DOI:
10.1039/C8SC02768D
(Edge Article)
Chem. Sci., 2018,
9, 7673-7680
A β-diketiminate manganese catalyst for alkene hydrosilylation: substrate scope, silicone preparation, and mechanistic insight†
Received
23rd June 2018
, Accepted 14th August 2018
First published on 15th August 2018
Abstract
The dimeric β-diketiminate manganese hydride compound, [(2,6-iPr2PhBDI)Mn(μ-H)]2, was prepared by treating [(2,6-iPr2PhBDI)Mn(μ-Cl)]2 with NaEt3BH. This compound was characterized by single crystal X-ray diffraction and found to feature high-spin Mn centres that exhibit strong magnetic coupling by EPR spectroscopy. Once characterized, [(2,6-iPr2PhBDI)Mn(μ-H)]2 was found to mediate the hydrosilylation of a broad scope of alkenes at elevated temperature. Aliphatic alkenes were found to undergo anti-Markovnikov hydrosilylation, while the hydrosilylation of styrenes using [(2,6-iPr2PhBDI)Mn(μ-H)]2 afforded Markovnikov's product. Importantly, this catalyst has also been employed for the cross-linking of industrially-relevant silicones derived from vinyl-terminated poly(dimethylsiloxane) and 1,2,4-trivinylcyclohexane with catalyst loadings as low as 0.05 mol%. To gain a mechanistic understanding of [(2,6-iPr2PhBDI)Mn(μ-H)]2-catalyzed olefin hydrosilylation, 4-tert-butylstyrene was added to [(2,6-iPr2PhBDI)Mn(μ-H)]2 and conversion to the monomeric Mn alkyl complex, (2,6-iPr2PhBDI)Mn(CH(CH3)(4-tBuPh)), was observed. Isolation of this secondary alkyl intermediate confirms that olefin insertion into the Mn–H bond dictates the observed regioselectivities. The importance of our mechanistic findings as they relate to recent advances in Mn hydrosilylation catalysis is described herein.
Introduction
The hydrosilylation of alkenes in the presence of hydride-functionalized siloxanes allows for the industrial preparation of silicones, which commonly serve as release coatings and water-repellent adhesives.1,2 Moreover, silicones are used to prepare a wide variety of consumer products3 and are of vital importance to the health care industry given their use in medical grade tubing.4 Industrial silicone synthesis is largely dependent on the application of Pt hydrosilylation catalysts; however, there are major disadvantages associated with using a precious metal for this purpose. Since Pt is not widely abundant in Earth's crust (0.005 mg kg−1),5 it is costly to obtain and its relative scarcity has led to wild fluctuations in Pt futures pricing.6 More importantly, after silicones cure, Pt remains in the product and cannot be easily recovered.6 In 2007 alone, it was estimated that 5.6 tonnes of Pt were consumed for the purpose of silicone cross-linking.7 Moreover, the residual Pt in silicones is toxic, which has caused side-effects in patients living with silicone implants.8
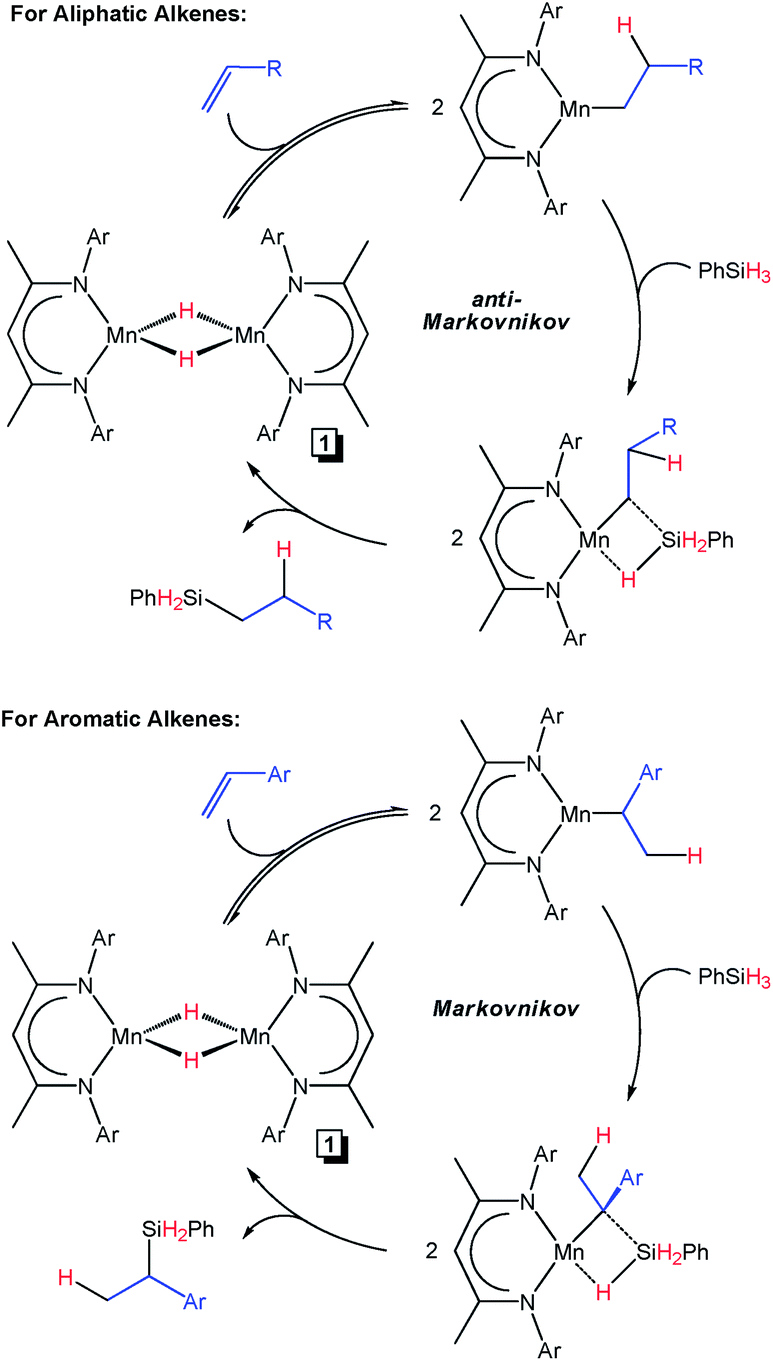
Given the Earth-abundance and biologically-benign nature of late first row transition metals, hydrosilylation catalysts featuring them have emerged as promising and sustainable substitutes.9,10 Significant progress has been made in the design of well-defined Fe,11 Co,12 and Ni13 olefin hydrosilylation catalysts, some of which have been shown to cure silicones of industrial importance.11e,12j,13e Although Mn is 38× more abundant than Co and 12× more abundant than Ni,5 Mn catalysts have not been widely employed for alkene hydrosilylation.14 In 1983, the thermal and photochemical hydrosilylation of 1-pentene using (CO)5MnSiPh3 was reported by researchers at GE.15 Hilal and co-workers subsequently showed that Mn2(CO)10 hydrosilylates 1-hexene in the presence of Et3SiH or (EtO)3SiH at 40 °C,16 and that polysiloxane-supported Mn2(CO)10 hydrosilylates 1-octene using (EtO)3SiH under a range of conditions.17 The same group also found that an intercalated Mn porphyrin compound catalyzes the same transformation at temperatures as low as 20 °C.18 In 2016, the Shenvi group observed methylenecyclohexane hydrosilylation in the presence of PhSiH3 and Mn(dpm)3 (dpm = dipivaloylmethane).19 Last year, the Mn silylene complex (dmpe)2MnH(Et2Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHMe) was found to achieve approximately 30 turnovers of ethylene hydrosilylation using Et2SiH2 after 24 h at 60 °C.20 Thomas and co-workers also reported that the 2,6-bis(imino)pyridine (or pyridine diimine, abbreviated as PDI in this manuscript) supported Mn precursor (2,6-Et2PhPDI)MnBr2 catalyzes 1-octene hydrosilylation in the presence of PhSiH3 following activation with NaOtBu.21 In a very recent follow-on manuscript,22 these researchers explored the role of chelate modification on hydrosilylation activity, finding that 2,6-iPr2Ph PDI substitution enhanced catalyst performance. For the first time, they were able to demonstrate a broad substrate scope for the ambient temperature Mn-catalyzed hydrosilylation of aliphatic alkenes to yield linear silanes selectively (Fig. 1). Although poor conversion was noted for the hydrosilylation of styrenes under the preferred reaction conditions (35–40% after 4 h at 25 °C), these substrates also yielded the anti-Markovnikov product. Importantly, Thomas and co-workers also described mechanistic experiments that suggest a Mn–H intermediate may be responsible for the observed catalysis.22
CHMe) was found to achieve approximately 30 turnovers of ethylene hydrosilylation using Et2SiH2 after 24 h at 60 °C.20 Thomas and co-workers also reported that the 2,6-bis(imino)pyridine (or pyridine diimine, abbreviated as PDI in this manuscript) supported Mn precursor (2,6-Et2PhPDI)MnBr2 catalyzes 1-octene hydrosilylation in the presence of PhSiH3 following activation with NaOtBu.21 In a very recent follow-on manuscript,22 these researchers explored the role of chelate modification on hydrosilylation activity, finding that 2,6-iPr2Ph PDI substitution enhanced catalyst performance. For the first time, they were able to demonstrate a broad substrate scope for the ambient temperature Mn-catalyzed hydrosilylation of aliphatic alkenes to yield linear silanes selectively (Fig. 1). Although poor conversion was noted for the hydrosilylation of styrenes under the preferred reaction conditions (35–40% after 4 h at 25 °C), these substrates also yielded the anti-Markovnikov product. Importantly, Thomas and co-workers also described mechanistic experiments that suggest a Mn–H intermediate may be responsible for the observed catalysis.22
 |
| | Fig. 1 Alkene hydrosilylation reported by Thomas and co-workers using (2,6-iPr2PhPDI)MnBr2.22 | |
Over the last five years, our group has explored the Mn-catalyzed hydrosilylation of carbonyl functionalities using donor-substituted chelates. The compounds (Ph2PPrPDI)Mn,23 (PyEtPDEA)Mn,24 [(Ph2PEtPDI)Mn]2,25 and (Ph2PPrPDI)MnH26 have all been found to exhibit exceptional carbonyl hydrosilylation activity; however, they are completely inactive for alkene hydrosilylation at temperatures as high as 120 °C.27 Considering the κ4- and κ5-chelate denticity that these catalysts feature, we have turned our focus to the development of Mn catalysts possessing a more open coordination environment. Herein, we describe the preparation of a β-diketiminate (BDI, often referred to as NacNac or β-diiminate) Mn hydride catalyst that has been found to hydrosilylate terminal, cyclic, and gem-alkenes at 130 °C. This catalyst can be used to prepare commercially relevant silicones and stoichiometric addition experiments have revealed that substrate-specific selectivity is dictated by alkene insertion into the Mn–H bond.
Results and discussion
Catalyst synthesis and characterization
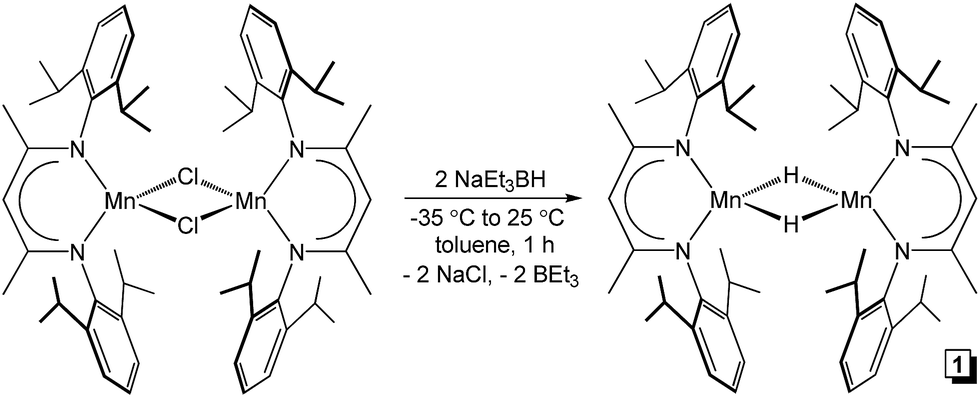
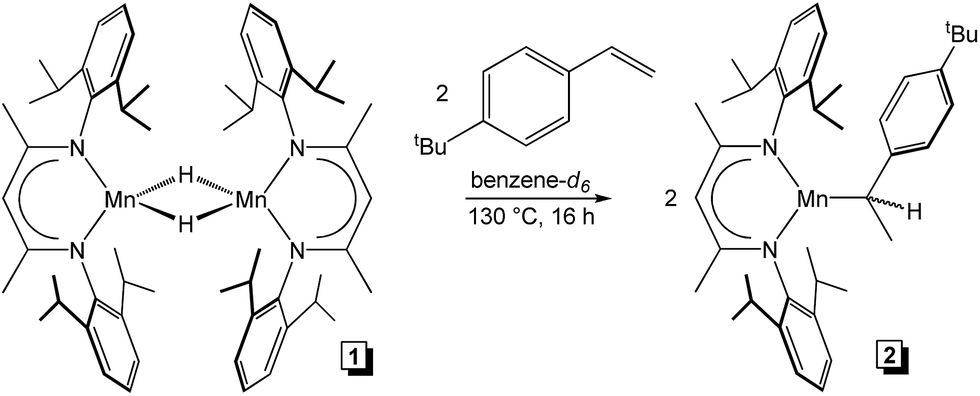
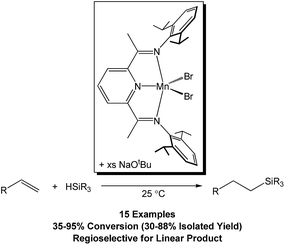
Although a handful of (BDI)Mn compounds have been reported,28 none have been evaluated for catalytic alkene hydrosilylation. Therefore, this study commenced with preparation of the bridging chloride complex [(2,6-iPr2PhBDI)Mn(μ-Cl)]2 (ref. 28b) (Scheme 1, left) upon reacting in situ generated [K][2,6-iPr2PhBDI] with (THF)2MnCl2. Adding one equivalent of NaEt3BH per Mn atom of [(2,6-iPr2PhBDI)Mn(μ-Cl)]2 afforded a yellowish-green complex upon warming from −35 to 25 °C over the course of 1 h, which was identified as [(2,6-iPr2PhBDI)Mn(μ-H)]2 (Scheme 1, 1). The 1H NMR spectrum of this product was found to exhibit broadened resonances over a 25 ppm range and its magnetic susceptibility was determined to be 5.2 μB at 298 K (Evans method), suggesting four unpaired electrons per dimer.
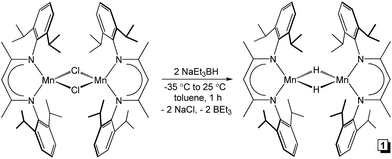 |
| | Scheme 1 Synthesis of [(2,6-iPr2PhBDI)Mn(μ-H)]2 (1). | |
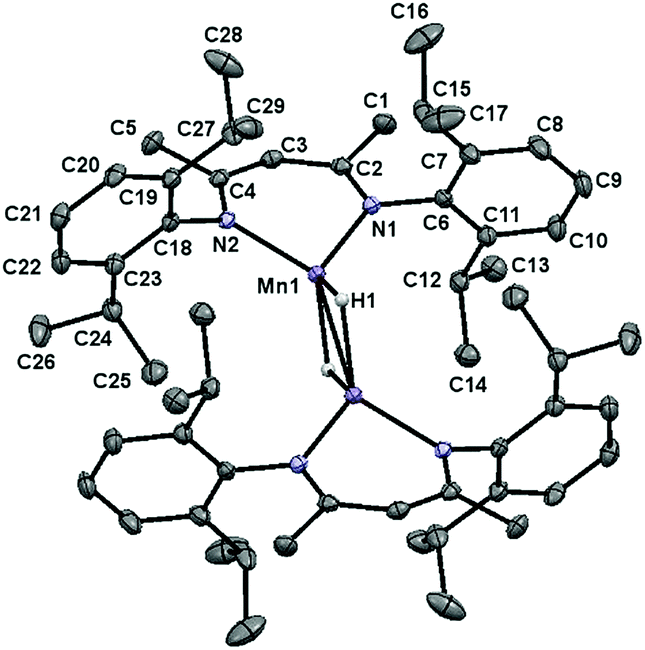
Single crystals of 1 suitable for X-ray diffraction were obtained by cooling a concentrated toluene solution layered with pentane to −35 °C. The solid-state structure of this compound (Fig. 2) was found to possess a near tetrahedral environment around each Mn centre, comprised of a κ2-BDI moiety and two bridging hydride ligands. Notably, the Mn–Mn distance of 2.8138(7) Å is longer than the distance of 2.7464(7) Å reported by Driess and co-workers for the ethyl-substituted variant, [(2,6-Et2PhBDI)Mn(μ-H)]2.28f This difference is likely due to greater steric repulsion between the isopropyl groups of 1; however, the Mn centres remain well within bonding distance (the covalent radius of high spin Mn(II) is approximately 1.61 Å).29 A complete list of metrical parameters is provided in Table S2.†
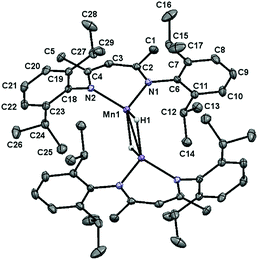 |
| | Fig. 2 The solid-state structure of 1 shown with 30% probability ellipsoids. | |
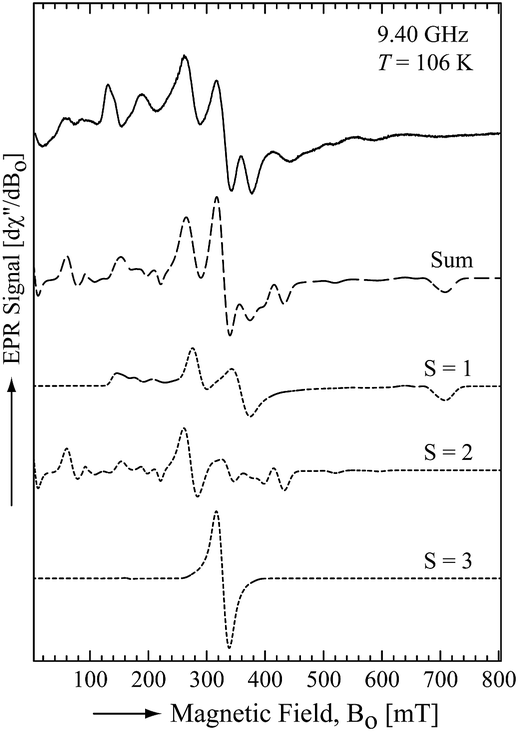
To obtain further electronic information, an X-band (9.40 GHz) electron paramagnetic resonance (EPR) spectrum of 1 was recorded in a toluene glass at 106 K. The EPR spectrum showed a broad and anisotropic signal extending over 800 mT (Fig. 3). Analogous spectra have previously been reported for dinuclear Mn(II) systems in both small molecules30 and proteins.31 The observed spectral features correspond to complexes in which the two Mn(II) centres are strongly antiferromagnetically coupled (i.e. |Jo| ≫ giβeBo). The EPR spectrum was simulated using a spin Hamiltonian that included the Zeeman and zero-field splitting (ZFS) interactions of the individual Mn(II) (Si = 5/2) sites within the dimer and the dipole–dipole interaction between the two Mn(II) sites of the dimer. The best fit was obtained considering the EPR transitions corresponding to the total spin manifolds S = 1, 2 and 3 (Fig. 3). The parameters obtained from the fit are summarized in Table 1. As expected for high-spin Mn(II) centres, an isotropic g-value (giso = 2.05) and small ZFS parameters (i.e. |D| < 0.1 cm−1) were obtained. The principal components of the dipole–dipole interaction tensor are relatively large and significantly deviated from axial symmetry due to the close proximity of the two Mn(II) centres.
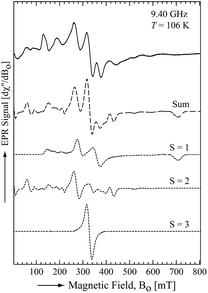 |
| | Fig. 3 The X-band EPR spectrum of 1 at 106 K. The solid line is the experimentally determined spectrum and the dashed line is the sum of the simulated spectra for different dimer spin states (dotted lines). | |
Table 1 Parameters used to fit the EPR spectrum of 1 in a toluene glass at 9.40 GHz and T = 106 K
| Parametera |
1
|
|
The fitting parameters were the isotropic g-value, giso, the zero-field splitting parameters, D and E, the principal components of the dipole–dipole interaction tensor J, (i.e. Jx′, Jy′, and Jz′), and isotropic line width, ΔB.
J
o is not a fitting parameter of a single spectrum but it can be determined by fitting the temperature dependence of the signal intensities corresponding to S = 1, S = 2, and S = 3.
|
|
g
iso
|
2.05 |
| |D| (cm−1) |
0.0932 |
| |E| (cm−1) |
0.0129 |
|
J
o (cm−1) |
n.d.b |
|
J
x′ (cm−1) |
−0.0089 |
|
J
y′ (cm−1) |
−0.0036 |
|
J
z′ (cm−1) |
0.0125 |
| ΔB (MHz) |
600 |
Alkene hydrosilylation
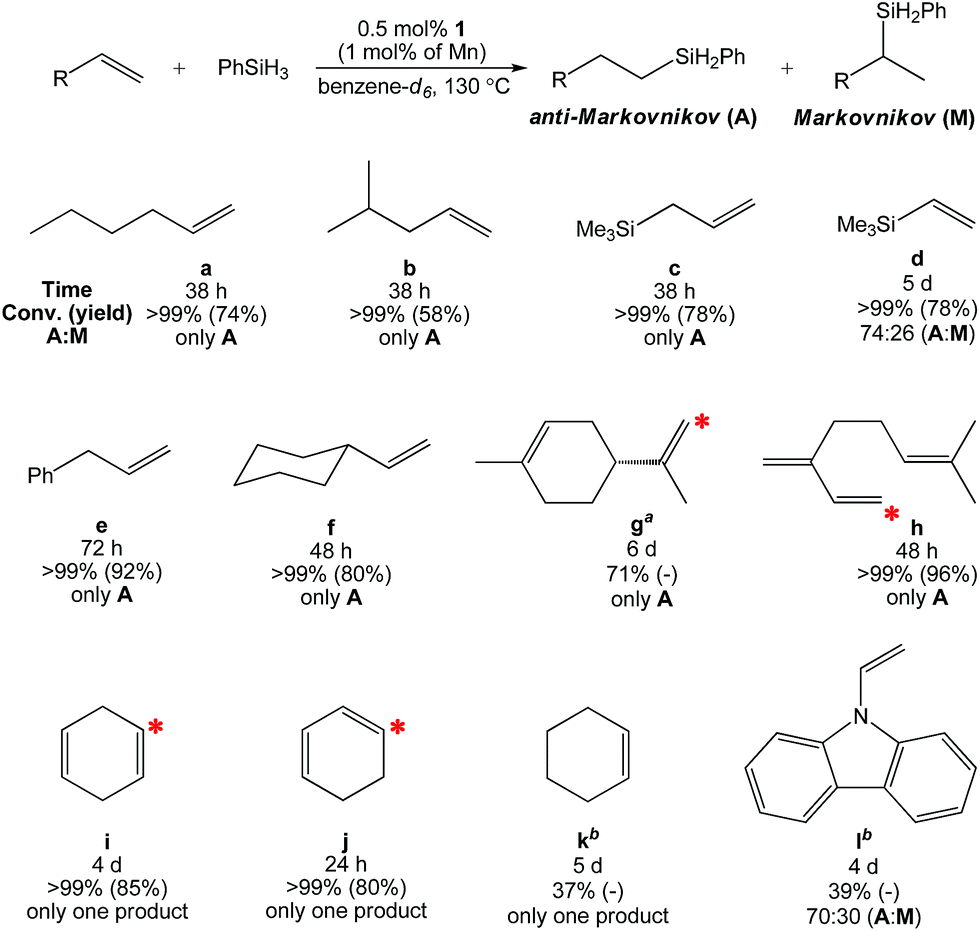
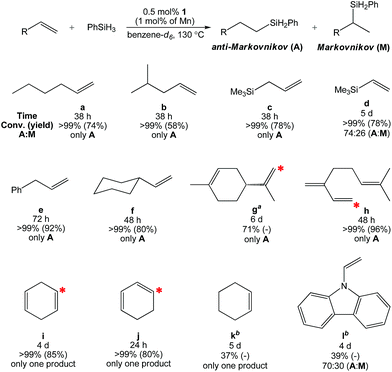
Having characterized 1, its ability to catalyze olefin hydrosilylation was evaluated. Although this catalyst was ineffective at lower temperatures, equimolar addition of 1-hexene (Table 2, a) and PhSiH3 to 0.5 mol% of 1 (1.0 mol% based on Mn) in benzene-d6 allowed for complete hydrosilylation to yield the anti-Markovnikov (A) product, n-C6H13SiH2Ph, after 38 h at 130 °C. Under these conditions, the use of Ph2SiH2, Ph3SiH, and (EtO)3SiH did not allow for conversion. In the presence of Et2SiH2, 1 was found to achieve 61% 1-hexene hydrosilylation after 48 h; however, longer reaction times did not allow for completion. Therefore, PhSiH3 was chosen as the Si–H source for further study.
Table 2 Hydrosilylation of alkenes with anti-Markovnikov selectivity using 0.5 mol% of 1 (1.0 mol% based on Mn)
| All trials were carried out under N2 atmosphere in a J. Young tube. The red star (*) shows the position of silane incorporation. The cyclic olefin moiety was untouched over the course of catalysis. The product was not isolated. |
|
Additional alkenes that lack aromatic substitution were screened for hydrosilylation under identical conditions (Table 2). The hydrosilylation of 4-methyl-1-pentene (b) and allyltrimethylsilane (c) required 38 h to reach completion, furnishing only the anti-Markovnikov product (A). The olefin functionalities of d-f required longer reaction times, and in the case of vinyltrimethylsilane (d) an A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M ratio of 74:26 was obtained. The gem-olefin of D-limonene (g) was 71% hydrosilylated after 6 d, while the cyclic olefin was left untouched. Likewise, the terminal olefin of myrcene (h) was selectively hydrosilylated over the gem and trisubstituted olefins after 48 h at 130 °C. Interestingly, one olefin of 1,4-cyclohexadiene (i) was hydrosilylated in 4 d to selectively yield cyclohex-3-en-1-yl(phenyl)silane. The conjugated regioisomer, 1,3-cyclohexadiene (j), furnished the same product after 24 h, suggesting that conjugation enhances the rate of hydrosilylation. We subsequently performed the hydrosilylation of i using PhSiD3 and observed methylene 2H NMR resonances at 1.62 and 1.82 ppm, indicating that isomerization occurs under catalytic conditions (Fig. S42†). Cyclohexene (k) afforded only 37% conversion to phenylcyclohexylsilane after 5 d along with products from dehydrogenative benzene-d6 silylation (see Fig. S43†). This substrate is less rigid than both 1,4-cyclohexadiene and 1,3-cyclohexadiene indicating that olefin coordination to Mn influences the rate of conversion. It is believed that entries i–k represent the first known examples of Mn-catalyzed cyclic olefin hydrosilylation. For each fully converted substrate, the alkylsilane products were isolated in good yield and purity following filtration and drying.
:M ratio of 74:26 was obtained. The gem-olefin of D-limonene (g) was 71% hydrosilylated after 6 d, while the cyclic olefin was left untouched. Likewise, the terminal olefin of myrcene (h) was selectively hydrosilylated over the gem and trisubstituted olefins after 48 h at 130 °C. Interestingly, one olefin of 1,4-cyclohexadiene (i) was hydrosilylated in 4 d to selectively yield cyclohex-3-en-1-yl(phenyl)silane. The conjugated regioisomer, 1,3-cyclohexadiene (j), furnished the same product after 24 h, suggesting that conjugation enhances the rate of hydrosilylation. We subsequently performed the hydrosilylation of i using PhSiD3 and observed methylene 2H NMR resonances at 1.62 and 1.82 ppm, indicating that isomerization occurs under catalytic conditions (Fig. S42†). Cyclohexene (k) afforded only 37% conversion to phenylcyclohexylsilane after 5 d along with products from dehydrogenative benzene-d6 silylation (see Fig. S43†). This substrate is less rigid than both 1,4-cyclohexadiene and 1,3-cyclohexadiene indicating that olefin coordination to Mn influences the rate of conversion. It is believed that entries i–k represent the first known examples of Mn-catalyzed cyclic olefin hydrosilylation. For each fully converted substrate, the alkylsilane products were isolated in good yield and purity following filtration and drying.
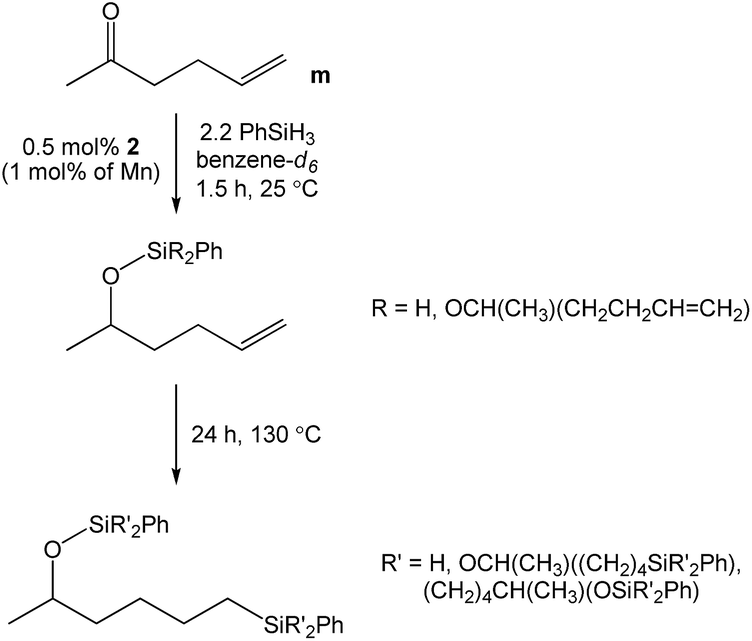
Given our prior efforts to evaluate Mn catalysts for carbonyl hydrosilylation,23–27 we sought to determine the chemoselectivity of 1-mediated 5-hexen-2-one (m) reduction. Immediately upon adding 2.2 equivalents of PhSiH3 to a benzene-d6 solution of this substrate and 0.5 mol% 1 (1.0 mol% based on Mn), an exothermic reaction ensued and complete carbonyl hydrosilylation was confirmed by 1H NMR spectroscopy after 1.5 h (Scheme 2). Heating to 130 °C allowed for greater than 99% alkene hydrosilylation after 24 h to yield a mixture of products (Fig. S45†). These results suggest that 1 is a highly active catalyst for carbonyl hydrosilylation and that it can selectively reduce carbonyl functionalities over olefins at ambient temperature.
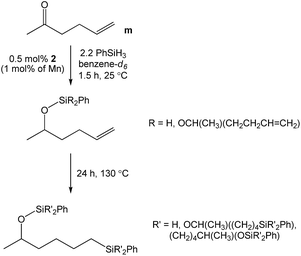 |
| | Scheme 2 The stepwise hydrosilylation of 5-hexen-2-one catalysed by 1. | |
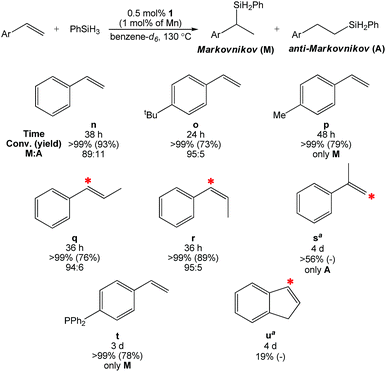
When styrenes were employed as substrates, 1 generated the Markovnikov hydrosilylation product in most cases (Table 3). When styrene (n) was heated to 130 °C with one equivalent of PhSiH3 and 0.5 mol% of 1 (1.0 mol% based on Mn) in benzene-d6, greater than 99% conversion was confirmed after 38 h by 1H NMR spectroscopy. Evaporation of the solvent and filtration with pentane allowed for catalyst removal, and drying the filtrate under vacuum yielded a pale yellowish oil containing 89% M and 11% A silane products. Six additional styrenes were screened under analogous conditions (o–t). Interestingly, trans-β-methylstyrene (q) and cis-β-methylstyrene (r) were successfully hydrosilylated to form the α-silyl product with good selectivity. In contrast, the 1-mediated hydrosilylation of α-methylstyrene (s) resulted in only 56% conversion after 4 d to form the β-silyl product. This trial suggests that gem-olefin substitution provides sufficient steric bulk at the α-carbon position to disfavor Markovnikov hydrosilylation (vide infra). Although substrates g and s were not fully hydrosilylated, they represent the second and third known examples of Mn-catalyzed gem-olefin hydrosilylation.19 Notably, 4-(diphenylphosphino)styrene (t) was completely hydrosilylated in 3 d, indicating that phosphine functionalities are tolerated by 1. It should be noted that 4-bromostyrene did not show any conversion after 4 d, while 4-fluorostyrene was completely consumed to yield a complex mixture of M, A, and silylated arene products following defluorination (multiple resonances were observed in the 19F spectrum, Fig. S76†).
Table 3 Hydrosilylation of styrenes with Markovnikov selectivity using 0.5 mol% of 1 (1.0 mol% based on Mn)
| All trials were carried out under N2 atmosphere in a J. Young tube. The red star (*) shows the position of silane incorporation. No further conversion was noted after prolonged heating and the products were not isolated. |
|
Silicone preparation
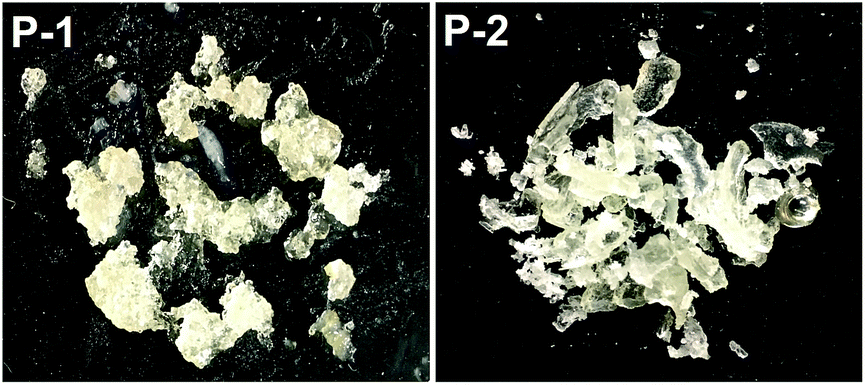
Although 1 requires higher temperatures and is less active than (2,6-iPr2PhPDI)MnBr2,22 attempts were made to utilize our catalyst to prepare silicones that are traditionally synthesized via Pt-mediated hydrosilylation. Given that the hydrosilylation of vinyl-terminated poly(dimethylsiloxane)s is conducted industrially to prepare a variety of silicone coatings and materials,13e 10 mol% of 1 relative to this substrate (5 mol% relative to olefin) was added in the presence of excess polymethylhydrosiloxane (PMHS). Heating this neat mixture at 130 °C for 5 d afforded a colourless, gummy silicone solid (Scheme 3, Fig. 4, P-1) after catalyst deactivation and washing with isopropanol.
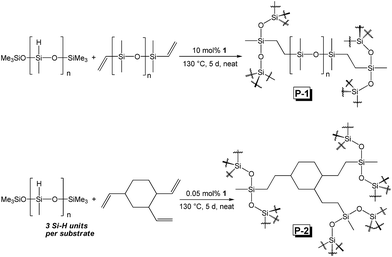 |
| | Scheme 3 The preparation of silicones following 1-catalyzed hydrosilylation. | |
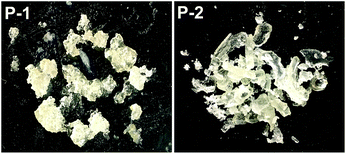 |
| | Fig. 4 Silicones prepared following the 1-catalyzed hydrosilylation of vinyl-terminated poly(dimethylsiloxane) (P-1) and 1,2,4-trivinylcyclohexane (P-2) using PMHS. | |
A polymeric product derived from 1,2,4-trivinylcyclohexane (mixture of isomers) was also targeted, since 1 has been shown to hydrosilylate vinylcyclohexane (Table 2, f) in anti-Markovnikov fashion. Notably, silicones prepared via the hydrosilylation of this substrate are used in oxygen permeable contact lenses32 and transparent LED screen compositions.33 Heating a neat mixture of PMHS and 1,2,4-trivinylcyclohexane (1:1 ratio of Si–H to olefin) in the presence of 0.05 mol% 1 (relative to substrate, 0.0167 mol% relative to total alkene count), afforded a colourless and transparent silicone after heating for 5 d in the absence of solvent (Scheme 3, Fig. 4, P-2). It should be noted that Du and co-workers have recently described the synthesis of silicones by way of Mn-mediated carbonyl hydrosilylation and dehydrogenative alcohol silylation;34 however, P-1 and P-2 are believed to be the first silicones featuring non-hydrolysable Si–C bonds prepared via Mn catalysis.
Mechanistic considerations
To rationalize the substrate-dependent regioselectivity observed for 1-catalysed alkene hydrosilylation, further mechanistic understanding was sought. First, 1 was heated with two equivalents of PhSiH3 (one equivalent per Mn) at 130 °C for 24 h. The reaction mixture was unchanged as judged by 1H NMR and 29Si NMR spectroscopy and 1 was recovered quantitatively following solvent and silane removal. This lack of reactivity suggests that σ-bond metathesis between the Si–H bond of PhSiH3 and the Mn–H bond of 1 does not occur during catalysis. However, heating 1 in the presence of two equivalents of 4-tert-butylstyrene at 130 °C for 16 h yielded a monomeric compound identified as (2,6-iPr2PhBDI)Mn(CH(CH3)(4-tBuPh)) (Scheme 4, 2). This compound was found to exhibit an ambient temperature magnetic moment of 6.0 μB, which is consistent with a high-spin Mn(II) electronic structure.
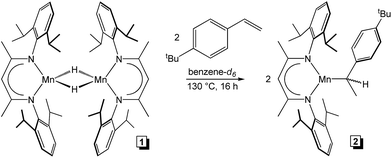 |
| | Scheme 4 Addition of 4-tert-butylstyrene to 1 to generate 2. | |
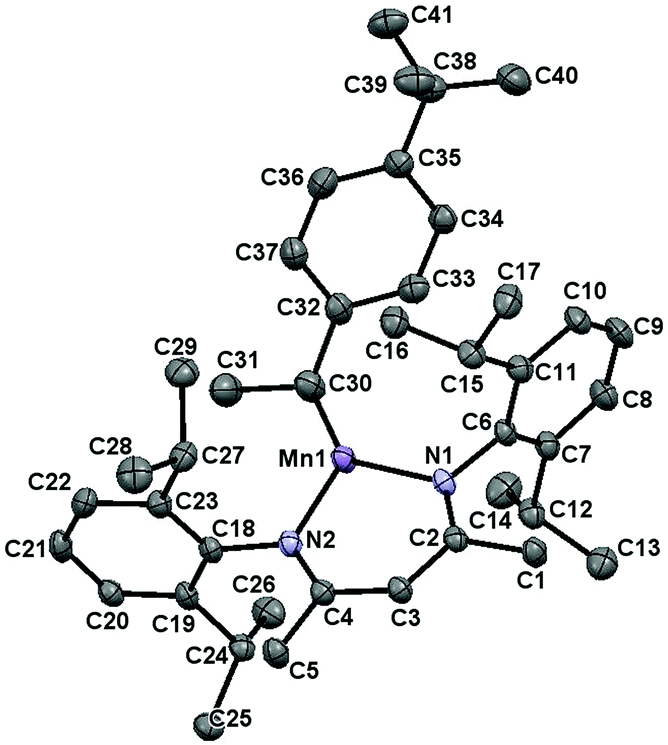
Complex 2 was found to exhibit a new set of paramagnetically broadened 1H NMR resonances over a 25 ppm range and cooling a concentrated Et2O solution at −35 °C afforded yellow crystals suitable for X-ray diffraction. Importantly, the solid-state structure of 2 possesses a secondary alkyl ligand (Fig. 5, for metrical parameters, see Table S3†). This observation suggests that 1-mediated alkene hydrosilylation proceeds via olefin coordination (with concurrent dimer dissociation) and subsequent insertion into the Mn–H bond. Moreover, the fact that 4-tert-butylstyrene insertion generates a secondary alkyl ligand rationalizes the Markovnikov selectivity observed for the styrene substrates in Table 3.
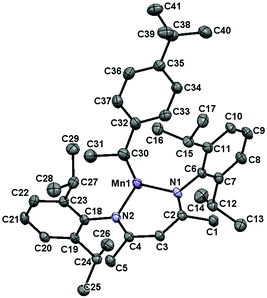 |
| | Fig. 5 The solid-state structure of 2 at 30% probability ellipsoids. | |
To confirm that 2 is a catalytic intermediate and not a deactivation product, the reactivity of this compound was evaluated. Under the conditions of catalysis, the addition of 1.5 equiv. of PhSiH3 to 2 afforded 1 and the respective Markovnikov silane product (Scheme 5). Moreover, compound 2 was evaluated for α-olefin and styrene hydrosilylation activity. Adding an equimolar quantity of 1-hexene and PhSiH3 to 1.0 mol% of 2 allowed for complete substrate hydrosilylation after 22 h at 130 °C. Importantly, only A product was observed, which is consistent with the selectivity reported in Table 2. Likewise, 2-mediated styrene hydrosilylation afforded a 91:9 ratio of M and A products after only 16 h at 130 °C. Taken together, these experiments indicate that alkyl intermediates such as 2 re-enter the catalytic cycle following reaction with PhSiH3 to regenerate 1.
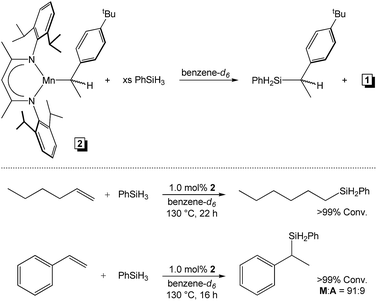 |
| | Scheme 5 Addition of PhSiH3 to 2 results in alkylsilane product formation and regeneration of 1 (top) and 2 catalyses 1-hexene and styrene hydrosilylation more efficiently than 1 (bottom). | |
With these considerations in mind, we propose concurrent mechanistic pathways that differ from the commonly evoked Chalk-Harrod hydrosilylation mechanism (which involves Si–H oxidative addition).35 Resonances associated with monomeric 1 have not been observed by 1H NMR spectroscopy at temperatures of up to 130 °C in the absence of substrate (Fig. S90†). Therefore, for the aliphatic alkenes in Table 1, alkene addition to 1 allows for dimer dissociation and insertion into the Mn–H bond to generate a primary alkyl intermediate (top right of Fig. 6). In the presence of PhSiH3, σ-bond metathesis occurs between the Si–H and Mn–C bonds to release the linear silane product and regenerate 1. A similar hydrosilylation cycle was reported by Chirik and co-workers for [(2,6-iPr2PhDI)NiH]2;13e however, it should be noted that Mn is believed to remain divalent throughout catalysis. In contrast, the styrene substrates in Table 3 (with the exception of α-methylstyrene) are electronically predisposed to insert at the benzylic position to generate a secondary alkyl intermediate.36 Due to steric constraints, α-methylstyrene (s) cannot insert to form a tertiary alkyl intermediate, and it is therefore hydrosilylated via the anti-Markovnikov pathway. The BDI ligand steric environment is less demanding than that of (2,6-iPr2PhPDI)MnBr2,22 which may explain why the in situ generated Mn–H proposed by Thomas and co-workers selectively yields linear hydrosilylation products.
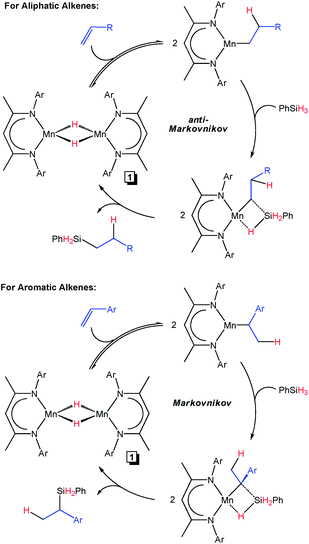 |
| | Fig. 6 Proposed mechanisms for 1-catalyzed alkene hydrosilylation. | |
Conclusions
Upon preparing and characterizing the well-defined β-diketiminate manganese hydride compound, [(2,6-iPr2PhBDI)Mn(μ-H)]2 (1), its ability to catalyse olefin hydrosilylation was explored. Low loadings of this catalyst (0.5 mol%) have been shown to hydrosilylate 20 different olefinic substrates at 130 °C, including some that are disubstituted. These hydrosilylation trials revealed substrate-specific regioselectivity; 1 was found to catalyse aliphatic alkene hydrosilylation in anti-Markovnikov fashion while styrenes underwent Markovnikov hydrosilylation. Two commercially relevant silicone formation reactions have also been described, both of which afforded colourless polymer following an isopropanol wash. Stoichiometric addition experiments revealed that 4-tert-butylstyrene reacts with 1 to yield a secondary alkyl product, (2,6-iPr2PhBDI)Mn(CH(CH3)(4-tBuPh)) (2), indicating that the regioselectivity of hydrosilylation is dictated by alkene insertion. This result is consistent with recent experiments by Thomas and co-workers22 that suggest alkene insertion into a Mn–H intermediate is responsible for (2,6-iPr2PhPDI)MnBr2-mediated olefin hydrosilylation. Taken together, these studies encourage the future utilization of inexpensive and non-toxic manganese catalysts for alkene hydrosilylation and silicone curing transformations.
Conflicts of interest
The authors declare the following competing financial interests: T. K. M. and R. J. T. retain rights to 1 through US Patent Application No. 62/678,624.
Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. 1651686.
Notes and references
-
B. Marciniec, Comprehensive Handbook on Hydrosilylation, Pergamon Press, New York, USA, 1st edn, 1992 Search PubMed.
- D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef.
- Y. Zhang, Adv. J. Food Sci. Technol., 2016, 12, 145–149 CrossRef.
- S. M. Bahal and J. M. Romansky, Pharm. Dev. Technol., 2002, 7, 317–323 CrossRef PubMed.
-
W. M. Haynes, CRC Handbook of Chemistry and Physics: A Ready-reference Book of Chemical and Physical Data, Taylor & Francis, Boca Raton, FL, 94th edn, 2013–2014 Search PubMed.
- The 10 year futures price for platinum (COMEX) has ranged between $800 and $1,900 USD per troy ounce. Accessed at: https://www.nasdaq.com/markets/platinum.aspx?timeframe=10y.
- A. J. Holwell, Platinum Met. Rev., 2008, 52, 243–246 CrossRef.
- E. D. Lykissa and S. V. M. Maharaj, Anal. Chem., 2006, 78, 2925–2933 CrossRef PubMed.
- X. Du and Z. Huang, ACS Catal., 2017, 7, 1227–1243 CrossRef.
- J. V. Obligacion and P. J. Chirik, Nat. Rev. Chem., 2018, 2, 15–34 CrossRef.
-
(a) G. Bellachioma, G. Cardaci, E. Colomer, R. J. P. Corriu and A. Vioux, Inorg. Chem., 1989, 28, 519–525 CrossRef;
(b) B. Marciniec and M. Majchrzak, Inorg. Chem. Commun., 2000, 3, 371–375 CrossRef;
(c) S. C. Bart, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2004, 126, 13794–13807 CrossRef PubMed;
(d) A. M. Archer, M. W. Bouwkamp, M.-P. Cortez, E. Lobkovsky and P. J. Chirik, Organometallics, 2006, 25, 4269–4278 CrossRef;
(e) A. M. Tondreau, C. C. H. Atienza, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis and P. J. Chirik, Science, 2012, 335, 567–570 CrossRef PubMed;
(f) K. Kamata, A. Suzuki, Y. Nakai and H. Nakazawa, Organometallics, 2012, 31, 3825–3828 CrossRef;
(g) A. M. Tondreau, C. C. H. Atienza, J. M. Darmon, C. Milsmann, H. M. Hoyt, K. J. Weller, S. A. Nye, K. M. Lewis, J. Boyer, J. G. P. Delis, E. Lobkovsky and P. J. Chirik, Organometallics, 2012, 31, 4886–4893 CrossRef;
(h) D. Peng, Y. Zhang, X. Du, L. Zhang, X. Leng, M. D. Walter and Z. Huang, J. Am. Chem. Soc., 2013, 135, 19154–19166 CrossRef PubMed;
(i) M. D. Greenhalgh, D. J. Frank and S. P. Thomas, Adv. Synth. Catal., 2014, 356, 584–590 CrossRef;
(j) J. Chen, B. Cheng, M. Cao and Z. Lu, Angew. Chem., Int. Ed., 2015, 54, 4661–4664 CrossRef PubMed;
(k) R. Gilbert-Wilson, W.-Y. Chu and T. B. Rauchfuss, Inorg. Chem., 2015, 54, 5596–5603 CrossRef PubMed;
(l) B. Marciniec, A. Kownacka, I. Kownacki, M. Hoffmann and R. Taylor, J. Organomet. Chem., 2015, 791, 58–65 CrossRef;
(m) Y. Sunada, D. Noda, H. Soejima, H. Tsutsumi and H. Nagashima, Organometallics, 2015, 34, 2896–2906 CrossRef;
(n) G. I. Nikonov, ChemCatChem, 2015, 7, 1918–1919 CrossRef;
(o) X. Jia and Z. Huang, Nat. Chem., 2016, 8, 157–161 CrossRef PubMed;
(p) D. Noda, A. Tahara, Y. Sunada and H. Nagashima, J. Am. Chem. Soc., 2016, 138, 2480–2483 CrossRef PubMed;
(q) X. Du, Y. Zhang, D. Peng and Z. Huang, Angew. Chem., Int. Ed., 2016, 55, 6671–6675 CrossRef PubMed;
(r) A. J. Challinor, M. Calin, G. S. Nichol, N. B. Carter and S. P. Thomas, Adv. Synth. Catal., 2016, 358, 2404–2409 CrossRef;
(s) K. Hayasaka, K. Kamata and H. Nakazawa, Nat. Chem., 2016, 89, 394–404 Search PubMed;
(t) Y. Toya, K. Hayasaka and H. Nakazawa, Organometallics, 2017, 36, 1727–1735 CrossRef;
(u) M.-Y. Hu, Q. He, S.-J. Fan, Z.-C. Wang, L.-Y. Liu, Y.-J. Mu, Q. Peng and S.-F. Zhu, Nat. Commun., 2018, 9, 221 CrossRef PubMed;
(v) B. Cheng, W. Liu and Z. Lu, J. Am. Chem. Soc., 2018, 140, 5014–5017 CrossRef PubMed.
-
(a) A. J. Chalk, J. Organomet. Chem., 1970, 21, 207–213 CrossRef;
(b) G. K. I. Magomedov, K. A. Andrianov, O. V. Shkolnik, B. A. Izmailov and V. N. Kalinin, J. Organomet. Chem., 1978, 149, 29–36 CrossRef;
(c) Y. Seki, K. Kawamoto, N. Chatani, A. Hidaka, N. Sonoda, K. Ohe, Y. Kawasaki and S. Murai, J. Organomet. Chem., 1991, 403, 73–84 CrossRef;
(d) M. Brookhart and B. E. Grant, J. Am. Chem. Soc., 1993, 115, 2151–2156 CrossRef;
(e) N. Chatani, T. Kodama, Y. Kajikawa, H. Murakami, F. Kakiuchi, S.-I. Ikeda and S. Murai, Chem. Lett., 2000, 29, 14–15 CrossRef;
(f) C. C. H. Atienza, T. Diao, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis, J. L. Boyer, A. K. Roy and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 12108–12118 CrossRef PubMed;
(g) C. Chen, M. B. Hecht, A. Kavara, W. W. Brennessel, B. Q. Mercado, D. J. Weix and P. L. Holland, J. Am. Chem. Soc., 2015, 137, 13244–13247 CrossRef PubMed;
(h) J. Sun and L. Deng, ACS Catal., 2016, 6, 290–300 CrossRef;
(i) A. Gorczyński, M. Zaranek, S. Witomska, A. Bocian, A. R. Stefankiewicz, M. Kubicki, V. Patroniak and P. Pawluć, Catal. Commun., 2016, 78, 71–74 CrossRef;
(j) C. H. Schuster, T. Diao, I. Pappas and P. J. Chirik, ACS Catal., 2016, 6, 2632–2636 CrossRef;
(k) A. D. Ibrahim, S. W. Entsminger, L. Zhu and A. R. Fout, ACS Catal., 2016, 6, 3589–3593 CrossRef;
(l) B. Raya, S. Biswas and T. V. RajanBabu, ACS Catal., 2016, 6, 6318–6323 CrossRef PubMed;
(m) W.-Y. Chu, R. Gilbert-Wilson and T. B. Rauchfuss, Organometallics, 2016, 35, 2900–2914 CrossRef;
(n) C. Wang, W. J. Teo and S. Ge, ACS Catal., 2017, 7, 855–863 CrossRef;
(o) B. Raya, S. Jing, V. Balasanthiran and T. V. RajanBabu, ACS Catal., 2017, 7, 2275–2283 CrossRef PubMed;
(p) Y. Liu and L. Deng, J. Am. Chem. Soc., 2017, 139, 1798–1801 CrossRef PubMed;
(q) C. Wang, W. J. Teo and S. Ge, Nat. Commun., 2017, 8, 2258 CrossRef PubMed;
(r) B. Cheng, P. Lu, H. Zhang, X. Cheng and Z. Lu, J. Am. Chem. Soc., 2017, 139, 9439–9442 CrossRef PubMed.
-
(a) A. Kuznetsov and V. Gevorgyan, Org. Lett., 2012, 14, 914–917 CrossRef PubMed;
(b) M. I. Lipschutz and T. D. Tilley, Chem. Commun., 2012, 48, 7146–7148 RSC;
(c) V. Srinivas, Y. Nakajima, W. Ando, K. Satoa and S. Shimada, Catal. Sci. Technol., 2015, 5, 2081–2084 RSC;
(d) V. Srinivas, Y. Nakajima, W. Ando, K. Sato and S. Shimada, J. Organomet. Chem., 2016, 809, 57–62 CrossRef;
(e) I. Pappas, S. Treacy and P. J. Chirik, ACS Catal., 2016, 6, 4105–4109 CrossRef.
-
(a) R. J. Trovitch, Synlett, 2014, 25, 1638–1642 CrossRef;
(b) X. Yang and C. Wang, Chem.–Asian J., 2018 DOI:10.1002/asia.201800618.
- S. L. Pratt and R. A. Faltynek, J. Organomet. Chem., 1983, 258, C5–C8 CrossRef.
- H. S. Hilal, M. Abu-Eid, M. Al-Subu and S. Khalaf, J. Mol. Catal., 1987, 39, 1–11 CrossRef.
- H. S. Hilal, M. A. Suleiman, W. J. Jondi, S. Khalaf and M. M. Masoud, J. Mol. Catal., 1999, 144, 47–59 CrossRef.
- W. Jondi, A. Zyoud, W. Mansour, A. Q. Husseinb and H. S. Hilal, React. Chem. Eng., 2016, 1, 194–203 RSC.
- C. Obradors, R. M. Martinez and R. A. Shenvi, J. Am. Chem. Soc., 2016, 138, 4962–4971 CrossRef PubMed.
- J. S. Price, D. J. H. Emslie and J. F. Britten, Angew. Chem., Int. Ed., 2017, 56, 6223–6227 CrossRef PubMed.
- J. H. Docherty, J. Peng, A. P. Dominey and S. P. Thomas, Nat. Chem., 2017, 9, 595–600 CrossRef PubMed.
- J. R. Carney, B. R. Dillon, L. Campbell and S. P. Thomas, Angew. Chem., Int. Ed., 2018, 57, 10620–10624 CrossRef PubMed.
- T. K. Mukhopadhyay, M. Flores, T. L. Groy and R. J. Trovitch, J. Am. Chem. Soc., 2014, 136, 882–885 CrossRef PubMed.
- C. Ghosh, T. K. Mukhopadhyay, M. Flores, T. L. Groy and R. J. Trovitch, Inorg. Chem., 2015, 54, 10398 CrossRef PubMed.
- T. K. Mukhopadhyay, C. Ghosh, M. Flores, T. L. Groy and R. J. Trovitch, Organometallics, 2017, 36, 3477–3483 CrossRef.
- T. K. Mukhopadhyay, C. L. Rock, M. Hong, D. C. Ashley, T. L. Groy, M.-H. Baik and R. J. Trovitch, J. Am. Chem. Soc., 2017, 139, 4901–4915 CrossRef PubMed.
- R. J. Trovitch, Acc. Chem. Res., 2017, 50, 2842–2852 CrossRef PubMed.
-
(a) J. Chai, H. Zhu, K. Most, H. W. Roesky, D. Vidovic, H.-G. Schmidt and M. Noltemeyer, Eur. J. Inorg. Chem., 2003, 4332–4337 CrossRef;
(b) J. Chai, H. Zhu, H. W. Roesky, C. He, H.-G. Schmidt and M. Noltemeyer, Organometallics, 2004, 23, 3284–3289 CrossRef;
(c) J. Chai, H. Zhu, H. Fan, H. W. Roesky and J. Magull, Organometallics, 2004, 23, 1177–1179 CrossRef;
(d) J. Chai, H. Zhu, A. C. Stückl, H. W. Roesky, J. Magull, A. Bencini, A. Caneschi and D. Gatteschi, J. Am. Chem. Soc., 2005, 127, 9201–9206 CrossRef PubMed;
(e) H. W. Roesky, M. S. Varonka and T. H. Warren, Inorg. Synth., 2010, 35, 34–38 Search PubMed;
(f) S. Yao, Y. Xiong and M. Driess, Chem.–Eur. J., 2012, 18, 11356–11361 CrossRef PubMed;
(g) V. Yempally, W. Y. Fan, B. A. Arndtsen and A. A. Bengali, Inorg. Chem., 2015, 54, 11441–11449 CrossRef PubMed;
(h) R. L. Webster, Dalton Trans., 2017, 46, 4483–4498 RSC;
(i) M. M. Stalzer, T. L. Lohr and T. J. Marks, Inorg. Chem., 2018, 57, 3017–3024 CrossRef PubMed.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- A. P. Golombek and M. P. Hendrich, J. Magn. Reson., 2003, 165, 33–48 CrossRef PubMed.
- B. Epel, K. O. Schäfer, A. Quentmeier, C. Friedrich and W. Lubitz, J. Biol. Inorg Chem., 2005, 10, 636–642 CrossRef PubMed.
- G. Friedmann, P. Sperry and J. Brossas, Eur. Polym. J., 1993, 39, 1197–1203 CrossRef.
-
K. Ouchi, M. Tsumura, H. Sakamoto, M. Fujita, M. Kuramoto and T. Miki, Jpn. Kokai Tokkyo Koho, JP2002341101A, 2002.
-
(a) S. Vijjamarri, V. K. Chidara and G. Du, ACS Omega, 2017, 2, 582–591 CrossRef;
(b) S. Vijjamarri, S. Streed, E. M. Serum, M. P. Sibi and G. Du, ACS Sustainable Chem. Eng., 2018, 6, 2491–2494 CrossRef.
- A. J. Chalk and J. F. Harrod, J. Am. Chem. Soc., 1965, 87, 16–21 CrossRef.
- Similar regioselectivity has been observed for Mn-catalyzed olefin hydroboration. See: G. Zhang, H. Zeng, J. Wu, Z. Yin, S. Zheng and J. C. Fettinger, Angew. Chem., Int. Ed., 2016, 55, 14369–14372 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Metrical parameters for 1 and 2, catalytic procedures, and NMR spectroscopic identification of products. CCDC 1851177 and 1851178 For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02768d |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Thomas L.
Groy
and
Ryan J.
Trovitch
,
Thomas L.
Groy
and
Ryan J.
Trovitch