
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorogenic thiazole orange TOTFO probes stabilise parallel DNA triplexes at pH 7 and above†
Sarah
Walsh
 ab,
Afaf Helmy
El-Sagheer
ac and
Tom
Brown
*a
ab,
Afaf Helmy
El-Sagheer
ac and
Tom
Brown
*a
aDepartment of Chemistry, University of Oxford, Oxford, OX1 3TA, UK. E-mail: tom.brown@chem.ox.ac.uk
bATDBio Ltd., Oxford Science Park, Oxford, UK
cChemistry Branch, Department of Science and Mathematics, Faculty of Petroleum and Mining Engineering, Suez University, Suez 43721, Egypt
First published on 1st August 2018
Abstract
The instability of DNA triplexes particularly at neutral pH and above severely limits their applications. Here, we demonstrate that the introduction of a thiazole orange (TO) intercalator onto a thymine nucleobase in triplex forming oligonucleotides (TFOs) resolves this problem. The stabilising effects are additive; multiple TO units produce nanomolar duplex binding and triplex stability can surpass that of the underlying duplex. In one example, a TFO containing three TO units increased the triplex melting temperature at pH 7 by a remarkable 50 °C relative to the unmodified triplex. Notably, TO intercalation promotes TFO binding to target sequences other than pure polypurine tracts by the use of 5-(1-propynyl)cytosine (pC) against C:G inversions. By overcoming the instability of triplexes across a broad range of pH and sequence contexts, these very simple ‘TOTFO’ probes could expand triplex applications into many areas including diagnostics and cell imaging.
Introduction
Triplex-forming oligonucleotides (TFOs) bind in the major groove of double stranded DNA (dsDNA) through Hoogsteen or reverse Hoogsteen hydrogen bonding.1,2 While most methods of sequence-specific DNA recognition rely on denaturation of dsDNA, TFOs do not require this, so they have great potential in areas such as gene inhibition,3–5 DNA repair and recombination studies,6 inhibition of protein-DNA binding,7,8 photo-crosslinking of duplex DNA,9 DNA mutagenesis,10 and nanotechnology.11 Unfortunately the most stabilising TFOs, those that form parallel triplexes, have much reduced duplex-binding affinity at neutral pH and above owing to the necessity for cytosine protonation. There have been many efforts to overcome the pH-related instability by introducing a variety of sugar, base and phosphate modifications into TFOs.12–16 Furthermore, the requirement for polypurine:polypyrimidine target duplexes is a significant limitation. In order to overcome this, a range of modified bases have been designed to stabilise pyrimidine:purine C:G and T:A inversions and thereby increase the range of accessible target duplexes.17,18Fluorescent TFOs have been used previously. One important application is visualisation of DNA for detection of genomic sequences and selective molecular recognition,19–21 but these applications require a mechanism upon triplex formation for fluorescence enhancement, usually through the use of both a fluorophore and quencher. In principle, this could instead be achieved via a fluorogenic intercalator which would simultaneously increase triplex stability, potentially across a wide pH-range. Indeed intercalators, both chemically attached to the TFO or untethered, have been shown to stabilise triplexes (and other non-canonical DNA structures).22–26 A suitable design for stabilized fluorescent triplexes could be achieved by labelling TFOs with an intercalative fluorophore such as TO (Fig. 1A). For many applications, the fluorogenic nature of TO is crucial.27,28 It acts as a “light switch”, experiencing a large enhancement in fluorescence emission upon intercalation into DNA due to inhibition of rotation of the dye around its methine bridge. In this context, a TO dimer has been incorporated into modified TFOs that are known as ‘ECHO probes’ (exciton controlled hybridization sensitive fluorescent oligonucleotide).29 However, no triplex formation was seen at pH 8 (essential for important in vitro enzymatic reactions such as PCR), and the stabilising effect was not additive; two TO dimers did not enhance triplex stability over that obtained from a single addition. Other studies have been carried out on parallel triplexes using terminal (3′ and 5′) or single internal TO units, but in neither case was stable parallel triplexes reported at pH 7.30,31 In these studies, only a limited range of target sequences was examined.
Here, we demonstrate that the chemical attachment of a very simple TOB6 modification (1) to the TFOs (TOTFO probes, Fig. 1A) significantly stabilises parallel triplexes at neutral pH and above with a large increase in fluorescence on triplex formation. We also show that this TO modification can be combined with a readily available modified cytosine base, 5-(1-propynyl)-dC (pC) to expand triplex target sequences to those containing pyrimidines (C:G inversions in the duplex, Fig. 1C), which are otherwise destabilising. Importantly, multiple additions of TO are shown to extend the duplex binding affinity of the TOTFO into the nanomolar region.
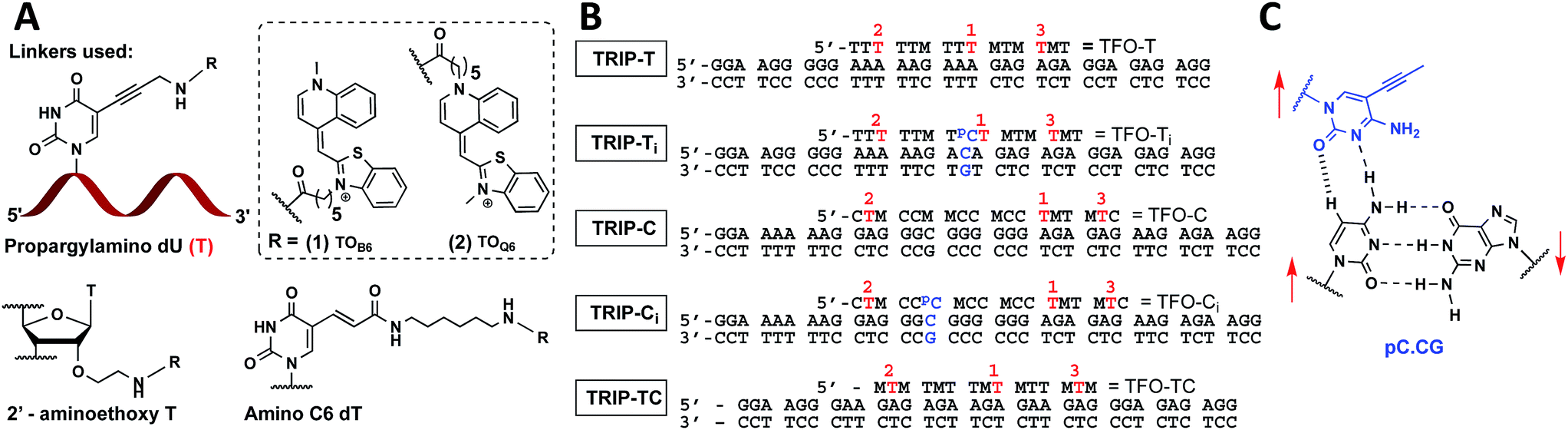 | ||
Fig. 1 (A) Structure of propargylamino deoxyuridine (pdU), 2′ aminoethoxy thymidine and amino-C6 dT scaffolds investigated for the attachment of TOB6(1) and TOQ6(2) to produce 1× TO, 2× TO and 3× TO TFOs. (B) Triplex sequences analysed in this study. Blue text = position of C:G inversion, M = 5-methyl cytosine (MeC), pC = 5-(1-propynyl)cytosine (pC),  = pdU linkage for attachment of TOB6(1) and TOQ6(2). In (B), the single-labelled TFOs have TO at position = pdU linkage for attachment of TOB6(1) and TOQ6(2). In (B), the single-labelled TFOs have TO at position  , the double-labelled TFOs have TO in positions , the double-labelled TFOs have TO in positions  and and  and the triple-labelled TFOs have TO in positions and the triple-labelled TFOs have TO in positions  , ,  and and 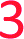 . (C) Pyrimidine:purine inversion triplet pC.CG. The pC base forms two H-bonds with cytosine of the C:G base pair; one is N–H⋯N and the other is C–H⋯O which is a weak H-bond. Red arrows indicate relative directionality of DNA strands. Other base triplets used in this study are shown in Fig. S6.† . (C) Pyrimidine:purine inversion triplet pC.CG. The pC base forms two H-bonds with cytosine of the C:G base pair; one is N–H⋯N and the other is C–H⋯O which is a weak H-bond. Red arrows indicate relative directionality of DNA strands. Other base triplets used in this study are shown in Fig. S6.† | ||
Results and discussion
Triplex design, synthesis and initial evaluation
With the aim of increasing the stability of parallel triplexes, a series of modified 15–18mer TFOs were designed to contain 5-(1-propargylamino)-dU (pdU) at various positions for internal attachment of TO. Since TO binds by intercalation, we chose well separated linker positions in the TFO to avoid hindrance of intercalation that might arise from clustered TO units. The triple bond in pdU is known to have a stabilising effect in triplexes due to increased base stacking.32,33 Other attachments such as 2′-aminoethoxy thymidine and a longer linker at the 5-position of thymine (amino C6-dT) (Fig. 1A) were also investigated, but pdU was ultimately the most effective (Fig. S1†). Importantly, the triplexes studied were chosen to cover a range of duplex sequence contexts and stacking environments (Fig. 1B). A ‘mixed’ sequence with 47% G:C duplex content was designed (TRIP-TC) as well as two T-rich sequences (TRIP-T and TRIP-Ti), and two C-rich sequences, (TRIP-C and TRIP-Ci). The subscript ‘i’ indicates the presence of a C:G inversion in the purine tract of duplex. 5-Methyl-cytosine (MeC) (M in Fig. 1B) was used at some loci in the TFOs because triplexes containing MeC are slightly more stable compared to those containing cytosine.34TOB6 and TOQ6 carboxylic acid active esters were synthesised according to protocol in Section SA.1.1† and were incorporated into the TFOs by labelling the aminopropargyl groups in the DNA strands using PyBOP coupling reagent (Section SA.2†). Efficient labelling was confirmed by mass spectrometry (Tables S1 and S2†). The melting temperatures (Tm values) of the triplexes were then analysed (Table 1 and S3†). The unlabelled T-rich TFO-T produced a triplex (TRIP-T) with a Tm of 43.2 °C in 10 mM sodium phosphate/10 mM MgCl2 buffer at pH 5.8 (‘high Mg2+ buffer’) (Entry 1, Table 1). Initially, TOQ6 was attached to the TFO (i.e. through the quinoline moiety) and this provided a large increase (+14.2 °C) in triplex stability in high Mg2+ buffer compared to the unlabelled triplex. However, as found previously for other TO modifications,29 a second TOQ6 unit did not confer any additional triplex stabilisation (Fig. S2†). When a single TOB6 unit was attached to the TFO, there was a huge increase in Tm compared to the unlabelled triplex (+23.3 °C) and fortunately, the stabilising effect of TOB6 increased with the addition of a second and third fluorophore, reaching 78.0 °C (ΔTm = +34.8 °C). Therefore, TOB6 was used for all remaining studies. The melting studies were also carried out in a buffer closer to cellular salt conditions but still at acidic pH and using Na+ ions instead of K+ ions (low Mg2+ buffer, 10 mM sodium phosphate/150 mM NaCl/2 mM MgCl2, pH 5.8). Strong stabilisation was observed with the addition of three TOB6 units giving a remarkable increase in Tm of 44.8 °C (Table 1, Entries 1–4). Stabilisation was also seen in the TRIP-TC triplex with the addition of three TO units in the TFO, but the overall enhancement in stability (up to 9.7 °C) was not as great, presumably because the unlabelled triplex is already very stable at this pH (Tm > 70 °C), surpassing the stability of the underlying duplex. However, at pH 7 when the unmodified triplex becomes unstable, the increase in Tm from TO incorporation is impressive (>20 °C) (Table 1, Entries 10 and 11). The effect of sequence on stability of triplexes can be observed in TRIP-C, which is highly unstable but exhibited major stabilisation with the introduction of 3× TO (+26.2 °C in low Mg2+ buffer) (Table 1, Entries 12–15). Melting curves and additional melting data are in Fig. S3 and Table S3.†
a
T
m values were obtained from the first derivatives of the melting curves (black text = UV melting analysis, blue text = fluorescence melting analysis). Fluorescence melting was used when the Tm could not be determined by UV melting. Buffer at pH 5.8 = 10 mM sodium phosphate and varying salt conditions according to the table header, buffer at pH 7 = 10 mM MOPS and varying salt conditions. Tm values are an average of two independent experiments, both of which are an average of 5 measurements. pC = pC, M = MeC,  = TO attached to pdU, nt = no triplex detected. Full triplex sequences are in Fig. 1B. Tm of the underlying duplexes are as follows (pH 5.8/pH 7): TRIP-T = 73.1/73.4 °C, TRIP-Ti = 73.9/74.3 °C, TRIP-TC = 73.2/74.1 °C, TRIP-C = 74.6/75.1 °C, TRIP-Ci = 77.3/77.5 °C. = TO attached to pdU, nt = no triplex detected. Full triplex sequences are in Fig. 1B. Tm of the underlying duplexes are as follows (pH 5.8/pH 7): TRIP-T = 73.1/73.4 °C, TRIP-Ti = 73.9/74.3 °C, TRIP-TC = 73.2/74.1 °C, TRIP-C = 74.6/75.1 °C, TRIP-Ci = 77.3/77.5 °C.
|
|---|
|
To further investigate the sequence specificity of TOTFOs, a scrambled duplex sequence was mixed with 1× TO TFO-T. No triplex formation was observed by UV melting, confirming that the stabilisation gained from intercalation of TO is coupled with retained sequence specificity (Fig. S4†). To confirm that the TO intercalator in the TFO does not reduce base pair selectivity, we investigated triplexes in which TFO-T (3× TO) was mixed with a duplex containing an incorrect base pair to give a T.CG, T.TA or T.GC base triplet instead of T.AT. As expected, there is a decrease in triplex stability (−10 to −14 °C) when a mismatched triplet is introduced, with triplet stability in the following order: T.AT > T.CG > T.TA > T.GC. This confirms that TO intercalation does not destroy the selectivity of the TFO for its fully complementary duplex (Fig. S5†).
Stabilisation of triplex inversions
Having established strong stabilisation of parallel triplexes at uninterrupted polypurine tracts, attention was focussed on triplexes in which the duplex contains a triplex-destabilising base pair inversion. To study this, a C:G base pair was inserted in the poly-A:T or poly-G:C tract of the duplex DNA target (TRIP-Ti and TRIP-Ci). Previous studies on modified bases that can recognise and stabilise C:G inversions require multi-step synthesis of special phosphoramidite monomers.17,35 Here, 5-(1-propynyl)-dC (pC), a commercially available monomer, was used to form a Hoogsteen interaction with the inverted cytosine base of the duplex (a pC.CG triplet). The pC nucleobase has been investigated in TFOs previously for binding to G:C base pairs, where it was destabilising due to its resistance to protonation at N3 (low pKaH).36 However, for C:G recognition, this is advantageous as protonation of pC is not required (Fig. 1C, S6†). A comparison to thymine, the most effective natural base to stabilise a C:G inversion (Fig. S5†),35 indicated that pC is slightly superior, giving a ΔTm = +1.2 °C and +1.6 °C in high and low Mg2+ buffers respectively (Table 1, Entry 7). To investigate the selectivity of pC for the C:G base pair, TFO-Ti was mixed with three different duplexes to form triplexes containing a mismatch triplet (pC.GC, pC.AT and pC.TA instead of pC.CG) (Fig. S7†). The pC.AT triplet is interesting because the A:T base pair would cause selectivity problems when using T to recognise C:G in a genomic context due to the fact that the T.AT triplet is more stable than T.CG. Fortunately, pC.AT produces a less stable triplex compared to pC.CG at pH 7 (ΔTm = −12.3 °C) and is much less stable than T.AT (ΔTm = −24.7 °C). pC.GC produces a triplex of similar stability to pC.CG in low pH but we see this stability is highly pH-dependent (ΔTm = −24.6 °C) as the pH is increased whilst pC.CG remains far less pH-dependent.Incorporation of the C:G inversion into the TRIP-Ti and TRIP-Ci sequence environments showed that the TRIP-Ci triplex is far less stable than TRIP-Ti (ΔTm = −10.8 °C, pH 5.8, high Mg2+ buffer, Table 1, Entries 6 and 17). This is expected as contiguous runs of cytosine bases destabilise triplexes because the pKaH of each individual C-nucleobase is reduced due to their close proximity.37,38 The inclusion of a single TO unit in TFO-Ti led to a large increase in the Tm of the TRIP-Ti triplex (ΔTm > 16.0 °C) and a remarkable +43.6 °C increase in low Mg2+ buffer for the triplex with three units of TO (Table 1, Entries 7–9). A smaller increase in stability was observed in the TRIP-Ci triplex with one TO-addition (ΔTm ∼ 3 °C, Table 1, Entry 18). However, the Tm increased much more (by 16 °C) when three TO units were introduced into the TFO-Ci (Table 1, Entry 20). Hence, in general TO intercalation and pC as a C:G recognition monomer more than compensates for the destabilising effect caused by the triplet inversion.
Fluorescence melting
As triplexes become more stable, UV monitoring of thermal denaturation is a less reliable determinant of Tm because melting of the triplex is obscured by the duplex melting transition (Fig. S8†) and this can also distort the triplex melting transition. In such cases, fluorescence melting can be used instead, because in this technique only the triplex to duplex transition is detectable.19 Fluorescence melting was therefore employed to analyse triplexes, and in cases where both UV and fluorescence melting could be measured the melting temperatures were in good agreement with slight differences due to the DNA concentrations used (1 μM for UV melting, 0.625 μM for fluorescence melting). Also, small differences in Tm result from the fact that the two techniques monitor different DNA melting-related processes. UV melting is sensitive to the dissociation of strands via the unstacking of bases whilst fluorescence melting reports on intercalation of the TO into the triplex.TOTFO triplex stabilisation at higher pH
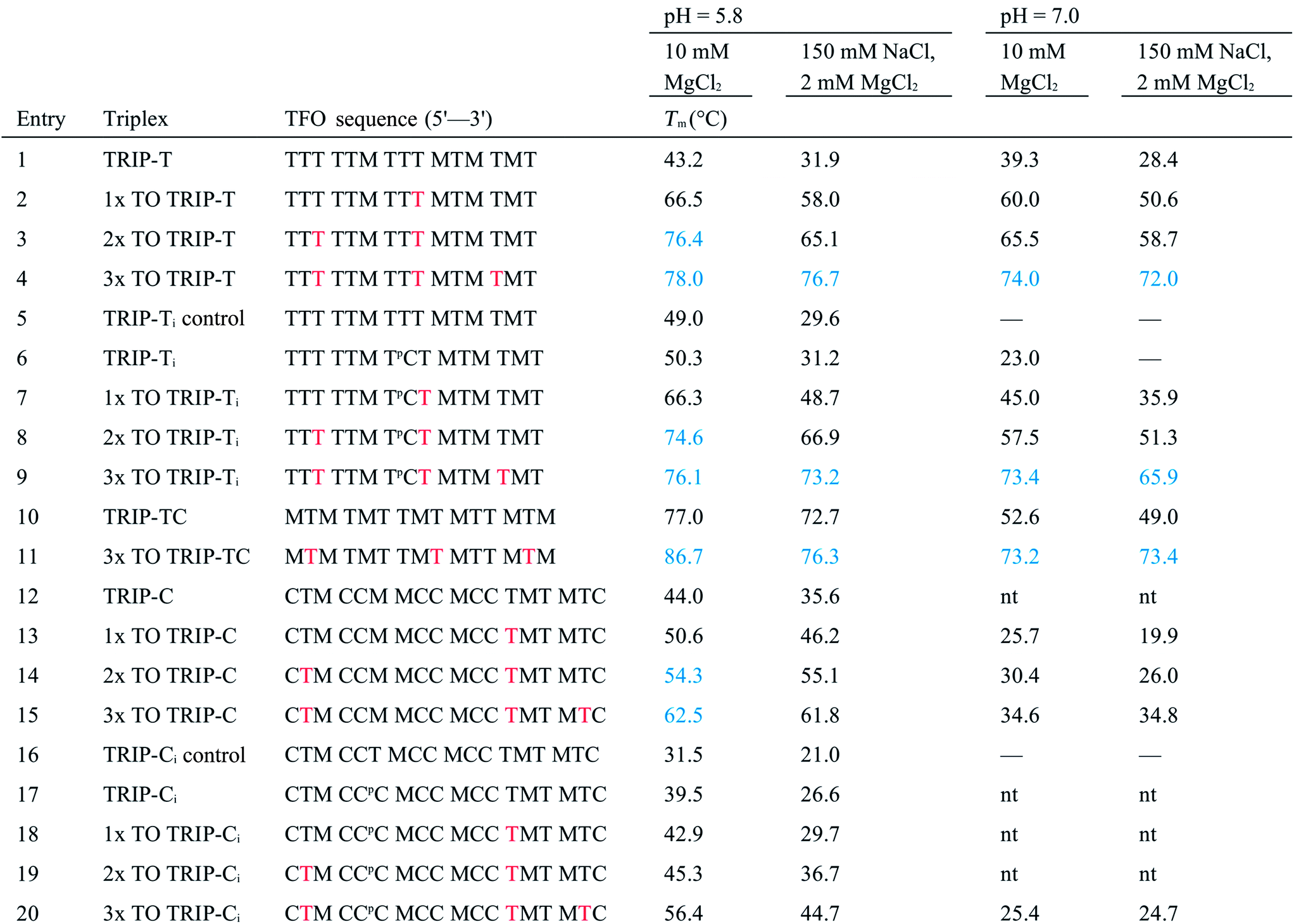
As discussed previously, one of the limitations of triplexes and their applications is the necessity to conduct experiments at low pH. With stabilisation from TO, it is possible to increase the pH and still form stable triplexes. 3× TO TRIP-Ti is one of the most stable triplex at pH 7, with a Tm > 70 °C (Fig. 2A and B) and the additive effective of TO is clear. Substantial stabilisation is also observed for TRIP-T and TRIP-TC compared to their unlabelled triplexes in low Mg2+ buffer (Fig. 2C). However, 3× TO TRIP-C is much less stable (Tm ∼ 35 °C). This is because it is rich in cytosine bases (78%) which require protonation to form stable C+.GC triplets and this will not occur at neutral pH. It is noteworthy that we could not observe any triplex formation for unmodified TRIP-C at pH 7 but could overcome this instability with TO attachment. In some sequence contexts, very stable triplexes can be produced at pH 7 with only one TO modification e.g. 1× TO TRIP-T, Tm = 60.0 °C and 50.6 °C in high and low Mg2+ buffer respectively (Table 1, Entry 2). Stable triplexes can even be formed above neutral pH; TRIP-TC with 3× TO has a Tm of 54 °C at pH 8 (Table S4†). Hence, these studies demonstrate that triplex formation in biologically compatible conditions is feasible using the simple TOB6 modification.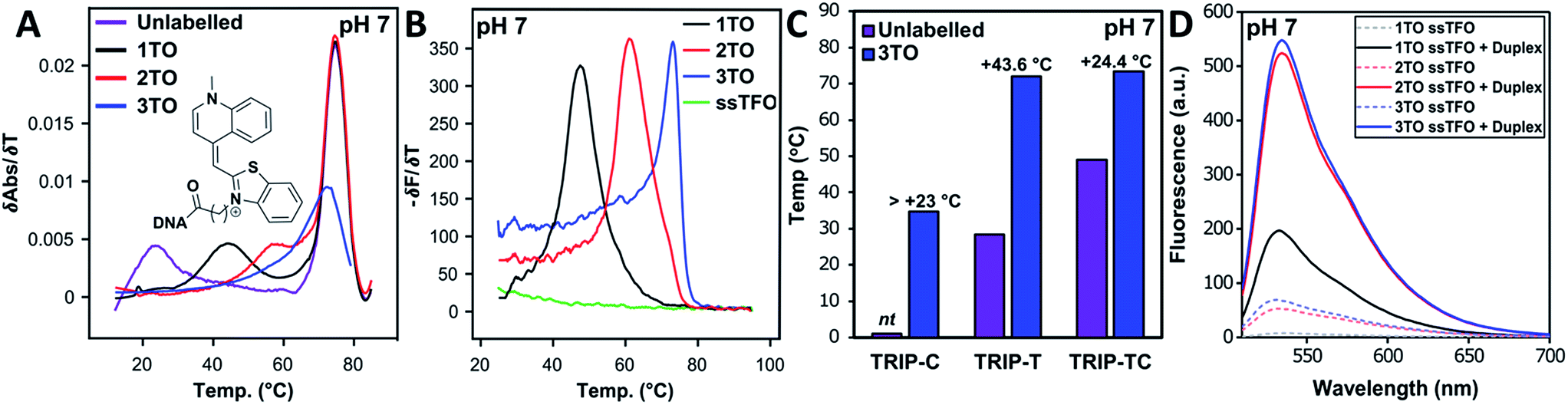 | ||
| Fig. 2 (A) First derivatives of UV absorption curves of TRIP-Ti triplex; unlabelled, 1×, 2× and 3× TO at pH 7. The first transition is triplex denaturation and the second (higher temperature) is duplex denaturation. Tm of 3× TO could not be determined by UV melting as the triplex Tm overlapped with the duplex transition. Duplex concentration is 1.0 μM and TFO concentration is 2.5 μM, λ = 260 nm. (B) First derivative of fluorescence melting curves of the 1×, 2× and 3× TO TRIP-Ti triplex with the ssTFO as a control at pH 7, 0.25 μM of duplex and 0.625 μM TFO, see Section SA.6† for excitation and emission channels. (C) Triplex stability vs. sequence environment. C+.GC-rich sequence TRIP-C, T.AT-rich sequence TRIP-T and mixed sequence TRIP-TC. nt = no triplex detected, therefore, accurate ΔTm could not be determined for the labelled triplex. Buffer contains 10 mM MOPS, 150 mM NaCl, 2 mM MgCl2, pH 7. All Tm values are an average of two independent experiments, both of which are an average of 5 measurements. (D) Fluorescence intensity of 1×, 2×, 3× TO (TRIP-T), ssTFO in the absence (dashed line) and presence (solid line) of the duplex at pH 7. Concentrations of 1.8 μM duplex and 1 μM TFO used. | ||
The influence of the T.AT/C+.GC triplet ratio on unlabelled and TO-labelled triplex stability at pH 7 is shown in Fig. 2C. C+.GC-rich triplexes are unstable, T.AT-rich triplexes are much more stable and triplexes with an even mixture of T.AT and C+.GC triplets are the most stable. All benefit from the presence of TO immensely (some with ΔTm > +40 °C).
Evaluation of fluorescence enhancement on triplex formation
To quantify the enhancement of fluorescence intensity upon triplex formation, the fluorescence spectra of the TFOs both in the absence and presence of the complementary duplex were measured (Fig. 2D). TFOs excited at 484 nm showed minimal fluorescence in single-strand form (ssTFO). Upon addition of the duplex, fluorescence intensity was greatly enhanced, 23-fold for the TRIP-T triplex at pH 5.8. At neutral pH, slightly greater enhancement (26-fold) was seen upon triplex formation (Table 2). The fluorescent intensity ratios between the free TFOs and the triplexes are given in Table 2. A decrease in the ratio is observed as additional TO units are introduced into the TFO, even though the triplexes with 2 and 3 TO units are much brighter than that with a single TO. This is due to the higher fluorescence of the single stranded TFOs that contain multiple TO units. Fluorescence increases slightly at higher pH, and the trends are similar. Further fluorescent analysis is provided in Fig. S9.† Quantum yield measurements were carried out on the triplexes (Table S5†) and lie mostly between 0.13 and 0.23 at pH 7, varying as a function of sequence context. This is in line with published data on thiazole orange in DNA duplexes and higher ordered structures.39–41| Triplex | pH = 5.8 | pH = 7.0 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| λ ssem nm | λ Tem nm | F ss@λTem a.u. | F T@λTem a.u. | F T/Fss | λ ssem nm | λ Tem nm | F ss@λTem a.u. | F T@λTem a.u. | F T/Fss | |
| a Buffer at pH 5.8 = 10 mM sodium phosphate, 10 mM MgCl2. Buffer at pH 7 = 10 mM MOPS, 10 mM MgCl2. Fluorescence values are averages of two independent measurements. λssem = max. emission wavelength of single strand TFO, λTem = max. emission wavelength of formed triplex, Fss@λTem = fluorescence intensity of ssTFO at the max. emission wavelength of the triplex, FT@λTem = fluorescence intensity of triplex at the max. emission wavelength of the triplex, FT/Fss = ratio of fluorescence intensity calculated by comparison of the ssTFO fluorescence intensity and the formed triplex fluorescence intensity at λTem. | ||||||||||
| 1× TO TRIP-T | 534 | 533 | 10.72 | 249.16 | 23.2 | 536 | 533 | 7.49 | 196.90 | 26.3 |
| 2× TO TRIP-T | 532 | 534.5 | 72.23 | 605.85 | 8.4 | 533 | 534.5 | 52.41 | 523.88 | 10.0 |
| 3× TO TRIP-T | 531.5 | 535 | 119.61 | 578.91 | 4.8 | 531 | 534.5 | 67.45 | 547.83 | 8.1 |
| 1× TO TRIP-Ti | 531.5 | 535 | 87.77 | 704.85 | 8.0 | 530 | 531.5 | 51.50 | 642.48 | 12.5 |
| 2× TO TRIP-Ti | 534.5 | 535 | 52.12 | 348.26 | 6.7 | 532 | 534.5 | 29.49 | 321.42 | 10.9 |
| 1× TO TRIP-C | 538 | 535.5 | 62.88 | 485.31 | 7.7 | 531.5 | 532 | 100.26 | 668.40 | 6.7 |
| 2× TO TRIP-C | 564 | 538 | 32.89 | 640.17 | 19.5 | 533 | 535 | 38.44 | 875.43 | 22.8 |
| 3× TO TRIP-C | 585 | 539 | 18.68 | 468.42 | 25.1 | 533.5 | 535 | 19.93 | 711.12 | 35.7 |
| 1× TO TRIP-Ci | 533 | 534.5 | 98.23 | 462.54 | 4.7 | 531.5 | 532.5 | 69.62 | 485.58 | 7.0 |
| 2× TO TRIP-Ci | 535.5 | 536 | 80.78 | 400.23 | 5.0 | 565.5 | 536 | 42.88 | 440.69 | 10.3 |
| 3× TO TRIP-Ci | 533 | 535.5 | 100.15 | 288.61 | 2.9 | 582.5 | 537.5 | 52.19 | 315.20 | 6.0 |
For many applications, the inherent fluorescence of thiazole orange will be adequate for detection purposes, but in specific cases a brighter and more stable fluorophore will be required e.g. for super-resolution imaging of duplex sequences in genomic DNA in live or fixed cells.42 Our TOTFO concept was designed with this in mind; the termini of the TFO are unmodified and therefore available for orthogonal fluorescent labelling. This was demonstrated by the synthesis of an ATTO 647N click-labelled TOTFO (TO-ATTO TRIP-Ti, Fig. S10†) which forms a stable triplex with its target duplex.
TOTFO triplex studies by other biophysical techniques
Triplex formation and stability were also monitored by other techniques. The TRIP-Ti triplex was investigated by native polyacrylamide gel-electrophoresis (PAGE) at pH 7 (Fig. 3). Triplexes were separated from the duplex by gel electrophoresis based on charge and size, then visualised by inherent TO fluorescence and SYBR Gold staining. The duplex can be identified from lane 1 and the triplex can be observed in lanes 3–7 where the TOTFOs were added to the duplexes. Under SYBR Gold staining it can be seen that the triplex band migrates slightly slower than the duplex band. As expected, decreasing the TFO:duplex ratio results in less intense triplex bands (compare lanes 5, 6, 7). The triplex band was extracted from the gel and analysed by mass spectrometry, which revealed three peaks of the expected masses, confirming the presence of each strand of the triplex (Fig. S11†).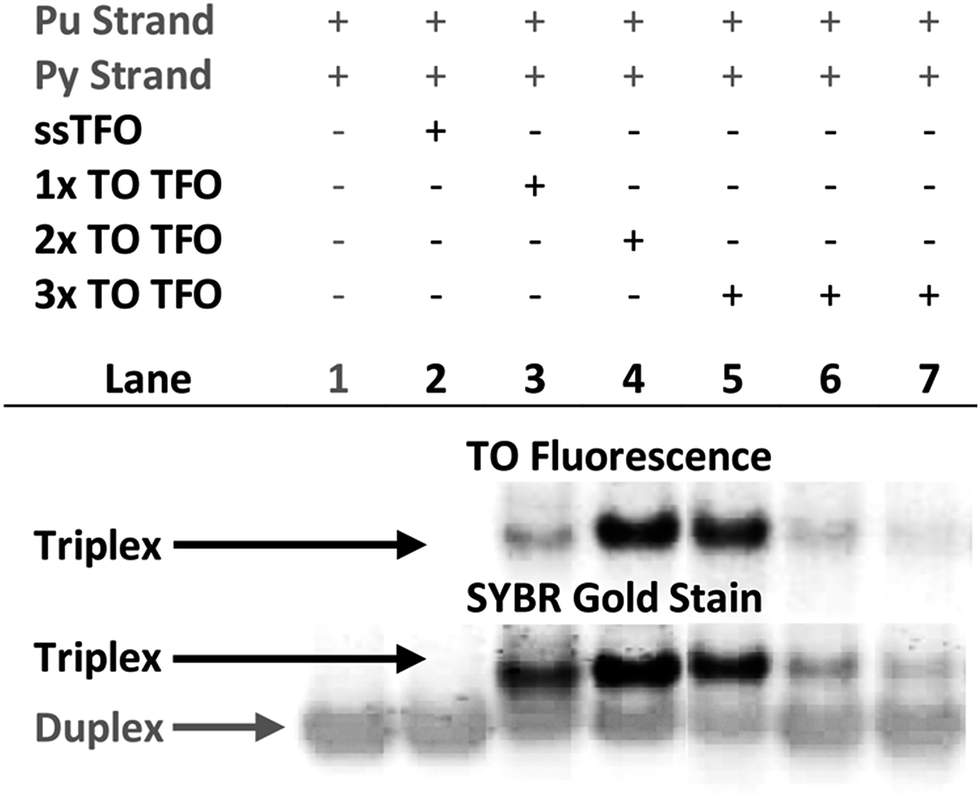 | ||
Fig. 3 Fluorescence and SYBR Gold staining, 15% non-denaturing polyacrylamide gels showing TRIP-Ti triplex formation. Buffer is 40 mM MOPS containing 10 mM NaOAc and 10 mM MgCl2 (pH 7). All duplex strand concentrations are 1.0 μM. Lane 2 – [10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1] unlabelled TFO:duplex, lane 3/4 – [10:1] TFO:duplex, lanes 5, 6, 7 – [10:1], [5:1], [2.5:1] TFO:duplex respectively. Lane 2 shows no triplex formation due to the low Tm of the unlabelled triplex at neutral pH (Tm = 23 °C). :1] unlabelled TFO:duplex, lane 3/4 – [10:1] TFO:duplex, lanes 5, 6, 7 – [10:1], [5:1], [2.5:1] TFO:duplex respectively. Lane 2 shows no triplex formation due to the low Tm of the unlabelled triplex at neutral pH (Tm = 23 °C). | ||
SPR analysis was carried out to corroborate the gel-electrophoresis and thermal denaturation studies. The unlabelled triplexes TRIP-Ti produced a KD of 1.5 μM in accordance with previous reports in which the dissociation constants of triplexes are generally within the micromolar range.43 A progressive increase in binding was observed with two then three additions of TO, three additions giving a binding constant of 3.9 nM (Fig. S12†). This data supports the trends observed in the triplex melting studies discussed above.
Conclusions
Triplex stability is greatly enhanced by incorporation of thiazole orange between the 5-position of thymine and the benzothiazole moiety of TO (TOB6) in triplex forming oligonucleotides (TOTFOs). Very stable triplexes are obtained at (and above) neutral pH using this straightforward methodology. The stabilising effects of TO are cumulative; the most stable triplexes at pH 7 was formed with three additions of TO in TFOs that target a A:T-rich and mixed-sequence duplexes, and Tm increases up to 45 °C were achieved. This greatly increased stability has enabled us to expand the range of target duplexes at neutral pH to polypurine sequences that contain a pyrimidine inversion using pC as a C:G recognition monomer. In one such case an increase in Tm of 50 °C was obtained at pH 7. TOTFOs undergo large enhancement in fluorescence upon triplex formation and this could be significant for many applications. TOTFOs are also substrates for dual dye labelling enabling the attachment of very bright fluorophores for stringent biological studies (ATTO-TOTFOs). This simple approach to triplex stabilisation could unlock many practical applications and might also be effective in the stabilisation of highly unstable antiparallel triplexes.Finally, the method used in this work for oligonucleotide labelling involved reaction of TO active esters with amino-modified oligonucleotides. Now that we have established which TO-nucleotide is the most effective (TOB6-pdU) it will be more efficient in future to introduce it directly into TFOs during solid-phase synthesis via its phosphoramidite monomer and equip future generations of the probes with substituents that interrogate the target sequence in both the major and minor groove.44
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project received funding from the European Union Horizon 2020 research and innovation programme under Marie Sklodowska Curie agreement 642023 (H2020-MSCA-ITN-2014-642023) and UK BBSRC grant BB/J001694/2 (extending the boundaries of nucleic acid chemistry).References
- H. E. Moser and P. B. Dervan, Science, 1987, 238, 645–650 CrossRef PubMed.
- K. Hoogsteen, Acta Crystallogr., 1959, 12, 822–823 CrossRef.
- A. R. Morgan and R. D. Wells, J. Mol. Biol., 1968, 37, 63–80 CrossRef PubMed.
- J. C. Hanvey, M. Shimizu and R. D. Wells, Nucleic Acids Res., 1990, 18, 157–161 CrossRef PubMed.
- Y. Taniguchi and S. Sasaki, Org. Biomol. Chem., 2012, 10, 8336 RSC.
- A. Mukherjee and K. M. Vasquez, Biochimie, 2011, 93, 1197–1208 CrossRef PubMed.
- L. Maher, B. Wold and P. Dervan, Science, 1989, 245, 725–730 CrossRef PubMed.
- S. B. Noonberg, G. K. Scott, C. A. Hunt, M. E. Hogan and C. C. Benz, Gene, 1994, 149, 123–126 CrossRef PubMed.
- K. M. Vasquez, T. G. Wensel, M. E. Hogan and J. H. Wilson, Biochemistry, 1996, 35, 10712–10719 CrossRef PubMed.
- R. Zain, C. Marchand, J. S. Sun, C. H. Nguyen, E. Bisagni, T. Garestier and C. Helene, Chem. Biol., 1999, 6, 771–777 CrossRef PubMed.
- Y. Hu, A. Cecconello, A. Idili, F. Ricci and I. Willner, Angew. Chem., Int. Ed., 2017, 56, 15210–15233 CrossRef PubMed.
- N. Ben Gaied, Z. Zhao, S. R. Gerrard, K. R. Fox and T. Brown, ChemBioChem, 2009, 10, 1839–1851 CrossRef PubMed.
- V. Kumar, V. Kesavan and K. V. Gothelf, Org. Biomol. Chem., 2015, 13, 2366–2374 RSC.
- T. Sato, Y. Sato and S. Nishizawa, J. Am. Chem. Soc., 2016, 138, 9397–9400 CrossRef PubMed.
- L. Lacroix, J. Lacoste, J. F. Reddoch, J. L. Mergny, D. D. Levy, M. M. Seidman, M. D. Matteucci and P. M. Glazer, Biochemistry, 1999, 38, 1893–1901 CrossRef PubMed.
- H. Torigoe, Y. Hari, M. Sekiguchi, S. Obika and T. Imanishi, J. Biol. Chem., 2001, 276, 2354–2360 CrossRef PubMed.
- V. Malnuit, M. Duca and R. Benhida, Org. Biomol. Chem., 2011, 9, 326–336 RSC.
- Y. Hari, S. Obika and T. Imanishi, Eur. J. Org. Chem., 2012, 2012, 2875–2887 CrossRef.
- R. J. Darby, M. Sollogoub, C. McKeen, L. Brown, A. Risitano, N. Brown, C. Barton, T. Brown and K. R. Fox, Nucleic Acids Res., 2002, 30, e39 CrossRef PubMed.
- Y. Wang, Q. Sun, L. Zhu, J. Zhang, F. Wang, L. Lu, H. Yu, Z. Xu and W. Zhang, Chem. Commun., 2015, 51, 7958–7961 RSC.
- T. N. Grossmann, L. Röglin and O. Seitz, Angew. Chem., Int. Ed., 2007, 46, 5223–5225 CrossRef PubMed.
- W. D. Wilson, F. Tanious, S. Mizan, S. Yao, A. Kiselyov, G. Zon and L. Strekowski, Biochemistry, 1993, 32, 10614–10621 CrossRef PubMed.
- K. R. Fox and T. Brown, in DNA Conjugates and Sensors, 2012, pp. 75–102 Search PubMed.
- J. S. Sun, J. C. François, T. Montenay-Garestier, T. Saison-Behmoaras, V. Roig, N. T. Thuong and C. Hélène, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 9198–9202 CrossRef.
- M. Grigoriev, D. Praseuth, P. Robin, A. Hemar, T. Saison-Behmoaras, A. Dautry-Varsat, N. T. Thuong, C. Helene and A. Harel-Bellan, J. Biol. Chem., 1992, 267, 3389–3395 Search PubMed.
- S. Bevers, S. Schutte and L. W. McLaughlin, J. Am. Chem. Soc., 2000, 122, 5905–5915 CrossRef.
- T. Sato, Y. Sato and S. Nishizawa, Chem. - Eur. J., 2017, 23, 4079–4088 CrossRef PubMed.
- I. Lubitz, D. Zikich and A. Kotlyar, Biochemistry, 2010, 49, 3567–3574 CrossRef PubMed.
- S. Ikeda, H. Yanagisawa, M. Yuki and A. Okamoto, Artif. DNA PNA XNA, 2013, 4, 19–27 CrossRef PubMed.
- O. Doluca, T. K. Hale, P. J. B. Edwards, C. Gonzalez and V. V. Filichev, ChemPlusChem, 2014, 79, 58–66 CrossRef.
- B. L. Renard, R. Lartia and U. Asseline, Org. Biomol. Chem., 2008, 6, 4413–4425 RSC.
- K. R. Fox, Curr. Med. Chem., 2000, 7, 17–37 CrossRef PubMed.
- J. Bijapur, M. D. Keppler, S. Bergqvist, T. Brown and K. R. Fox, Nucleic Acids Res., 1999, 27, 1802–1809 CrossRef PubMed.
- O. A. Amosova and J. R. Fresco, Nucleic Acids Res., 1999, 27, 4632–4635 CrossRef PubMed.
- S. R. Gerrard, M. M. Edrees, I. Bouamaied, K. R. Fox and T. Brown, Org. Biomol. Chem., 2010, 8, 5087–5096 RSC.
- B. C. Froehler, S. Wadwani, T. J. Terhorst and S. R. Gerrard, Tetrahedron Lett., 1992, 33, 5307–5310 CrossRef.
- A. M. Soto, J. Loo and L. A. Marky, J. Am. Chem. Soc., 2002, 124, 14355–14363 CrossRef PubMed.
- J. S. Lee, M. L. Woodsworth, L. J. Latimer and A. R. Morgan, Nucleic Acids Res., 1984, 12, 6603–6614 CrossRef PubMed.
- E. Privat, T. Melvin, F. Mérola, G. Schweizer, S. Prodhomme, U. Asseline and P. Vigny, Photochem. Photobiol., 2002, 75, 201–210 CrossRef PubMed.
- J. Nygren, N. Svanvik and M. Kubista, Biopolymers, 1998, 46, 39–51 CrossRef PubMed.
- I. Lubitz, D. Zikich and A. Kotlyar, Biochemistry, 2010, 49, 3567–3574 CrossRef PubMed.
- S. Beater, P. Holzmeister, E. Pibiri, B. Lalkens and P. Tinnefeld, Phys. Chem. Chem. Phys., 2014, 16, 6990–6996 RSC.
- P. J. Bates, H. S. Dosanjh, S. Kumar, T. C. Jenkins, C. A. Laughton and S. Neidle, Nucleic Acids Res., 1995, 23, 3627–3632 CrossRef PubMed.
- R. Haug, M. Kramer and C. Richert, Chem.–Eur. J., 2013, 19, 15822–15826 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02418a |
| This journal is © The Royal Society of Chemistry 2018 |