
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diarylmethane synthesis through Re2O7-catalyzed bimolecular dehydrative Friedel–Crafts reactions†
Qi
Qin
b,
Youwei
Xie
*a and
Paul E.
Floreancig
 *b
*b
aSchool of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, P. R. China. E-mail: xieyw@hust.edu.cn
bDepartment of Chemistry, University of Pittsburgh, Pittsburgh, Pennsylvania 15260, USA. E-mail: florean@pitt.edu
First published on 13th September 2018
Abstract
This manuscript describes the application of Re2O7 to the syntheses of diarylmethanes from benzylic alcohols through solvolysis followed by Friedel–Crafts alkylation. The reactions are characterized by broad substrate scope, low catalyst loadings, high chemical yields, and minimal waste generation. The intermediate perrhenate esters are superior leaving groups to chlorides and bromides in these reactions. The polarity and water sequestering capacity of hexafluoroisopropyl alcohol are critical to the success of these processes. Re2O7 is a precatalyst for HOReO3, which serves as a less costly and easily handled promoter for these reactions. Oxorhenium catalysts selectively activate alcohols in the presence of similarly substituted acetates, indicating a unique chemoselectivity and mechanism in comparison to Brønsted acid catalysis.
Introduction
A report in 2007 from the American Chemical Society Green Chemistry Institute Pharmaceutical Roundtable Research Grant Program identified “OH activation for nucleophilic substitution” as a process for which reactions are currently used but better reagents are preferred.1 This can be achieved by transiently enhancing the hydroxy group's nucleofugacity through association with an electrophilic catalyst. The benefit of this approach to substitution chemistry lies in the avoidance of a distinct step to generate a leaving group and through the generation of water as an environmentally benign by-product. A number of recent reviews have highlighted the benefits of dehydrative coupling reactions in complex molecule synthesis.2 However, alcohol activation to generate minimally-stabilized carbocations remains rare. The Hall group made a notable advance in this area by showing3 that benzylic alcohols bearing electron-withdrawing groups such as –CF3 or –CO2Me ionize efficiently in the presence of a ferrocenyl boronic acid catalyst in (CF3)2CHOH (HFIP) and CH3NO2 to form benzylic cations that react with arenes in Friedel–Crafts reactions. The Moran group subsequently showed4 that previously inaccessible benzylic cations can be formed through TfOH-mediated benzylic alcohol ionization in HFIP at elevated temperatures.Our studies in the applications of Re2O7 to allylic alcohol transposition reactions5,6 resulted in a new dehydrative cyclization protocol in which allylic cations react with pendent hydroxy groups.7,8 Re2O7 proved to be superior to Brønsted acids for promoting these processes, leading us to speculate that it could be an effective catalyst for enabling bimolecular dehydrative coupling reactions. This manuscript describes the realization of this objective by demonstrating that Re2O7 acts as an exceptional catalyst for converting benzylic alcohols to carbocations. This serves as an entry to efficient, green Friedel–Crafts reactions with broad substrate scope at low catalyst loadings, which is another transformation in the “reactions are currently used but better reagents are preferred” list. Experiments highlight the role of HFIP in promoting efficient reactivity and define the reactivity of perrhenate as a leaving group in ionization processes.
Results and discussion
p-Fluorobenzyl alcohol (1) served as the initial substrate for this study (Scheme 1) since a fluoro group (σp = +0.06)9 will provide modest destabilization for the intermediate benzylic cation. Exposing 1 to 10% (w/w) Re2O7·SiO2 (ref. 5c) (1 mol%) and p-xylene (3 equiv.) in HFIP (0.5 M) provided diarylmethane 3 in 85% yield within 6 h at rt. Re2O7·SiO2 was used as the catalyst rather than crystalline Re2O7 because it can be measured more easily for reactions in which low quantities of catalyst are required. Conducting the reaction at 80 °C resulted in the reaction being complete within 2 h while delivering 3 in 89% yield. A reasonable pathway for this process involves the conversion of 1 to perrhenate ester 4 by reaction with Re2O7 or HOReO3. The ionization of 4 to form cation 5 followed by the addition of 2 yields diarylmethane 3. The weak nucleophilicity arenes and the substituent effects shown below strongly implicate an ionization-based mechanism.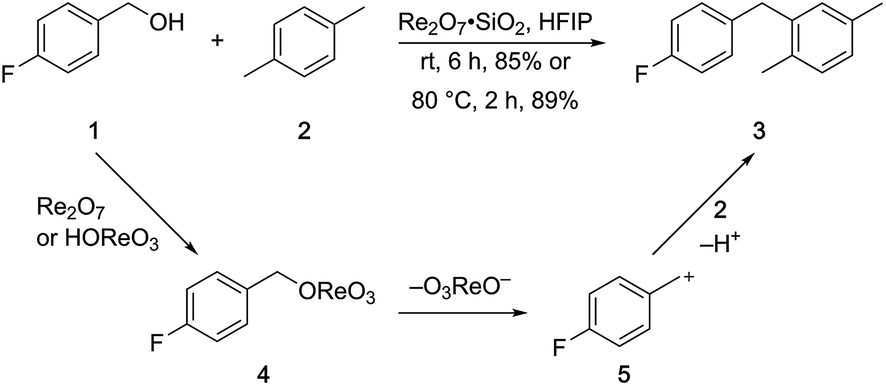 | ||
| Scheme 1 Re2O7-mediated dehydrative Friedel–Crafts alkylation. | ||
A partial scope of the process is shown in Fig. 1. These transformations use p-xylene as the nucleophile (3 equiv.), HFIP as the solvent (used as supplied from the source), and Re2O7·SiO2 as the catalyst (1 mol%) with the alcohol concentration being 0.5 M. The reactions were conducted in a sealed vial at 80 °C unless otherwise noted (oil bath temperature) and were stopped after 2 h without monitoring progress.
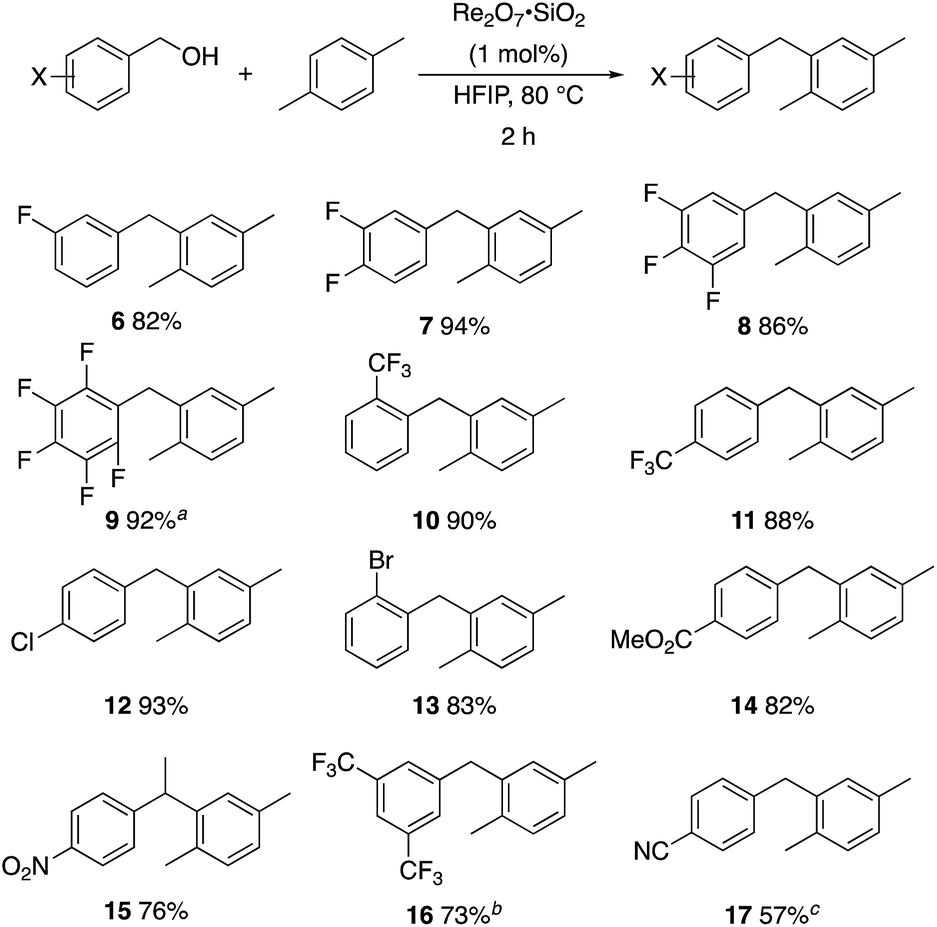 | ||
| Fig. 1 Representative scope of the dehydrative diarylmethane synthesis. aReaction was conducted at 100 °C for 24 h. The product was isolated in 95% yield when the reaction was conducted at 80 °C for 5 h with crystalline Re2O7. bReaction was conducted with crystalline Re2O7 for 23 h. cReaction was conducted with crystalline Re2O7 for 48 h. | ||
The reaction is effective for regioisomeric fluorobenzylic alcohols and polyfluorinated substrates. This is noteworthy because of the strong inductive cation destabilization displayed by m-fluoro groups (σμ = +0.34). Pentafluorobenzyl alcohol reacts somewhat more sluggishly but it can be converted to the diarylmethane efficiently at slightly higher temperature and over a longer reaction time. Other inductively electron-withdrawing moieties, such as trifluoromethyl groups (σp = +0.54) do not suppress ionization. Chloro and bromo groups (σp = +0.23) are also tolerated in the processes. Groups that moderately destabilize the intermediate cation through conjugation, such as the carbomethoxy group (σp = +0.45), do not suppress ionization. p-Cyanobenzyl alcohol is not a suitable substrate for this process under the standard conditions (σp = +0.66 for the cyano group), but can be employed if crystalline Re2O7 is employed rather than Re2O7·SiO2. We postulate that the SiO2, while facilitating reagent handling, provides a buffering effect that diminishes reactivity. Crystalline Re2O7 can also be used to effect ionization of 3,5-bis-trifluoromethylbenzyl alcohol (σμ = +0.43 for the CF3 group), though this reaction requires heating for 2 d to achieve a moderate product yield. p-Nitrobenzyl alcohol (σp = +0.78 for the nitro group) reacts very slowly (39% after 2 d at 100 °C, not shown), though p-nitrophenethyl alcohol reacts smoothly under the standard reaction conditions due to the cation stabilizing effect of the additional methyl group. Small amounts of dialkylated products are also formed in these reactions though generally in less than 10% yield.
A study of initial reaction rates showed that Re2O7·SiO2 is slightly less reactive than Re2O7 in promoting reactions with most substrates. Our prior studies on the use of Re2O7·SiO2,5c which is simply prepared from stirring Re2O7 with SiO2 in Et2O followed by solvent evaporation, showed that the reclaimed catalyst was less potent in subsequent transformations. This indicates that catalytic activity arises from Re2O7 leaching from the SiO2 surface. This was tested (Scheme 2) by stirring Re2O7·SiO2 in HFIP at 80 °C for 30 min and removing the silica gel by filtration (PTFE filter). Compounds 1 and 2 were then added to the filtrate to generate 3 in 82% yield, indicating that Re2O7 is released from SiO2 into solution to a sufficient degree to promote the reaction of a moderately reactive substrate. This behavior is distinct from agents in which Re2O7 is grafted onto oxygenated surfaces at high temperature.10 We therefore postulate that the difference in reactivity between crystalline Re2O7 and Re2O7·SiO2 arises from the lower concentrations of the relevant catalyst in solution due to incomplete release from or readsorption onto the SiO2 surface.
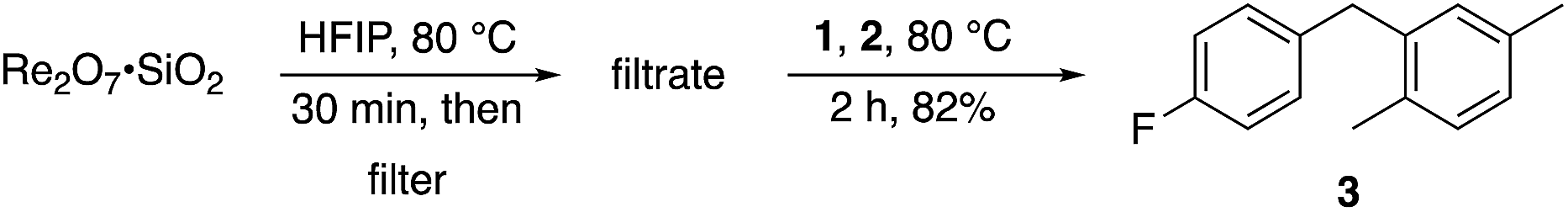 | ||
| Scheme 2 Demonstration of catalyst release from SiO2. | ||
We next studied the reactivity of various simple arene nucleophiles in this reaction. This study utilized pentafluorobenzyl alcohol (18) as a relatively unreactive substrate. The results are shown in Table 1. Reaction rates generally correlated with expected nucleophilicity trends. Thus benzene reacts efficiently but significantly more slowly than p-xylene. Increasing the number of methyl groups leads to faster reactions. Reactions with nucleophiles that have more than one reactive site lead to regioisomeric product mixtures. Naphthalene also serves as a competent nucleophile, with reaction rates that are comparable to methylated benzene derivatives. Anisole reacts at a rate that is comparable to toluene, despite the superior electron donating ability of the methoxy group in comparison to a methyl group. We postulate that the HFIP forms hydrogen bonds to the oxygen of the methoxy group, thereby inhibiting its capacity to act as an electron donor. Regiocontrol was not high in these processes, as expected for electrophilic aromatic substitution reactions with extremely reactive electrophiles.
|
|
|||
|---|---|---|---|
| Entry | Product | Yieldb | Temp, t |
| a Reactions proceeded with pentafluorobenzyl alcohol (1 equiv.), the nucleophilic arene (3–5 equiv.), and crystalline Re2O7 in HFIP at the indicated temperatures and for the indicated times. b Yields refer to purified materials. Ratios were determined by 1H NMR. | |||
| 1 |
|
92% | 100 °C, 72 h |
| 2 |
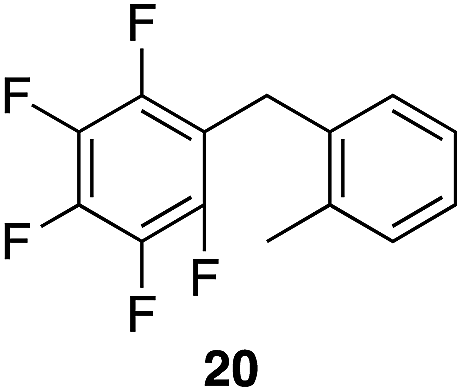
|
99%, 1.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0:1.7, o:m:p :1.0:1.7, o:m:p |
80 °C, 24 h |
| 3 |
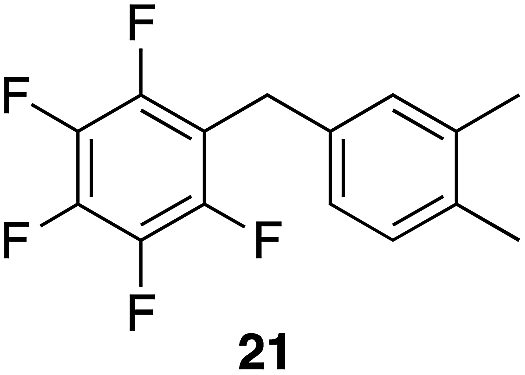
|
97%, 3:2, 1,2,4:1,2,3 |
80 °C, 5 h |
| 4 |
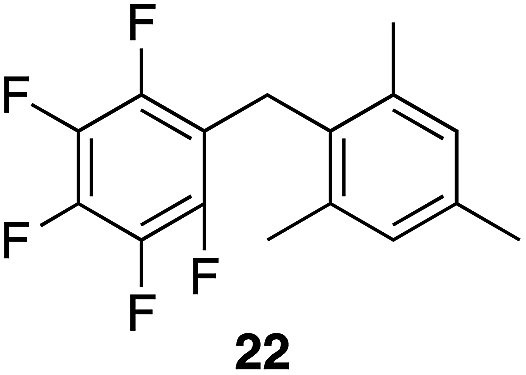
|
99% | 80 °C, 4 h |
| 5 |
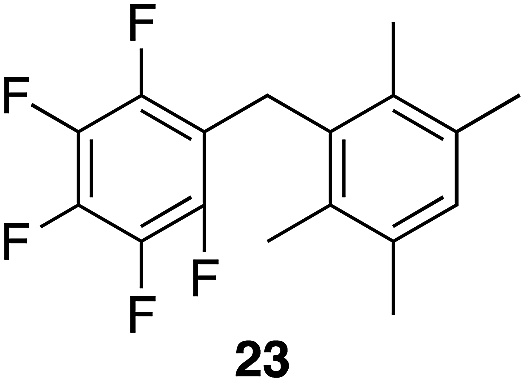
|
97% | 80 °C, 4 h |
| 6 |
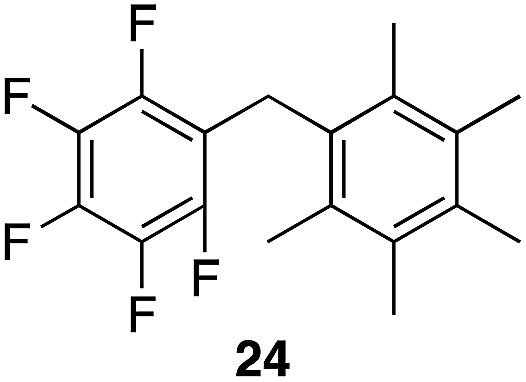
|
94% | 80 °C, 4 h |
| 7 |
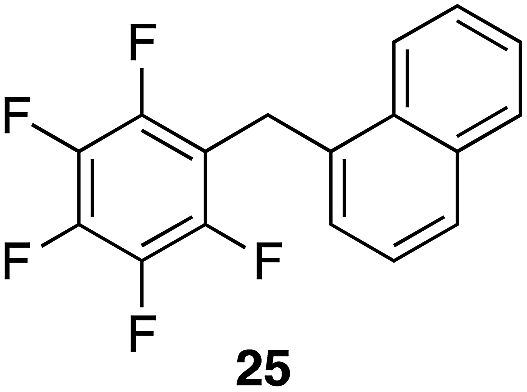
|
97%, 3:1, α:β |
80 °C, 4 h |
| 8 |
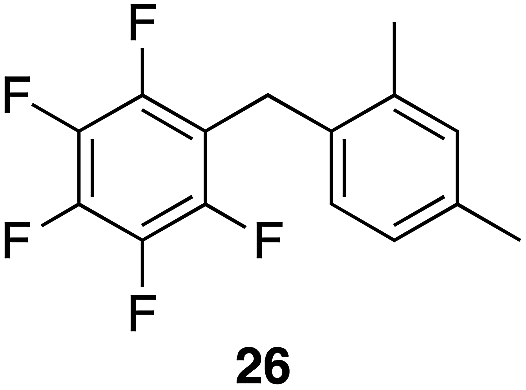
|
97%, 8.4:2.0:1.0, 1,2,4:1,2,6:1,3,5 |
80 °C, 5 h |
| 9 |
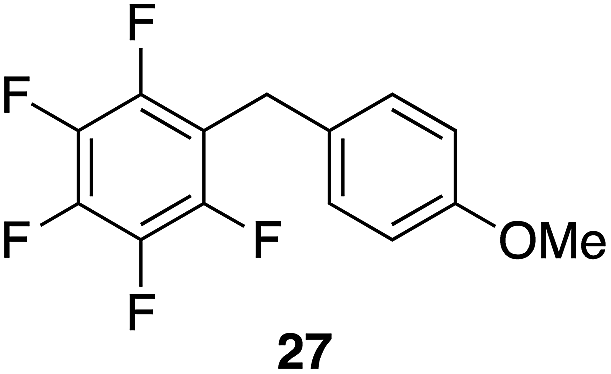
|
98%, 2.8:1, p:o |
80 °C, 24 h |
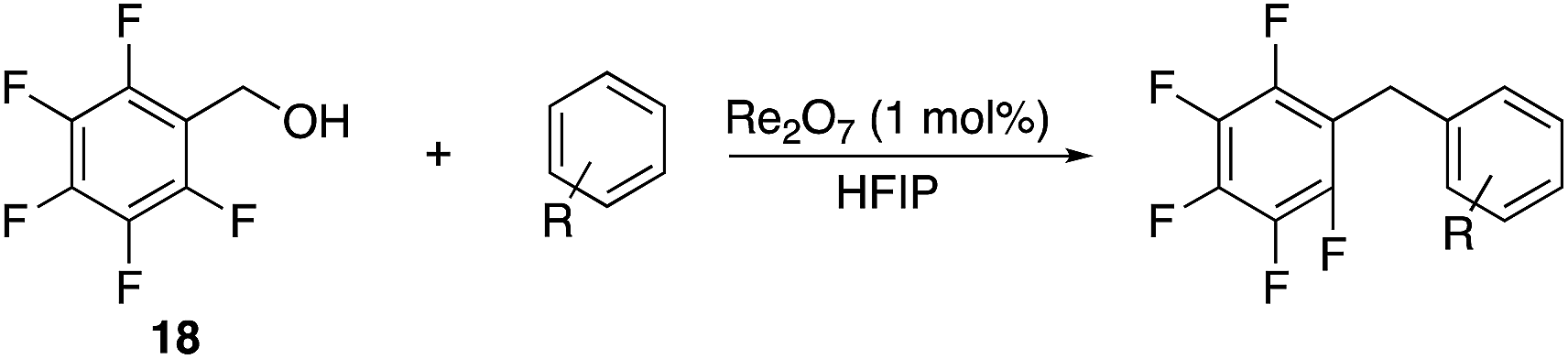
The high electrophilicity of the intermediate carbocations in these processes led us to explore haloarene nucleophiles (Table 2). Haloarenes were predominantly used as the nucleophiles in these reactions due to the capacity to convert the products to other materials through cross-coupling reactions and because of the established benefits of incorporating fluorine into pharmaceutical agents.11 The reactions with weaker nucleophiles were somewhat less efficient, due to increased polyalkylation resulting from the formation of products that are less sterically hindered than those derived from p-xylene, regioisomeric mixtures were produced,12 as expected for Friedel–Crafts reactions on halobenzenes, with fluorobenzene being more selective for para-additions than the other halobenzenes. Despite these issues the reactions delivered useful yields of the desired products. Mayr proposed that arene nucleophilicity correlates with σ+ Hammett values.13 The success of fluoro- (σ+ = −0.07, entries 1, 4, and 5), chloro- (σ+ = +0.12), bromo- (σ+ = +0.15), and iodo- (σ+ = +0.14) benzenes, coupled with our observation of only trace amounts of the adduct with methyl benzoate (σ+ = +0.32) by LC-MS (not shown), suggest that the σ+ cut-off for substituents to show useful reactivity is approximately +0.2. Regiocontrol can be dictated by avoiding steric interactions, as seen in the selective formation of 33 (entry 8).12 The capacity to remove the tert-butyl group through retro Friedel–Crafts chemistry14 make this a potentially powerful approach to preparing ortho-disubstituted products.15
| Entry | Arene | Alcohol | Product | Yieldb, p:o |
|---|---|---|---|---|
| a All reactions were run with 10% Re2O7·SiO2 (1 mol%) in HFIP (0.5 M) with three equiv. of the nucleophile. b Isolated yields of regioisomeric mixtures. Ratios were determined by 1H NMR. | ||||
| 1 |
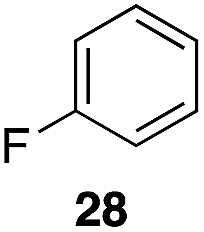
|
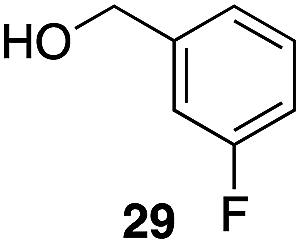
|
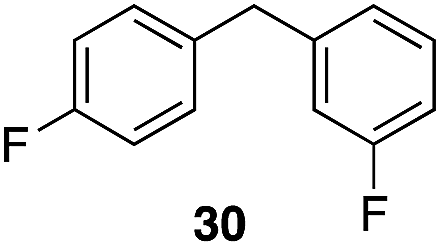
|
40%, 1.6:1 |
| 2 |
|
29 |
|
46%, 1:1 |
| 3 |
|
29 |
|
62%, 1.2:1 |
| 4 | 28 |
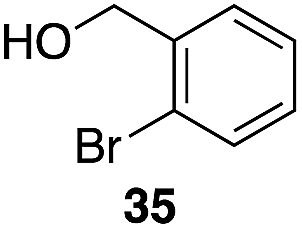
|
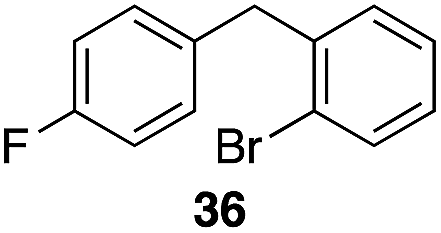
|
55%, 2.8:1 |
| 5 |
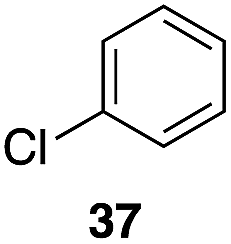
|
35 |
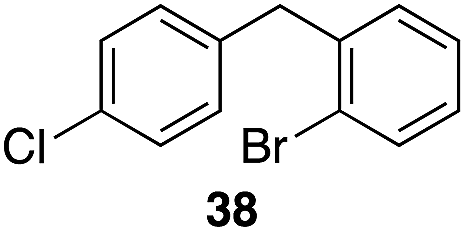
|
56%, 2.2:1 |
| 6 | 31 | 1 |
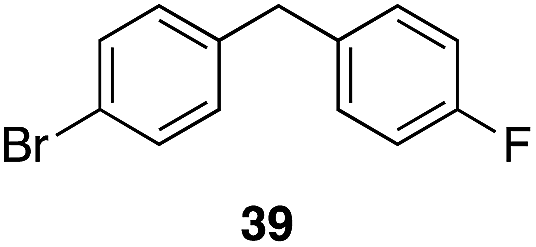
|
56%, 2:1 |
| 7 | 31 |
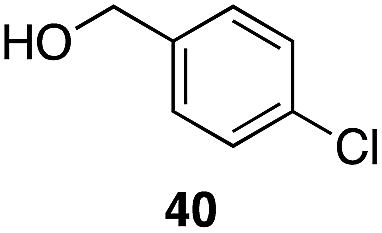
|
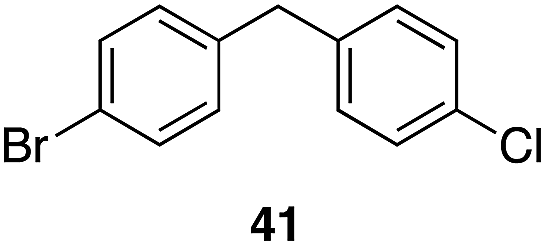
|
53%, 1.8:1 |
| 8 |
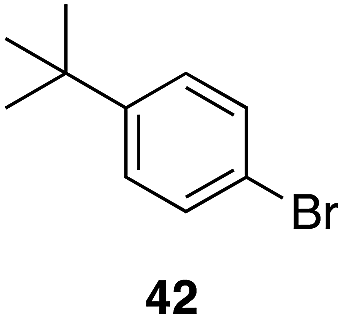
|
29 |
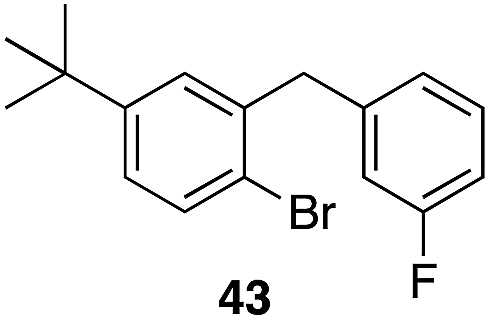
|
43%, 1:0 |
We next tested whether the catalyst loading could be lowered below 1 mol% (Scheme 3). Initial experiments using 0.1 mol% Re2O7·SiO2 showed that m-fluorobenzyl alcohol reacted with p-xylene to yield 6 in 73% yield after 3.5 h. The less reactive substrate p-trifluoromethyl benzyl alcohol (44), however, only provided diarylmethane 11 in 9% yield after 24 h. These results indicate that success with very low catalyst loading is dependent upon the ease of carbocation formation. This led us to investigate the limits of catalyst loading with a substrate that readily undergoes ionization and an active nucleophile. The reaction of p-methoxybenzyl alcohol (45) with mesitylene (46) proceeds to completion in <20 min at rt to yield 47 in 90% yield in the presence of 1 mol% Re2O7·SiO2, in accord with predictions about the influence of cation stability on reaction rates. Lowering the catalyst loading by a factor of 100 (0.01 mol%) resulted in the reaction proceeding to complete conversion after 7 h at rt and a 93% isolated yield of 47. Lowering the catalyst loading by another factor of 3 (0.0033 mol%, 33 ppm) resulted in the need for gentle heating, with the reaction requiring 9 h at 45 °C to proceed to completion with a 96% yield of 47. The concentration of the reaction was increased in consideration of large scale reactions being the likely applications of extremely low catalyst loading. Conducting the reaction at 2.5 M and 45 °C with 33 ppm Re2O7 provided results that were essentially identical to those from the corresponding reaction at 0.5 M. Notably, increasing the concentration further to 5.0 M led to a slightly less efficient process, presumably due to the change in solvent properties as the reaction approaches being solvent free, and dialkylation became a minor competitive pathway. Although HFIP is not an inherently green solvent,16 the capacity to conduct these reactions at high concentrations and removing the solvent through distillation without an aqueous work-up improves the process mass intensity17 and mitigates concerns of using a halogenated solvent for these processes.
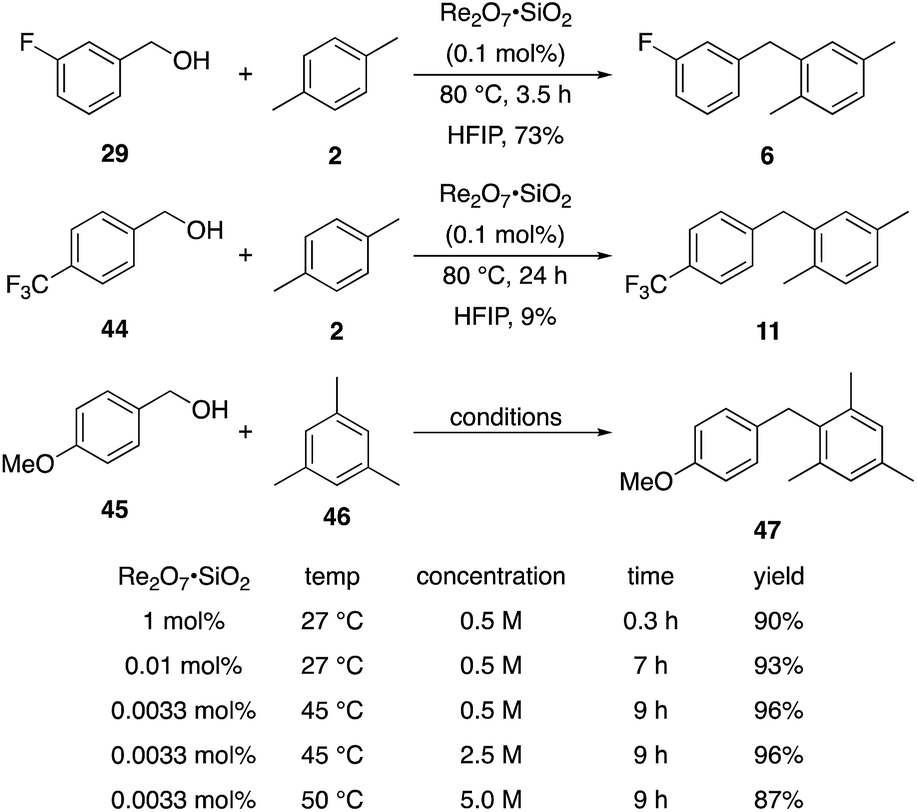 | ||
| Scheme 3 Reactions at lower catalyst loading. | ||
The concentration studies led us to study the role of HFIP in promoting the transformation. Ghorai and co-workers showed18 in related processes that Re2O7-mediated Friedel–Crafts reactions with highly activated benzylic alcohols in CH3CN are significantly slower than the reactions that we have studied, indicating that the efficiencies of our processes can be attributed to the solvent. The roles of fluorinated alcohols in promoting chemical reactions is a topic of significant interest, and has been reviewed thoroughly.19 HFIP is an extremely polar solvent as determined by solvatochromism studies.20 This can be attributed to its exceptional capacity as a hydrogen bond donor.21 HFIP is also a highly effective sequestering agent for H2O,22 which can have a strong influence on equilibrium concentrations of the intermediate benzylic cations. We conducted the reaction of highly reactive alcohol 45 with 46 in solvent mixtures containing varying fractions of HFIP to determine its role in promoting the coupling reactions (Scheme 4). We observed no qualitative difference in moving from 100% HFIP to 75% HFIP:25% 1,2-dichloroethane (DCE), 50% HFIP:50% DCE, and 25% HFIP:75% DCE. A reaction in 90% DCE:10% HFIP proceeded rapidly, but provided dialkylated product 48 in 14% yield in addition to 83% of 47, indicating that this solvent system actually enhances reactivity relative to pure HFIP. Notably, conducting the reaction in pure DCE resulted in the formation of 47 in 68% yield and ether 49 in 29% yield. Moderately reactive alcohol 29 reacted with 2 in 25% DCE:75% HFIP with at 80 °C to yield 6 in 64% yield after 17 h, and somewhat unreactive alcohol 44 reacted with 2 under these conditions to yield 11 in just 14% yield. These results indicate that the polarity of HFIP is very important for reactions in which the intermediate carbocation is not highly stabilized, and slight changes in the solvent mixture can have a pronounced effect on reaction efficiency. Additionally HFIP's hydrogen bonding capacity is important for blunting the nucleophilicity of the hydroxy groups in the substrates, consistent with Mayr's studies on the nucleophilicity of HFIP/H2O mixtures,23 and eliminating dehydrative ether formation.
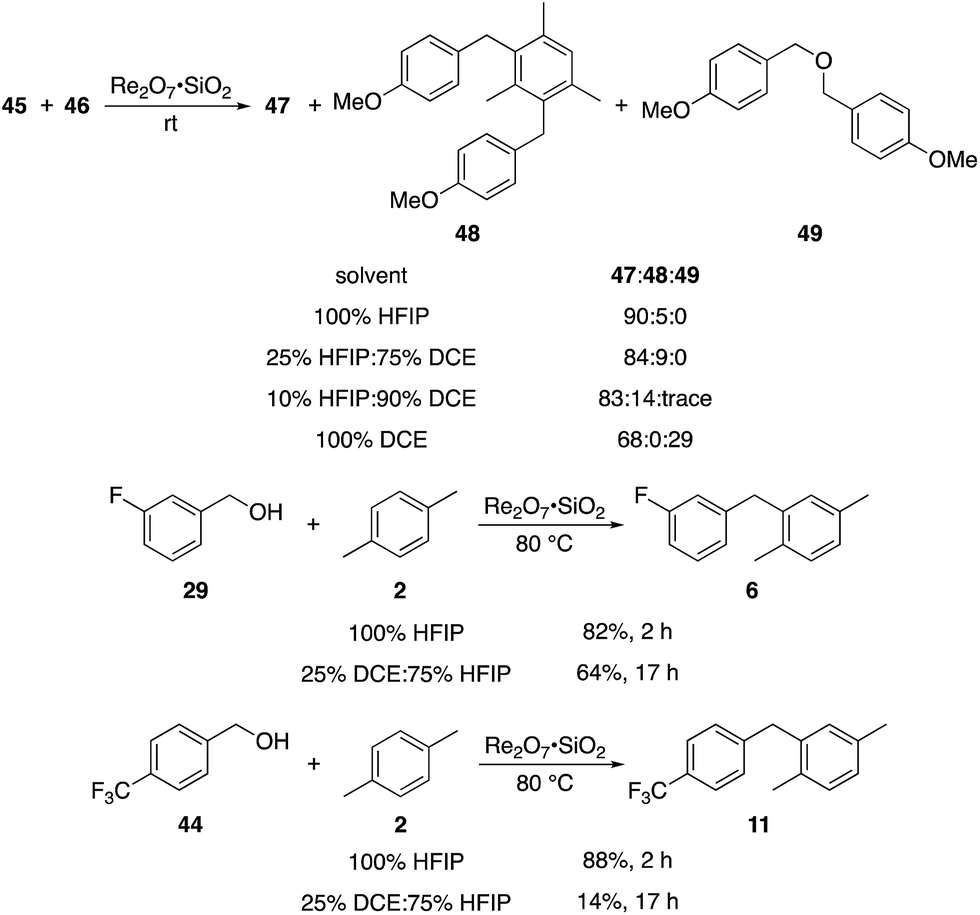 | ||
| Scheme 4 Solvent effects. | ||
Additionally we conducted studies to determine whether the unusual efficiency of these reactions could be attributed to the reaction medium or to the nucleofugacity of perrhenate. The pKa of perrhenic acid is −1.25 in water, which is significantly higher than those of HCl and HBr. Therefore carbocation formation through the loss of perrhenate should be less efficient than through the loss of chloride or bromide. We compared (Scheme 5) the Friedel–Crafts reactions of p-fluorobenzyl alcohol (1) with p-xylene (2) in the presence and absence of Re2O7·SiO2 to the reactions of the corresponding benzylic chloride (50) and bromide (51). The reaction of 1 with 2 in the presence of 1 mol% Re2O7·SiO2 in HFIP at rt, as described above, delivered diarylmethane 3 in 85% isolated yield. No reaction was observed in the absence of Re2O7·SiO2 or in the presence of SiO2, thereby confirming the need for Re2O7. Only 60% of chloride 50 was consumed after 6 h at rt, giving 3 in 43% yield. The reaction of bromide 51 with 2 in HFIP provided 3 in 69% yield after 6 h. Therefore the Re2O7-mediated conditions generate a significantly better leaving group than bromide or chloride. The rate difference is particularly striking in consideration of the low concentrations of the intermediate perrhenate esters and indicates that these species are unexpectedly effective leaving groups.
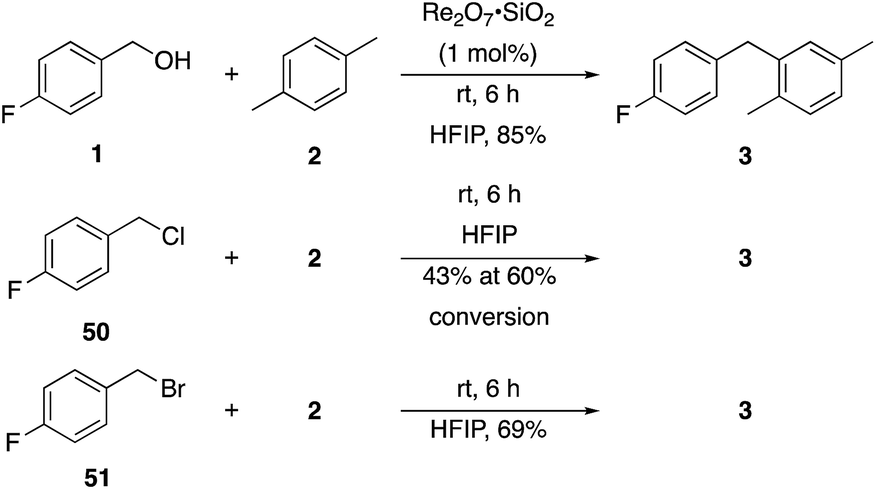 | ||
| Scheme 5 Leaving group comparison. | ||
2,6-Di-tert-butylpyridine completely suppresses these reactions. This led us to speculate that HOReO3 is the relevant catalyst. We tested the viability of perrhenic acid (76.5 weight% in H2O) as a catalyst by comparing its efficacy in the coupling of the slow-reacting substrate pentafluorobenzyl alcohol (18) and p-xylene with crystalline Re2O7 and Re2O7·SiO2 (Scheme 6) to yield 9. This study showed that HOReO3 is comparable to crystalline Re2O7 and is significantly superior to Re2O7·SiO2 in promoting these reactions. The highly deactivated substrate 3,5-bis-trifluoromethylbenzyl alcohol (52) provided a 51% yield of 16 after 23 h with 2 mol% HOReO3, which is somewhat less efficient than the result shown in Fig. 1 for Re2O7. Several aspects of these results are noteworthy. The loading of rhenium with HOReO3 is half of what is required for the Re2O7 reactions for most substrates, making its use significantly more economical since Re2O7 is approximately 5 times more expensive than HOReO3 on a per mole comparison.24 Additionally, HOReO3 is a liquid that can be handled readily. Thus utilizing HOReO3 provides the reactivity of crystalline Re2O7 and the handling ease of Re2O7·SiO2 while being much less expensive. We conclude based on these results and those described later that reactions with Re2O7 and HOReO3 most likely proceed through the same fundamental mechanism. HOReO3 is also an extremely effective catalyst at low loadings, with 0.01 mol% sufficing to provide 47 from 45 and 46 in 92% yield within 9 h at rt.
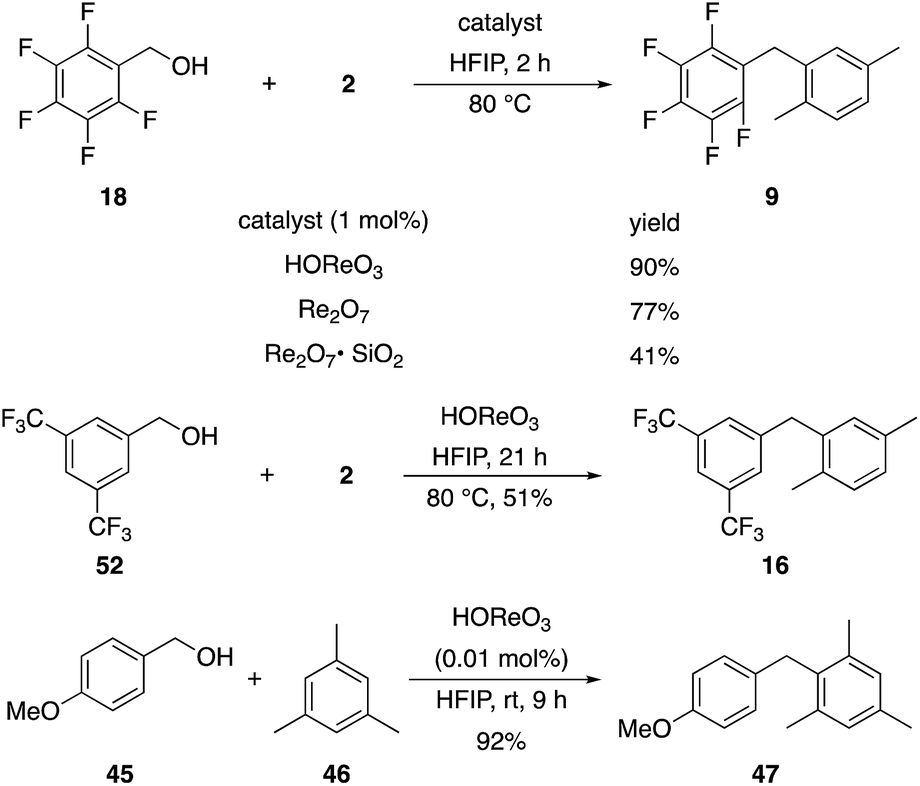 | ||
| Scheme 6 Catalyst comparison. | ||
These results, coupled with Moran's observation of catalysis by TfOH activation,4 lead to questions about the mechanism of the alcohol activation. Although HOReO3 is a much weaker Brønsted acid (pKa of −14 for TfOH) we cannot eliminate the possibility that HFIP causes an anomalous increase in the acidity of HOReO3, resulting in these reactions proceeding through protonation rather than perrhenate ester formation. We addressed this issue (Fig. 2) initially through comparing the rates of Re2O7 (1 mol%), TfOH (10 mol% and 2 mol%), HOReO3 (2 mol% and 1 mol%), and Re2O7·SiO2 (1 mol%) in the conversion of 53 to 11. These experiments showed that 1 mol% Re2O7, 10 mol% TfOH, and 2 mol% HOReO3 promoted the transformation at approximately the same rate. Lowering the loading of TfOH to 2 mol% resulted in a reaction that was even slower than the Re2O7·SiO2 catalyzed process. Thus if the oxorhenium catalysts act solely as Brønsted acids then the HFIP environment must cause HOReO3 to be a stronger acid than TfOH. Notably, p-TsOH (pKa = −2.8) does not promote the coupling reaction, indicating that the potential acidity enhancement by HFIP is not a general phenomenon.
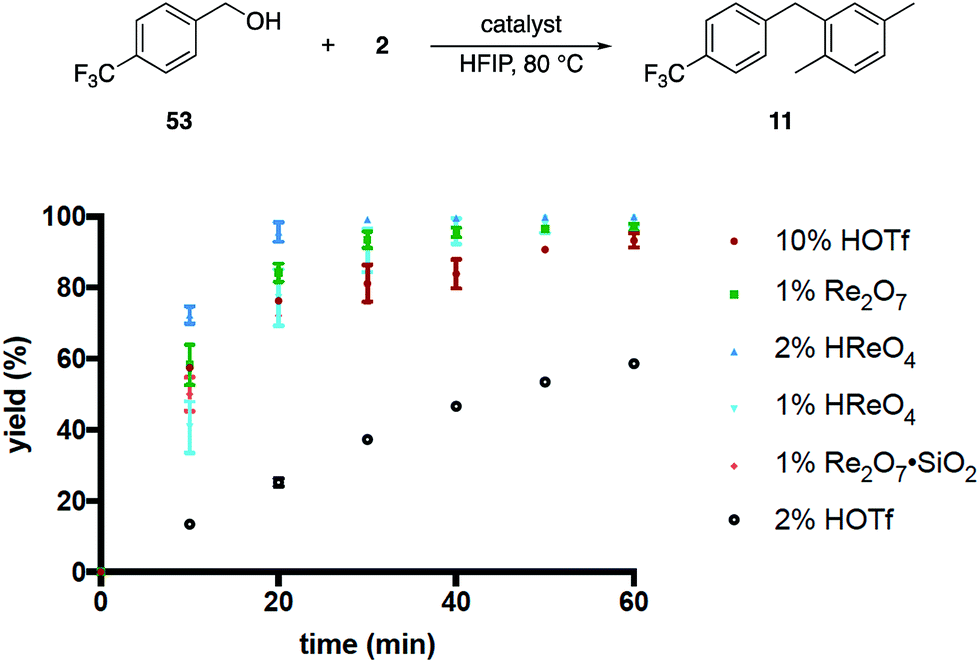 | ||
| Fig. 2 Reaction kinetics as a function of catalyst. | ||
We then compared the rates of HOReO3, Re2O7 and TfOH in the conversion of acetate 54 to 11 (Fig. 3) under the assumption that Brønsted acid catalysis will promote ionization even more effectively for this substrate relative to the corresponding alcohol while perrhenate ester formation would be suppressed. The reaction with TfOH proceeded to a significant extent within 2 h but, surprisingly, was slower than the reaction of the corresponding alcohol. We attribute this to acetic acid being sequestered less effectively than water, leading to lower concentrations of the intermediate benzylic cation. The reactions with HOReO3 and Re2O7, however, proceeded to <5% conversion at the same time and temperature. This result is inconsistent with a simple solvent-mediated acidity enhancement since the oxorhenium catalysts are approximately equivalent to TfOH in their abilities to promote alcohol ionization.
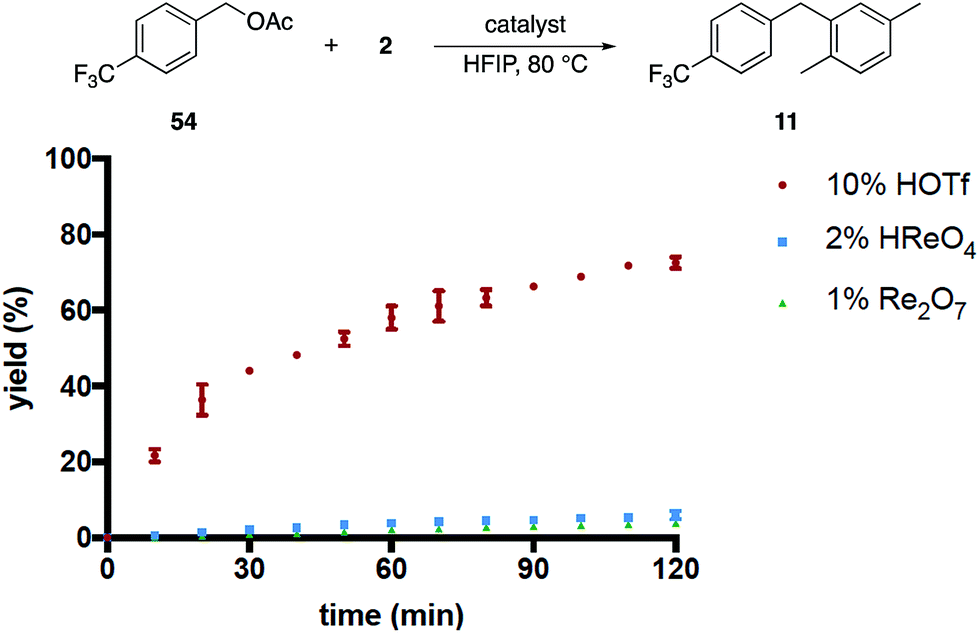 | ||
| Fig. 3 Acetate ionization by different catalysts. | ||
Additionally we exposed difunctional compound 55 and 2 to Re2O7 and TfOH at partial (1 h) and nearly complete (2.25 h) conversion (Scheme 7). We observed selective formation of 56, the product of alcohol activation, in 52% yield with Re2O7 at 54% conversion, with only 2% of the disubstitution product 57. TfOH produced a 21% yield of 56 and a 14% yield of 47 at 36% conversion. At 95% conversion the reaction with Re2O7 provided a 78% yield of 56 and only a 17% yield of 57, while TfOH produced a 5% yield of 56 and a 75% yield of 57 at 90% conversion. These results are consistent with Re2O7 showing a strong preference for alcohol ionization over acetate ionization while TfOH shows little to no selectivity. This system provides a very stringent challenge for the systems because the cation arising from product ionization is more stable than that derived from the starting material, as σm values for benzylic groups increase as the electronegativity of the substituent increases.9 Thus monosubstitution at high conversion is a challenging transformation that requires a selective catalyst. The absence of the monoalkylation product resulting from preferential acetate ionization suggests that this species is significantly more reactive than the starting material, which is consistent with the more positive σm value for the acetoxymethyl group relative to the hydroxymethyl group.9 This study further supports our hypothesis that the oxorhenium-catalyzed reactions do not proceed by simple alcohol protonation by a Brønsted acid with solvent-promoted enhanced acidity, and suggest that a perrhenate ester is likely to be relevant intermediate. A secondary role for a Brønsted acid cannot be discarded by our studies, however, with the highly non-basic nature of HFIP potentially allowing for protonated perrhenate esters to be the direct precursors to benzylic cations. The heightened selectivity for activating hydroxy groups by oxorhenium catalysts, however, demonstrates an advantage for these agents over strong Brønsted acids with respect to chemoselectivity.
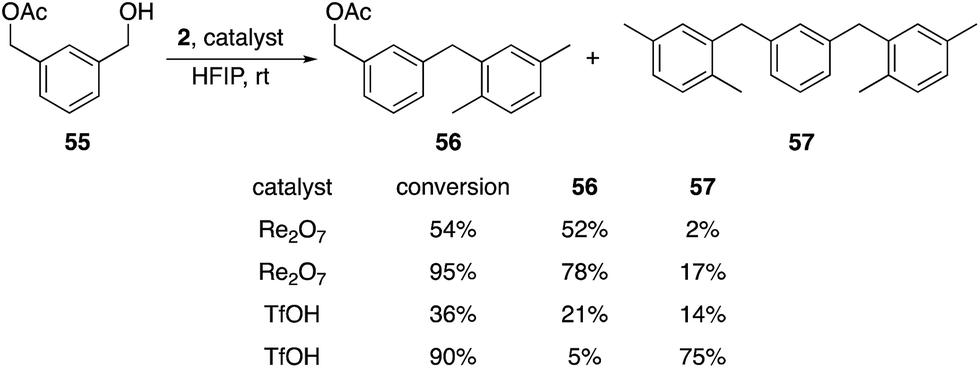 | ||
| Scheme 7 Reactions with a difunctionalized substrate. | ||
Conclusions
We have described an efficient protocol for diarylmethane synthesis through Friedel–Crafts reactions between benzene derivatives and benzylic alcohols. Re2O7, either in its crystalline form or adsorbed onto silica gel, is an exceptionally effective catalyst for these processes. The transformation has a very broad scope, including electron deficient benzylic alcohols, and requires low catalyst loading. The use of highly polar HFIP as the reaction solvent is important for enhancing rates and for minimizing the nucleophilicity of the hydroxy groups of the benzylic alcohols. The intermediates in these reactions are exceptional leaving groups, proving to be superior to chlorides and bromides. Re2O7 can be replaced by perrhenic acid to minimize cost and facilitate handling. Mechanistic studies confirmed that the mechanism of these reactions does not proceed through simple protonation, with the perrhenate ester or protonated perrhenate ester being the most likely reactive intermediate. This leads to unique chemoselectivity in which alcohols can be activated in preference to superior leaving groups. This work shows the capacity of oxorhenium species to promote important carbon–carbon bond forming reactions from readily available substrates while mitigating environmental impact.25Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank the National Institutes of Health (R01GM103886) for generous funding of this project.Notes and references
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC.
- (a) M. Dryzhakov, E. Richmond and J. Moran, Synthesis, 2016, 48, 935 CrossRef; (b) L. Chen, A.-P. Yin, C.-H. Wang and J. Zhou, Org. Biomol. Chem., 2014, 12, 6033 RSC; (c) A. Baeza and C. Nájera, Synthesis, 2014, 25 Search PubMed; (d) R. R. Naredla and D. A. Klumpp, Chem. Rev., 2013, 113, 6905 CrossRef PubMed.
- X. Mo, J. Yakiwchuk, J. Dansereau, J. A. McCubbin and D. G. Hall, J. Am. Chem. Soc., 2015, 137, 9694 CrossRef PubMed.
- V. D. Vuković, E. Richmond, E. Wolf and J. Moran, Angew. Chem., Int. Ed., 2017, 56, 3085 CrossRef PubMed.
- (a) H. H. Jung, J. R. Seiders II and P. E. Floreancig, Angew. Chem., Int. Ed., 2007, 46, 8464 CrossRef PubMed; (b) Y. Xie and P. E. Floreancig, Chem. Sci., 2011, 2, 2423 RSC; (c) Y. Xie and P. E. Floreancig, Angew. Chem., Int. Ed., 2013, 52, 625 CrossRef PubMed; (d) Y. Xie and P. E. Floreancig, Angew. Chem., Int. Ed., 2014, 53, 4926 CrossRef PubMed.
- For reviews of Re2O7-mediated transformations, see: (a) I. Volchkov and D. Lee, Chem. Soc. Rev., 2014, 43, 4381 RSC; (b) S. Bellemin-Laponnaz, ChemCatChem, 2009, 1, 357 CrossRef. For relevant examples, see: (c) K. Narasaka, H. Kusama and Y. Hiyashi, Tetrahedron, 1992, 48, 2059 CrossRef; (d) S. Bellemin-Laponnaz, H. Gisie, J. P. Le Ny and J. A. Osborn, Angew. Chem., Int. Ed., 1997, 36, 976 CrossRef; (e) C. Morrill and R. H. Grubbs, J. Am. Chem. Soc., 2006, 128, 8142 CrossRef PubMed; (f) B. M. Trost and F. D. Toste, J. Am. Chem. Soc., 2000, 122, 11262 CrossRef; (g) J. M. Hutchison, H. A. Lindsay, S. S. Dormi, G. D. Jones, D. A. Vivic and M. C. McIntosh, Org. Lett., 2006, 8, 3663 CrossRef PubMed; (h) E. C. Hansen and D. Lee, J. Am. Chem. Soc., 2006, 128, 8142 CrossRef PubMed; (i) A. T. Herrmann, T. Saito, C. E. Stivala, J. Tom and A. Zakarian, J. Am. Chem. Soc., 2010, 132, 5962 CrossRef PubMed; (j) S. Y. Yun, E. C. Hansen, I. Volchkov, W. Y. Lo and D. Lee, Angew. Chem., Int. Ed., 2010, 49, 4261 CrossRef PubMed; (k) J. Hu, D. Xu, Q. Zhang, Y. Shang, M. Shi, Y. Huangfu, L. Liu, R. Liang, Y. Lai, Y. He, J.-m. Gao and W. Xie, RSC Adv., 2016, 6, 52583 RSC.
- T. M. Rohrs, Q. Qin and P. E. Floreancig, Angew. Chem., Int. Ed., 2017, 56, 10090 CrossRef PubMed.
- For a related example, see: (a) X. Wan, J. Hu, D. Xu, Y. Shang, Y. Zhen, C. Hu, F. Xiao, Y.-P. He, Y. Lai and W. Xie, Tetrahedron Lett., 2017, 58, 1090 CrossRef. For dehydrative bond-forming processes using a different oxorhenium catalyst, see: (b) B. D. Sherry, A. T. Radosevich and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 6076 CrossRef PubMed; (c) M. R. Luzung and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 15760 CrossRef PubMed.
- While σ+ values would be more appropriate for several of the substituents we use σ values to remain consistent between substituents that can influence cation stability through resonance and induction. For a comprehensive listing of σ and σ+ values, see C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef.
- S. L. Scott and J.-M. Basset, J. Am. Chem. Soc., 1994, 116, 12069 CrossRef.
- J. Wang, M. Sánchez-Roselló, J. L. Aceñal, C. del Pozo, A. E. Sorochinsky, S. Fuestro, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef PubMed.
- Please see the ESI† for experimental validation of regioisomer assignments.
- M. F. Gotta and H. Mayr, J. Org. Chem., 1998, 63, 9769 CrossRef.
- (a) V. N. Ipatieff and H. Pines, J. Am. Chem. Soc., 1937, 59, 56 CrossRef; (b) R. M. Roberts, E. K. Baylis and G. J. Fonken, J. Am. Chem. Soc., 1963, 85, 3454 CrossRef.
- For an elegant recent example of this strategy, see: S. Jaracz, M. Kozlowski, L. Y. Eun and S. M. Kim, US Pat. 2016058258, Colgate-Palmolive Company and the Trustees of the University of Pennsylvania, 2016.
- L. J. Diorazio, D. R. J. Hose and N. K. Adlington, Org. Process Res. Dev., 2016, 20, 760 CrossRef.
- C. Jiminez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912 CrossRef.
- R. Nallagonda, M. Rehan and P. Ghorai, J. Org. Chem., 2014, 79, 2934 CrossRef PubMed.
- (a) I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 0088 CrossRef; (b) I. A. Shuklov, N. V. Dubrovina and A. Börner, Synthesis, 2007, 2925 CrossRef; (c) J.-P. Bégué, D. Bonnet-Delphon and B. Crouse, Synlett, 2004, 18 Search PubMed.
- C. Reichert, Chem. Rev., 1994, 94, 2319 CrossRef.
- L. Eberson, M. P. Hartshorn, O. Persson and F. Radner, Chem. Commun., 1996, 2105 RSC.
- K. P. Dockery, J. P. Dinnocanzo, S. Farid, J. L. Goodman, I. R. Gould and W. P. Todd, J. Am. Chem. Soc., 1997, 119, 1876 CrossRef.
- S. Minegishi, S. Kobayashi and H. Mayr, J. Am. Chem. Soc., 2004, 126, 5174 CrossRef PubMed.
- Based on current pricing from Sigma-Aldrich.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols and spectral data. See DOI: 10.1039/c8sc03570a |
| This journal is © The Royal Society of Chemistry 2018 |