DOI:
10.1039/C8SC03339K
(Edge Article)
Chem. Sci., 2018,
9, 8253-8259
Redox-triggered cascade dearomative cyclizations enabled by hexafluoroisopropanol†
Received
27th July 2018
, Accepted 31st August 2018
First published on 13th September 2018
Abstract
An unprecedented cascade dearomative cyclization through hydrogen-bonding-assisted hydride transfer is realized. The aggregate effect of HFIP enables the rapid buildup of polycyclic amines directly from phenols and o-aminobenzaldehydes via a cascade dearomatization/rearomatization/dearomatization sequence. This unique transformation addressed the drawbacks of hydride transfer-involved redox-neutral reactions.
Introduction
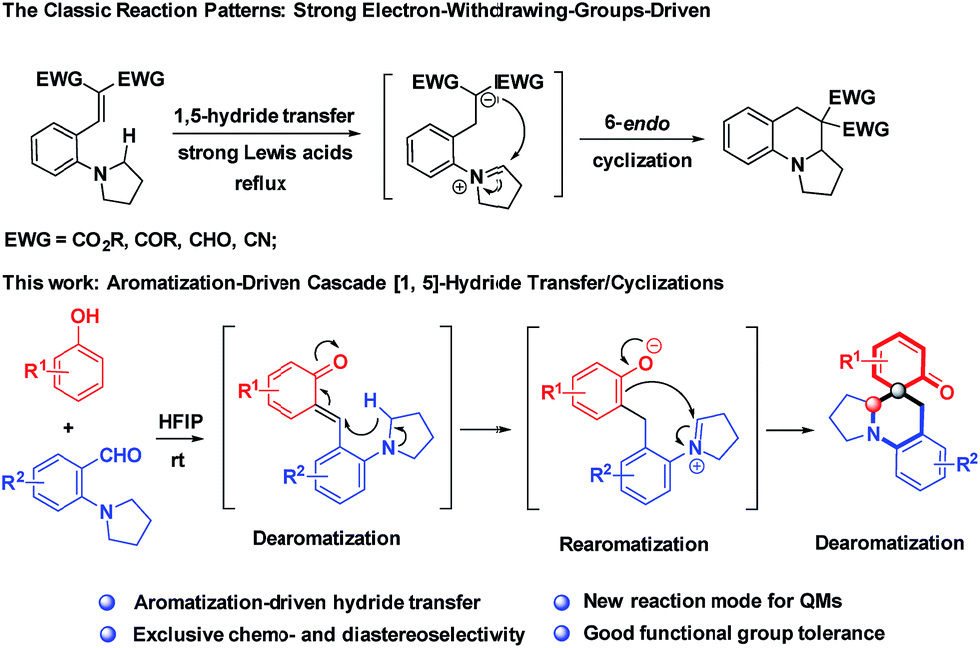
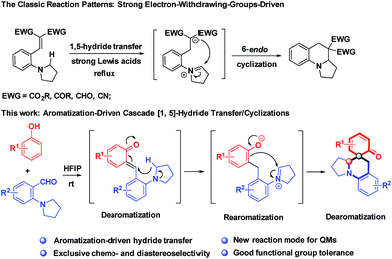
The direct α-C–H functionalization of amines provides the most powerful toolbox for the synthesis of highly functionalized cyclic amines, which are increasingly momentous scaffolds in the exploration of new pharmaceuticals.1 The desire for an oxidant-free and green process has brought about the renaissance of intramolecular hydride transfer as a key step for α-C–H functionalization of amines.2 The atom-economic and redox-neutral nature of this transformation makes it ideal for sustainable and complexity-enhancing amine synthesis. However, for a long time, hydride acceptors have been dominated by electrophilic alkenes, typically activated by double electron-withdrawing groups,3 whereas the utilization of other types of hydride acceptors remains elusive (Scheme 1).4,5 So far, this type of reaction has generally relied on substrate engineering, and hydride acceptors and donors have to be installed into one substrate beforehand via synthetic operations, which are often dissimilar from the targets. Furthermore, the typical protocols necessitate strong Lewis acids or Brønsted acids for decent yields, and high temperature is demanded in most cases. These disadvantages restrict the applicability of this method for the synthesis of structurally diverse cyclic amines as well as their utility for late-stage functionalizations.
 |
| | Scheme 1 Cascade dearomative cyclizations via a dearomatization/rearomatization/dearomatization sequence. | |
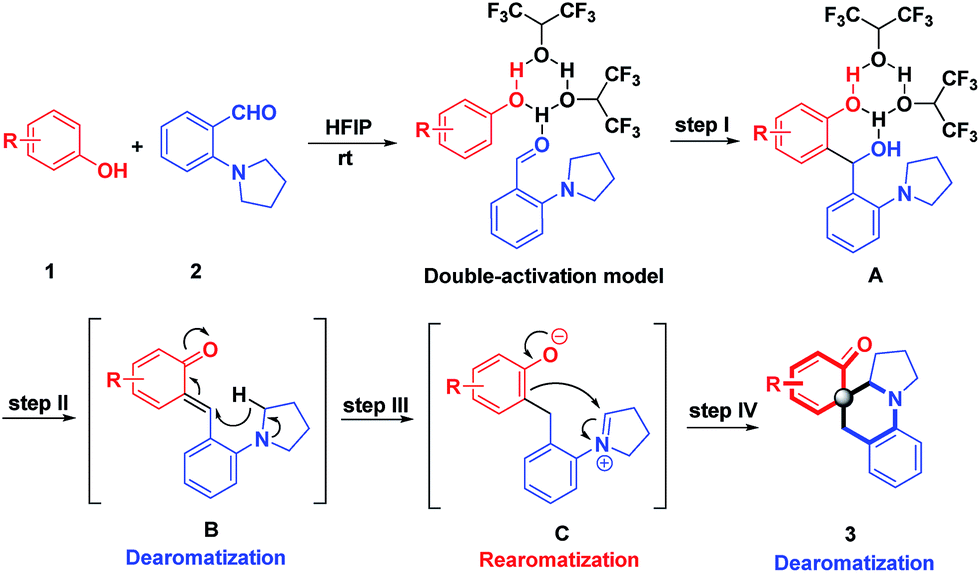
Probably, the high energy barrier for the cleavages of α-C–H bonds accounts for the current situations.6 To address these challenges, we propose the use of highly reactive species, ortho-quinone methides (o-QMs), to trigger hydride transfer reactions, offsetting the high activation energy of C–H bond cleavages. Although o-QMs have been extensively used as synthetic intermediates with wide-ranging applications in constructing useful chemical structures,7 to the best of our knowledge, there is no example for the hydride transfer-involved cascade reactions of o-QMs.8 Moreover, all the reactions involving o-QMs ended up with thermodynamically stable phenol derivatives and no report was available for dearomative cyclohexaketenes as final products.9 As part of our continuing interest in one-step assembly of molecular complexity via hydride transfer reactions,5d,10 herein, we report a conceptually distinct strategy that is the employment of aromatization of QMs as a driving force to initiate hydride transfer-involved cascade reactions, realizing redox-neutral cascade dearomative cyclizations with cyclohexaketene-embedded polycyclic amines as final products (Scheme 1). It is noteworthy that these architecturally complex structures were inaccessible via a one-step reaction, despite their prevalence in a number of natural products.11
Results and discussion
Optimization of HFIP-promoted dearomatization of sesamol with aminobenzaldehyde
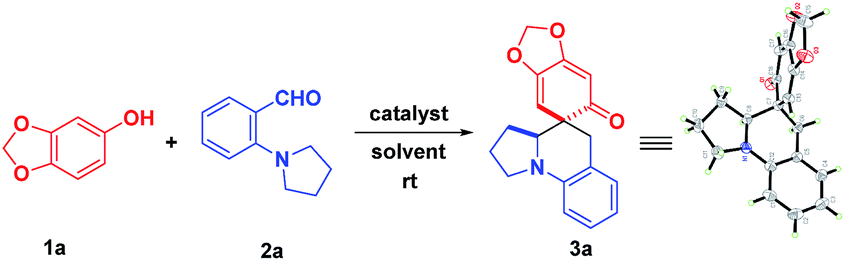
Undoubtedly, the in situ generation of precursors of o-QMs from readily accessible starting materials would be an attractive strategy for future application of o-QMs-involved reactions. Thus, we embarked on our research through screening the reaction between sesamol 1a and 2-(pyrrolidin-1-yl)benzaldehyde 2a, as shown in Table 1.
Table 1 Optimization of cascade dearomative cyclizationsa
Initially, 10 mol% protonic acids were employed in DCE to catalyze this reaction at room temperature (entries 1–3). To our delight, the desired product 3a was obtained when BINOL-derived phosphoric acid and (−)-CSA were used, albeit with low productivities (entries 1–2). Unfortunately, the strong trifluoromethanesulfonic acid (TfOH) did not produce the desired product at all. The structure of 3a was unambiguously confirmed by X-ray crystallographic analysis (see the ESI†). Next, various Lewis acid catalysts were evaluated to improve the reaction efficiency (entries 4–8). It was found that only InCl3 could afford the desired product in satisfying yield after 8 days (entry 4). In view of the unique properties of fluorinated alcohols12 and our recent success in hydrogen-bonding activation by these solvents,13 subsequently, trifluoroethanol (TFE) and hexafluoro-2-propanol (HFIP) were examined at room temperature acting as both promoters and solvents (entries 9–10). Disappointingly, the product was yielded only in 32% yield in TFE (entry 9). Nevertheless, HFIP exhibited good productivity, promoting this cascade reaction to deliver 3a in 81% yield within 12 hours (entry 10). Additionally, exclusive chemoselectivity and excellent diastereoselectivity (>20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were also observed. However, when H2O and a non-fluorinated alcohol such as i-PrOH were used as solvents, they did not take effect at all (entries 11–12). Further screenings with 10 mol% HFIP in i-PrOH (entry 13) or acid catalysts in MeOH (entries 14–16) were unsuccessful, demonstrating that the use of HFIP as a solvent was important. This result implied that the hydrogen bonding network and the hydrogen bond donor ability of HFIP were important for the success of this reaction. With this result in hand, the reaction conditions by varying the concentrations and ratio of starting materials was further investigated. It was found that decreasing concentration was beneficial to this transformation, affording 3a in 88% yield (entries 17–18). However, an attempt to improve the reaction efficiency through adjusting the ratio of starting materials met with failure (entries 19–21). Consequently, the most effective reaction conditions were using HFIP as both the reaction promoter and the solvent, with the ratio of 1a and 2a being 1.3:1.
:1) were also observed. However, when H2O and a non-fluorinated alcohol such as i-PrOH were used as solvents, they did not take effect at all (entries 11–12). Further screenings with 10 mol% HFIP in i-PrOH (entry 13) or acid catalysts in MeOH (entries 14–16) were unsuccessful, demonstrating that the use of HFIP as a solvent was important. This result implied that the hydrogen bonding network and the hydrogen bond donor ability of HFIP were important for the success of this reaction. With this result in hand, the reaction conditions by varying the concentrations and ratio of starting materials was further investigated. It was found that decreasing concentration was beneficial to this transformation, affording 3a in 88% yield (entries 17–18). However, an attempt to improve the reaction efficiency through adjusting the ratio of starting materials met with failure (entries 19–21). Consequently, the most effective reaction conditions were using HFIP as both the reaction promoter and the solvent, with the ratio of 1a and 2a being 1.3:1.
Substrate scope of HFIP-promoted cascade reactions of phenols with aminobenzaldehydes
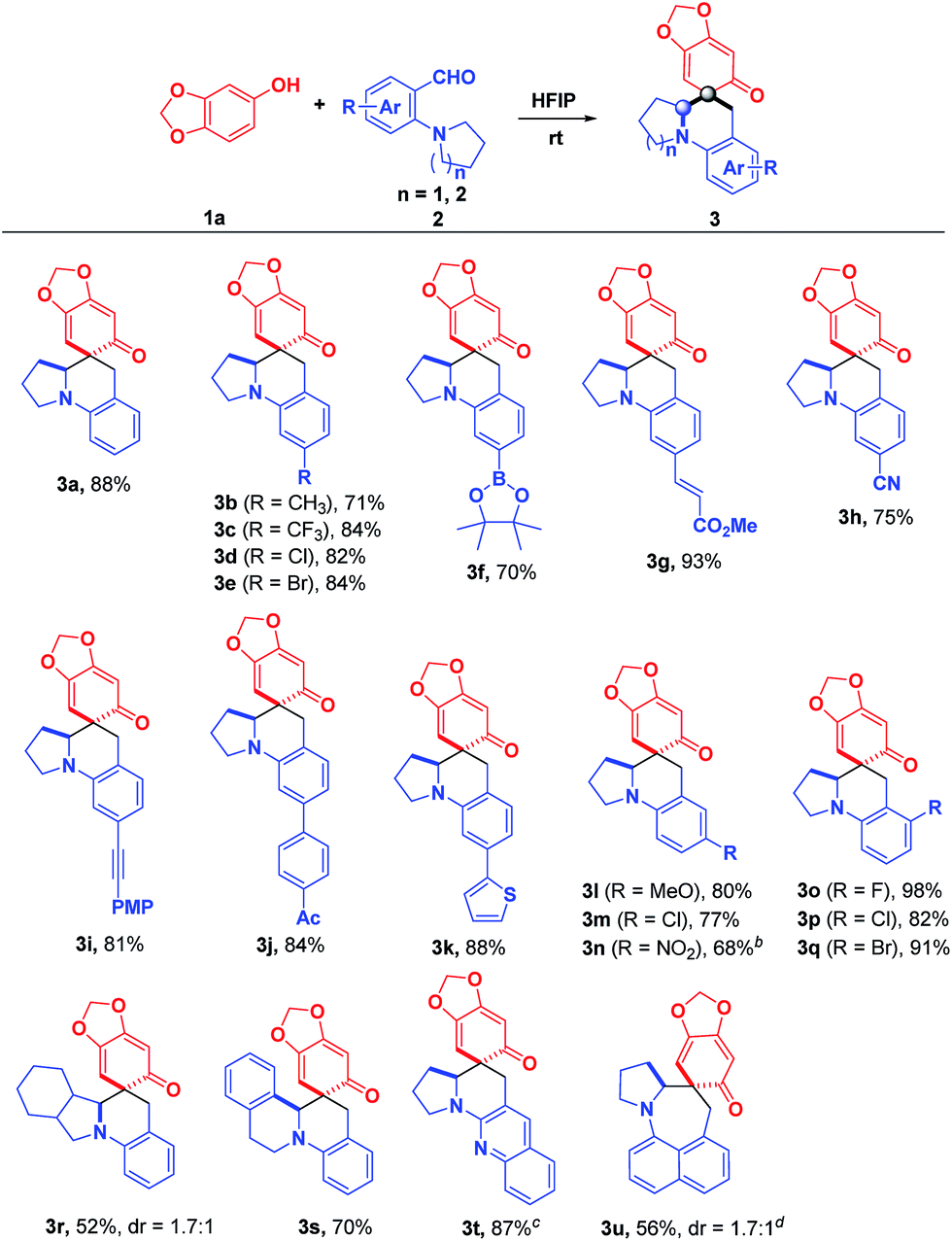
Under the optimized reaction conditions, the substrate scope with regard to o-aminobenzaldehydes 2 was firstly investigated. As shown in Scheme 2, a variety of electronically and sterically diverse substituents were installed on the aromatic ring of 2-(pyrrolidin-1-yl)benzaldehydes to evaluate the generality of this methodology. To our delight, these reactions proceeded smoothly, yielding the corresponding products in moderate to good yields with high stereocontrol (>20:1 dr) in most cases. Notably, the substrates bearing either electron-deficient or electron-rich substituents at the para-position of the formyl group reacted smoothly with sesamol, affording 3b–3k in good to excellent yields. Particularly, a broad range of functional groups, including boronate (3f), alkenyl (3g), cyano (3h), alkynyl (3i), acetyl (3j) and thienyl (3k), were all well tolerated, demonstrating its robust compatibility for preparing diversely functionalized target molecules. Further investigation of the impact of the substituent positions indicated that the substituents at the meta-position of the formyl group also worked nicely, affording the corresponding products 3l–3n in good yields. The sharp contrast between methoxyl and nitro groups illustrated that the strong electron-withdrawing substituent diminished the reactivity, which requires slightly higher reaction temperature and longer reaction time (3l and 3n). Satisfyingly, the substrates 2 with substituents at the ortho-position of the formyl group were also totally compatible, furnishing the desired products 3o–3q in almost quantitative yields. Not surprisingly, a further modulation of the pyrrolidine ring was feasible, affording the privileged polycyclic products 3r and 3s in 52% and 70% yields, respectively. In addition to pyrrolidine and octahydro-isoindole rings, tetrahydroisoquinoline was also a good candidate for this reaction, giving rise to the tetrahydroisoquinoline-fused polycyclic compound 3s in good yield, which is a privileged moiety for medicinal chemistry. It was worth mentioning that the replacement of the phenyl skeleton by a quinoline framework was also feasible, furnishing 3t in 87% yield, albeit with higher temperature. More importantly, the cascade reaction of 1a with 8-(pyrrolidin-1-yl)-1-naphthaldehyde, which involved a 1,6-hydride transfer through space, was also applicable to afford the product 3u in 56% yield.
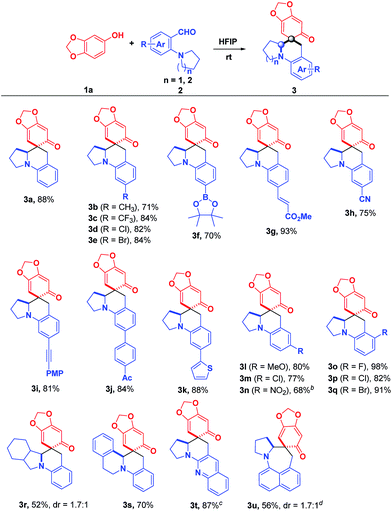 |
| | Scheme 2 Substrate scope: o-aminobenzaldehydes. aReaction conditions: 1a (0.13 mmol) and 2 (0.1 mmol) in 2.0 mL HFIP at room temperature under air; isolated yield; dr > 20:1; dr was determined by 1H NMR spectroscopy. bAt 40 °C. cIn 1.0 mL toluene and 1.0 mL HFIP at 120 °C. d1a (0.13 mmol), 2 (0.1 mmol), and 2.0 equiv. of piperidine were added to 1.0 mL toluene with stirring for 12 h at 120 °C, and then cooled to room temperature followed by the addition of 1.0 mL HFIP and stirred for another 10 min. | |
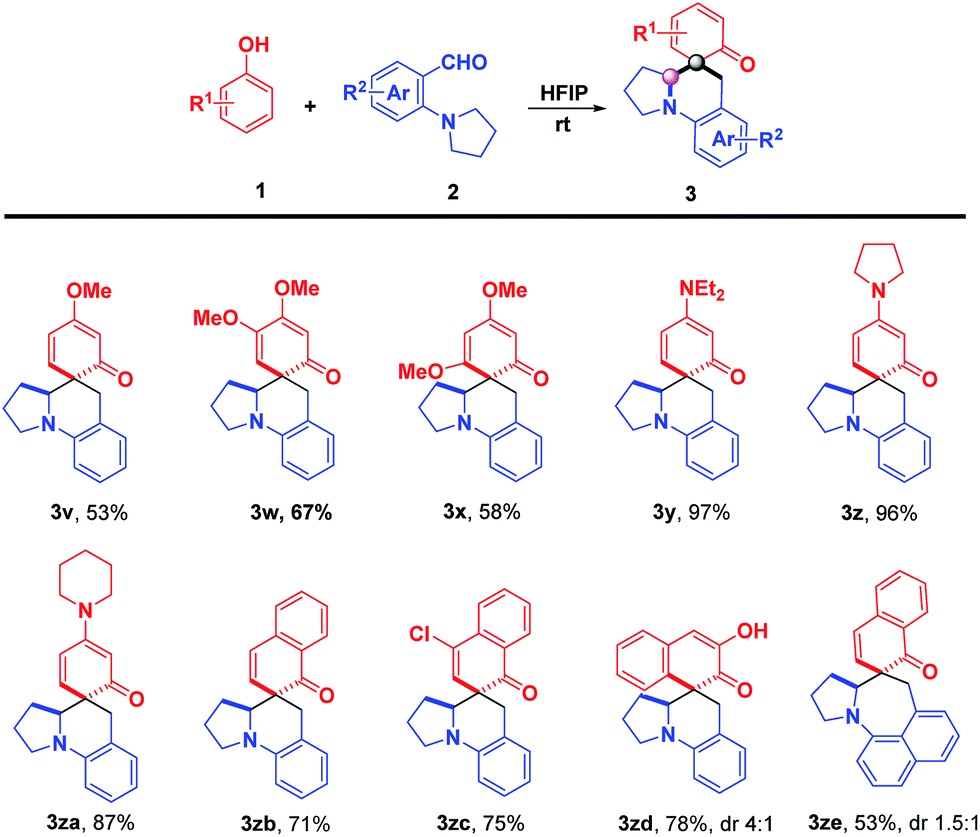
Subsequently, the substrate scope of phenols and their derivatives was tested to examine the generality of the strategy (Scheme 3). Encouragingly, the phenols substituted by electron-donating groups reacted smoothly with 2-(pyrrolidin-1-yl)benzaldehyde 2a, providing spirocyclic amines 3v–3za in moderate to excellent yields with exclusive chemoselectivity and excellent diastereoselectivities (>20:1 dr). Apparently, the electronic characteristics had a significant impact on the reaction efficiency. For instance, substrates with amino groups (3y–3za) led to much higher yields than methoxyl groups (3v–3x). Apart from phenols, 1-naphthols and 2,3-naphthalenediol could also be transformed into α-spirocyclic dearomative products 3zb–3ze in good yields. Unfortunately, 2-naphthols were inert reaction partners, even under reflux temperature. In previous reports, the auxiliary side chain has to be pre-installed at the ortho-position of the hydroxyl group of phenols or naphthols to achieve dearomative 2,4-cyclohexadienone motifs.14 However, in our cases, phenols or naphthols could be directly converted to various dearomative products.
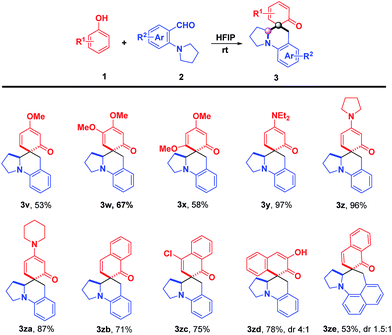 |
| | Scheme 3 Substrate scope: phenols. Reaction conditions: 1 (0.13 mmol) and 2 (0.1 mmol) in 2.0 mL of HFIP at room temperature under air; isolated yield; dr > 20:1; dr was determined by 1H NMR spectroscopy. | |
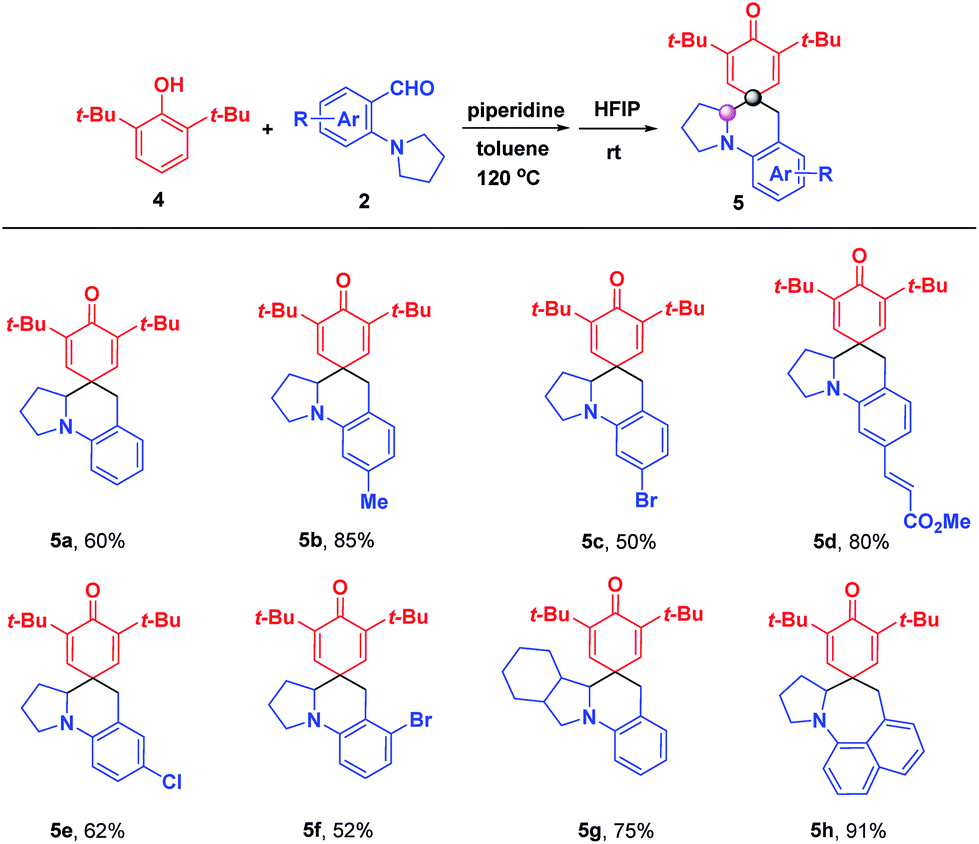
In order to further expand the universality of this strategy, 2,6-di-tert-butylphenol 4 was investigated to synthesize para-spirocyclic ketenes, aiming to solve the scope limitations of ortho-spirocyclic ketene products (Scheme 4). Gratifyingly, the desired reactions occurred smoothly, yielding the corresponding para-spirocyclic ketenes 5a–5h without an adverse effect on the efficiency. In contrast to ortho-spirocyclic ketenes, the addition of piperidine was necessary to accelerate the formation of para-quinone methides (p-QMs).
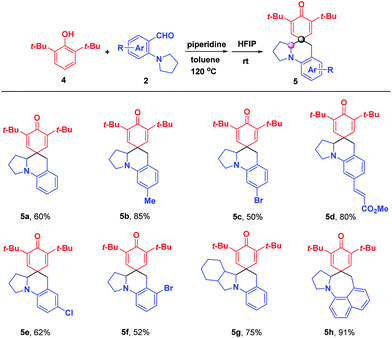 |
| | Scheme 4 Substrate scope for the formation of para-spirocyclic ketenes. 4 (0.13 mmol), 2 (0.1 mmol), and piperidine (2.0 equiv.) were added to 1.0 mL toluene with stirring for 12 h at 120 °C, and then cooled to room temperature followed by the addition of 1.0 mL HFIP and stirred for another 10 min; isolated yield. | |
The synthetic elaboration of this method
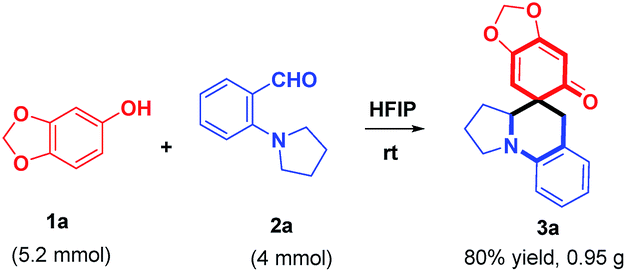
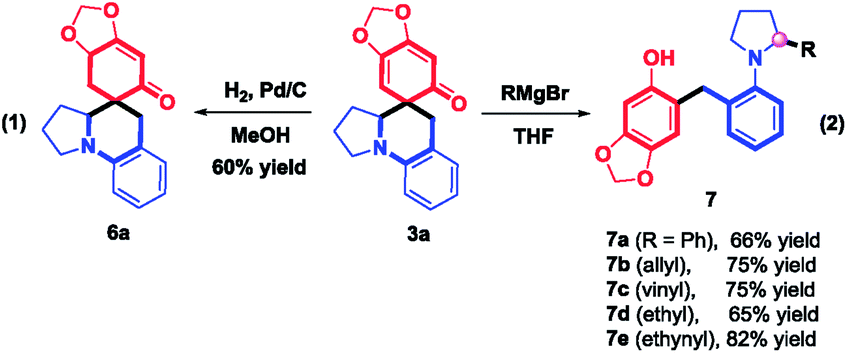
To explore the synthetic utility of this method, a gram-scale reaction between 1a and 2a was carried out in HFIP at room temperature, furnishing 3a in 80% yield (Scheme 5). Further structural manipulation of 3a is summarized in Scheme 6. Interestingly, partially hydrogenated product 6a was produced under an atmosphere of H2 (5% Pd/C). When Grignard reagents were subjected to the THF solution of 3a, benzylated aniline derivatives 7a–e were furnished in 65–82% yields, including the arylation, allylation, vinylation, alkylation, and alkynylation reactions, as different adducts of α-C–H functionalization of amines. The formation of 7a–e might be attributed to the aromatization-driven sequential C–C bond cleavage/iminium trapping, which clearly demonstrated the power of rearomatization. It should be noted that the preparation of benzylated aniline in a regioselective manner is challenging,15 considering that compound 7 is a structural hybrid between ortho-benzylated phenol and ortho-benzylated aniline motifs, which is the core structure of natural products, dyes, medicines and materials.16
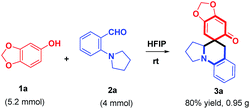 |
| | Scheme 5 Gram scale reaction. | |
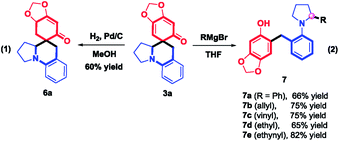 |
| | Scheme 6 Derivatization of products. | |
Mechanistic study
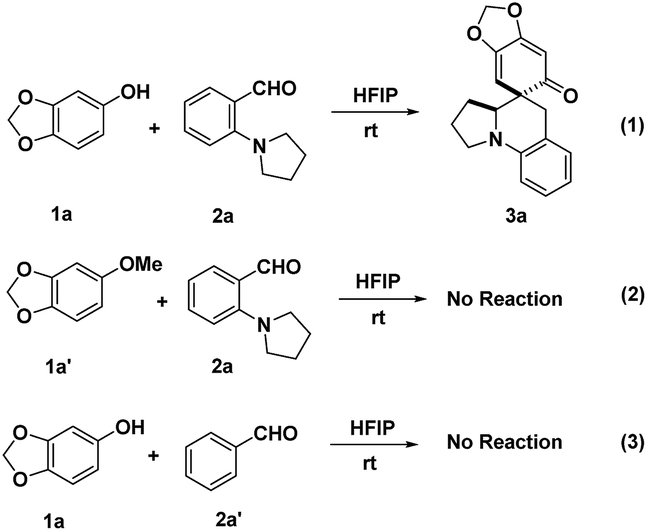
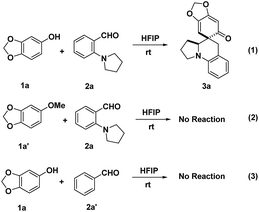
In order to probe into the mechanism of this transformation, a deuterated substrate [D]-2a was prepared and subjected to the optimal conditions (Scheme 7). The deuteration at the benzylic position of product [D]-3a fully corroborated the occurrence of intramolecular [1,5]-hydride transfer (eqn (1)). Furthermore, a deuterium kinetic isotope effect (DKIE) of 1.22 was obtained through a competitive reaction between substrate 2a and [D]-2a, implying that an intramolecular [1,5]-hydride transfer might not be involved in the rate-determining step (eqn (2)). These results illuminated that the in situ generated o-QMs offered a powerful driving force to initiate the intramolecular [1,5]-hydride transfer, which was consistent with the fact that this cascade dearomatization/rearomatization/dearomatization proceeded under really mild conditions. Subsequently, some control experiments were conducted to gain more mechanistic insights (Scheme 8). When substrate 1a′ containing protected hydroxyl group was subjected to the reaction, no desired product was observed, even under elevated temperature (eqn (2)). This result elucidated the indispensability of the hydrogen bonding interaction between the free hydroxyl group and HFIP in facilitating this reaction. More interestingly, the absence of a hydride donor rendered this reaction unproductive, indicating the significance of hydride transfer in triggering the whole sequential process (eqn (3)).
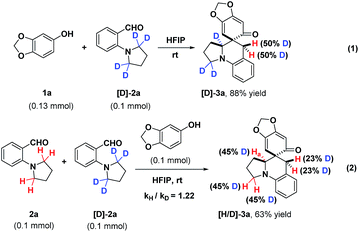 |
| | Scheme 7 Deuterium labeling experiments. | |
 |
| | Scheme 8 Control experiments. | |
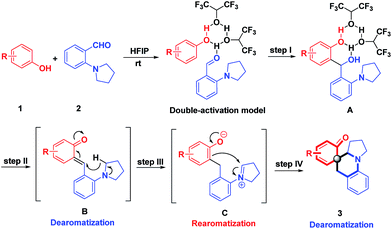
On the basis of these understandings, a plausible mechanism was proposed as shown in Scheme 9. In view of the fascinating properties of HFIP with strong hydrogen bond donor ability and the cationic species stabilization ability,17 we propose that HFIP aggregates reactants 1 and 2 through hydrogen bonding via the double-activation model to generate the nucleophilic addition product A. Assisted by the hydrogen-bonding cluster of HFIP, the benzyl alcohol A undergoes dehydration and dearomatization immediately to furnish o-QMs. The propensity of o-QMs to rearomatize acts as a powerful driving force to trigger [1,5]-hydride transfer to afford the rearomatic intermediate C, which is followed by nucleophilic dearomatization, generating dearomative cyclohexaketenes as final products. One of the major challenges in this reaction is that three possible reaction pathways might operate: the Friedel–Crafts reaction between phenol and A, O-alkylation of intermediate C, and a C-Pictet–Spengler type reaction of intermediate C. Notably, the chemospecificity increases its practical application in medicinal chemistry. In the whole process, the hydrogen bonding network of HFIP plays the key role in generating and stabilizing all the intermediates as well as the well-organized transition states. Recently, combined computational and experimental evidence has revealed that the higher-order aggregates of HFIP are decisive factors to promote the reactions.18
 |
| | Scheme 9 Plausible reaction mechanism. | |
Conclusions
In conclusion, we have developed cascade hydride transfer/dearomative cyclizations for one-step construction of structurally diverse polycyclic amines in good yields with good functional group compatibilities and high diastereoselectivities. This redox-neutral cascade process addressed several formidable challenges of cascade hydride transfer/cyclization with the following prominent advantages: (1) in situ generated o-QMs as novel hydride acceptors and the use of aromatization as the driving force to initiate the hydride transfer without resorting to electron-withdrawing groups; (2) this reaction offers a new reaction mode of o-QMs, which were trapped by a hydride and for the first time provides dearomative cyclohexaketenes as final products; (3) a 4-step cascade dearomatization/rearomatization/dearomatization process with high atom- and step economy for efficient construction of architecturally complex polycyclic amines; (4) mild reaction conditions with operational simplicity, readily available reactants and good functional group tolerance. We are optimistic that this reaction will not only offer synthetic chemists a distinctive toolbox for the generation of diverse polycyclic amine libraries in drug discovery, but also facilitate the one-step assembly of complex molecules via a redox-neutral strategy.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for financial support from the NSFC (No. 21702117, 21878167, U1706210) and the Natural Science Foundation of Shandong Province for Distinguished Young Scholars (JQ201604) and the General Project (ZR2017BB005). Financial support from the Key Research and Development Program of Shandong Province (2017GSF218073) is also gratefully acknowledged. We thank Prof. Xingwei Li (Shanxi Normal University), Prof. Bin Tan (Southern University of Science and Technology) and Prof. Mengchun Ye (Nankai University) for valuable suggestions.
Notes and references
-
(a) K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069 RSC;
(b) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902 RSC;
(c) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC;
(d) E. A. Mitchell, A. Peschiulli, N. Lefevre, L. Meerpoel, B. Maes and U. W. Chem, Chem.–Eur. J., 2012, 18, 10092 CrossRef CAS PubMed;
(e) C. V. T. Vo and J. W. Bode, J. Org. Chem., 2014, 79, 2809 CrossRef CAS PubMed;
(f) D. Seidel, Acc. Chem. Res., 2015, 48, 317 CrossRef CAS PubMed;
(g) S. Mahato and C. K. Jana, Chem. Rec., 2016, 16, 1477 CrossRef CAS PubMed.
-
(a) M. Tobisu and N. Chatani, Angew. Chem., Int. Ed., 2006, 45, 1683 CrossRef CAS PubMed;
(b) B. Peng and N. Maulide, Chem.–Eur. J., 2013, 19, 13274 CrossRef CAS PubMed;
(c) L. Wang and J. Xiao, Adv. Synth. Catal., 2014, 356, 1137 CrossRef CAS;
(d) L. Wang and J. Xiao, Top. Curr. Chem., 2016, 374, 17 CrossRef PubMed;
(e) L. Wang and J. Xiao, Org. Chem. Front., 2016, 3, 635 RSC;
(f) S. C. Pan, Beilstein J. Org. Chem., 2012, 8, 1374 CrossRef CAS PubMed;
(g) M. C. Haibach and D. Seidel, Angew. Chem., Int. Ed., 2014, 53, 5010 CrossRef CAS PubMed;
(h) M. Xiao, S. Zhu, Y. Shen, L. Wang and J. Xiao, Chin. J. Org. Chem., 2018, 38, 328 CrossRef;
(i) S. J. Kwon and D. Y. Kim, Chem. Rec., 2016, 16, 1191 CrossRef CAS PubMed.
- For examples of electrophilic alkenes, see:
(a) S. J. Pastine, K. M. McQuaid and D. Sames, J. Am. Chem. Soc., 2005, 127, 12180 CrossRef CAS PubMed;
(b) S. Murarka, C. Zhang, M. D. Konieczynska and D. Seidel, Org. Lett., 2009, 11, 129 CrossRef CAS PubMed;
(c) Y. Han, W. Han, X. Hou, X. Zhang and W. Yuan, Org. Lett., 2012, 14, 4054 CrossRef CAS PubMed;
(d) T. Yoshida and K. Mori, Chem. Commun., 2017, 53, 4319 RSC;
(e) J. C. Ruble, A. R. Hurd, T. A. Johnson, D. A. Sherry, M. R. Barbachyn, P. L. Toogood, G. L. Bundy, D. R. Graber and G. M. Kamilar, J. Am. Chem. Soc., 2009, 131, 3991 CrossRef CAS PubMed;
(f) K. Mori, K. Ehara, K. Kurihara and T. Akiyama, J. Am. Chem. Soc., 2011, 133, 6166 CrossRef CAS PubMed;
(g) C. W. Suh and D. Y. Kim, Org. Lett., 2014, 16, 5374 CrossRef CAS PubMed;
(h) K. Mori, K. Kurihara, S. Yabe, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2014, 136, 3744 CrossRef CAS PubMed;
(i) K. Mori, R. Isogai, Y. Kamei, M. Yamanaka and T. Akiyama, J. Am. Chem. Soc., 2018, 140, 6203 CrossRef CAS PubMed.
- For representative examples, see:
(a) I. D. Jurberg, B. Peng, E. Woestefeld, M. Wasserloos and N. Maulide, Angew. Chem., Int. Ed., 2012, 51, 1950 CrossRef CAS PubMed;
(b) D. Chen, Z. Han, Y. He, J. Yu and L.-Z. Gong, Angew. Chem., Int. Ed., 2012, 51, 12307 CrossRef CAS PubMed;
(c) P. A. Vadola and D. Sames, J. Am. Chem. Soc., 2009, 131, 16525 CrossRef CAS PubMed;
(d) J. Barluenga, M. Fananas-Mastral, F. Aznar and C. Valdes, Angew. Chem., Int. Ed., 2008, 47, 6594 CrossRef CAS PubMed;
(e) T. Sugiishi and H. Nakamura, J. Am. Chem. Soc., 2012, 134, 2504 CrossRef CAS PubMed;
(f) C. Theunissen, B. Metayer, N. Henry, G. Compain, J. Marrot, A. Martin-Mingot, S. Thibaudeau and G. Evano, J. Am. Chem. Soc., 2014, 136, 12528 CrossRef CAS PubMed;
(g) L. Cui, Y. Peng and L. M. Zhang, J. Am. Chem. Soc., 2009, 131, 8394 CrossRef CAS PubMed;
(h) Y. K. Kang, S. M. Kim and D. Y. Kim, J. Am. Chem. Soc., 2012, 132, 11847 CrossRef PubMed.
- For recently developed hydride acceptors, see:
(a) Y.-Z. Chang, M.-L. Li, W.-F. Zhao, X. Wen, H. Sun and Q.-L. Xu, J. Org. Chem., 2015, 80, 9620 CrossRef CAS PubMed;
(b) C. W. Suh, S. J. Kwon and D. Y. Kim, Org. Lett., 2017, 19, 1334 CrossRef CAS PubMed;
(c) S. Liu, J. Qu and B. Wang, Chem. Commun., 2018, 54, 7928 RSC;
(d) S.-S. Li, L. Zhou, L. Wang, H. Zhao, L. Yu and J. Xiao, Org. Lett., 2018, 20, 138 CrossRef CAS PubMed;
(e) F. I. M. Idiris, C. E. Majesté, G. B. Craven and C. R. Jones, Chem. Sci., 2018, 9, 2873 RSC ; For an intermolecular hydride transfer, see: ;
(f) W. Chen, L. Ma, A. Paul and D. Seidel, Nat. Chem., 2018, 10, 165 CrossRef CAS PubMed.
-
(a) X.-S. Xue, P. Ji, B. Zhou and J.-P. Cheng, Chem. Rev., 2017, 117, 8622 CrossRef CAS PubMed;
(b) Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433 CrossRef CAS PubMed.
- For selected recent reviews, see:
(a) W.-J. Bai, J. G. David, Z.-G. Feng, M. G. Weaver, K.-L. Wu and T. R. R. Pettus, Acc. Chem. Res., 2014, 47, 3655 CrossRef CAS PubMed;
(b) W. Ai, D. Liao and X. Lei, Chin. J. Org. Chem., 2015, 35, 1615 CrossRef CAS;
(c) Z. Wang and J. Sun, Synthesis, 2015, 47, 3629 CrossRef;
(d) T. P. Pathak and M. S. Sigman, J. Org. Chem., 2011, 76, 9210 CrossRef CAS PubMed;
(e) N. J. Willis and C. D. Bray, Chem.–Eur. J., 2012, 18, 9160 CrossRef CAS PubMed.
- For the only example of transfer hydrogenation of o-QMs, see: Z. Wang, F. Ai, Z. Wang, W. Zhao, G. Zhu, Z. Lin and J. Sun, J. Am. Chem. Soc., 2015, 137, 383 CrossRef CAS PubMed.
- For selected examples, see:
(a) J. Izquierdo, A. Orue and K. A. Scheidt, J. Am. Chem. Soc., 2013, 135, 10634 CrossRef CAS PubMed;
(b) H. Lv, W.-Q. Jia, L.-H. Sun and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 8607 CrossRef CAS PubMed;
(c) O. El-Sepelgy, S. Haseloff, S. K. Alamsetti and C. Schneider, Angew. Chem., Int. Ed., 2014, 53, 7923 CrossRef CAS PubMed;
(d) C.-C. Hsiao, H.-H. Liao and M. Rueping, Angew. Chem., Int. Ed., 2014, 53, 13258 CrossRef CAS PubMed;
(e) J.-J. Zhao, S.-B. Sun, S.-H. He, Q. Wu and F. Shi, Angew. Chem., Int. Ed., 2015, 54, 5460 CrossRef CAS PubMed;
(f) S. K. Alamsetti, M. Spanka and C. Schneider, Angew. Chem., Int. Ed., 2016, 55, 2392 CrossRef CAS PubMed;
(g) P. Chen, K. Wang, W. Guo, X. Liu, Y. Liu and C. Li, Angew. Chem., Int. Ed., 2017, 56, 3689 CrossRef CAS PubMed;
(h) X. Wu, L. Xue, D. Li, S. Jia, J. Ao, J. Deng and H. Yan, Angew. Chem., Int. Ed., 2017, 56, 13722 CrossRef CAS PubMed;
(i) Y. Xie and B. List, Angew. Chem., Int. Ed., 2017, 56, 4936 CrossRef CAS PubMed.
- S. Zhu, C. Chen, M. Xiao, L. Yu, L. Wang and J. Xiao, Green Chem., 2017, 19, 5653 RSC.
-
(a) Q. Zhu, C.-P. Tang, C.-Q. Ke, X.-Q. Li, J. Liu, L.-S. Gan, H.-C. Weiss, E.-R. Gesing and Y. Ye, J. Nat. Prod., 2010, 73, 40 CrossRef CAS PubMed;
(b) C. Rouger, S. Derbre, B. Charreau, A. Pabois, T. Cauchy, M. Litaudon, K. Awang and P. Richomme, J. Nat. Prod., 2015, 78, 2187 CrossRef CAS PubMed.
- For recent reviews, see:
(a) I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 88 CrossRef CAS;
(b) J. Wencel-Delord and F. Colobert, Org. Chem. Front., 2016, 3, 394 RSC.
-
(a) H. Wen, L. Wang, L. Xu, Z. Hao, C.-L. Shao, C.-Y. Wang and J. Xiao, Adv. Synth. Catal., 2015, 357, 4023 CrossRef CAS;
(b) J. Liu, L. Wang, X. Wang, L. Xu, Z. Hao and J. Xiao, Org. Biomol. Chem., 2016, 14, 11510 RSC.
-
(a) A. R. Pape, K. P. Kaliappan and E. P. Kundig, Chem. Rev., 2000, 100, 2917 CrossRef CAS PubMed;
(b) W.-T. Wu, L. Zhang and S.-L. You, Chem. Soc. Rev., 2016, 45, 1570 RSC.
-
(a) L. L. Anderson, J. Arnold and R. G. Bergman, J. Am. Chem. Soc., 2005, 127, 14542 CrossRef CAS PubMed;
(b) X. Hu, D. Martin, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2014, 136, 13594 CrossRef CAS PubMed;
(c) J. Zhu, M. Perez, C. B. Caputo and D. W. Stephan, Angew. Chem., Int. Ed., 2016, 55, 1417 CrossRef CAS PubMed;
(d) M. Amezquita-Valencia and H. Alper, Chem.–Eur. J., 2016, 22, 16774 CrossRef CAS PubMed.
-
(a) T. Kimura, J. Hosokawa-Muto, Y. O. Kamatari and K. Kuwata, Bioorg. Med. Chem. Lett., 2011, 21, 1502 CrossRef CAS PubMed;
(b) S. Hosoda, A. Tanatani, K. Wakabayashi, Y. Nakano, H. Miyachi, K. Nagasawa and Y. Hashimoto, Bioorg. Med. Chem. Lett., 2005, 15, 4327 CrossRef CAS PubMed.
- For selected examples of HFIP-promoted reactions, see:
(a) M. C. DiPoto, R. P. Hughes and J. Wu, J. Am. Chem. Soc., 2015, 137, 14861 CrossRef CAS PubMed;
(b) P. A. Champagne, Y. Benhassine, J. Desroches and J.-F. Paquin, Angew. Chem., Int. Ed., 2014, 53, 13835 CrossRef CAS PubMed;
(c) V. D. Vukovic, E. Richmond, E. Wolf and J. Moran, Angew. Chem., Int. Ed., 2017, 56, 3085 CrossRef CAS PubMed;
(d) A. M. Mfuh, V. T. Nguyen, B. J. Chhetri, E. Burch, J. D. Doyle, V. N. Nesterov, H. D. Arman and O. V. Larionov, J. Am. Chem. Soc., 2016, 138, 8408 CrossRef CAS PubMed;
(e) I. Colomer, C. Batchelor-McAuley, B. Odell, T. J. Donohoe and R. G. Compton, J. Am. Chem. Soc., 2016, 138, 8855 CrossRef CAS PubMed;
(f) A. Libman, H. Shalit, Y. Vainer, S. Narute, S. Kozuch and D. Pappo, J. Am. Chem. Soc., 2015, 137, 11453 CrossRef CAS PubMed;
(g) X. Zeng, J. Li, C. K. Ng, G. B. Hammond and B. Xu, Angew. Chem., Int. Ed., 2018, 57, 2924 CrossRef CAS PubMed;
(h) A. M. Arnold, A. Pöthig, M. Drees and T. Gulder, J. Am. Chem. Soc., 2018, 140, 4344 CrossRef CAS PubMed.
- A. Berkessel, J. A. Adrio, D. Hüttenhain and J. M. Neudörfl, J. Am. Chem. Soc., 2006, 128, 8421 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1829304. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03339k |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Xintong
Lv
a,
Didi
Ren
a,
Chang-Lun
Shao
b,
Qing
Liu
c and
Jian
Xiao
a,
Xintong
Lv
a,
Didi
Ren
a,
Chang-Lun
Shao
b,
Qing
Liu
c and
Jian
Xiao