DOI:
10.1039/C8SC03267J
(Edge Article)
Chem. Sci., 2018,
9, 8883-8889
Phenolation of cyclodextrin polymers controls their lead and organic micropollutant adsorption†
Received
24th July 2018
, Accepted 23rd September 2018
First published on 24th September 2018
Abstract
Porous β-cyclodextrin polymers linked with tetrafluoroterephthalonitrile (TFN-CDPs) have shown promise for adsorbing organic micropollutants (MPs) more quickly and effectively than conventional adsorbents. Prior to their discovery, the nucleophilic aromatic substitution (SNAr) reaction used to prepare TFN-CDP was nearly unknown for the aliphatic alcohol nucleophiles, and the low isolated yields of TFN-CDP motivated model studies of the reaction between TFN and n-butanol. These experiments reveal a previously undescribed substitution reaction of TFN in which a fluorine is substituted by a hydroxyl group. This process is responsible for the low yields of the polymerization and incorporates phenolate groups into the polymer network. Phenolation and polymerization (etherification) are competing processes, and the level of phenolate incorporation was controlled by varying the rate of base addition and initial monomer concentrations. TFN-CDPs with varying phenolate content were prepared and evaluated as adsorbents for both Pb2+ ions and 83 MPs. More heavily phenolated polymers showed increased capacity to bind Pb2+ ions. Phenolation was also correlated with increased binding affinity for almost all of the 83 MPs tested, including neutral, cationic, and anionic substances. These results leverage a newly discovered side reaction during SNAr reactions of electron-poor aryl fluorides to improve both the yield and the uptake affinity for both lead and organic MPs of TFN-CDPs.
Introduction
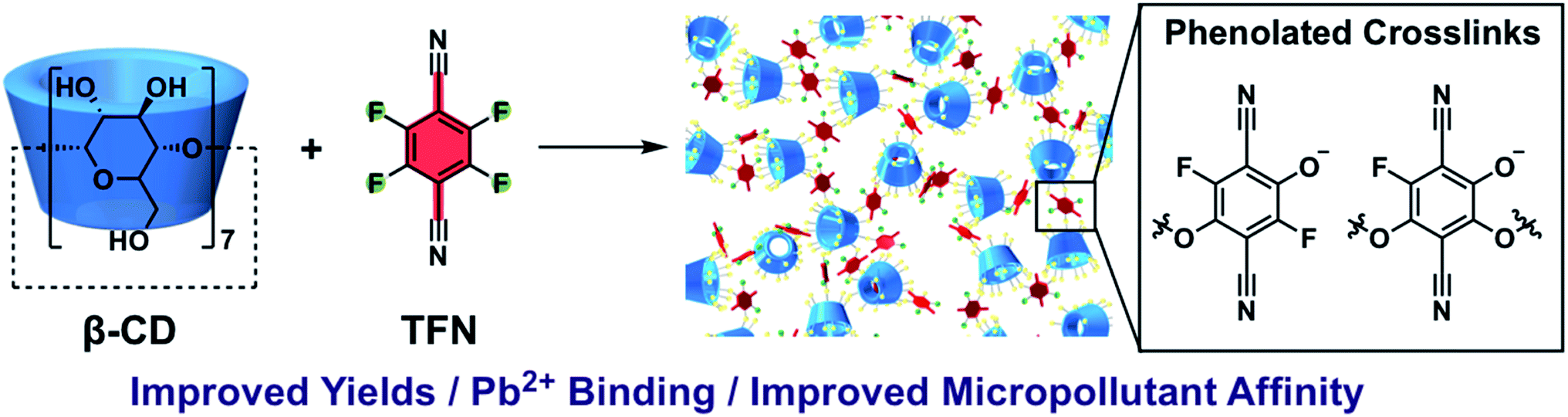
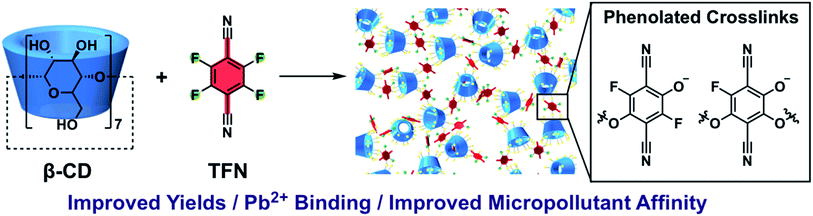
The contamination of groundwater and surface water by heavy metals and organic micropollutants (MPs) is a world-wide problem.1–4 There is no safe level for lead in drinking water.5 Low concentrations of MPs (μg to ng L−1) can have deleterious effects in aquatic ecosystems,6–8 and long-term exposure to complex MP mixtures in drinking water may contribute to health and behavioral problems in humans.9 Currently, the economically scalable methods of water treatment for organic MPs are advanced oxidation10 and activated carbon (AC) adsorption.11 Advanced oxidation effectively reduces the concentration of organic MPs in water but can leave behind partially oxidized byproducts that retain toxic activity.12,13 AC adsorption effectively removes organic MPs but requires energy-intensive regeneration14 and can be fouled by natural organic matter.15 These problems might be addressed by using cyclodextrin (CD)-based adsorbents, which form well-defined host–guest complexes,16–18 are readily regenerated,19 and can exhibit rapid micropollutant removal.20 Cyclodextrin polymers and other porous, bio-derived materials have been used in water purification to remove pollutants including: phenolic molecules,21,22 dyes,23–25 naphthenic acids,26 perfluoroalkylated acids,27 and heavy metals.28,29 We previously reported porous polymer networks prepared via a nucleophilic aromatic substitution (SNAr) polyaddition between β-CD and tetrafluoroterephthalonitrile (TFN-CDP-1).30TFN-CDP-1 had a Brunauer–Emmett–Teller surface area of 260 m2 g−1 and exhibited more rapid uptake for many MPs compared to leading ACs and low-surface-area CDPs. The adsorbent was regenerated easily, was not fouled by humic acid in simulated surface water,20 and was further developed as a promising resin for solid-phase microextraction.19 However, the yield of the material obtained from the initial polymerization conditions was low (18%), and the TFN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β-CD ratio of the isolated polymer (ca. 6:1) did not match the monomer feed ratio (3:1).30 Furthermore, while polycondensations of TFN with catechols provide high molecular weight polymers that indicate efficient and selective reactions,31 there was only a single prior example of an SNAr reaction between TFN and an aliphatic alcohol.32 Here we report a phenolation side-reaction of TFN that competes with its SNAr reactions with aliphatic alcohols. Although this process contributed to the low yields of TFN-CDP in our previous report, we now balance the rates of phenolation and productive etherification reactions to improve polymerization yields and tune adsorbent performance (Fig. 1). Phenolated TFN-CDPs bind both organic micropollutants and Pb2+ ions more effectively than similar polymers with fewer phenolates. These findings demonstrate the promise and tunability of CD polymer networks to purify contaminated water. Identifying and controlling the loading of these phenolate functional groups in TFN-CDPs is therefore important for maximizing their performance and manufacturability. Furthermore, TFN is a common monomer in other classes of porous polymers31,33–36 and understanding its reactivity with aliphatic hydroxyl groups will significantly broaden the scope of potential comonomers.
:β-CD ratio of the isolated polymer (ca. 6:1) did not match the monomer feed ratio (3:1).30 Furthermore, while polycondensations of TFN with catechols provide high molecular weight polymers that indicate efficient and selective reactions,31 there was only a single prior example of an SNAr reaction between TFN and an aliphatic alcohol.32 Here we report a phenolation side-reaction of TFN that competes with its SNAr reactions with aliphatic alcohols. Although this process contributed to the low yields of TFN-CDP in our previous report, we now balance the rates of phenolation and productive etherification reactions to improve polymerization yields and tune adsorbent performance (Fig. 1). Phenolated TFN-CDPs bind both organic micropollutants and Pb2+ ions more effectively than similar polymers with fewer phenolates. These findings demonstrate the promise and tunability of CD polymer networks to purify contaminated water. Identifying and controlling the loading of these phenolate functional groups in TFN-CDPs is therefore important for maximizing their performance and manufacturability. Furthermore, TFN is a common monomer in other classes of porous polymers31,33–36 and understanding its reactivity with aliphatic hydroxyl groups will significantly broaden the scope of potential comonomers.
 |
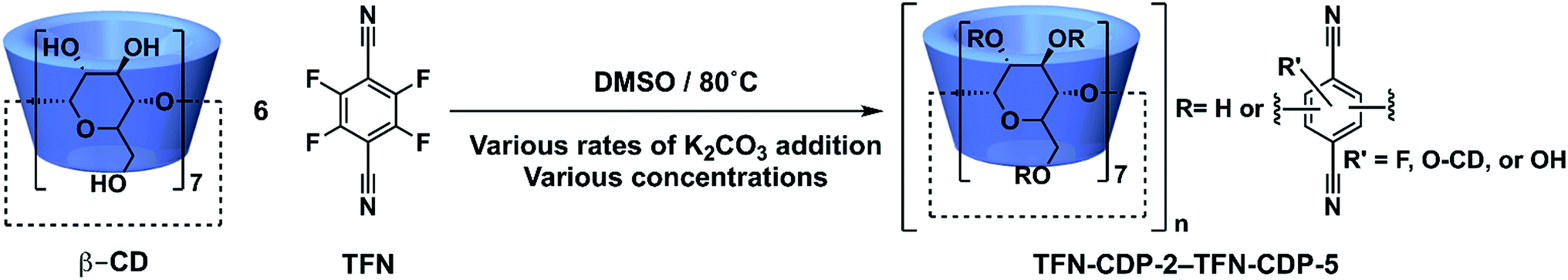
| | Fig. 1 Synthesis of β-cyclodextrin (β-CD) polymers linked with tetrafluoroterephthalonitrile. This study identifies phenolated TFN-derived species incorporated into the polymer and characterizes their effect on Pb2+ and organic micropollutant binding. | |
Results and discussion
Model studies
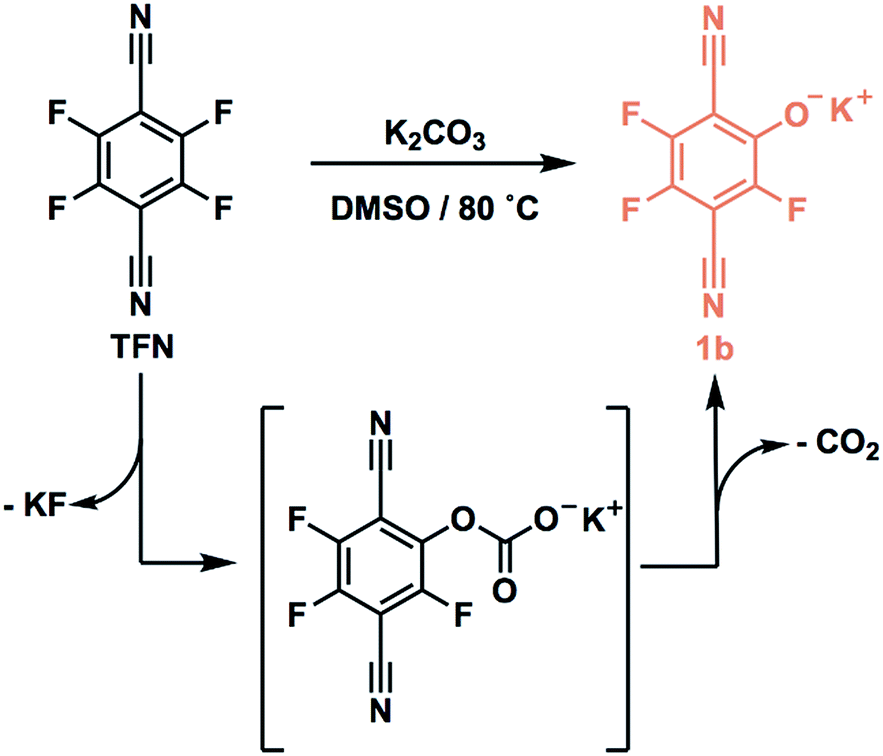
TFN-CDP-1 was prepared in THF in 18% isolated yield under similar conditions as our previous report;30 it is synthesized via a SNAr polycondensation in which the alcohol groups of β-CD are deprotonated by K2CO3. Yield is defined throughout this work as (mass of isolated TFN-CDP)/(mass of the monomers), because the theoretical yield depends on both the efficiency of monomer incorporation and the number of SNAr reactions that occur. Furthermore, the ratio of TFN:β-CD (6:1) found in the isolated polymer deviated from the monomer feed ratio (3:1 TFN:β-CD). The low yield and difference between the monomer feed and incorporation ratios were suggestive of side reactions, which motivated us to study the reactivity of TFN. Polymerizations in DMSO (TFN-CDP-2) provided higher yields (64%) compared to TFN-CDP-1, even at shorter reaction times (18 h vs. 48 h). A monomer feed ratio of 6:1 TFN:β-CD provided an incorporation ratio of 5.2:1, corresponding to a loss of 0.8 equiv. (13 mol%) of the TFN. After isolating the insoluble polymer, analysis of the soluble fraction by 19F and 13C NMR spectroscopy and high-resolution mass spectrometry indicated the formation of phenolate 1b as a major side product. We hypothesized that 1b is formed via the SNAr of TFN and K2CO3, followed by decarboxylation. Indeed, 1b is formed quantitatively within 10 min when TFN (0.3 mM) and K2CO3 (3.3 equiv.) are combined in anhydrous DMSO at 80 °C (Scheme 1), as measured by 19F NMR spectroscopy. Evidence for this side reaction under the polymerization conditions, combined with the rapid formation of 1b in the absence of other nucleophiles, suggested that further study of the phenolation of TFN might improve the yield of the polymerization.
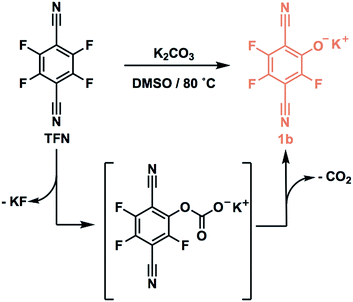 |
| | Scheme 1 | |
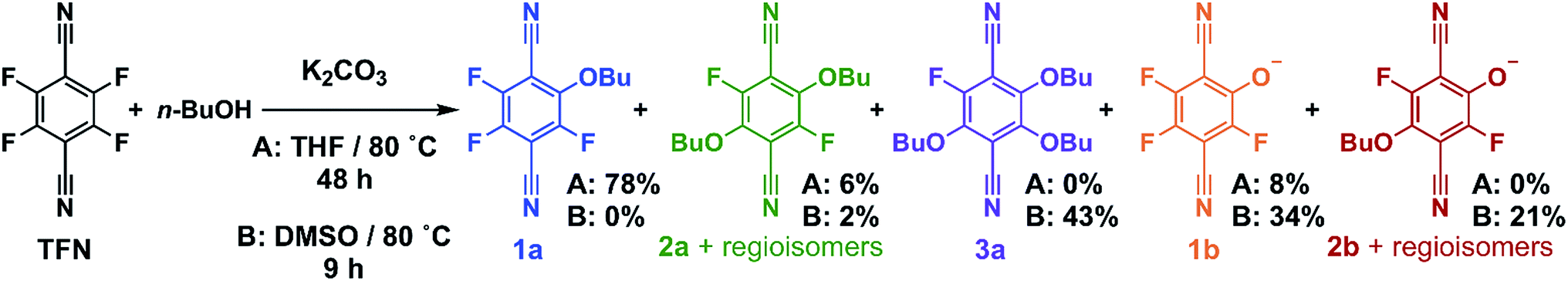
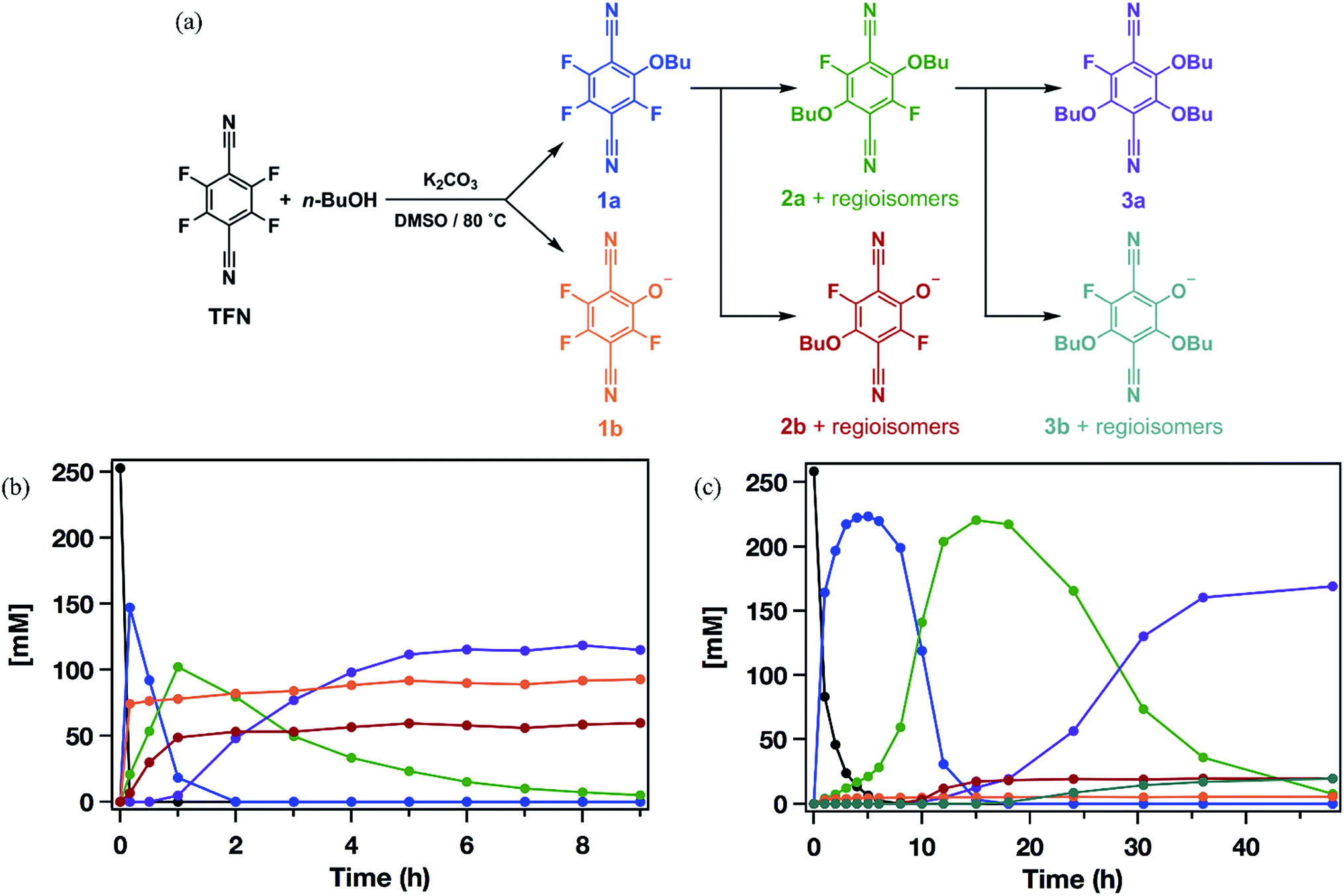
Model SNAr reactions between TFN and n-butanol show that phenolation and etherification reactions are in competition. Etherification and phenolation both proceed slowly in THF over 48 h. The dominant product observed by 19F NMR spectroscopy was the monosubstituted ether 1a (78%). Minor products included a mixture of dibutyl ethers 2a (6%) and phenolate 1b (8%). More highly substituted ethers and phenolate products were observed when the reaction was run in DMSO for 9 h. These higher substituted products may explain why the polymerization is both faster and higher yielding in DMSO. The major products (Scheme 2) were the trisubstituted ether 3a (43%), 1b (34%), a mixture of regioisomers containing one phenolate and one ether (2b, 21%), and a mixture of disubstituted ethers (2a, 2%). Phenolation is enhanced in this solvent, and 1b is much less active for further substitutions as compared to the etherification products. When a pure sample of phenolate 1b was subjected to these reaction conditions, only 4% conversion to the 1,2-substituted regioisomer of 2b was observed after 24 h, indicating that 2b forms via1a. A model reaction in DMSO monitored by 19F NMR spectroscopy as a function of reaction time also demonstrates that phenolated products essentially do not undergo further reactions in the presence of competing electrophiles (Fig. 2a). Most of the TFN is consumed within 10 min, with 59% conversion to 1a and 30% conversion to 1b. 1a continues to react to form more highly substituted products, whereas the concentration of 1b remains approximately constant over 9 h. Most of 1a reacts within 2 h to provide dibutyl ethers 2a and phenolates 2b, which also persist for the remainder of the reaction. 2a reacts further to provide 3a over the next several hours, and no evidence for the formation of dibutyl-ether/monophenolates (3b) was observed (Fig. 2b). These results provide significant insight into the formation of TFN-CD polymers. First, the competition between the first etherification and phenolation process in the first few minutes of the reaction are likely to influence yield and total incorporation of TFN derivatives in the resulting polymer since 1b is mostly unreactive. Furthermore, the formation of 2b demonstrates a process by which phenolate groups might be incorporated into the polymer.
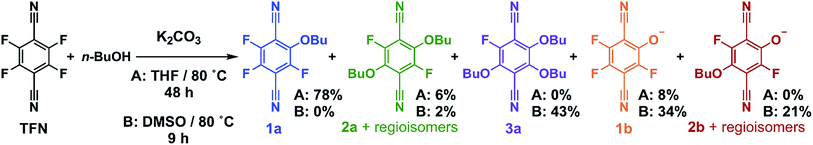 |
| | Scheme 2 | |
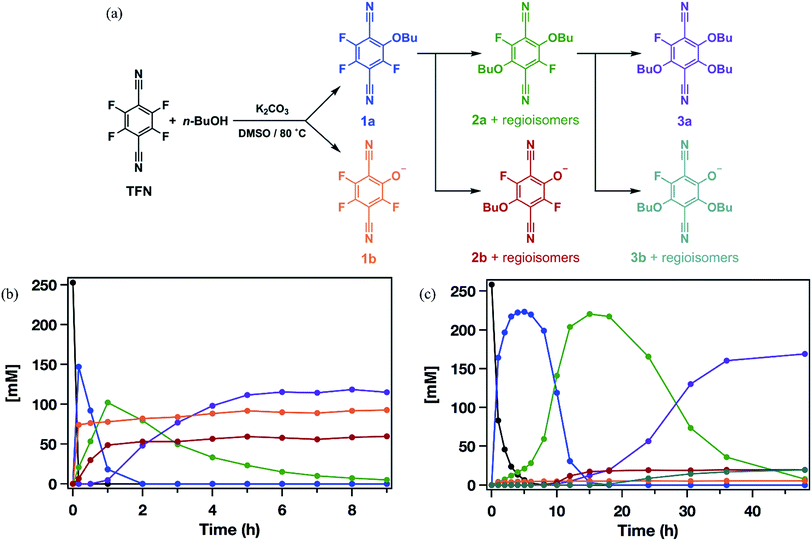 |
| | Fig. 2 (a) Reaction pathways for the formation of n-butoxy and phenolate-substituted TFN derivatives. (b) Concentrations of each species as a function of reaction time observed when 3.3 equiv. K2CO3 is added at t = 0. (c) Concentrations of each species as a function of reaction time observed when 3.3 equiv. K2CO3 was added at 0.1 equiv. per h. | |
The apparent competition between etherification and phenolation suggested that slow addition of the K2CO3 might suppress phenolation. A model reaction in DMSO in which K2CO3 was added gradually (3.3 equiv., 0.1 equiv. per h) provided sequential etherification from 1a to 2a to 3a (Fig. 2c) as the dominant reaction pathway. After 6 h, 87% of the TFN was converted to 1a, and only 2% proceeded to 1b. This selectivity corresponds to a 94% reduction in phenolation relative to when K2CO3 is added at once. As before, 1a reacts to form more substituted products, and the concentration of 1b does not decrease at longer reaction times. After 15 h, the conversion to 2b was only 7%, and the combined regioisomers of 2a, represented 85% of the TFN-derived species. The concentration of the 2b regioisomers also remain approximately constant for the remainder of the reaction, and 2a proceeds to the trisubstituted product, 3a, and the dibutoxy phenolate products, 3b. After 48 h, 3a made up 76% of the TFN-derived species, as compared to 43% when the base is present throughout the reaction. We attribute the formation of 3b to the near-complete consumption of n-butanol at the later stages of the reaction. These experiments demonstrate that phenolation occurs readily in the presence of excess K2CO3 and that etherification is favored when the concentration of base is kept low. Therefore, the rate of K2CO3 addition might influence both the yield and degree of phenolate incorporation into TFN-CDP polymers.
Polymer synthesis and characterization
The above findings suggest that the K2CO3 addition rate will also affect the yield and properties of TFN-CDP (Scheme 3). For example, the formation of 1b during the polymerization would decrease the yield as further substitution reactions on this species are slow. Later phenolation processes, analogous to the formation of 2b or 3b, do not decrease yield but incorporate phenolated TFN groups into the polymer network. We performed two polymerizations, either at [TFN] = 0.33 M and 0.17 equiv. β-CD, to which 3.3 equiv. K2CO3 were added all at once (TFN-CDP-2) or at 0.1 equiv. per h (TFN-CDP-3). The suppression of 1b is reflected in the combustion analysis of the two polymers. The C:N ratio was used to determine the number of TFN per β-CD. TFN-CDP-2 exhibits a TFN:β-CD ratio of 5.2, corresponding to a loss of 0.8 equiv. (13 mol%) TFN to 1b. TFN-CDP-3 had a TFN:β-CD ratio of 5.9, which indicates that only 0.1 equiv. (2 mol%) of the TFN was lost to the first phenolation process. TFN-CDP-2 and TFN-CDP-3 also had a different concentration of phenolate incorporated into the polymers (Table 1). The phenolate concentration in the polymers was determined by deprotonating the phenolates using Li2CO3 and determining the amount of bound Li ions using inductively coupled plasma optical emission spectroscopy (ICP-OES, see ESI† for detailed analysis procedures). To further validate this method, we performed a similar analysis of K+ content in the polymers by using K2CO3 in place of Li2CO3 and found similar phenolate loadings (Table S1†). TFN-CDP-2 had a higher phenolate concentration (0.44 mmol g−1) compared to TFN-CDP-3 (0.22 mmol g−1). These results are consistent with the model studies and demonstrate a means to control the phenolate concentration within the polymer networks.
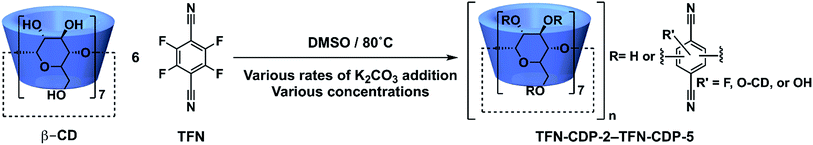 |
| | Scheme 3 | |
Table 1 Polymerization conditions, bound phenolate concentrations, and TFN:CD ratio in polymer samplesa
| Polymer |
[Phenolate] (mmol g−1) |
K2CO3 addition rate |
[TFN]0 (M) |
TFN:CD ratio |
S
BET (m2 g−1) |
Yield (%) |
|
TFN-CDP refers to tetrafluoroterephthalonitrile-linked β-cyclodextrin polymer.
|
|
TFN-CDP-2
|
0.44 (+/− 0.01) |
At once |
0.3 |
5.2 |
346 (+/− 113) |
64 |
|
TFN-CDP-3
|
0.22 (+/− 0.02) |
0.1 equiv. per h |
0.3 |
5.9 |
Non-porous |
67 |
|
TFN-CDP-4
|
0.14 (+/− <0.01) |
At once |
1.2 |
5.2 |
Non-porous |
54 |
|
TFN-CDP-5
|
0.08 (+/− <0.01) |
0.1 equiv. per h |
1.2 |
5.9 |
Non-porous |
54 |
Further manipulation of the phenolate loading was achieved by using higher monomer concentration in the polymerization. TFN is no longer fully soluble under the reaction conditions at a concentration of 1.2 M. A TFN-CDP formulation was prepared (TFN-CDP-4) using a concentration of 1.2 M and by adding the K2CO3 all at once. TFN-CDP-4 has fewer phenolates (0.14 mmol g−1) compared to TFN-CDP-2 (0.44 mmol g−1), which is formed under similar conditions but lower initial monomer concentration. In the synthesis of TFN-CDP-4, the volume of DMSO was decreased four-fold, which renders a portion of the TFN undissolved and effectively increases the ratio of β-CD to dissolved TFN compared to reactions run at lower concentration. This higher effective ratio of β-CD alcohols to TFN-fluorides favors etherification over phenolation. Finally, combining both effects that provide lower phenolate concentration, TFN-CDP-5 was synthesized using gradual K2CO3 addition (0.1 equiv. per h) and high concentration (1.2 M), which provided the lowest concentration of bound phenolate (0.08 mmol g−1) (Scheme 3). The results of these polymerizations demonstrate that CD-TFN polymers with varying phenolate concentrations can be accessed by exploiting the competition between etherification and phenolation.
Pb2+ capacity
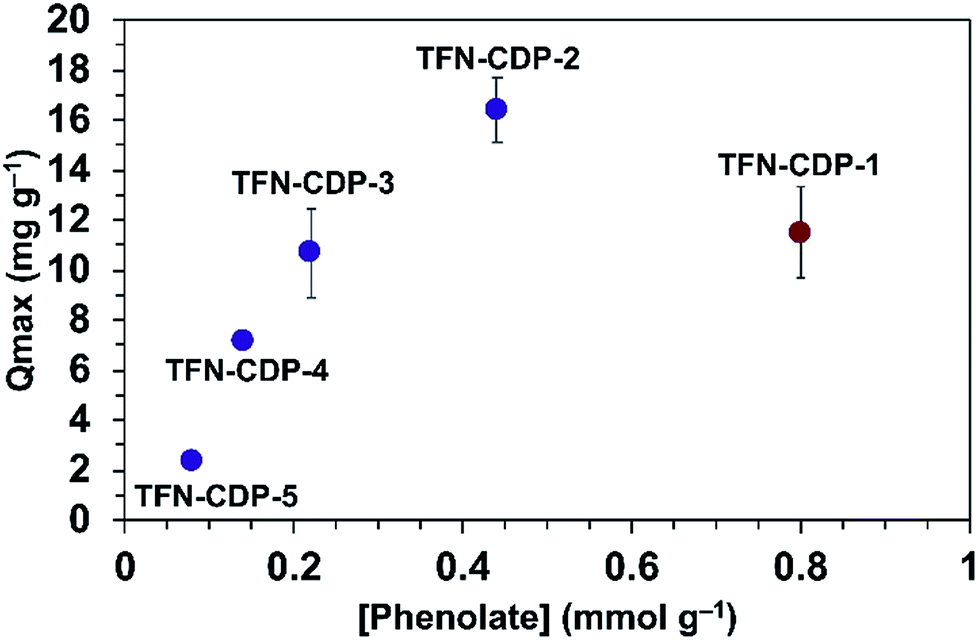
He and coworkers recently reported that a polymer synthesized under similar conditions as TFN-CDP-1 binds Pb2+ ions with a high capacity (Qmax = 215 mg g−1).37 They attributed Pb2+ binding to interactions with the hydroxyl groups of β-CD, but the possible presence of phenolates in the polymers was not yet recognized. We evaluated TFN-CDP polymer samples of varying phenolate content to determine their role in sequestering Pb2+ ions. The Qmax of each polymer was determined by batch experiments in which the polymers were exposed to Pb(NO3)2 solutions (1–50 ppm, pH = 5) for 24 h. After removing the polymers by filtration, the Pb2+ concentrations were determined via ICP-OES. The Pb2+ removal was determined by the concentration difference between the polymer treated solution and control solutions not exposed to polymers. Qmax was determined by building an isotherm of the average solid phase concentration of Pb2+ verses the aqueous concentration of Pb2+ and fitting this to the Langmuir model. The Qmax for TFN-CDP-1–TFN-CDP-5 was 16.4–2.4 mg g−1 (Fig. 3). Polymers prepared in DMSO (TFN-CDP-2–TFN-CDP-5) with varying phenolate content showed a strong positive correlation between Pb2+ capacity and phenolation, TFN-CDP-1 was also found to have a high phenolate content and exhibited a reasonable capacity for Pb2+, but not as high as TFN-CDP-2. Unlike TFN-CDP-2–TFN-CDP-5, TFN-CDP-1 was synthesized under different conditions (THF) under which β-CD is insoluble. Therefore, it may have other structural differences, such as variations in local TFN functionalization density or regioselectivity in its reactions with β-CD that also influence its Pb2+ binding. We have not explored these differences further because TFN-CDP-1 is difficult to scale up and shows lower Pb2+ binding capacity than TFN-CDP-2. Although the SBET of the materials are different, it is likely that the polymer is fully accessible to pollutants at equilibrium. This point is consistent with bisphenol-A (BPA) isotherms generated for each of the polymers (Fig. S4†), from which Langmuir fits provided high Qmax (>1 BPA:β-CD) for both porous and non-porous polymers. Therefore, the differences in Qmax for Pb2+ binding likely arise from the variation in phenolate loading of each polymer. Finally, it is important to note that all of our polymers show approximately 20 fold inferior capacity to the very high value (215 mg g−1) reported by He and coworkers37 for a polymer prepared similarly to TFN-CDP-1. The discrepancy between our measured values and this report cannot be readily explained.
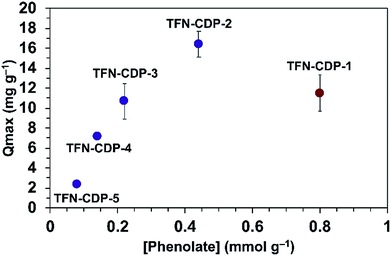 |
| | Fig. 3
Q
max for Pb2+ as a function of bound phenolate functionalities in polymer samples TFN-CDP-1–TFN-CDP-5. | |
The rate of Pb2+ removal is dependent on the porosity in these polymers. To probe the importance of porosity to Pb2+ removal, the rate of Pb2+ removal (Pb2+kobs) was determined for all polymers (Fig. S6†). TFN-CDP-2 has a higher surface area than non-porous TFN-CDP-3–TFN-CDP-5 and has superior Pb2+kobs compared to the other polymers. Among the non-porous polymers, the phenolate content did not have a pronounced effect on Pb2+kobs but did have an effect on the capacity at equilibrium (qe). These results further suggest that phenolate concentration is correlated with affinity to Pb2+ ions at low concentrations.
Micropollutant affinity
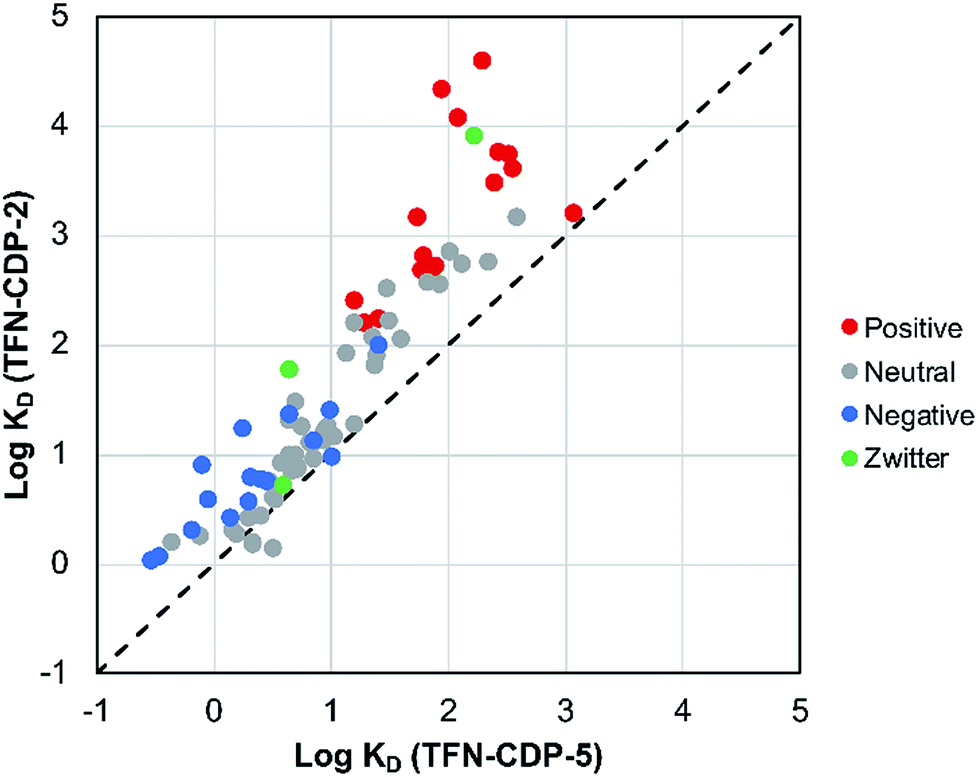
MP affinity testing on all polymer samples indicate that phenolation is desirable in that these charged groups in the polymer predictably affect polymer affinity to positively charged MPs. MP affinity was determined by measuring the adsorption of a mixture of 83 MPs at low concentrations (1 μg L−1; 1 ppb) by means of HPLC-MS/MS (Table S2†). The adsorption was used to determine the affinity of each MP (KD) for each polymer. To compare the MP affinity of highly phenolated TFN-CDP-2 with weakly phenolated TFN-CDP-5 the logKD for each MP for the two polymer samples was plotted on the binary chart in Fig. 4. Of the 83 micropollutants, 81 bind more strongly to the more heavily phenolated polymer. Not surprisingly, cationic substances bind with the highest KD and show the largest differential affinity for the more heavily phenolated polymer. However, although anionic substances bind with lower affinity, they still bind to the more heavily phenolated polymer TFN-CDP-2 more strongly than TFN-CDP-5. Similar affinity trends for both cationic and anionic substances were observed in pairwise comparisons of the polymers with intermediate phenolate content (Fig. S3†). These findings indicate that TFN phenolation is correlated with improved affinity for cationic MPs yet has a smaller and often non-deleterious impact on binding of anionic and uncharged substances.
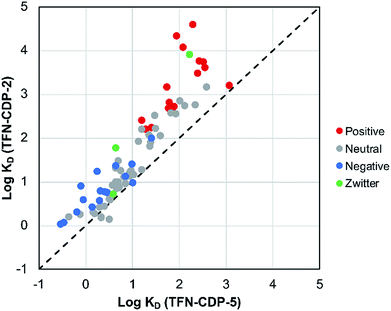 |
| | Fig. 4 Affinity experiment comparing higher-phenolate-content TFN-CDP-2 and lower-phenolate-content TFN-CDP-5. This experiment shows that TFN-CDP-2 has higher affinities for 81 of 83 micropollutants tested. | |
The phenolate content of the non-porous samples (TFN-CDP-3 through TFN-CDP-5) also influences their rates of MP removal. To probe this effect, the pseudo 2nd-order rate constant for BPA removal (BPA kobs) was determined for all polymers under identical conditions (Fig. S5†). Among these non-porous samples, the BPA kobs is correlated to phenolate concentration, even though BPA is a neutral molecule. We speculate that the phenolated polymers are more hydrophilic, which lead to faster diffusion of BPA-contaminated water into the polymer network. In addition, the differences in SBET also affect BPA kobs. Porous TFN-CDP-2 has a significantly higher surface area than non-porous TFN-CDP-3 through TFN-CDP-5 and has a BPA kobs 1–2 orders of magnitude higher than the other polymers (1.61 and 0.02–0.23 g mg−1 min−1, respectively). These experiments demonstrate that both phenolation and porosity affect the rate of BPA removal.
Conclusions
An improved understanding of the SNAr polycondensation of aliphatic alcohols and TFN was achieved using model etherification reactions, the first in depth such studies for TFN and aliphatic alcohols. These findings were adapted to synthesize TFN-CDPs with varying phenolate contents. The results show that the main factors in controlling the inclusion of phenolic functional groups are concentration of the reaction and rate of base addition. Polymers with higher concentrations of phenolic functionalities had higher affinity for 81 MPs, particularly those that are positively charged, as well as higher Pb2+ binding capacity. These results suggest that high phenolate content in TFN-CDP is key for Pb2+ ion capacity and MP affinity. Finally, this study demonstrates that the rational synthesis of TFN-CDPs can be achieved by controlling the rate of K2CO3 addition and the concentration of reactants. Future studies will focus on the incorporation of chelating groups to increase the capacity of CDPs for lead ion binding and expand the application of CDPs to the removal of other heavy metal ions.
Conflicts of interest
D. E. H. and W. R. D. serve on the scientific advisory board and own equity and/or stock options in CycloPure, Inc., which is commercializing related cyclodextrin polymers.
Acknowledgements
M. J. K., A. A., and W. R. D. were supported by the Center for Sustainable Polymers (CSP) at the University of Minnesota, a National Science Foundation (NSF) supported Center for Chemical Innovation (CHE-1413862). Y. L. and D. E. H. were supported by an NSF EAGER grant (grant number CHE-1541820) and by Cornell University's David R. Atkinson Center for a Sustainable Future (ACSF). This research made use of the Cornell Center for Materials Research User Facilities, which are supported through the NSF MRSEC program (DMR-1120296), the IMSERC at Northwestern University, which has received support from the NSF (CHE-1048773); Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the State of Illinois and International Institute for Nanotechnology (IIN). Metal analysis was performed at the Northwestern University Quantitative Bio-element Imaging Center (QBIC).
References
- C. M. G. Carpenter and D. E. Helbling, Environ. Sci. Technol., 2018, 52, 6187–6196 CrossRef CAS PubMed.
- S. D. Richardson and T. A. Ternes, Anal. Chem., 2018, 90, 398–428 CrossRef CAS PubMed.
- S. Masters, G. J. Welter and M. Edwards, Environ. Sci. Technol., 2016, 50, 5269–5277 CrossRef CAS PubMed.
- J. St Clair, C. Cartier, S. Triantafyllidou, B. Clark and M. Edwards, Environ. Eng. Sci., 2016, 33, 53–64 CrossRef CAS PubMed.
- T. Vorvolakos, S. Arseniou and M. Samakouri, Psychiatriki, 2016, 27, 204–214 CrossRef PubMed.
- A. M. Vajda, L. B. Barber, J. L. Gray, E. M. Lopez, J. D. Woodling and D. O. Norris, Environ. Sci. Technol., 2008, 42, 3407–3414 CrossRef CAS PubMed.
- G. R. Tetreault, C. J. Bennett, K. Shires, B. Knight, M. R. Servos and M. E. McMaster, Aquat. Toxicol., 2011, 104, 278–290 CrossRef CAS PubMed.
- F. Gagne, B. Bouchard, C. Andre, E. Farcy and M. Fournier, Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol., 2011, 153, 99–106 CAS.
- E. Diamanti-Kandarakis, J. P. Bourguignon, L. C. Giudice, R. Hauser, G. S. Prins, A. M. Soto, R. T. Zoeller and A. C. Gore, Endocr. Rev., 2009, 30, 293–342 CrossRef CAS PubMed.
- S. G. Zimmermann, M. Wittenwiler, J. Hollender, M. Krauss, C. Ort, H. Siegrist and U. von Gunten, Water Res., 2011, 45, 605–617 CrossRef CAS PubMed.
- A. Joss, H. Siegrist and T. A. Ternes, Water Sci. Technol., 2008, 57, 251–255 CrossRef CAS PubMed.
- C. Prasse, M. Wagner, R. Schulz and T. A. Ternes, Environ. Sci. Technol., 2012, 46, 2169–2178 CrossRef CAS PubMed.
- J. Hollender, S. G. Zimmermann, S. Koepke, M. Krauss, C. S. McArdell, C. Ort, H. Singer, U. von Gunten and H. Siegrist, Environ. Sci. Technol., 2009, 43, 7862–7869 CrossRef CAS PubMed.
- J. Margot, C. Kienle, A. Magnet, M. Weil, L. Rossi, L. F. de Alencastro, C. Abegglen, D. Thonney, N. Chevre, M. Scharer and D. A. Barry, Sci. Total Environ., 2013, 461–462, 480–498 CrossRef CAS PubMed.
- P. A. Quinlivan, L. Li and D. R. Knappe, Water Res., 2005, 39, 1663–1673 CrossRef CAS PubMed.
- G. Crini, Chem. Rev., 2014, 114, 10940–10975 CrossRef CAS PubMed.
- H.-J. Schneider, Angew. Chem., Int. Ed. Engl., 1991, 30, 1417–1436 CrossRef.
- L. Liu and Q. X. Guo, J. Inclusion Phenom. Macrocyclic Chem., 2002, 42, 1–14 CrossRef CAS.
- C. Li, M. J. Klemes, W. R. Dichtel and D. E. Helbling, J. Chromatogr. A, 2018, 1541, 52–56 CrossRef CAS PubMed.
- Y. Ling, M. J. Klemes, L. Xiao, A. Alsbaiee, W. R. Dichtel and D. E. Helbling, Environ. Sci. Technol., 2017, 51, 7590–7598 CrossRef CAS PubMed.
- H. Yamasaki, Y. Makihata and K. Fukunaga, J. Chem. Technol. Biotechnol., 2008, 83, 991–997 CrossRef CAS.
- J. Liu, Y. Yang, J. Bai, H. Wen, F. Chen and B. Wang, Anal. Chem., 2018, 90, 3621–3627 CrossRef CAS PubMed.
- Y. Yang, G. Hu, F. Chen, J. Liu, W. Liu, H. Zhang and B. Wang, Chem. Commun., 2015, 51, 14405–14408 RSC.
- E. Yilmaz, S. Memon and M. Yilmaz, J. Hazard. Mater., 2010, 174, 592–597 CrossRef CAS PubMed.
- G. Crini, Dyes Pigm., 2008, 77, 415–426 CrossRef CAS.
- M. H. Mohamed, L. D. Wilson, J. V. Headley and K. M. Peru, Process Saf. Environ. Prot., 2008, 86, 237–243 CrossRef CAS.
- L. Xiao, Y. Ling, A. Alsbaiee, C. Li, D. E. Helbling and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 7689–7692 CrossRef CAS PubMed.
- F. Zhao, E. Repo, D. Yin, L. Chen, S. Kalliola, J. Tang, E. Iakovleva, K. C. Tam and M. Sillanpaa, Sci. Rep., 2017, 7, 15811 CrossRef PubMed.
- J. Hai, F. Chen, J. Su, F. Xu and B. Wang, Anal. Chem., 2018, 90, 4909–4915 CrossRef CAS PubMed.
- A. Alsbaiee, B. J. Smith, L. Xiao, Y. Ling, D. E. Helbling and W. R. Dichtel, Nature, 2016, 529, 190–194 CrossRef CAS PubMed.
- P. M. Budd, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib and C. E. Tattershall, Chem. Commun., 2004, 230–231 RSC.
- L. Wang, T. Chen, S. C. Chen, Q. Chen and M. Y. He, J. Heterocycl. Chem., 2014, 51, 1536–1540 CrossRef CAS.
- H. J. Mackintosh, P. M. Budd and N. B. McKeown, J. Mater. Chem., 2008, 18, 573–578 RSC.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163–5176 CrossRef CAS.
- N. B. McKeown, P. M. Budd, K. J. Msayib, B. S. Ghanem, H. J. Kingston, C. E. Tattershall, S. Makhseed, K. J. Reynolds and D. Fritsch, Chem.–Eur. J., 2005, 11, 2610–2620 CrossRef CAS PubMed.
- I. Rose, C. G. Bezzu, M. Carta, B. Comesana-Gandara, E. Lasseuguette, M. C. Ferrari, P. Bernardo, G. Clarizia, A. Fuoco, J. C. Jansen, K. E. Hart, T. P. Liyana-Arachchi, C. M. Colina and N. B. McKeown, Nat. Mater., 2017, 16, 932–938 CrossRef CAS PubMed.
- J. He, Y. Li, C. Wang, K. Zhang, D. Lin, L. Kong and J. Liu, Appl. Surf. Sci., 2017, 426, 29–39 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, Pb2+ binding isotherms and affinity plots, 1H, 13C, and 19F NMR spectra, FT-IR Spectra. See DOI: 10.1039/c8sc03267j |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Yuhan
Ling
a,
Yuhan
Ling