
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly effective photoreduction of CO2 to CO promoted by integration of CdS with molecular redox catalysts through metal–organic frameworks†
Chunjun
Chen
ab,
Tianbin
Wu
*a,
Haihong
Wu
 c,
Huizhen
Liu
ab,
Qingli
Qian
a,
Zhimin
Liu
ab,
Guanying
Yang
a and
Buxing
Han
*ab
c,
Huizhen
Liu
ab,
Qingli
Qian
a,
Zhimin
Liu
ab,
Guanying
Yang
a and
Buxing
Han
*ab
aBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid and Interface and Thermodynamics, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China. E-mail: hanbx@iccas.ac.cn; wtb@iccas.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cShanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
First published on 24th September 2018
Abstract
We propose a strategy to synthesize ternary CdS/UiO-bpy/Co composites by integration of CdS and molecular redox catalysts through metal–organic framework (MOF) UiO-bpy. The CdS/UiO-bpy/Co composites were very effective for photocatalytic conversion of CO2 to CO under visible light irradiation. The evolution rate of CO could reach 235 μmol g−1 h−1, which was a 10.2-fold improvement compared to that for the parent CdS, and selectivity for CO was 85%. The outstanding performance of the CdS/UiO-bpy/Co composites resulted from promoting the separation and migration of photo-induced charge carriers, enhancing the adsorption of CO2 molecules, and offering abundant active sites for CO2 reduction. As far as we know, this is the first study to integrate inorganic semiconductors and molecular redox catalysts through MOFs.
Introduction
Global energy demand and climate change have underpinned broad interest in the sustainable reduction of CO2 into value added carbon products, such as CO, alcohols, and hydrocarbons. Photocatalytic reduction of CO2 is regarded as an essential and important technology for mitigation of the greenhouse effect and generation of renewable transportation fuels.1–3 Although great effort has been devoted to exploring various photocatalysts,4–6 the photocatalytic efficiency is far from satisfactory, mainly because of fast recombination of the photogenerated electron–hole pairs, low specific surface area and confined active sites.7–9 Increasing the CO2 adsorption ability of photocatalysts is an effective strategy to improve the CO2 conversion efficiency.10,11 Due to their large surface area and well-ordered porous structure, metal–organic frameworks (MOFs) are considered to be promising candidates for gas adsorption, and can facilitate CO2 adsorption.12,13 Nevertheless, the performance of MOFs in photocatalytic reduction of CO2 is obstructed by the low efficiency of exciton generation and charge separation in MOFs, as well as inconsistencies between catalytic and adsorption sites.14,15Cadmium sulfide (CdS), with an excellent visible-light response and appropriately positioned conduction band, has been investigated extensively as a photocatalyst for photocatalytic reduction of CO2, whose applications have unfortunately been limited due to the fast recombination of photogenerated electron–hole pairs, the serious photocorrosion, the low CO2 adsorption, and the lack of catalytic sites.16–18 It has been reported that the combination of CdS and MOFs can help to surmount their respective weak points by increasing the visible-light response and facilitating CO2 adsorption.19 However, it is difficult for CdS/MOF composites to achieve high photocatalytic performance for CO2 reduction due to the large size of CdS and lack of catalytic sites.20
Some molecular redox catalysts containing metals such as Re, Co, Ni, and Fe are capable of reducing CO2 to CO photochemically.21,22 However, they need to be fed with electrons by a photosensitizer. So far, a number of studies have been proposed to utilize inorganic semiconductors as photosensitizers,23–25 but the interaction between inorganic semiconductors and complexes is very poor, leading to difficulty in cycling experiments. It was reported that bipyridine cobalt complexes can efficiently promote the photoreduction of CO2 to CO when combined with these inorganic semiconductors.26 However, bipyridine cobalt complexes can dissolve in the solution, which is unfavorable for recovery. Although a few studies have been reported on combining MOFs and molecular redox catalysts for photocatalytic reduction of CO2, either precious metals or complex ligands were used.27,28 UiO-bpy MOFs, with exceptional CO2 adsorption capacity and stability,29 have been constructed from linear 2,2′-bipyridine-5,5′-dicarboxylate (bpydc) bridging ligands and Zr6(μ3-O)4(μ3-OH)4 SBUs. Although the bridging ligands of bpydc can coordinate with some transition metal (e.g. Co2+, Cu2+ and Ni2+), the CO2 photoreduction is also obstructed by the low efficiency of visible light exciton generation.
In this work, we designed ternary CdS/UiO-bpy/Co composites. The inorganic semiconductors and the molecular redox catalysts were integrated through UiO-bpy. The CdS/UiO-bpy/Co composites showed outstanding performance for photocatalytic conversion of CO2 to CO under visible light irradiation. To the best of our knowledge, there is no report on the combination of inorganic semiconductors and molecular redox catalysts through MOFs for photoreduction of CO2.
Results and discussion
The method proposed to synthesize the ternary CdS/UiO-bpy/Co composites is shown schematically in Fig. 1. UiO-bpy was firstly prepared from ZrCl4 and bpydc following a reported method.30 Then, the vacuum-activated sample of UiO-bpy was treated with Cd(CH3COO)2·2H2O and dimethylsulfoxide (DMSO) at 180 °C for 12 h to form CdS/UiO-bpy. Because the bpydc bridging ligands can coordinate with Cd2+ in DMSO, and the Cd2+ can slowly react with DMSO to form CdS,31 small CdS nanoparticles were generated. The synthesized CdS/UiO-bpy samples and cobalt chloride were added into tetrahydrofuran (THF) solvent at room temperature and stirred for 12 h. Finally, the CoCl2 was integrated into CdS/UiO-bpy by coordinating with bpydc and the ternary CdS/UiO-bpy/Co composites were obtained in this way.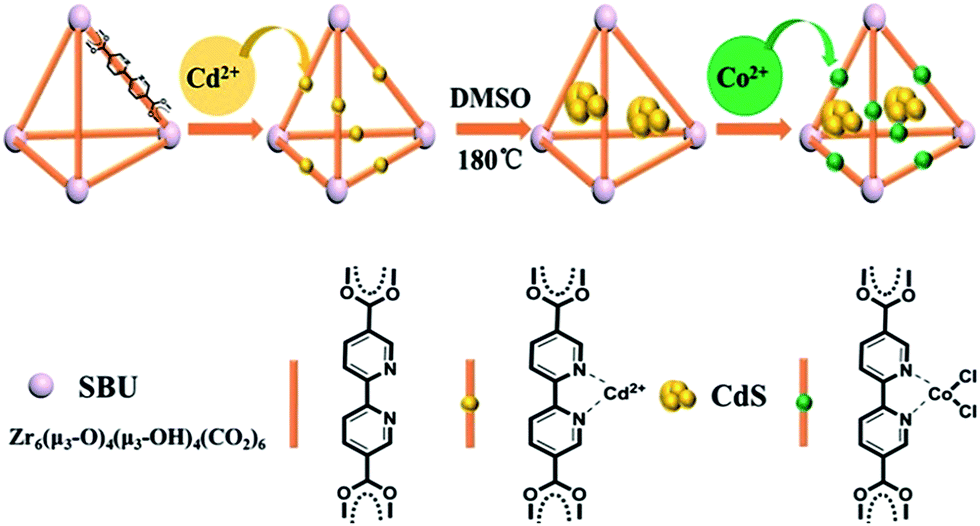 | ||
| Fig. 1 The route for the synthesis of the CdS/UiO-bpy/Co composites. | ||
The obtained CdS, UiO-bpy, CdS/UiO-bpy and CdS/UiO-bpy/Co composites were characterized by X-ray diffraction (XRD), as shown in Fig. S1.† For these samples, a group of strong diffraction peaks before 20° matched with the simulated UiO-bpy signals,32 proving that CdS and Co loading did not change the structure of UiO-bpy. In addition, the three diffraction peaks with 2θ values of 26.5°, 44.0, and 52.1° are related to the (111), (220), and (311) crystal planes of hawleyite CdS, respectively. The contents of CdS and Co2+ were determined by ICP-AES (VISTA-MPX) and the results are shown in Table S1.† The loading amount of Co in the CdS/UiO-bpy/Co composites was determined to be 6.18 wt%, indicating that 50.8% of the bpy units were metalated with Co in the UiO-bpy framework.
The scanning electron microscopy (SEM), transmission electron microscopy (TEM), and high-resolution TEM (HR-TEM) images of the as-synthesized UiO-bpy and CdS/UiO-bpy/Co composites are shown in Fig. 2. We can observe that the UiO-bpy is a regular octahedron, and the morphology of UiO-bpy was retained after the UiO-bpy was loaded with CdS and Co, as shown in Fig. 2A and B. It is found that the CdS nanoparticles with a diameter of about 10 nm were well dispersed on the UiO-68 framework in the CdS/UiO-bpy/Co composites, as shown in Fig. 2C and D. The lattice fringes of individual CdS particles with an interplanar spacing of ≈0.176 nm can be assigned to the (311) lattice plane of hawleyite (Fig. 2C), which is in good agreement with XRD analysis (Fig. S1†). Energy dispersive X-ray (EDX) elemental mapping of the CdS/UiO-bpy/Co composites in Fig. 2E shows that Cd, S, Co and Cl elements were dispersed uniformly in the composites.
 | ||
| Fig. 2 (A) SEM image of UiO-bpy. (B) SEM image of CdS/UiO-bpy/Co. (C) TEM image of CdS/UiO-bpy/Co and the high-resolution TEM image of the selected area. (D) The high-magnification TEM image of CdS/UiO-bpy/Co and the particle size distribution of CdS. (E) EDX mapping of CdS/UiO-bpy/Co. | ||
The CdS, UiO-bpy, CdS/UiO-bpy and CdS/UiO-bpy/Co composites were characterized using the N2 adsorption–desorption method. As shown in Fig. S2,† pure UiO-bpy had a total surface area (SBET) of 1323 m2 g−1, while the SBET value based on the total weight of CdS/UiO-bpy was 52.6 m2 g−1. The decrease in SBET after incorporation of CdS is due to the fact that non-porous CdS nanoparticles blocked some pores of UiO-bpy. The SBET of the CdS/UiO-bpy/Co composites decreased to 25.5 m2 g−1 due to high metal loading and the potential blocking of pore entrances by the sterically demanding CoCl2. Despite the fact that the BET surface area of the CdS/UiO-bpy/Co composites was smaller than that of pure CdS, the CO2 adsorption amount was larger than that of pure CdS, as shown in Fig. S3,† which indicates that the UiO-bpy can facilitate CO2 adsorption.
The size of CdS in the CdS/UiO-bpy was smaller than that of the pure CdS, as can be seen from Fig. S4,† mainly resulting from the coordination of bpydc with Cd2+. In order to elucidate the role of bpydc, we replaced the bpydc with biphenyl-4,4′-dicarboxylic acid. Similar to bpydc, biphenyl-4,4′-dicarboxylic acid can also react with ZrCl4 to produce UiO-67, and the morphology of UiO-67 was similar to that of the UiO-bpy (Fig. S5a†). The preparation conditions for CdS/UiO-67 were the same as those for CdS/UiO-bpy. However, as shown in Fig. S5b,† the size of CdS particles in CdS/UiO-67 was about 50 nm, which was larger than that in CdS/UiO-bpy (10 nm, Fig. 2), mainly because biphenyl-4,4′-dicarboxylic acid does not coordinate with Cd2+. So we can conclude that the bridging ligands of bpydc played a key role in obtaining small size CdS nanoparticles. Furthermore, we also found that the amount of cadmium acetate had an important effect on the size of CdS, as shown in Fig. S6.†
In order to elucidate the local coordination environment of the Co atom in the CdS/UiO-bpy/Co composites, X-ray absorption near-edge structure (XANES) spectra and Fourier-transformed Co K-edge extended X-ray absorption fine structure (EXAFS) spectra were recorded. As shown in Fig. 3A, the absorption edge position of the CdS/UiO-bpy/Co composites is located between that of Co foil and Co3O4 and is close to that of CoS, suggesting that the Co atom carries positive charge and the valence state of Co is between +1 and +3. As shown in the Fourier transform of the EXAFS spectrum of CdS/UiO-bpy/Co composites (Fig. 3B), the peaks at around 1.6 Å can be attributed to the Co–N bond and the Co–Cl bond. The quantitative coordination configuration of the Co atom can be obtained by EXAFS fitting (Fig. S7 and Table S2†). Both the coordination numbers of Co–N and Co–Cl are 2. These results indicate that the coordination structure of the Co atom is consistent with the schematic diagram in Fig. 1. Diffuse reflectance UV/Vis spectroscopy and X-ray photoelectron spectroscopy (XPS) were also used to elucidate the local coordination environment of the Co atom in the CdS/UiO-bpy/Co composites. A typical absorption band in the region of 600–800 nm corresponding to the Co complex in the UV/vis spectrum was observed, as shown in Fig. S8.† It was also confirmed that the absorption properties are similar to those of the corresponding homogeneous complex Co-bpy, indicating that the Co atoms were coordinated with the ligands of bpydc, which was further supported by the XPS survey spectra in Fig. S9.† The N 1s XPS spectra of CdS/UiO-bpy shifted to higher binding energies by 0.16 eV after the Co atoms were loaded into the CdS/UiO-bpy, suggesting that the surface chemical states of N atoms were changed by the loaded Co atoms, due to the interaction between the N atoms and Co atoms in the CdS/UiO-bpy/Co composites. XPS spectra of the Co 2p and Cl 2p also indicated the introduction of Co and Cl elements into the framework (Fig. S10†).
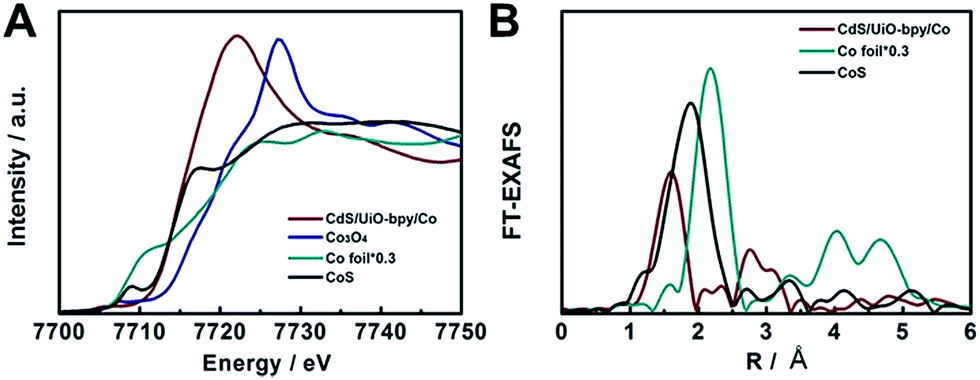 | ||
| Fig. 3 (A) XANES spectra at the Co K-edge. (B) Fourier-transformed Co K-edge EXAFS spectra. | ||
To evaluate the photocatalytic applications of CdS, UiO-bpy, CdS/UiO-bpy, and CdS/UiO-bpy/Co composites, their optical absorption properties were investigated by UV-vis diffuse reflectance spectroscopy, as shown in Fig. S8.† It is shown that the UiO-bpy can only absorb ultraviolet light. However, CdS, CdS/UiO-bpy and CdS/UiO-bpy/Co composites exhibited strong optical absorption extending to the visible light region, suggesting that these materials can be excited by visible light irradiation to generate electron–hole pairs for redox reactions. To gain a deeper understanding of the efficacy of photoexcited charge separation, the photo-current responses of CdS, CdS/UiO-bpy and CdS/UiO-bpy/Co composites were tested, as shown in Fig. 4A. The transient photocurrent results provide clear evidence for electron transfer within these catalysts. It is clear that the photocurrent intensity of CdS/UiO-bpy/Co was higher than that of both CdS and CdS/UiO-bpy under visible light irradiation, indicating more effective charge separation in the CdS/UiO-bpy/Co composites.
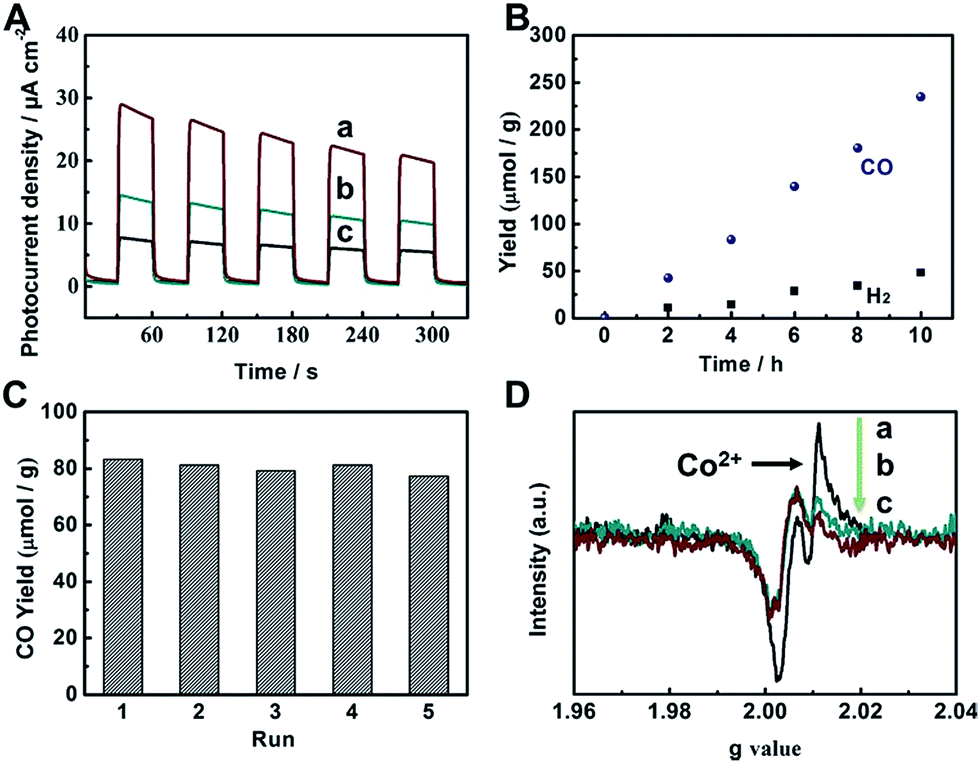 | ||
| Fig. 4 (A) Transient photocurrent responses under visible light irradiation (λ ≥ 420 nm): CdS/UiO-bpy/Co (a), CdS/UiO-bpy (b), and CdS (c). (B) Yields of CO and H2 over CdS/UiO-bpy/Co. (C) Reusability of CdS/UiO-bpy/Co with a reaction time of 4 h for each cycle. (D) EPR spectra of CdS/UiO-bpy/Co under different conditions. Key: without light and N2 (a), with light and CO2 (b), with light and N2 (c). | ||
The catalytic behaviors of the CdS/UiO-bpy/Co composites were examined for the photochemical reduction of CO2 and compared with those of CdS and CdS/UiO-bpy. The CO2 reduction was conducted under visible light irradiation, using triethanolamine (TEOA) as an electron donor and acetonitrile as the solvent. As shown in Fig. 4B, the amount of CO generated increased almost linearly with irradiation time. The CdS/UiO-bpy/Co composites showed a high CO evolution rate of 235 μmol g−1 h−1 under light illumination for 10 hours, which was higher than that of CdS (CO, 23 μmol g−1 h−1) and CdS/UiO-bpy (CO, 0 μmol g−1 h−1), as shown in Table 1. The turnover number of CO formation using the CdS/UiO-bpy/Co composites was 2.24. The CO evolution rate is 195 μmol g−1 h−1 for the CdS/UiO-bpy/Co composites under ambient pressure of CO2 (Table 1). And the CO evolution rate was 110 μmol g−1 h−1 for CdS with the aid of Co-bpy as a cocatalyst, which was lower than that of the CdS/UiO-bpy/Co composites (Table 1). These results reveal that CdS/UiO-bpy/Co composites have comparable activity for CO2 reduction compared with previously reported catalysts under comparative conditions, as can be seen from Table S3.† The yield and the morphology of the catalyst had no obvious change after five cycles, as shown in Fig. 4C and S11,† indicating the excellent stability of the CdS/UiO-bpy/Co composites. Our experiments showed that in the absence of photocatalysts or light illumination, no products were detected in the reaction, implying that CO was generated by the photocatalytic reduction of CO2 on the CdS/UiO-bpy/Co composites, as shown in Table 1. In order to confirm that the reaction is induced by the light absorption of CdS, the action spectrum for the photocatalytic reaction was determined, as shown in Fig. S12.† Furthermore, the calculated apparent quantum yield of the CdS/UiO-bpy/Co composites at 420 nm was 0.65%, which was higher than that of CdS (0.063%) and CdS@Co-bpy (0.302%). Comparable AQY was previously reported for CO2 to CO conversion with a CdS-[Ni(terpyS)2]2+ hybrid catalyst (0.28 ± 0.04%)33 and CO2 to formate conversion with a Ru–Ag–TaON hybrid catalyst (0.48%).34 It is also found that higher efficiencies can be achieved by using a RuP/C3N4 hybrid catalyst with a suitable solvent of N,N-dimethylacetamide (5.7%).35
| Entry | CO (μmol g−1 h−1) | H2 (μmol g−1 h−1) | Selectivity of COb (%) | AQYCOc (%) |
|---|---|---|---|---|
| a Reaction conditions: CdS/UiO-bpy/Co composites (10 mg), CH3CN (3 mL), TEOA (0.5 mL), CO2 (0.8 MPa), 300 W Xe lamp, λ ≥ 420 nm, 10 h. b Selectivity of CO = nCO/(nCO + nH2). c Apparent Quantum Yield (AQY): AQYCO = 2 × the number of evolved CO molecules/the number of incident photons. d Using CdS instead of CdS/UiO-bpy/Co composites. e Using CdS/UiO-bpy instead of CdS/UiO-bpy/Co composites. f Using CdS/UiO-bpy/Ni composites instead of CdS/UiO-bpy/Co composites. g In the dark. h Using N2 instead of CO2. nd: not detected. i Using CdS and Co-bpy instead of CdS/UiO-bpy/Co composites. j The pressure of CO2 was 0.1 MPa. | ||||
| 1 | 235 | 41 | 85 | 0.65 |
| 2d | 23 | 248 | 8 | 0.063 |
| 3e | nd | 275 | 0 | — |
| 4f | nd | 580 | 0 | — |
| 5g | nd | nd | — | — |
| 6h | nd | nd | — | — |
| 7i | 110 | 132 | 45 | 0.302 |
| 8j | 195 | 45 | 81 | 0.54 |
To further validate the source of the generated CO, an isotopic experiment using 13CO2 as a substrate was performed under identical photocatalytic reaction conditions, and the products were analyzed by gas chromatography and mass spectrometry. As shown in Fig. S13,† the peak at m/z = 29 could be assigned to 13CO, indicating that the carbon source of CO was the CO2 used. When the CO2 was replaced by N2, no detectable products were formed, as shown in Table 1.
In order to clarify the role of Co in the CdS/UiO-bpy/Co composites, we also loaded Ni atoms into the preformed CdS/UiO-bpy composites to form ternary CdS/UiO-bpy/Ni composites, which were similar to CdS/UiO-bpy/Co composites, and the results are shown in Fig. S14.† The CdS/UiO-bpy/Ni composites displayed 2.1-fold higher photocatalytic activity for H2 evolution compared with CdS/UiO-bpy, but no CO was generated, as shown in Table 1. This indicated that the Co in the composites was crucial for the photoreduction of CO2 to CO. Electron paramagnetic resonance (EPR) spectroscopy was used to further study the function of Co atoms during the CO2 photoreduction process. As shown in Fig. 4D(a), a strong signal for high-spin state Co2+ was observed for the CdS/UiO-bpy/Co composite without irradiation in a nitrogen atmosphere.36 However, the peak intensity of Co2+ was greatly weakened with light irradiation, which clearly indicates that the Co valence states transformed from high-spin state Co2+ to Co+ in a low-spin state (Fig. 4D(c)). The EPR signal corresponding to Co2+ was enhanced when CO2 was introduced into the irradiated CdS/UiO-bpy/Co composites, implying that some of the Co+ was oxidized back to Co2+ species during the CO2 photoreduction process (Fig. 4D(b)).37 This kinetic behavior also indicates that photoexcited electrons are transferred to the Co center,38,39 and the photoreduction of CO2 was enhanced by the valence transformation of the Co.
Conclusions
In conclusion, we demonstrate the design and fabrication of ternary CdS/UiO-bpy/Co composites, which combined inorganic semiconductors with molecular redox catalysts through MOFs using the function of the ligands. CdS nanoparticles of small size were obtained due to the presence of bridging ligands of bpydc. The ternary CdS/UiO-bpy/Co composites exhibited very high efficiency for photoreduction of CO2, affording CO as the sole carbonaceous product with an evolution rate of 235 μmol g−1 h−1, and the selectivity was over 85%. We anticipate that the implantation strategy, combining inorganic semiconductors with molecular redox catalysts through MOFs, will be applicable for designing many other efficient photocatalysts for CO2 reduction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Key Research and Development Program of China (2017YFA0403102 and 2017YFA0403003), National Natural Science Foundation of China (21733011, 21533011, and 21373230), and Chinese Academy of Sciences (QYZDY-SSW-SLH013).Notes and references
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS PubMed.
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 1704649 CrossRef PubMed.
- Y. Gao, L. Ye, S. Cao, H. Chen, Y. Yao, J. Jiang and L. Sun, ACS Sustainable Chem. Eng., 2017, 6, 781–786 CrossRef.
- X. Chang, T. Wang, P. Zhang, Y. Wei, J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2016, 55, 8840–8845 CrossRef CAS PubMed.
- I. Shown, S. Samireddi, Y. C. Chang, R. Putikam, P. H. Chang, A. Sabbah, F. Y. Fu, W. F. Chen, C. I. Wu, T. Y. Yu, P. W. Chung, M. C. Lin, L. C. Chen and K. H. Chen, Nat. Commun., 2018, 9, 169 CrossRef PubMed.
- X. Jiao, X. Li, X. Jin, Y. Sun, J. Xu, L. Liang, H. Ju, J. Zhu, Y. Pan, W. Yan, Y. Lin and Y. Xie, J. Am. Chem. Soc., 2017, 139, 18044–18051 CrossRef CAS PubMed.
- R. L. Siegelman, T. M. McDonald, M. I. Gonzalez, J. D. Martell, P. J. Milner, J. A. Mason, A. H. Berger, A. S. Bhown and J. R. Long, J. Am. Chem. Soc., 2017, 139, 10526–10538 CrossRef CAS PubMed.
- L.-L. Long, J.-J. Chen, X. Zhang, A.-Y. Zhang, Y.-X. Huang, Q. Rong and H.-Q. Yu, NPG Asia Mater., 2016, 8, e263 CrossRef CAS.
- R. K. Yadav, G. H. Oh, N. J. Park, A. Kumar, K. J. Kong and J. O. Baeg, J. Am. Chem. Soc., 2014, 136, 16728–16731 CrossRef CAS PubMed.
- J. Park, D. Yuan, K. T. Pham, J. R. Li, A. Yakovenko and H. C. Zhou, J. Am. Chem. Soc., 2012, 134, 99–102 CrossRef CAS PubMed.
- W. J. Yin, B. Wen, S. Bandaru, M. Krack, M. W. Lau and L. M. Liu, Sci. Rep., 2016, 6, 23298 CrossRef CAS PubMed.
- P. J. Milner, R. L. Siegelman, A. C. Forse, M. I. Gonzalez, T. Runcevski, J. D. Martell, J. A. Reimer and J. R. Long, J. Am. Chem. Soc., 2017, 139, 13541–13553 CrossRef CAS PubMed.
- C. Chen, B. Li, L. Zhou, Z. Xia, N. Feng, J. Ding, L. Wang, H. Wan and G. Guan, ACS Appl. Mater. Interfaces, 2017, 9, 23060 CrossRef CAS PubMed.
- S. Wang, W. Yao, J. Lin, Z. Ding and X. Wang, Angew. Chem., Int. Ed., 2014, 53, 1034–1038 CrossRef CAS PubMed.
- H. Q. Xu, J. Hu, D. Wang, Z. Li, Q. Zhang, Y. Luo, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2015, 137, 13440–13443 CrossRef CAS PubMed.
- K. M. Cho, K. H. Kim, K. Park, C. Kim, S. Kim, A. Al-Saggaf, I. Gereige and H.-T. Jung, ACS Catal., 2017, 7, 7064–7069 CrossRef CAS.
- J. Jin, J. Yu, D. Guo, C. Cui and W. Ho, Small, 2015, 11, 5262–5271 CrossRef CAS PubMed.
- H. Park, H. H. Ou, A. J. Colussi and M. R. Hoffmann, J. Mater. Chem. A, 2015, 119, 4658–4666 CAS.
- S. Wang and X. Wang, Appl. Catal., B, 2015, 162, 494–500 CrossRef CAS.
- F. Ke, L. Wang and J. Zhu, Nano Res., 2015, 8, 1834–1846 CrossRef CAS.
- Y. Kou, Y. Nabetani, D. Masui, T. Shimada, S. Takagi, H. Tachibana and H. Inoue, J. Am. Chem. Soc., 2014, 136, 6021–6030 CrossRef CAS PubMed.
- J. Agarwal, E. Fujita, H. F. Schaefer 3rd and J. T. Muckerman, J. Am. Chem. Soc., 2012, 134, 5180–5186 CrossRef CAS PubMed.
- S. Lian, M. S. Kodaimati, D. S. Dolzhnikov, R. Calzada and E. A. Weiss, J. Am. Chem. Soc., 2017, 139, 8931–8938 CrossRef CAS PubMed.
- R. Kuriki, H. Matsunaga, T. Nakashima, K. Wada, A. Yamakata, O. Ishitani and K. Maeda, J. Am. Chem. Soc., 2016, 138, 5159–5170 CrossRef CAS PubMed.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS PubMed.
- S. Wang, B. Y. Guan, Y. Lu and X. W. D. Lou, J. Am. Chem. Soc., 2017, 139, 17305–17308 CrossRef CAS PubMed.
- K. M. Choi, D. Kim, B. Rungtaweevoranit, C. A. Trickett, J. T. Barmanbek, A. S. Alshammari, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 356–362 CrossRef CAS PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- H. Wu, Y. S. Chua, V. Krungleviciute, M. Tyagi, P. Chen, T. Yildirim and W. Zhou, J. Am. Chem. Soc., 2013, 135, 10525–10532 CrossRef CAS PubMed.
- B. An, J. Zhang, K. Cheng, P. Ji, C. Wang and W. Lin, J. Am. Chem. Soc., 2017, 139, 3834–3840 CrossRef CAS PubMed.
- K. T. Yong, Y. Sahoo, M. T. Swihart and P. N. Prasad, J. Phys. Chem. C, 2007, 111, 2447–2458 CrossRef CAS.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W. C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- M. F. Kuehnel, K. L. Orchard, K. E. Dalle and E. Reisner, J. Am. Chem. Soc., 2017, 139, 7217–7223 CrossRef CAS PubMed.
- A. Nakada, T. Nakashima, K. Sekizawa, K. Maeda and O. Ishitani, Chem. Sci., 2016, 7, 4364–4371 RSC.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS PubMed.
- J. McAlpin, G. Surendranath, M. Dinca, T. A. Stich, S. A. Stoian, W. H. Casey, D. G. Nocera and R. D. Britt, J. Am. Chem. Soc., 2010, 132, 6882–6883 CrossRef CAS PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- H. Zhang, J. Wei, J. Dong, G. Liu, L. Shi, P. An, G. Zhao, J. Kong, X. Wang, X. Meng, J. Zhang and J. Ye, Angew. Chem., Int. Ed., 2016, 55, 14310–14314 CrossRef CAS PubMed.
- C. Chen, T. Wu, D. Yang, P. Zhang, H. Liu, Y. Yang, G. Yang and B. Han, Chem. Commun., 2018, 54, 5984–5987 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc02809e |
| This journal is © The Royal Society of Chemistry 2018 |