
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gating the electron transfer at a monocopper centre through the supramolecular coordination of water molecules within a protein chamber mimic†
Nicolas
Le Poul
 a,
Benoit
Colasson
b,
Grégory
Thiabaud
b,
Dany Jeanne
Dit Fouque
a,
Claudio
Iacobucci
a,
Antony
Memboeuf
a,
Bénédicte
Douziech
a,
Jan
Řezáč
c,
Thierry
Prangé
d,
Aurélien de la
Lande
e,
Olivia
Reinaud
*b and
Yves
Le Mest
*a
a,
Benoit
Colasson
b,
Grégory
Thiabaud
b,
Dany Jeanne
Dit Fouque
a,
Claudio
Iacobucci
a,
Antony
Memboeuf
a,
Bénédicte
Douziech
a,
Jan
Řezáč
c,
Thierry
Prangé
d,
Aurélien de la
Lande
e,
Olivia
Reinaud
*b and
Yves
Le Mest
*a
aLaboratoire de Chimie, Electrochimie Moléculaires et Chimie Analytique, UMR CNRS 6521, Université de Brest, 29238 Brest, France. E-mail: yves.lemest@univ-brest.fr; nicolas.lepoul@univ-brest.fr; dany.jeanneditfouque@univ-brest.fr; iacobucci.claudio@gmail.com; antony.memboeuf@univ-brest.fr; benedicte.douziech@univ-brest.fr
bLaboratoire de Chimie et Biochimie Pharmacologiques et Toxicologiques, UMR CNRS 8601, Université Paris Descartes, 75006 Paris, France. E-mail: olivia.reinaud@parisdescartes.fr; benoit.colasson@parisdescartes.fr
cInstitute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Flemingovonám. 2, 166 10 Prague 6, Czech Republic. E-mail: rezac@uochb.cas.cz
dLaboratoire de Cristallographie et de Résonance Magnétique Nucléaire, Biologiques (CNRS UMR 8015), Université Paris Descartes, 4, Avenue de l’Observatoire, 75006 Paris, France. E-mail: thierry.prange@parisdescartes.fr
eLaboratoire de Chimie Physique, UMR CNRS 8000, Université Paris Sud, 91405 Orsay, France. E-mail: aurelien.de-la-lande@u-psud.fr
First published on 30th August 2018
Abstract
Functionality of enzymes is strongly related to water dynamic processes. The control of the redox potential for metallo-enzymes is intimately linked to the mediation of water molecules in the first and second coordination spheres. Here, we report a unique example of supramolecular control of the redox properties of a biomimetic monocopper complex by water molecules. It is shown that the copper complex based on a calix[6]arene covalently capped with a tetradentate [tris(2-methylpyridyl)amine] (tmpa) core, embedding the metal ion in a hydrophobic cavity, can exist in three different states. The first system displays a totally irreversible redox behaviour. It corresponds to the reduction of the 5-coordinate mono-aqua-CuII complex, which is the thermodynamic species in the +II state. The second system is detected at a high redox potential. It is ascribed to an “empty cavity” or “water-free” state, where the CuI ion sits in a 4-coordinate trigonal environment provided by the tmpa cap. This complex is the thermodynamic species in the +I state under “dry conditions”. Surprisingly, a third redox system appears as the water concentration is increased. Under water-saturation conditions, it displays a pseudo-reversible behaviour at a low scan rate at the mid-point from the water-free and aqua species. This third system is not observed with the Cu-tmpa complex deprived of a cavity. In the calix[6]cavity environment, it is ascribed to a species where a pair of water molecules is hosted by the calixarene cavity. A molecular mechanism for the CuII/CuI redox process with an interplay of (H2O)x (x = 0, 1, 2) hosting is proposed on the basis of computational studies. Such an unusual behaviour is ascribed to the unexpected stabilization of the CuI state by inclusion of the pair of water molecules. This phenomenon strongly evidences the drastic influence of the interaction between water molecules and a hydrophobic cavity on controlling the thermodynamics and kinetics of the CuII/CuI electron transfer process.
Introduction
The role of water in biochemical events has been for a long time neglected, water being considered only for its solvation properties. Recently, more attention has been paid to its involvement in a large number of biological processes.1,2 It is now widely accepted that water has a crucial effect on both the structure1,3 and functionality of proteins (folding,4–7 molecular recognition,8 electron transfer,9–11 proton delivery12 and catalysis13,14). Its action covers a wide range of localizations, from cavities and distal pockets15,16 to inter-protein spaces.17 Water has been recognized as a particularly good mediator for transferring electrons between remote biological redox cofactors by quantum tunneling,10,18,19 thanks to the existence of multiple strong hydrogen bonding channels favoured by a constrained environment.20 For metallo-enzymes requiring dioxygen binding, it has been well recognized that water hosting in the hydrophobic pocket defining the active site plays key roles in the catalytic activity and control. For example, water release is a pre-requirement for the reduction of the resting FeIII state in P-450 enzymes, and consequently for dioxygen binding.21 It has also been demonstrated that a pair of water molecules plays a key role in O–O bond heterolytic cleavage leading to the active species.22 More recently, it has been proposed that embedment of a water-cluster in the active site of cytochrome c-oxidase has a strong impact on its redox activity.14,16 Water release from the metal centre is also a necessary step for α-ketoglutarate-dependent non-heme mono-oxygenases. In copper enzymes, water molecules are suspected to play key roles in their catalytic cycles as well. For instance, a rare example of a bis-aqua adduct was recently reported for a CuII site of a Cu nitrite reductase (RpNiR).23 Water molecules were shown to specifically protect it from nitrite coordination at CuII and favour catalytic activation at CuI by displacement of the water ligand. In copper mono-oxygenases, water access or gating in the active site pocket may be key for generating a CuI state prone to O2 activation. This may also be implied in the fast reduction of the CuII state at the end of the catalytic cycle to regenerate the CuI active species. So far, the influence of water on the thermodynamics of redox biological systems has been little investigated.24 Most of the recent studies related to this subject have been devoted to the modification of the second coordination sphere in metalloproteins (i.e. peptidic residues) with the aim of tuning the redox properties and functionalities.25–27On the chemistry side, several examples of supramolecular objects such as organic cages or cavities able to encapsulate one or several water molecules have been described.28–32 Such systems are based on the ability of water molecules to form hydrogen-bonded clusters, hence neutralizing their intrinsic high polarity. Only few studies in solution have reported the encapsulation of a definite number of water molecules.33,34 Remarkably, for one of them, the water molecule was trapped in apolar C60 under high-pressure conditions.33 However, the electrochemical properties of the fullerene were not modified by the inner H2O molecule, probably due to the absence of electronic host/guest coupling. Another interesting redox-inactive example was obtained with ZnII complexes based on calix[6]arenes. According to the substitution pattern at the large rim of the calixarene ligand, the number of water molecules inside the funnel could be varied. As shown in Fig. 1, only one H2O molecule coordinated to the metal centre was trapped when three amino and three tBu groups were present at the large rim of the calixarene, whereas two water molecules were identified for the complex-bearing six tBu moieties (Fig. 1).28,29
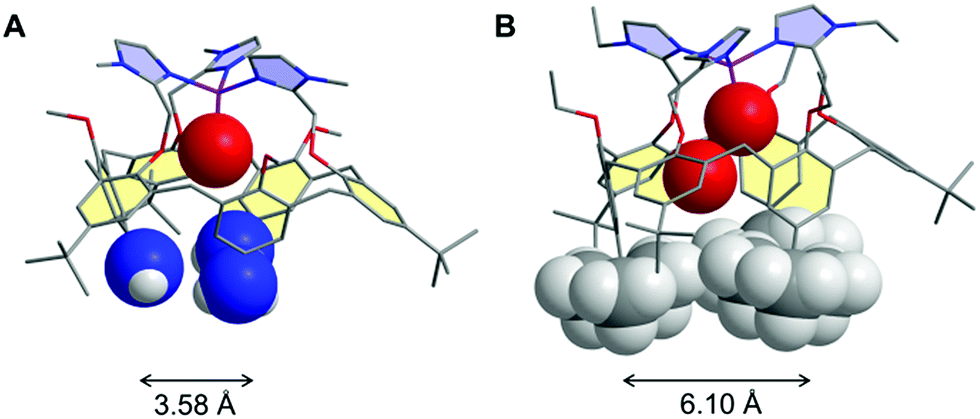 | ||
| Fig. 1 Comparison of the XRD structures of the tris-amino, tris-tBu (A) and hexa-tBu (B) calixarene-based ZnII–aqua complexes with a space-filling representation of the guest water molecules and the large rim inner substituents evidencing a different cavity space. Hydrogen atoms and counter anions have been omitted for clarity. | ||
It is noteworthy that the trapping of a pair of water molecules inside the calix[6]cone was established in the solid state (XRD analysis) as well as in solution (by 1H NMR spectroscopy in CDCl3). As previously discussed,29 such a difference between these two complexes is due to the substitution at the large rim: for the tris-amino derivative, the OH–π interaction between the bent aniline unit and the guest water molecule shrinks the cavity size, thus adapting it to the smallness of the guest ligand (the average distance between the nitrogen atoms is 3.58 Å, Fig. 1A). For the hexa-tBu complex, the bulky tBu groups define a larger cavity space (the average distance between the central tBu carbon atoms is 6.10 Å, Fig. 1B) hence allowing the inclusion of a second guest water molecule which is in interaction with the first one via a strong hydrogen bond.
Being interested in cavity effects associated with biomimetic redox centres, we thought of exploring the redox properties of a redox-active metal ion embedded in the calix[6]arene cone, knowing that the latter defines an open space prone for hosting a well-defined number of water molecules.
Among the different systems that we have described up to now,35 our choice was a calix[6]azacryptand featuring a [tris(2-methylpyridyl)amine] (tmpa) cap (Fig. 2). Indeed, a recent study by some of us concerning water coordination associated with the CuII/CuI electron transfer within the tmpa ligand showed decoordination of the water molecule upon monoelectronic reduction of the CuII complex. Moreover, back-coordination of the water ligand in the CuI state was detected when increasing the water content and/or decreasing the timescale of the experiment.36 In the present study, we focus on water inclusion inside the hydrophobic calix[6]arene cavity and its effects on the redox properties of the copper centre bound to the tmpa core covalently attached to the macrocyclic conic scaffold.
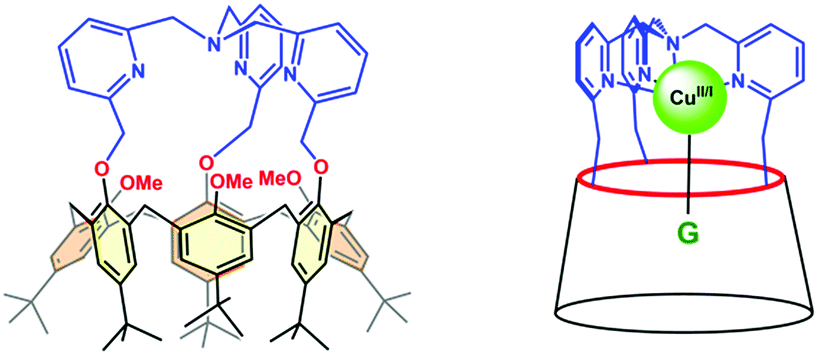 | ||
| Fig. 2 Schematic of the calix[6]tmpa ligand and the corresponding host–guest (G) Cu complex. | ||
Results and discussion
Description of the calix[6]tmpa copper system
The structure of the calix[6]tmpa ligand is displayed in Fig. 2. The corresponding CuII and CuI complexes were synthesized as previously reported37,38 by reacting one equiv. of metal salts, CuII(OH2)6(ClO4)2 and CuI(CF3SO3), respectively, in a non-coordinating solvent. In this system, the tmpa unit closes the small rim of the cone-shape cavity, precluding coordination to ligands in the exo position. The metal ion is embedded at the top of the half-open cavity that acts as a funnel controlling guest ligand exchange. Importantly, the small rim of the calixarene, which defines the second coordination sphere of the embedded metal ion, prevents simultaneous coordination of two guest ligands, thus leaving a single coordination site accessible to a ligand.39 The cavity can open and close (breathing), and adapt to the size of the entrapped guest (induced-fit).29Such supramolecular features mimic the hydrophobic pocket in Cu enzymes. In the presence of coordinating molecules, the Cu complexes readily form host–guest adducts, provided the guest can fit into the calixarene cavity. As illustrated in Fig. 3 displaying the XRD structures of the two CuII nitrilo complexes, this cavity can host guest ligands of different sizes without dramatic modifications (see ESI†). In chloroform or dichloromethane, and in the strict absence of any trace of the coordinating solvent, these copper complexes were identified as a monocationic “empty cavity”40 4-coordinate species, [CuI(calix[6]tmpa)]+ in the +I oxidation state,38 and a dicationic 5-coordinate mono-aqua complex, [CuII(calix[6]tmpa)(H2O)]2+ in the +II oxidation state.37 Previous electrochemical studies by cyclic voltammetry (CV) showed that these Cu complexes display an irreversible redox behaviour due to the interconversion between a water-free CuI species and a mono-aqua CuII species.41 The aqua ligand is readily exchanged for small neutral donors such as MeCN and DMF to give rise to the corresponding host–guest dicationic CuII complexes that display reversible redox behaviours. Very interestingly, CV studies evidenced, during the electron exchange at the +II state, the kinetic trapping of a transient species, different from the thermodynamic species. This very unusual phenomenon is a direct consequence of the control exerted by the calixarene cavity. The latter, imposing a dissociative process at the CuII state, precludes the more favoured associative pathway and blocks the guest interconversion at the CV time scale. This study thus highlighted the crucial role that embedment of a reactive redox metal ion in a funnel-like cavity has in ligand exchange associated with electron transfer. We now report another impressive phenomenon related to the water content in the cavity of the CuI and CuII funnel complexes. It highlights the key role water molecules have in the control of the redox potential of the embedded copper ion and thus, possibly, in the redox activity of electron transfer metalloproteins and enzymes.
 | ||
| Fig. 3 XRD structures of two CuII complexes based on the calix[6]tmpa scaffold. The guest ligand bound in the calixarene cavity: (A) PhCN and (B) MeCN. In both cases, the metal centre sits in a strongly axial trigonal bipyramidal environment (see ref. 37 and the ESI†) (hydrogen atoms and counterions are omitted for clarity). | ||
Studies under dry conditions
The redox behaviour of the aqua CuII complex, [CuII(calix[6]tmpa)(H2O)]2+, was first investigated under dry and inert conditions in CH2Cl2.42 An irreversible cathodic peak at Epc(1) = −0.35 V vs. Fc (E0(Fc+/Fc) = +0.64 V vs. NHE under the present conditions), associated with an irreversible anodic peak at Epa(2) = +0.75 V, was observed at 0.02 < v < 5 V s−1 by CV (Fig. 4A). Rotating disk electrode voltammetry (RDEV) before and after electrolysis (n = 1 e−) showed the cathodic and anodic waves, respectively, corresponding to both CV peaks (inset, Fig. 4A). A counter electrolysis restored the initial RDEV cathodic wave. This observation revealed a chemically reversible process as a square scheme mechanism with two interchangeable species stable at different redox potentials. Indeed, the CV in CH2Cl2 of the synthesized CuI complex displays irreversible peaks at Epa(2) = +0.75 V and Epc(1) = −0.20 V (Fig. 4B). The reverse oxidation peak at Epa(1) could not be detected even at high scan rates (Fig. 4C and S1†). Conversely, the reverse peak Epc(2) was detected at a high scan rate (100 V s−1) while ipc(1) decreased (Fig. 4C). In the second scan, the oxidation peak at Epa(1) was not observed. In agreement with the spectroscopic data of the CuII and CuI-(calix[6]tmpa) complexes,37,38 and analogous with previous electrochemical studies with the related calix[6]tren-Cu system,43 the peak at Epc(1) is ascribed to the reduction of the aqua CuII complex (H2O coordinated to CuII inside the calixarene cone), whereas the anodic peak at Epa(2) corresponds to the oxidation of the water-free CuI complex (no H2O coordination to CuI).37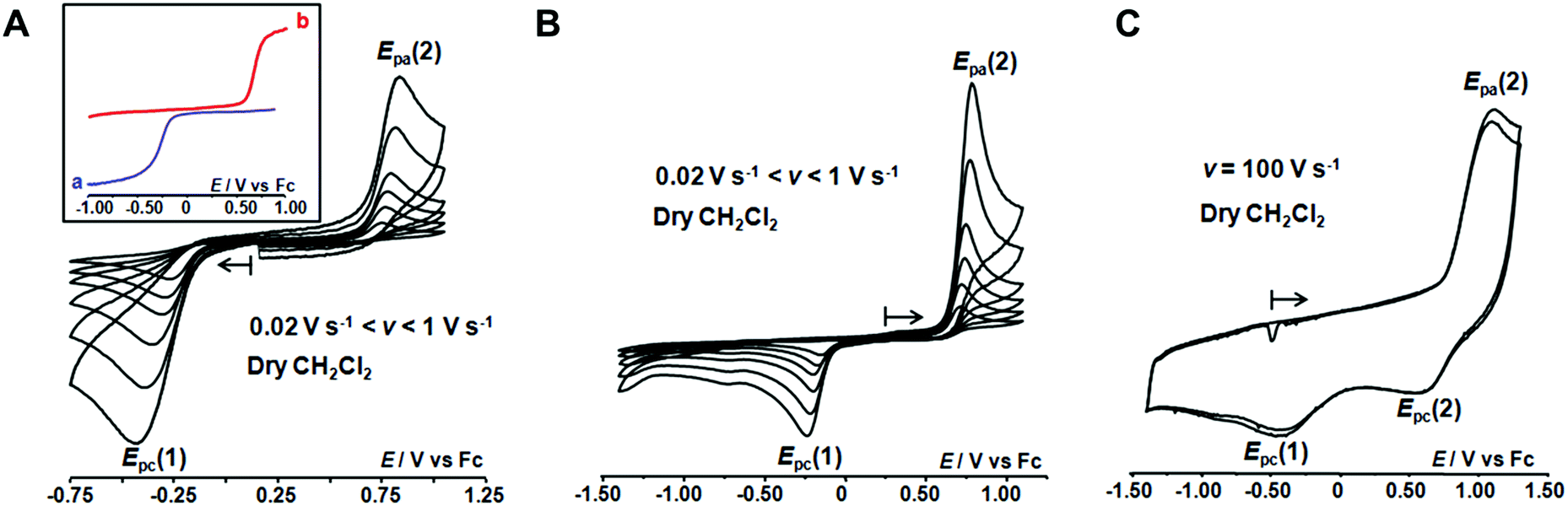 | ||
| Fig. 4 CVs at a Pt electrode in anhydrous CH2Cl2/NBu4PF6 (0.1 M) at [complex] = 10−3 M: (A) [CuII(calix[6]tmpa)(H2O)]2+ and (B) [CuI(calix[6]tmpa)]+ at varying scan rates, 0.02 V s−1 < v < 1 V s−1. Inset in (A): RDEV (a) before and (b) after electrolysis of [CuII(calix[6]tmpa)(H2O)]2+ at −0.50 V vs. Fc. (C) CV at v = 100 V s−1 of [CuI(calix[6]tmpa)]+ (2 cycles). | ||
The detection of the Epc(2) peak only at a high scan rate (100 V s−1) further supports the ejection of water out of the calixarene cavity at the CuI state, leading to the water-free [CuI(calix[6]tmpa)]+ species, and shows that water coordination at the CuII redox state is a very fast process (Fig. 4C). This redox behaviour can be visualized according to the mechanism depicted in Scheme 1 (middle and bottom parts).
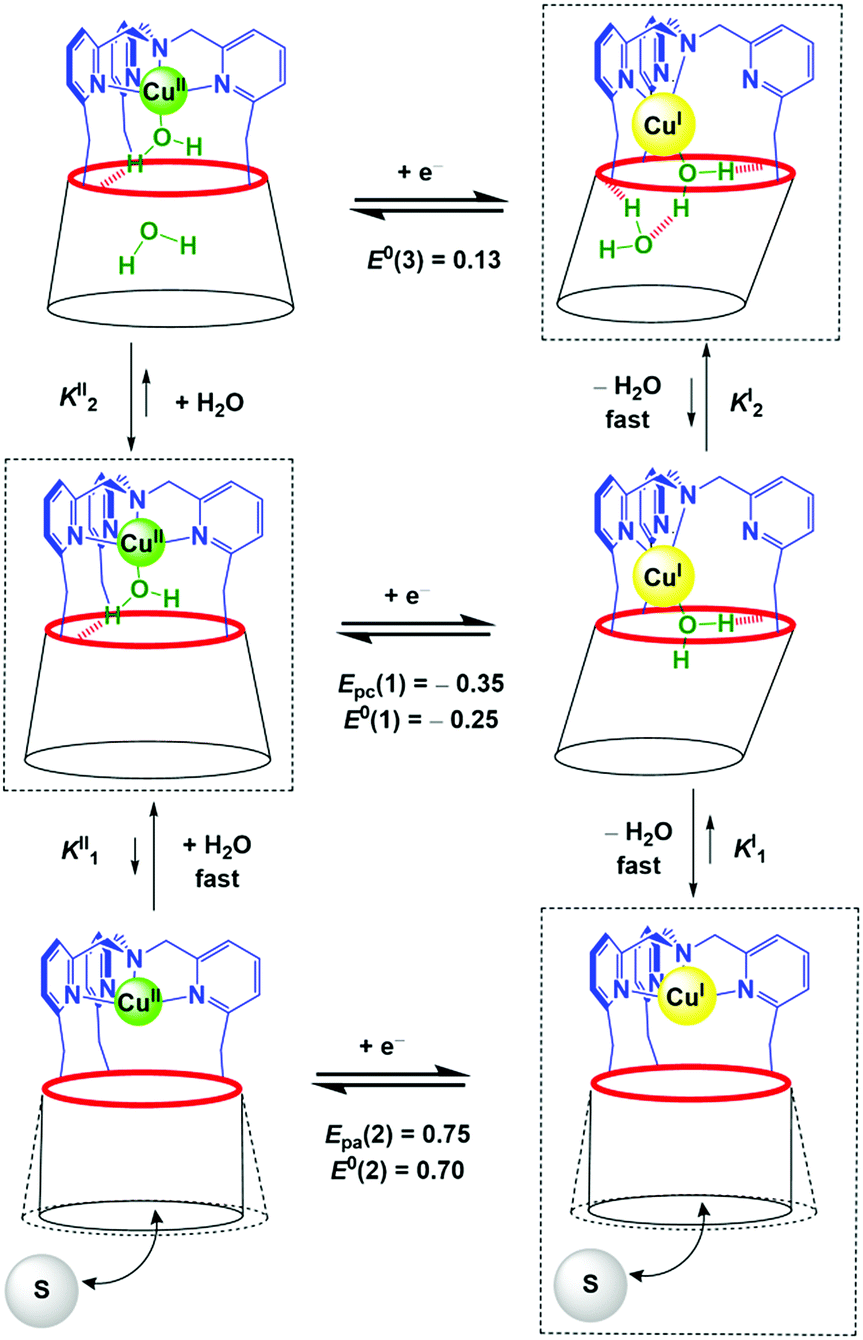 | ||
| Scheme 1 Proposed ladder mechanism of electron transfer pathways for the [Cu(calix[6]tmpa)(H2O)x]n+ complexes (x = 0, 1, 2) in CH2Cl2 under variation of the water content. The rectangles depict the thermodynamic species that have been observed by electrochemistry (E/V vs. Fc). | ||
Addition of water
Addition of H2O to CH2Cl2 solutions of either CuII or CuI complexes up to saturation revealed a strong influence of the water content on the electron transfer process. As shown in Fig. 5A by CV for the CuI complex (see also Fig. S2A† for RDEV), the oxidation peak corresponding to the system at Epa(2) = +0.75 V shifted negatively with the addition of H2O. Upon water content increase up to saturation, it ultimately evolved to a new peak at Epa(3), corresponding to a quasi-reversible process at E0(3) = +0.13 V (ΔEp = 135 mV at v = 0.1 V s−1; see Fig. 5B for CV and inset curve a for RDEV). The increase of the scan rate revealed a re-appearance of the reduction peak at Epc(1) at the expense of Epc(3) on the back scan (Fig. 5C). For the CuII complex, the same trend but less clear-cut was observed (ESI†). CV at various scan rates of a water-saturated solution containing the CuII complex also denoted a balance between the peaks at Epc(3) and Epc(1) for its reduction (Fig. S2B†). However Epc(1) corresponded to the stable species as confirmed by RDEV (Fig. 5B, inset curve b). After reduction down to CuI, the back scan emphasized a single species by its reoxidation at Epa(3). Hence, at water saturation, the presence of a thermodynamically stable CuI redox species is observed at E0(3), and the corresponding CuII species is only transiently detected by CV (vide infra for kinetics). Considering that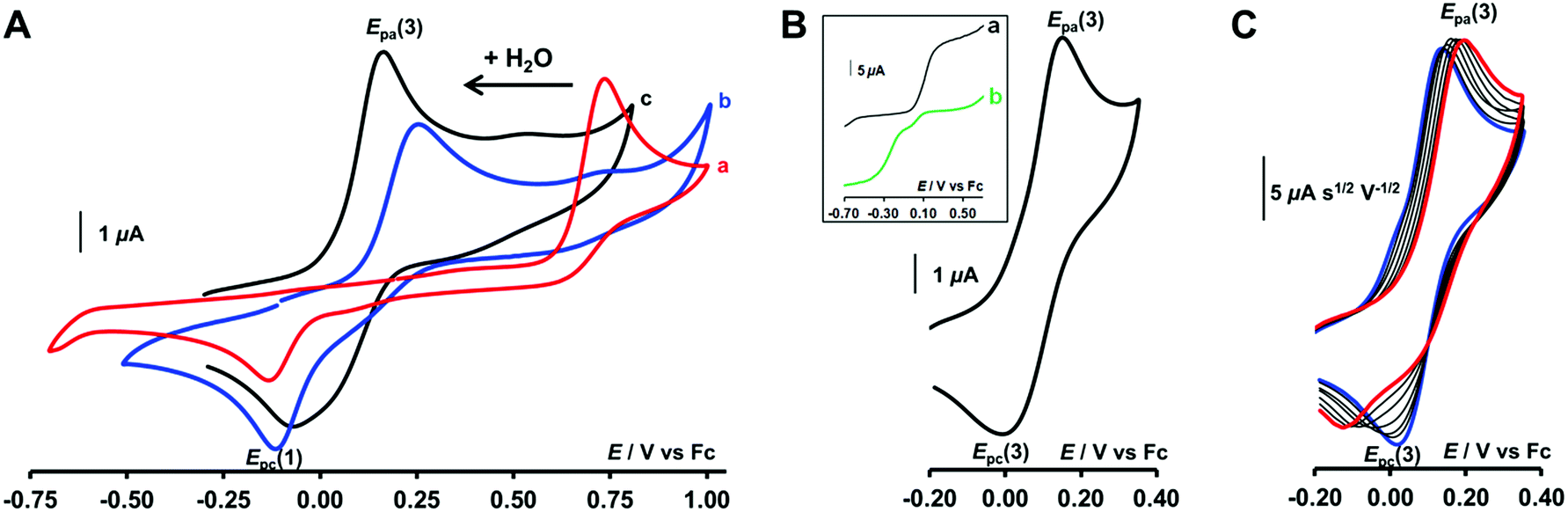 | ||
| Fig. 5 CVs at a Pt electrode in CH2Cl2/NBu4PF6 (0.1 M) at [complex] = 10−3 M: (A) CV at v = 0.1 V s−1 of [CuI(calix[6]tmpa)]+ after successive addition of H2O: (a) 0 equiv (b), 14 equiv, and (c) 56 equiv. (B and C): CVs of [CuI(calix[6]tmpa)]+ under H2O-saturated conditions (B) at v = 0.1 V s−1 and (C) for 0.02 V s−1 (blue) < v < 2 V s−1 (red) (i/v1/2 CV curves). Inset in (B): RDEV for water-saturated solutions of (a) CuI and (b) CuII complexes. | ||
(i) the redox systems (1) and (2) correspond to the mono-aqua and water-free complexes, respectively,
(ii) system (3) does not correspond to the hydroxo complex, which displays an irreversible reduction peak at a much lower potential (Epc = - 1.34 V vs. Fc at 0.1 V s−1)37 (furthermore, the latter, which has been previously fully characterized by UV-vis and EPR spectroscopies in the CuII state,37 is obtained only after addition of a base),
(iii) system (3) is observed only at a high water content and
(iv) the hexa-tBu calix[6]cone can trap and stabilize either one or two H2O molecules,28
we hypothesize that (3) corresponds to a Cu complex hosting two water molecules inside the cavity. The associated redox potentials lie in the following order: (H2O)1 < (H2O)2 < (H2O)0 (Scheme 2).
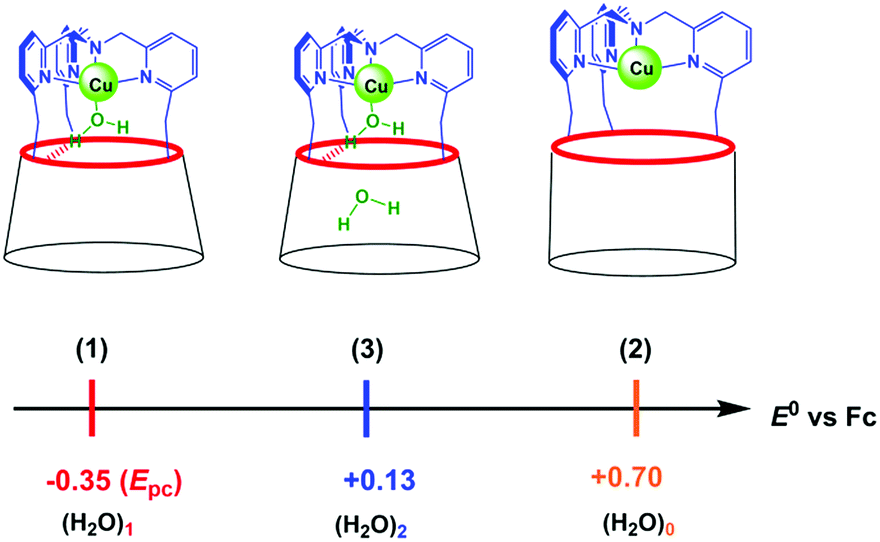 | ||
| Scheme 2 Redox potentials for [CuII/I(calix[6]tmpa)(H2O)x]2+/+ in CH2Cl2/NBu4PF6. (x = 0, 1, 2). | ||
In regard to the interaction with water, the behaviour of the tmpa complexes deprived of a cavity, [CuII/I(tmpa)(H2O)]2+/+, differs radically. Under dry conditions and in non-coordinating solvents, a pseudo-reversible redox process corresponding to the reduction of the 5-coordinate aqua-complex is observed at E0(1) = −0.33 V.36 Importantly, the associated redox system was essentially not affected by the addition of water. This indicates that the effects observed with the calixarene copper complex are not due to modifications of the medium such as the net dielectric constant. This also highlights the specific interactions between water and the calixarene cavity leading to system (3), which is not observed with the Cu-tmpa complex deprived of cavity.
Insights into the kinetics of water exchange: ladder mechanism
Monitoring the redox process by CV at various scan rates gave further insights into the in and out mechanism for water exchange. As described above, under water-saturated conditions, the CuI state exists as a stable water-cluster adduct defined by a reversible process at E0(3). Accordingly, for a water-saturated solution of the CuI complex, only the oxidation peak of the bis-aqua species [Epa(3)] was detected at any scan rates (Fig. 5C), whereas the corresponding reversible reduction peak Epc(3) is observed at a low scan rate. For the highest scan rates, a second cathodic peak appears at the reduction potential of the CuII-aqua complex [Epc(1)]. Such a behaviour characterizes a rate-limiting reaction in the CuII redox state between the two reducible species, i.e. a CE (Chemical–Electrochemical) mechanism:44 the oxidation of the CuI bis-aqua complex leads to the very fast release of the clustered guest water molecules to yield the thermodynamic CuII species, the aqua complex, that is reducible only at a low potential (Epc(1)). At a low scan rate, the timescale of the CV is long enough to allow the back-hosting of the water molecules, leading to the observation of the bis-aqua reduction peak at a higher potential [Epc(3)]. As the scan rate is increased, the back-hosting process becomes rate-limiting and the reduction of both mono-aqua and bis-aqua CuII species is observed. The situation looks a little different when starting from the CuII mono-aqua complex, which is the thermodynamic species in the +II state (Fig. S2B†). In this case, even at low scan rates, the reduction of the aqua complex detected at Epc(1) remains the major redox process observed by CV, indicating that the CE process that would lead to Epc(2) is less effective. This can result from a slow entrance step of the water molecules due to a closed conformation of the hexa-tBu calix[6]cone in the CuII resting state, which slows down the mono-aqua![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) bis-aqua equilibrium at the CuII state. Remarkably, after the reduction step to CuI, the bis-aqua cluster CuI, detected at Epa(3), is observed even at high scan rates. From the electrochemical behaviour, the water-dependent redox behaviour of the cavity complexes can be rationalized by a ladder mechanism (Scheme 1). Digital voltammetric simulations based on a square scheme mechanism involving interchange between two systems allow reproducing the experimental trends and yielded the kinetic constants (see ESI†).
bis-aqua equilibrium at the CuII state. Remarkably, after the reduction step to CuI, the bis-aqua cluster CuI, detected at Epa(3), is observed even at high scan rates. From the electrochemical behaviour, the water-dependent redox behaviour of the cavity complexes can be rationalized by a ladder mechanism (Scheme 1). Digital voltammetric simulations based on a square scheme mechanism involving interchange between two systems allow reproducing the experimental trends and yielded the kinetic constants (see ESI†).
Assuming that E0(3) = +0.13 V, E0(1) = −0.25 V and E0(2) = +0.70 V, numerical values for KxI/KxII can be obtained by using eqn (1) and (2), where x depicts the number of water molecules (x = 0, 1, 2):
| E0(1) − E0(2) = RT/nF ln (K1I/K1II) | (1) |
| E0(3) − E0(1) = RT/nF ln (K2I/K2II) | (2) |
These simple calculations clearly show a strong difference between K1I/K1II (≈10−16) and K2I/K2II (≈106). Thus, the addition of the first water molecule to the “empty cavity” complex is much more favoured in the CuII redox state than in the CuI state. This is an expected behaviour since the CuII complex is obviously highly stabilized by catching the fifth ligand (H2O) in its trigonal coordination sphere, whereas CuI is already stabilized by the tmpa core when the cavity is empty. Interestingly, this ratio is reversed when more water is added: this indicates that embedment of a bis-aqua cluster stabilizes more CuIvs. CuII. At first, this seemed to be unexpected, in view of the classical behaviour of copper complexes. In fact, the presence of an additional water molecule connected to the first one through hydrogen bonding, is expected to induce an increase of the charge density on the CuII centre (which is a stronger Lewis acid than CuI) and hence a negative shift of the redox system.45
Mass spectrometry analysis
Dry and wet dichloromethane solutions of CuI complexes were mass-analysed by using an ion trap mass spectrometer. In each case, the expected empty-cavity CuI complex was observed at m/z 1403.7 with the correct relative intensities for the isotopologues (Fig. S5 and S6†). Additional clusters of peaks were detected around m/z 1422 and 1439, the relative intensity of which significantly increased with the water content (2 to 6-fold). MS2 experiments were then performed on the species isolated at m/z 1421.6 and m/z 1439.6 (Fig. 6).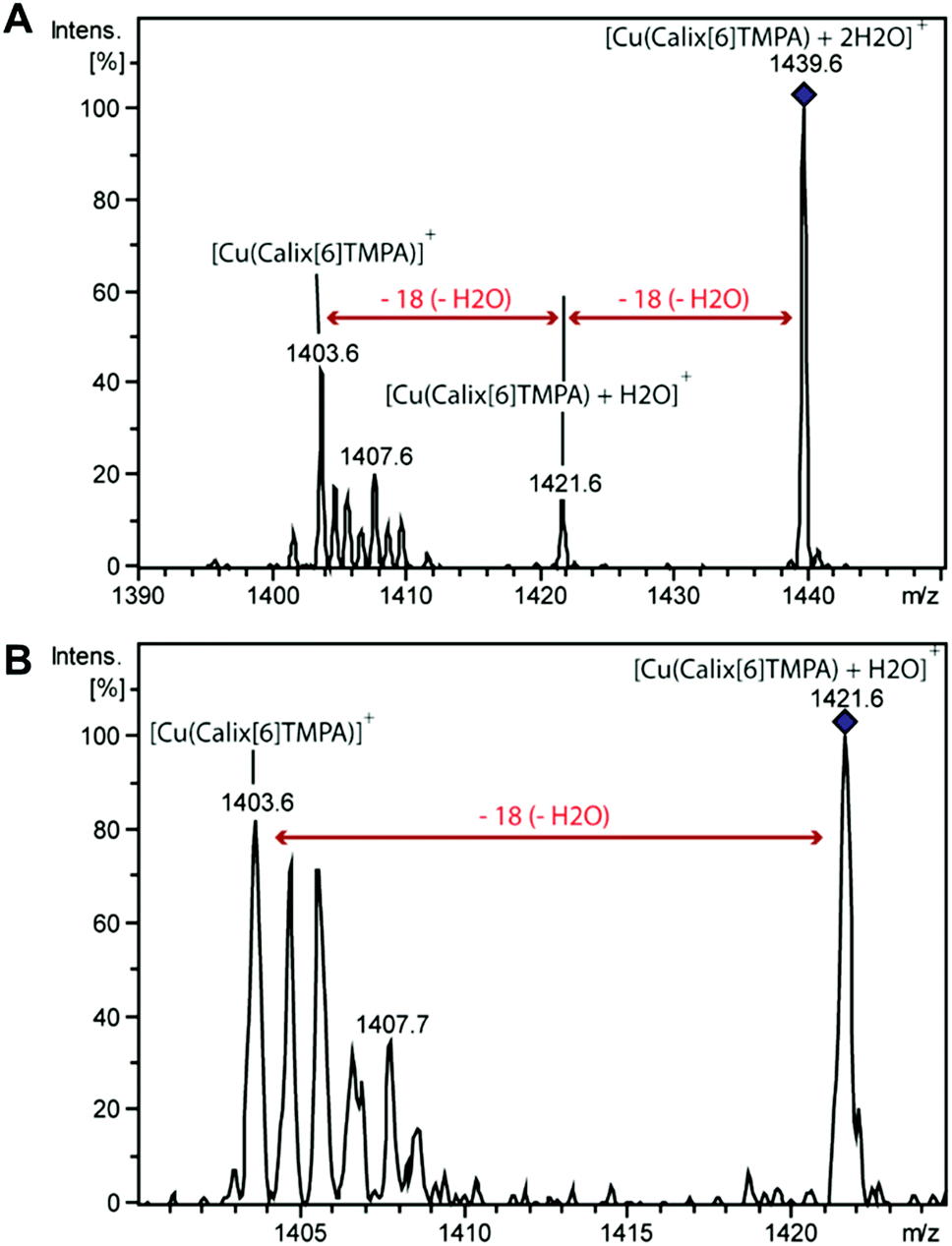 | ||
| Fig. 6 MS2 spectrum of [Cu(calix[6]tmpa) + 2H2O]+ at m/z 1439.6 (A) and MS3 spectrum of [Cu(calix[6]tmpa) + H2O]+ at m/z 1421.6 (B) obtained as a fragment ion of [Cu(calix[6]tmpa) + 2H2O]+ after the MS2 stage (cf. the spectrum above). | ||
In both cases, a neutral loss of one water molecule (−18 Da) was observed leading to the peaks at m/z 1403.7 and m/z 1421.3, respectively (along with other fragment ions from contaminants of parent ions). The fragment ion at m/z 1421.3 was further mass-selected and subjected to an additional CID stage (MS3 experiments, see Fig. 6): an additional 18 Da neutral loss was observed. These results confirm that the fragment ions at m/z 1421.6 and 1439.6 correspond to the mono-aqua ([CuI(calix[6]tmpa)(H2O)]+) and bis-aqua ([CuI(calix[6]tmpa)(H2O)2]+) complexes. Hence, these mass analyses demonstrate the existence of mono and bis-aqua copper complexes provided the calixarene cavity is not occupied by a bulky ligand. This further substantiates the positioning of the water ligands in the endo position, as observed for the related hexa-tBu calix[6]arene-based ZnII complexes displayed in Fig. 1. In order to localize the position of the bound water molecules (inside or outside the calix[6]arene cavity), the same experiment was carried out starting with the benzonitrile complex. Indeed, as shown by its X-ray structure (Fig. 3), the benzonitrile ligand fully occupies the calixarene cavity, leaving no inner space for water coordination. Mass-analysis of the complex [CuII(Calix[6]TMPA)(PhCN)]2+ in a water-saturated dichloromethane solution showed a peak at m/z 1506.9 corresponding to the [CuI(calix[6]tmpa)(PhCN)]+ complex with, however, no evidence of the corresponding hydrated species [CuI(calix[6]tmpa)(PhCN)(H2O)x]+ (x = 1 or 2, see Fig. S7†). MS2 experiments only led to the neutral loss of PhCN at m/z 1507.9 (−103 Da).
Insights into the structures of the aqua-Cu complexes
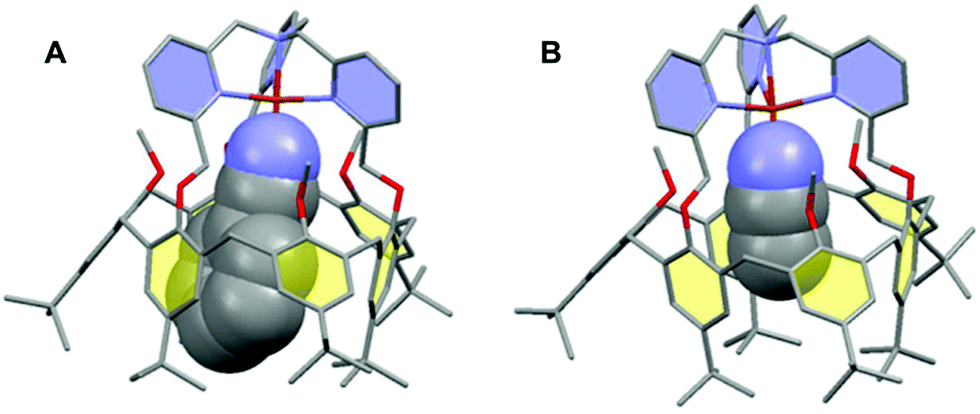
The interplay between the inclusion of water molecules inside the cavity and the CuII/I complex stabilities was scrutinized by the Molecular Dynamics Simulations (MDS) of the [CuII/I(calix[6]tmpa)(H2O)x]2+/+ complexes (x = 0–2) in explicit solvent (CH2Cl2). Our MDS are based on hybrid Density Functional Theory/Molecular Mechanics (DFT/MM) which enables the modifications of the coordination spheres of the Cu cations during the simulations. Details of the computational protocol can be found in the ESI.† Throughout the MDS, the water-free CuI and CuII complexes exhibited similar trigonal pyramidal geometries around the copper ion with C3v symmetrical cavities (Fig. 7A and S17 and Table S3†). However, the introduction of one water molecule led to different behaviours for each redox state.In the +II redox state, the water molecule binds to the copper ion in a TBP (trigonal bipyramidal) geometry, imposing a C3v symmetry to the supramolecular system (Fig. 7B). A weak hydrogen bond between one methoxy group of the small rim and the water molecule is only occasionally formed (<50%, Table S5†). Hence, the principal source of stabilization when introducing a water molecule is the formation of the CuII–OH2 bond.
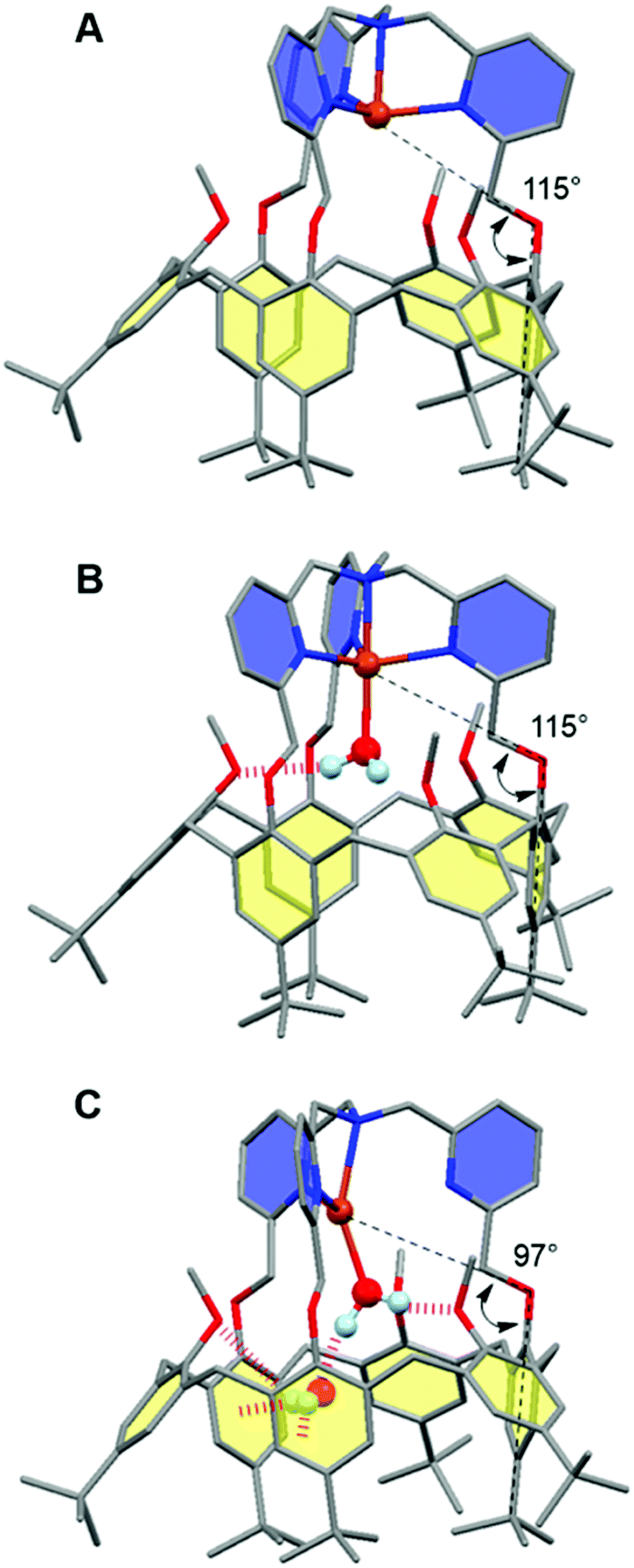 | ||
| Fig. 7 Representative snapshots extracted from the hybrid DFT/MM MDS for the [CuII/I(calix[6]tmpa)(H2O)x]2+/+ complexes (A: CuI, x = 0; B: CuII, x = 1; C: CuI, x = 2). For clarity, solvent molecules (chloroform) and hydrogen atoms are not represented except for the water molecules. The copper ion, the water molecules and the poly-aza cap were treated at the DFT level of theory, and the remaining ones were modelled using Molecular Mechanics. The black dashed lines illustrate the definition of the angles highlighting the partial inclusion of one tBu group for the [CuI(calix[6]tmpa)(H2O)x]+ complexes (Tables S3 and S4†). The red dotted lines indicate hydrogen and OH–π bonds. Colour code: grey = carbon, dark blue = nitrogen, red = oxygen, orange = copper, and pale blue = hydrogen. | ||
In the +I redox state, two coordination modes involving the loose interaction of one out of the five donors (either a pyridine residue or water) are observed during the MDS with frequent interchanges (Fig. S14†). This competition suggests that a single water molecule is not particularly stabilized within the [CuI(calix[6]tmpa)]+ complex, which is interpreted as a consequence of the well-known preference of CuI complexes for a 4-coordination mode and soft donors. Hence, the water-free CuI species is favoured over a mono-aqua adduct.
Similar features were found regarding the copper coordination sphere of the bis-aqua systems, as compared to the mono-aqua situation examined above. A TBP 5-coordination is maintained during the MDS at the +II state (〈τ〉 = 0.89 ± 0.02, Tables S3 and S5†) (ESI†). However, a 4-coordination mode was found for the +I state with the inclusion of one tBu group inside the cavity and disruption of the C3v symmetry (Table S3† and Fig. 7C). The presence of the second water molecule implies, for the +I state, (i) decoordination of a pyridine arm, (ii) strengthening of the hydrogen bond between the higher water molecule and one methoxy group compared to the mono-aqua system (Table S5†), (iii) a moderate hydrogen bond between the lower water molecule and another methoxy group, and (iv) a strong hydrogen bond between the two water molecules formed at almost 100% of the time during the MDS. The binding energy of the second water molecule amounts to −60 kJ mol−1 (Fig. S15†), a number that is higher in absolute values than that associated with the inclusion of the first water molecule (−42 kJ mol−1). Lastly, it was found that the inclusion of the third water molecule gives back a binding energy of −42 kJ mol−1 (Fig. S16†).
This series of values reflects the fact that a synergistic network of supramolecular interactions favours the inclusion of the second water molecule inside the cavity (Fig. 7C). In contrast, at the CuII state, the coordination of all four nitrogen atoms of the tmpa cap maintains the copper cation high in the cavity. Consequently the bound water molecule sits further away from the methoxy groups with which it only weakly interacts. In addition, the lower water molecule is not as firmly hydrogen bonded to the higher one as that in the +I state, and the second water molecule is frequently found lower in the cavity during the simulations.
In fact when DFT geometry optimizations were carried out on the [CuII(calix[6]tmpa)(H2O)2]2+ complex, the resulting structures did not present hydrogen bonds between the water molecules (Fig. S19†). As a consequence, the energy change associated with the inclusion of the second water molecule at the +II state is almost zero. In view of the loss of symmetry suggested by molecular modelling at a high water content for the CuI state, NMR studies were conducted in CDCl3 with various water contents (ESI, Fig. S8–S12†). Under dry conditions, whatever the temperature is, the spectrum remained characteristic of C3v symmetrical species. Variation of the peak widths and shifts may well be related to solvent hosting, as classically observed. Very interestingly, addition of water until saturation to the CuI complex induced a change of the spectrum towards a profile that, at low T, evidenced the formation of a new, non-symmetrical species for which the protons of the three pyridines are not equivalent anymore. This information is fully consistent with the hypothesized decoordination of a pyridine arm associated with the coordination of a water molecule to the CuI state. It is worth noting that decoordination of a pyridyl arm associated with the binding of an exogenous donor has been experimentally observed in the specific case of a CuI-tmpa complex46 and that previously published theoretical calculations also predicted the decoordination of a pyridyl arm upon water binding to the CuI-tmpa core.47 All attempts to directly or indirectly evidence a guest water molecule failed, possibly due to fast exchange with free water vs. the NMR time scale.48 Finally, the fact that this non-symmetrical species is observed only at low T by 1H NMR analysis is also consistent with an enthalpically driven water binding process as reported for the bis-aqua ZnII complex.28
Conclusions
Hence, on fundamental aspects, important findings emerge from the present study with regard to the modulation of the thermodynamics and kinetics of the CuII/CuI electron exchange by water hosting and binding through cavity effects. The switch from an electrochemically irreversible (OFF) to a reversible (ON) state of the complex is fully controlled by the water content in the space next to the metal centre defined by the calixarene cone. Interestingly, the control of the redox potential for the bis-aqua system is essentially due to CuI stabilization. This result runs against the accepted rule, i.e. control by CuII stabilization.42 Such a supramolecular control of the redox activity by water molecules entrapped in a hydrophobic cavity actually finds echoes in biological systems. Indeed, many intermediates in biological cycles cannot be isolated, and their redox potentials are often not known, being only estimated from the resting states of the enzymes. It is shown here that the insulating effect of the cavity can yield transiently very strong oxidants and reductants under dry conditions. Hence, the specific design of the coordination spheres within a cavity surrounding the copper ion allows the control of the redox properties of the system by simple variation of the water content.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the Centre National de la Recherche Scientifique (CNRS), the Ministère de l'Enseignement Supérieur et de la Recherche, and the Agence Nationale pour la Recherche (ANR) [Cavity-zyme(Cu) project, number ANR-2010-BLAN-7141]. Calculations have been performed at the CINES computer centre (project number c2014076913).Notes and references
- P. Ball, Chem. Rev., 2008, 108, 74–108 CrossRef PubMed.
- F. Yu, V. M. Cangelosi, M. L. Zastrow, M. Tegoni, J. S. Plegaria, A. G. Tebo, C. S. Mocny, L. Ruckthong, H. Qayyum and V. L. Pecoraro, Chem. Rev., 2014, 114, 3495–3578 CrossRef PubMed.
- G. A. Papoian, J. Ulander, M. P. Eastwood, Z. Luthey-Schulten and P. G. Wolynes, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 3352–3357 CrossRef PubMed.
- S. Sharma and P. G. Debenedetti, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4365–4370 CrossRef PubMed.
- C. Zong, G. A. Papoian, J. Ulander and P. G. Wolynes, J. Am. Chem. Soc., 2006, 128, 5168–5176 CrossRef PubMed.
- J. Roche, J. A. Caro, D. R. Norberto, P. Barthe, C. Roumestand, J. L. Schlessman, A. E. Garcia, B. E. Garcia-Moreno and C. A. Royer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6945–6950 CrossRef PubMed.
- S. H. Chong and S. Ham, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7636–7641 CrossRef PubMed.
- R. Talhout, A. Villa, A. E. Mark and J. B. Engberts, J. Am. Chem. Soc., 2003, 125, 10570–10579 CrossRef PubMed.
- D. R. Martin and D. V. Matyushov, Sci. Rep., 2017, 7, 5495 CrossRef PubMed.
- J. Lin, I. A. Balabin and D. N. Beratan, Science, 2005, 310, 1311–1313 CrossRef PubMed.
- F. A. Tezcan, B. R. Crane, J. R. Winkler and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5002–5006 CrossRef PubMed.
- T. M. Makris, K. von Koenig, I. Schlichting and S. G. Sligar, Biochemistry, 2007, 46, 14129–14140 CrossRef PubMed.
- B. Zhao, F. P. Guengerich, M. Voehler and M. R. Waterman, J. Biol. Chem., 2005, 280, 42188–42197 CrossRef PubMed.
- J. P. Collman, R. A. Decreau, A. Dey and Y. Yang, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4101–4105 CrossRef PubMed.
- R. A. Goldbeck, M. L. Pillsbury, R. A. Jensen, J. L. Mendoza, R. L. Nguyen, J. S. Olson, J. Soman, D. S. Kliger and R. M. Esquerra, J. Am. Chem. Soc., 2009, 131, 12265–12272 CrossRef PubMed.
- J. P. Collman, R. A. Decreau, H. Lin, A. Hosseini, Y. Yang, A. Dey and T. A. Eberspacher, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7320–7323 CrossRef PubMed.
- A. Fernandez and H. A. Scheraga, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 113–118 CrossRef PubMed.
- I. M. van Amsterdam, M. Ubbink, O. Einsle, A. Messerschmidt, A. Merli, D. Cavazzini, G. L. Rossi and G. W. Canters, Nat. Struct. Biol., 2002, 9, 48–52 CrossRef PubMed.
- O. Miyashita, M. Y. Okamura and J. N. Onuchic, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3558–3563 CrossRef PubMed.
- A. de la Lande, N. S. Babcock, J. Rezac, B. C. Sanders and D. R. Salahub, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11799–11804 CrossRef PubMed.
- I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253–2277 CrossRef PubMed.
- T. L. Poulos, Biochem. Biophys. Res. Commun., 2005, 338, 337–345 CrossRef PubMed.
- S. V. Antonyuk, C. Han, R. R. Eady and S. S. Hasnain, Nature, 2013, 496, 123–126 CrossRef PubMed.
- P. Hosseinzadeh and Y. Lu, Biochim. Biophys. Acta, 2016, 1857, 557–581 CrossRef PubMed.
- N. M. Marshall, D. K. Garner, T. D. Wilson, Y. G. Gao, H. Robinson, M. J. Nilges and Y. Lu, Nature, 2009, 462, 113–116 CrossRef PubMed.
- A. Dey, F. E. Jenney Jr, M. W. Adams, E. Babini, Y. Takahashi, K. Fukuyama, K. O. Hodgson, B. Hedman and E. I. Solomon, Science, 2007, 318, 1464–1468 CrossRef PubMed.
- C. A. Bortolotti, A. Amadei, M. Aschi, M. Borsari, S. Corni, M. Sola and I. Daidone, J. Am. Chem. Soc., 2012, 134, 13670–13678 CrossRef PubMed.
- O. Sénèque, M.-N. Rager, M. Giorgi and O. Reinaud, J. Am. Chem. Soc., 2001, 123, 8442–8443 CrossRef.
- D. Coquiere, J. Marrot and O. Reinaud, Org. Biomol. Chem., 2008, 6, 3930–3934 RSC.
- P. S. Lakshminarayanan, E. Suresh and P. Ghosh, J. Am. Chem. Soc., 2005, 127, 13132–13133 CrossRef PubMed.
- M. Yoshizawa, T. Kusukawa, M. Kawano, T. Ohhara, I. Tanaka, K. Kurihara, N. Niimura and M. Fujita, J. Am. Chem. Soc., 2005, 127, 2798–2799 CrossRef PubMed.
- Q. Q. Wang, V. W. Day and K. Bowman-James, Angew. Chem., Int. Ed., 2012, 51, 2119–2123 CrossRef PubMed.
- K. Kurotobi and Y. Murata, Science, 2011, 333, 613–616 CrossRef PubMed.
- J. Garric, J. M. Leger and I. Huc, Chem.–Eur. J., 2007, 13, 8454–8462 CrossRef PubMed.
- N. Le Poul, Y. Le Mest, I. Jabin and O. Reinaud, Acc. Chem. Res., 2015, 48, 2097–2106 CrossRef PubMed.
- A. G. Porras Gutierrez, J. Zeitouny, A. Gomila, B. Douziech, N. Cosquer, F. Conan, O. Reinaud, P. Hapiot, Y. Le Mest, C. Lagrost and N. Le Poul, Dalton Trans., 2014, 43, 6436–6445 RSC.
- G. Izzet, X. Zeng, H. Akdas, J. Marrot and O. Reinaud, Chem. Commun., 2007, 810–812 RSC.
- G. Thiabaud, G. Guillemot, I. Schmitz-Afonso, B. Colasson and O. Reinaud, Angew. Chem., Int. Ed., 2009, 48, 7383–7386 CrossRef PubMed.
- Y. Rondelez, A. Duprat and O. Reinaud, J. Am. Chem. Soc., 2002, 124, 1334–1340 CrossRef PubMed.
- The term “empty cavity” will be employed throughout the article to design 4-coordinate complexes with no coordinated guest as the 5th donor. Depending on the temperature and the nature of the solvent, the cavity may actually host a solvent molecule.
- N. Le Poul, B. Douziech, J. Zeitouny, G. Thiabaud, H. Colas, F. Conan, N. Cosquer, I. Jabin, C. Lagrost, P. Hapiot, O. Reinaud and Y. Le Mest, J. Am. Chem. Soc., 2009, 131, 17800–17807 CrossRef PubMed.
- “Extra-dry” dichloromethane (H2O < 50 ppm) from Acros was used as received and kept under N2 in the glovebox (H2O content <1 ppm). However, due to the high “coordination” demand of CuII, the residual H2O content in non-coordinating solvents such as CH2Cl2 is always sufficient for generating the mono-aqua CuII adduct (typically in “dry” CH2Cl2/NBu4PF6, [H2O] = 5 10−3 M as measured by Karl-Fischer coulometry) (see ref. 36).
- G. Izzet, B. Douziech, T. Prange, A. Tomas, I. Jabin, Y. Le Mest and O. Reinaud, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6831–6836 CrossRef PubMed.
- J.-M. Savéant, Elements of Molecular and Biomolecular Electrochemistry: An Electrochemical Approach to Electron Transfer Chemistry, 2006,pp. 92–95 Search PubMed.
- D. B. Rorabacher, Chem. Rev., 2004, 104, 651–697 CrossRef PubMed.
- D.-H. Lee, N. Wei, N. N. Murthy, Z. Tyeklar, K. D. Karlin, S. Kaderli, B. Jung and A. D. Zuberbuehler, J. Am. Chem. Soc., 1995, 117, 12498–12513 CrossRef.
- L. Bonniard, A. de la Lande, S. Ulmer, J.-P. Piquemal, O. Parisel and H. Gérard, Catal. Today, 2011, 177, 79–86 CrossRef.
- In the case of the calix[6]tris(imidazole) bis-aqua ZnII complex (ref. 28), the embedded pair of water molecules could be characterized indirectly by curve fitting of the equilibrium constant between the bis(aqua) adduct and the DMF adduct. This strategy could not be applied in the case of the present CuI complex in view of the affinity for nitrilo guests that is far too high, DMF being not coordinated in the CuI redox state.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, details on electrochemical experiments and simulations, 1H NMR data, mass analysis, computational studies, and X-ray data. See DOI: 10.1039/c8sc03124j |
| This journal is © The Royal Society of Chemistry 2018 |