrophobic MMA units in the vicinity of the Gem-polymer linker, but also by the nanoparticulate nature of the polymer prodrug itself, both preventing extensive water uptake and/or enzyme access.
Altogether, these results showed that Gem release was independently governed by: (i) the MPDL content – the lower the MPDL content, the greater the Gem release; (ii) the nature of the linker – the presence of the diglycolate linker enabled a greater Gem release compared to a simple amide bond and (iii) the hydrophilicity of the methacrylate monomer–OEGMA enabled a greater Gem release compared to MMA. It is therefore suggested that increasing the hydrophilicity in the vicinity of the drug-polymer linkage, by using OEGMA and/or by decreasing the amount of inserted MPDL, which is a strongly hydrophobic monomer, had a beneficial influence on the Gem release. Regarding the diglycolate linker, its beneficial impact may be explained by: (i) its higher lability compared to a single amide bond that may enable its rapid cleavage and (ii) its connection to the amide bond that may promote the amide bond accessibility to enzymes.
In vitro anticancer activity
A crucial question is whether the above-mentioned drug release trends observed in human serum directly correlate with the anticancer activity of the polymer prodrugs. The cell viability of two cancer cell lines corresponding to clinically relevant cancer models for Gem, human lung carcinoma (A549) and human pancreatic cancer (MiaPaCa-2), was then determined after incubation with the different polymer prodrugs at various concentrations.Gem-free control copolymers, P(OEGMA-co-MPDL) P7 and P(MMA-co-MPDL) P8, were not cytotoxic for all concentrations tested while free Gem exhibited half maximal inhibitory concentrations (IC50) of 4 nM and 14 nM for A549 and MiaPaCa-2 cells, respectively. When increasing the MPDL fraction, IC50 values of Gem-P(OEGMA-co-MPDL) P1–P3 increased from 0.30 to 2.14 μM for A549 cells and from 0.13 to 1.07 μM for MiaPaCa-2 cells (Fig. 8), which is totally in line with the previously mentioned trends observed from drug release experiments. As expected from drug release experiments, diglycolate-containing polymer prodrugs (P1d–P3d) gave the same trend but were significantly more cytotoxic, leading to IC50 values 5–8-fold lower for A549 cells (Fig. 8a and b) to 3–4-fold lower for MiaPaCa-2 cells (Fig. 8c and d) compared to those obtained from P1–P3. Note that the small fraction of Gem-P(OEGMA-co-MPDL) nanoparticles present along with the soluble Gem-P(OEGMA-co-MPDL) copolymer did not affect the MTT results as a purified aqueous solution of copolymers P2 gave the same cytotoxicity profile (Fig. S19†).
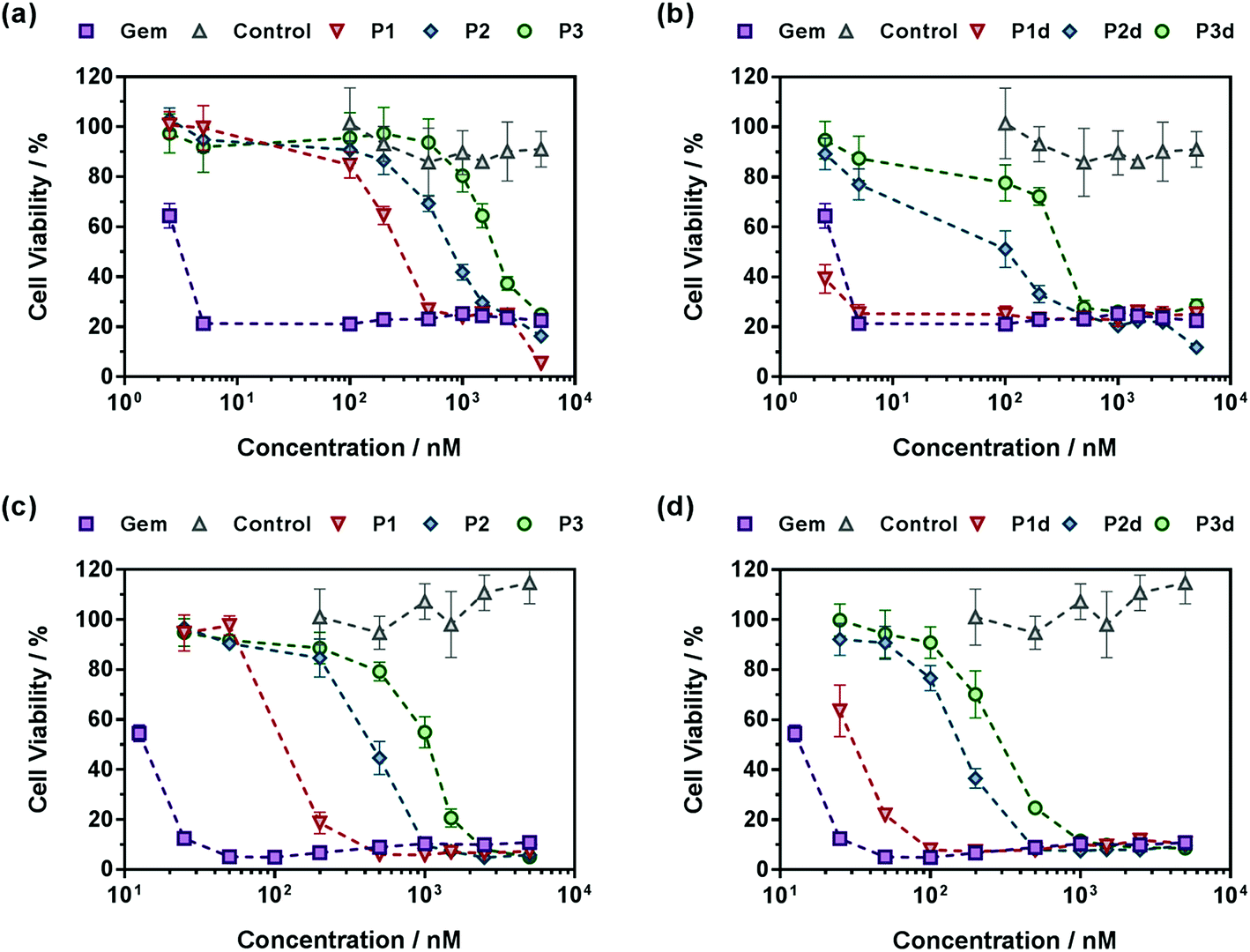 | ||
| Fig. 8 Cell viability (MTT test) with increasing concentrations of (a and c) Gem, Gem-P(OEGMA-co-MPDL) (P1–P3) and P(OEGMA-co-MPDL) (P7, control) or (b and d) Gem, Gem-digly-P(OEGMA-co-MPDL) (P1d–P3d) and P(OEGMA-co-MPDL) (P7, control) on (a and b) A549 cells and (c and d) MiaPaCa-2 cells. | ||
Also, in agreement with drug release experiments, Gem-P(MMA-co-MPDL) P4–P6 and Gem-digly-P(MMA-co-MPDL) P4d–P6d polymer prodrug nanoparticles (Fig. 9) were always less cytotoxic than OEGMA-based counterparts. Importantly, whatever the cell line, no decrease in cell viability was observed for P4–P6 even at the highest concentrations, whereas the use of the diglycolate linker made the corresponding prodrugs cytotoxic, especially for those containing less MPDL (P5d and P6d, see Fig. 9b and d). As suggested from drug release experiments, this trend may be correlated with a too high hydrophobicity in the vicinity of the Gem-polymer linker, but also with the nanoparticulate nature of the polymer prodrugs that, conversely to fully water-soluble counterparts, were less accessible to water/enzymes, thus preventing efficient release of Gem (only surface exposed Gem-digly moieties were likely accessible for cleavage). Note that, in general, higher cytotoxicity was observed against MiaPaCa-2 cells, with nearly complete cell death. This is explained by the fact that A549 cells are known to exhibit some resistance against Gem, as evidenced by a plateau at 20% of cell viability.67 Even though results with Gem-P(MMA-co-MPDL) and Gem-digly-P(MMA-co-MPDL) nanoparticles may appear somewhat disappointing, they are crucial to identify key structural parameters to improve the cytotoxicity of the materials.
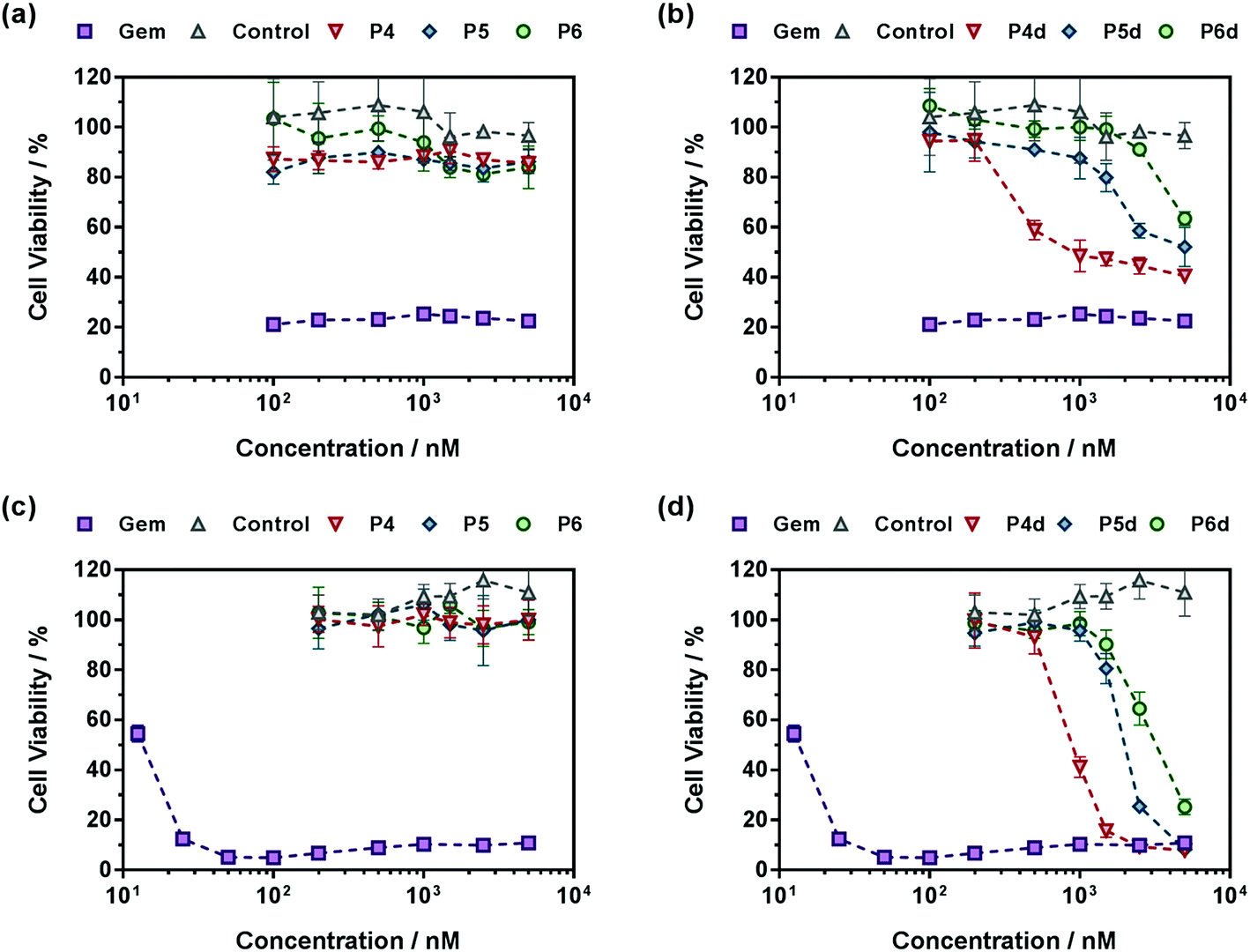 | ||
| Fig. 9 Cell viability (MTT test) with increasing concentrations of (a and c) Gem, Gem-P(MMA-co-MPDL) (P4–P6) and P(MMA-co-MPDL) (P8, control) or (b and d) Gem, Gem-digly-P(MMA-co-MPDL) (P4d–P6d) and P(MMA-co-MPDL) (P8, Control) on (a and b) A549 cells and (c and d) MiaPaCa-2 cells. | ||
In a nutshell, the results for both cell lines were in excellent agreement with those obtained from drug release experiments: (i) polymer prodrugs based on P(OEGMA-co-MPDL) were more cytotoxic than those based on P(MMA-co-MPDL); (ii) increasing the MPDL fraction in the copolymers led to a decrease in cytotoxicity and (iii) polymer prodrugs based on the diglycolate linker were significantly more cytotoxic than those based on a single amide linkage (see Table S4† for all IC50 values). Remarkably, the best candidate, Gem-digly-P(OEGMA-co-MPDL) P1d enabled reaching the cytotoxic activity of free Gem.
Conclusion
Degradable vinyl polymer prodrugs were designed by “drug-initiated” NMrROP of a methacrylic ester monomer with MPDL from an alkoxyamine derivatized with Gem as an anticancer drug. Two libraries of polymer prodrugs differing by the nature of the methacrylic ester monomer (OEGMA or MMA), the nature of the drug-polymer linker and the MPDL content were prepared. Whereas MMA-based prodrugs formed highly stable nanoparticles upon nanoprecipitation, OEGMA-based prodrugs were water-soluble. The degradation of the copolymer prodrugs was proved under accelerated conditions (i.e., basic hydrolysis) and the degradation level was finely tuned by adjusting the MPDL content. Drug-release profiles in human serum and in vitro anticancer activity against two different cancer cell lines helped to establish structure/activity relationships and select the most favorable structural parameters for having the best activity. We demonstrated that three structural parameters independently governed the anticancer activity: (i) soluble OEGMA-based prodrugs were more cytotoxic than MMA-based counterparts; (ii) the lower the MPDL content, the greater the anticancer activity and (iii) a diglycolate linker afforded greater activity compared to a simple amide bond.Overall, this unique approach enabled us to combine the best of different worlds: (i) degradability from ring-opening polymerization; (ii) ease of synthesis by a radical polymerization method and (iii) sustained drug release and high anticancer activity by a prodrug approach. Additionally, this approach is versatile and general as it could easily be extended to other polymers, to other pathologies by using different drugs, and to other biologically relevant molecules (e.g., fluorescent dyes, imaging agents) for theranostic applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the French Ministry of Research for the financial support of the PhD thesis of EG and the French National Research Agency (ANR-11-JS08-0005 and ANR-15-CE08-0019) for the financial support of the Master internship and PhD of JT, respectively. The authors thank Julie Mougin and Stéphanie Denis (Institut Galien Paris-Sud) for technical assistance in HPLC and cell culture, respectively. Arkema is warmly acknowledged for kindly providing the BlocBuilder MA™ alkoxyamine and the SG1 nitroxide. The CNRS is also acknowledged for financial support.References
- J. Nicolas, S. Mura, D. Brambilla, N. Mackiewicz and P. Couvreur, Chem. Soc. Rev., 2013, 42, 1147–1235 RSC.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef PubMed.
- V. Delplace, P. Couvreur and J. Nicolas, Polym. Chem., 2014, 5, 1529–1544 RSC.
- J. Nicolas, Chem. Mater., 2016, 28, 1591–1606 CrossRef PubMed.
- R. Tong and J. Cheng, Angew. Chem., Int. Ed., 2008, 47, 4830–4834 CrossRef PubMed.
- R. Tong and J. Cheng, J. Am. Chem. Soc., 2009, 131, 4744–4754 CrossRef PubMed.
- R. Tong and J. Cheng, Bioconjugate Chem., 2009, 21, 111–121 CrossRef PubMed.
- R. Tong and J. Cheng, Macromolecules, 2012, 45, 2225–2232 CrossRef PubMed.
- Q. Yin, R. Tong, Y. Xu, K. Baek, L. W. Dobrucki, T. M. Fan and J. Cheng, Biomacromolecules, 2013, 14, 920–929 CrossRef PubMed.
- S. Harrisson, J. Nicolas, A. Maksimenko, D. T. Bui, J. Mougin and P. Couvreur, Angew. Chem., Int. Ed., 2013, 52, 1678–1682 CrossRef PubMed.
- Y. Bao, T. Boissenot, E. Guégain, D. Desmaële, S. Mura, P. Couvreur and J. Nicolas, Chem. Mater., 2016, 28, 6266–6275 CrossRef.
- Y. Bao, E. Guegain, J. Mougin and J. Nicolas, Polym. Chem., 2018, 9, 687–698 RSC.
- Y. Bao and J. Nicolas, Polym. Chem., 2017, 8, 5174–5184 RSC.
- Y. Bao, V. Nicolas and J. Nicolas, Chem. Commun., 2017, 53, 4489–4492 RSC.
- D. T. Bui, A. Maksimenko, D. Desmaele, S. Harrisson, C. Vauthier, P. Couvreur and J. Nicolas, Biomacromolecules, 2013, 14, 2837–2847 CrossRef PubMed.
- A. Maksimenko, D. T. Bui, D. Desmaële, P. Couvreur and J. Nicolas, Chem. Mater., 2014, 26, 3606–3609 CrossRef.
- C. C. Williams, S. H. Thang, T. Hantke, U. Vogel, P. H. Seeberger, J. Tsanaktsidis and B. Lepenies, ChemMedChem, 2012, 7, 281–291 CrossRef PubMed.
- B. Louage, L. Nuhn, M. D. P. Risseeuw, N. Vanparijs, R. DeCoen, I. Karalic, S. VanCalenbergh and B. G. DeGeest, Angew. Chem., Int. Ed., 2016, 55, 11791–11796 CrossRef PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402–1472 CrossRef.
- V. Delplace and J. Nicolas, Nat. Chem., 2015, 7, 771–784 CrossRef PubMed.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Chem. Rev., 2017, 117, 1319–1406 CrossRef PubMed.
- S. Agarwal, Polym. Chem., 2010, 1, 953–964 RSC.
- W. J. Bailey, S. R. Wu and Z. Ni, Makromol. Chem., 1982, 183, 1913–1920 CrossRef.
- W. J. Bailey, Z. Ni and S. R. Wu, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 3021–3030 CrossRef.
- W. J. Bailey, Z. Ni and S. R. Wu, Macromolecules, 1982, 15, 711–714 CrossRef.
- W. J. Bailey and L.-L. Zhou, Tetrahedron Lett., 1991, 32, 1539–1540 CrossRef.
- S. Agarwal and L. Ren, Macromolecules, 2009, 42, 1574–1579 CrossRef.
- L. Ren, C. Speyerer and S. Agarwal, Macromolecules, 2007, 40, 7834–7841 CrossRef.
- J. Undin, A. Finne-Wistrand and A.-C. Albertsson, Biomacromolecules, 2013, 14, 2095–2102 CrossRef PubMed.
- J. Undin, T. Illanes, A. Finne-Wistrand and A.-C. Albertsson, Polym. Chem., 2012, 3, 1260–1266 RSC.
- H. Wickel and S. Agarwal, Macromolecules, 2003, 36, 6152–6159 CrossRef.
- H. Wickel, S. Agarwal and A. Greiner, Macromolecules, 2003, 36, 2397–2403 CrossRef.
- H. Shi, L. Liu, X. Wang and J. Li, Polym. Chem., 2012, 3, 1182–1188 RSC.
- J. Huang, R. Gil and K. Matyjaszewski, Polymer, 2005, 46, 11698–11706 CrossRef.
- J.-F. Lutz, J. Andrieu, S. Üzgün, C. Rudolph and S. Agarwal, Macromolecules, 2007, 40, 8540–8543 CrossRef.
- C. Riachi, N. Schüwer and H.-A. Klok, Macromolecules, 2009, 42, 8076–8081 CrossRef.
- D. J. Siegwart, S. A. Bencherif, A. Srinivasan, J. O. Hollinger and K. Matyjaszewski, J. Biomed. Mater. Res., Part A, 2008, 87A, 345–358 CrossRef PubMed.
- J.-Y. Yuan and C.-Y. Pan, Eur. Polym. J., 2002, 38, 1565–1571 CrossRef.
- G. G. Hedir, C. A. Bell, N. S. Ieong, E. Chapman, I. R. Collins, R. K. O'Reilly and A. P. Dove, Macromolecules, 2014, 47, 2847–2852 CrossRef.
- J. M. J. Paulusse, R. J. Amir, R. A. Evans and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 9805–9812 CrossRef PubMed.
- Q. Smith, J. Huang, K. Matyjaszewski and Y.-L. Loo, Macromolecules, 2005, 38, 5581–5586 CrossRef.
- S. Ganda, Y. Jiang, D. S. Thomas, J. Eliezar and M. H. Stenzel, Macromolecules, 2016, 49, 4136–4146 CrossRef.
- S. Kobben, A. Ethirajan and T. Junkers, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1633–1641 CrossRef.
- G. G. Hedir, C. A. Bell, R. K. O'Reilly and A. P. Dove, Biomacromolecules, 2015, 16, 2049–2058 CrossRef PubMed.
- S. Harrisson, T. P. Davis, R. A. Evans and E. Rizzardo, Macromolecules, 2001, 34, 3869–3876 CrossRef.
- S. Harrisson, T. P. Davis, R. A. Evans and E. Rizzardo, Macromolecules, 2001, 34, 3869–3876 CrossRef.
- R. A. Evans and E. Rizzardo, Macromolecules, 1996, 29, 6983–6989 CrossRef.
- A.-C. Albertsson and I. K. Varma, Biomacromolecules, 2003, 4, 1466–1486 CrossRef PubMed.
- N. Grabe, Y. Zhang and S. Agarwal, Macromol. Chem. Phys., 2011, 212, 1327–1334 CrossRef.
- Q. Jin, F. Mitschang and S. Agarwal, Biomacromolecules, 2011, 12, 3684–3691 CrossRef PubMed.
- T. Cai, Y. Chen, Y. Wang, H. Wang, X. Liu, Q. Jin, S. Agarwal and J. Ji, Polym. Chem., 2014, 5, 4061–4068 RSC.
- T. Cai, Y. Chen, Y. Wang, H. Wang, X. Liu, Q. Jin, S. Agarwal and J. Ji, Macromol. Chem. Phys., 2014, 215, 1848–1854 CrossRef.
- J. Tran, E. Guegain, N. Ibrahim, S. Harrisson and J. Nicolas, Polym. Chem., 2016, 7, 4427–4435 RSC.
- H. Fessi, F. Puisieux, J. P. Devissaguet, N. Ammoury and S. Benita, Int. J. Pharm., 1989, 55, R1–R4 CrossRef.
- S. Mura, E. Buchy, G. Askin, F. Cayre, J. Mougin, S. Gouazou, D. Sobot, S. Valetti, B. Stella and D. Desmaele, Biochimie, 2016, 130, 4–13 CrossRef PubMed.
- A. Maksimenko, J. Mougin, S. Mura, E. Sliwinski, E. Lepeltier, C. Bourgaux, S. Lepêtre, F. Zouhiri, D. Desmaële and P. Couvreur, Cancer Lett., 2013, 334, 346–353 CrossRef PubMed.
- L. W. Hertel, G. B. Boder, J. S. Kroin, S. M. Rinzel, G. A. Poore, G. C. Todd and G. B. Grindey, Cancer Res., 1990, 50, 4417–4422 Search PubMed.
- V. Delplace, E. Guegain, S. Harrisson, D. Gigmes, Y. Guillaneuf and J. Nicolas, Chem. Commun., 2015, 51, 12847–12850 RSC.
- V. Delplace, A. Tardy, S. Harrisson, S. Mura, D. Gigmes, Y. Guillaneuf and J. Nicolas, Biomacromolecules, 2013, 14, 2837–2847 CrossRef PubMed.
- V. Delplace, S. Harrisson, A. Tardy, D. Gigmes, Y. Guillaneuf and J. Nicolas, Macromol. Rapid Commun., 2014, 35, 484–491 CrossRef PubMed.
- V. Heinemann, Y.-Z. Xu, S. Chubb, A. Sen, L. W. Hertel, G. B. Grindey and W. Plunkett, Cancer Res., 1992, 52, 533–539 Search PubMed.
- E. Guegain, V. Delplace, T. Trimaille, D. Gigmes, D. Siri, S. R. A. Marque, Y. Guillaneuf and J. Nicolas, Polym. Chem., 2015, 6, 5693–5704 RSC.
- D. Gigmes, P. H. M. Van Steenberge, D. Siri, D. R. D'hooge, Y. Guillaneuf and C. Lefay, Macromol. Rapid Commun., 2018, 1800193 CrossRef PubMed.
- M. Gooyit, M. Lee, V. A. Schroeder, M. Ikejiri, M. A. Suckow, S. Mobashery and M. Chang, J. Med. Chem., 2011, 54, 6676–6690 CrossRef PubMed.
- E. Guégain, J.-P. Michel, T. Boissenot and J. Nicolas, Macromolecules, 2018, 51, 724–736 CrossRef.
- R. Ikeda, L. C. Vermeulen, E. Lau, Z. Jiang, K. Sachidanandam, K. Yamada and J. M. Kolesar, Int. J. Oncol., 2011, 38, 513–519 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02256a |
| This journal is © The Royal Society of Chemistry 2018 |