
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Merohedral icosahedral M48 (M = CoII, NiII) cage clusters supported by thiacalix[4]arene†
Dantong
Geng‡
a,
Xu
Han‡
a,
Yanfeng
Bi
 *a,
Yucai
Qin
a,
Qiang
Li
a,
Liangliang
Huang
a,
Kun
Zhou
a,
Lijuan
Song
*a and
Zhiping
Zheng
*ab
*a,
Yucai
Qin
a,
Qiang
Li
a,
Liangliang
Huang
a,
Kun
Zhou
a,
Lijuan
Song
*a and
Zhiping
Zheng
*ab
aCollege of Chemistry, Chemical Engineering and Environmental Engineering, Liaoning Shihua University, Fushun 113001, P. R. China. E-mail: biyanfeng@lnpu.edu.cn; lsong56@263.net
bShenzhen Grubbs Institute and Department of Chemistry, Southern University of Science and Technology, Shenzhen, Guangdong 518000, China. E-mail: zhengzp@sustc.edu.cn
First published on 12th September 2018
Abstract
Cage clusters are a discrete chemically and topologically diverse family of molecule-based functional materials. Presented here are two isostructural M48 (M = CoII for LSHU01, NiII for LSHU02) cage clusters with a merohedral icosahedral cage structure featuring 12 M4-TC4A (H4TC4A, p-tert-butylthiacalix[4]arene) second building units as vertices and 18 asymmetric 5-(1H-tetrazol-1-yl)isophthalate ligands as faces. They are the highest-nuclearity cage compounds of CoII and NiII. The activated Co48 cage exhibited high selectivity in the sorption of C3H8 over CH4 under ambient conditions. Frequency response experiments indicated that the extrinsic voids and matrix interface of the activated crystalline samples are primarily responsible for the observed gas adsorption performance.
Introduction
Polyhedral coordination clusters (PCCs) are an intriguing class of compounds because of their structural aesthetics, interesting physical and chemical properties, and potential application as molecule-based functional materials.1–4 Numerous such species varying in both structure and composition have been reported. Although their nuclearities and overall structures generally cannot be predicted a priori, in many of such species smaller and structurally well-defined complex units are clearly recognizable.5–10 For example, the Mn84 torus can be formally built by alternating linear [Mn3O4] and cubic [Mn4O2(OMe)2] units;11 the record-setting Fe64 cage consists of Fe8 units linked by H3tea (triethanolamine) ligands,12 while the spheroidal Cu36 cluster can be formally constructed from metal–organic units.13,14 The structural modularity of such PCCs suggests the possibility of assembling even larger polynuclear species by using preformed cluster building units in combination with various bridging ligands, in a stepwise fashion or formally from a one-pot reaction.15–17 Efforts along this line have indeed produced much success. For example, the largest CoII cluster known (Co36) can be viewed as an assembly of a cubane core of Co12 clamped by two identical Co12 wings via a 2,3-dicarboxypyrazine ligand.18Typical calixarenes and their derivatives are used as multidentate ligands for assembly of cage clusters, e.g. Co8/Ni8 and Ni24/Co24 cage clusters, that have been reported by Atwood.19–23 In comparison, p-tert-butylthiacalix[4]arene (H4TC4A, Scheme S1a, ESI†) and its derivatives24–26 with four potentially bridging coordinating groups (S, SO, and SO2) and four OH groups form tetranuclear MII-thiacalixarene compounds that have been used as secondary building units (SBUs) with appropriate bridging ligands for the construction of more higher nuclearity PCCs including the highest nuclearity Co32 and Ni40 cages.27–30 Recently, these PCCs were also found to have new applications in molecule-based materials, and it was demonstrated that the obtained properties highly rely on the cluster shapes and components.31–36 One interesting observation is that when bridging units of an apparent structural symmetry were used, clusters of regular polyhedral structures including tetrahedral,37 cubic,29 and octahedral27,28,32–34,38–45 platonic solids were obtained. In comparison, the use of a less symmetric bridging ligand afforded an even larger cage cluster such as the largest known Ni40 cage containing 10 Ni4-TC4A units.30 Although the profound ligand effect is clear, how exactly the ligand dictates the assembly of the clusters eventually formed remains unclear. More such species with various compositions and shapes will help understand how these giant cage clusters may have been assembled. With such an understanding, we report here two isostructural M48 cage clusters of the general formula [M48(TC4A)12(L)18Cl12(H2O)6]·(+solvents) (LSHU01, M = Co; LSHU02, M = Ni) consisting of 12 M4-TC4A units and 18 deprotonated 5-(1H-tetrazol-1-yl)isophthalic acid (H2L, Scheme S1b, ESI†) ligands. They represent the largest known PCCs of CoII and NiII.
Results and discussion
Structure of M48 cage clusters
Crystallographic studies (Table S1, ESI†) revealed that LSHU01 and LSHU01 are isomers and there are 2 M4-TC4A units, 3 L2− ligands, 2Cl− ions, and an aqua ligand in the asymmetric unit. The tetranuclear unit of M4-TC4A, with its TC4A ligand adopting a cone conformation, is capped at the bottom by μ4-Cl− showing a C4v symmetry (Fig. 1a). This unit structure is the same as previously reported.24 The expected metric values of the M–O, M–N, and M–S bond lengths (Table S2, ESI†), the charge-balanced composition as determined crystallographically, and the bond valence sum calculations (BVS) indicate that the cobalt and nickel ions are divalent. Both magnetic susceptibility measurements and XPS investigations are consistent with this conclusion (Fig. S1, ESI†).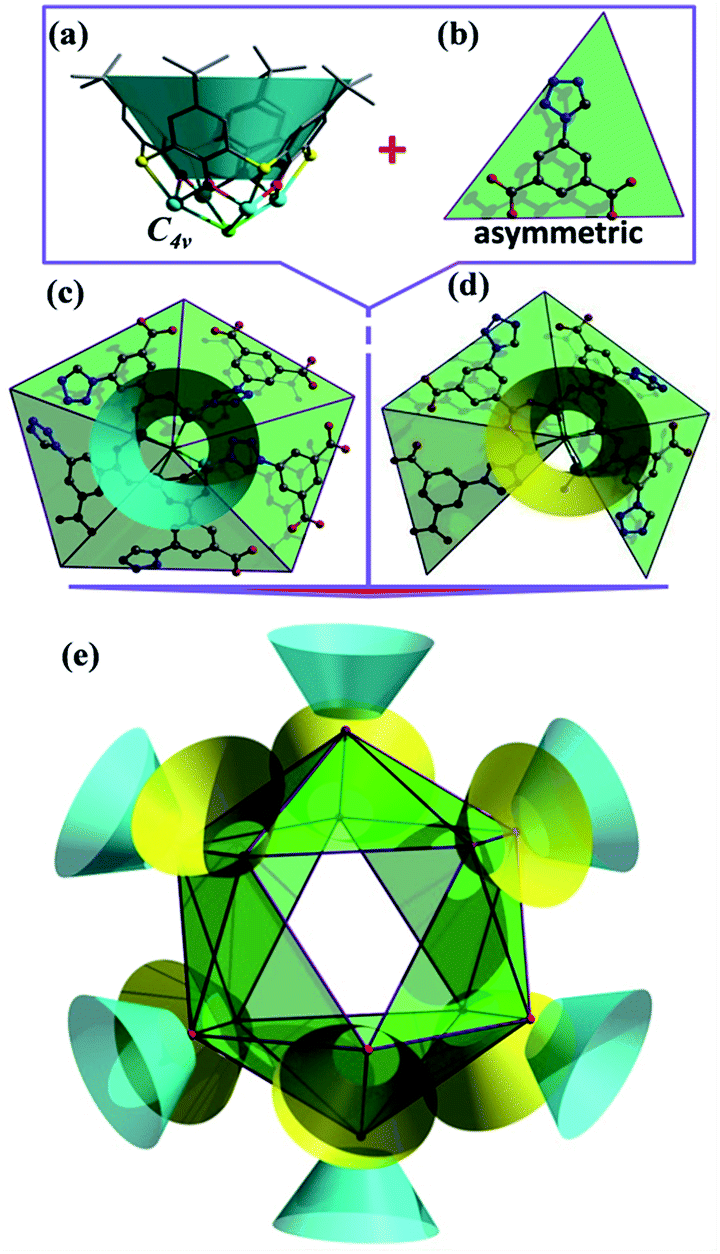 | ||
| Fig. 1 The structure of the M48 (M = Co, Ni) cage cluster determined by X-ray crystallography: (a) representations of the [(M4-TC4A)Cl] SBU with an approximate C4v symmetry (all hydrogen atoms omitted for clarity), (b) the asymmetric 5-(1H-tetrazol-1-yl)isophthalate (L2−) ligand, (c) SBU-1 with five L2− ligands, (d) SBU-2 with four L2− ligands and (e) the M48 cage cluster showing the merohedral icosahedral arrangement of 12[(M4-TC4A)Cl] units. Color code: M turquoise, S yellow, Cl green, O red, N blue, C gray, TC4A in SBU-1 turquoise and in SBU-2 yellow, and merohedral icosahedron green. | ||
Further structural analysis revealed subtle differences between the two M4-TC4A units in the asymmetric unit with the one (SBU-1, Fig. 1c) containing five L2− ligands while the other (SBU-2, Fig. 1d) has four L2− ligands. The L2− ligands in SBU-1 exhibit two different coordination modes: three adjacent L2− ligands each use one of its two carboxylate groups to bridge the four metal ions, while the remaining two L2− ligands each use one of the N atoms in its tetrazolyl group to coordinate in a monodentate fashion as reported in the literature (Fig. S2, ESI†).46 In comparison, SBU-2 can be viewed as being derived from SBU-1 by replacing one of the two L2− ligands that use the tetrazolyl N atom for coordination with an aqua ligand. SBU-1 and SBU-2 units, 6 of each kind, are organized into an icosahedron with the tetranuclear units occupying its 12 vertices (Fig. 1e). A more careful analysis of the structure revealed that the SBU-2 units are in an octahedral arrangement, while the SBU-1 units are in a chair-like arrangement, much like the more stable conformation of cyclohexane. Unlike in the arrangement of the SBU-1 units, the three adjacent SBU-2 units are not directly linked by an L2− ligand. Rather, they are hinged by three L2− ligands to form a metallamacrocycle with three aqua ligands, one on each SBU-2 unit, disposed within the ring structure (Fig. S3, ESI†). As such, the overall structure of the cage cluster can be best described as a merohedral convex icosahedron with 18 triangular faces rather than a regular icosahedron with 20 triangular faces (Fig. 1e). We note that this structure type is rarely observed in metal–organic systems despite its common existence in pure inorganic compounds including B12, Keggin-type polyoxometalates, and metal-centered endohedral clusters.47–50 With the assembly of UO22+ and calix[5]arene-carboxylate being viewed as a dodecahedron,51 the present M48 cage clusters complete the five structural types of platonic solids in metal-calixarene chemistry (Fig. S4, ESI†).19,24–26 MALDI-TOF mass spectra confirmed the presence of the Co48 cluster (Fig. S5, ESI†).
The cage cluster has an outside dimension of 37.3 × 35.8 × 33.8 Å3 (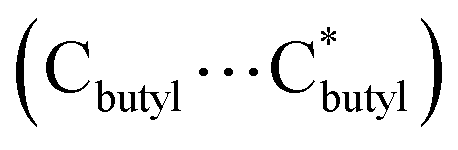 ) with an inner cavity (Cl⋯Cl*) of 22.3 × 20.8 × 20.8 Å3; the latter is ca. 1.5 times larger than that of the Ni40 cage,30 although the former is similar. The solvent-accessible voids of LSHU01 and LSHU02 were estimated to be 54
) with an inner cavity (Cl⋯Cl*) of 22.3 × 20.8 × 20.8 Å3; the latter is ca. 1.5 times larger than that of the Ni40 cage,30 although the former is similar. The solvent-accessible voids of LSHU01 and LSHU02 were estimated to be 54![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 653.5 Å3/64029.6 Å3 per cell 96152.0 Å3 and 106649.0 Å3, respectively, corresponding to 56.8%/60.0% of the total crystal volume. It is plausible that the different estimated solvent-accessible voids between the two isostructural cage clusters are due to the presence of different solvent molecules as well as the different collecting temperatures of the crystallographic data. That the hollow cages have two opposite triangle windows between two metallomacrocyclic rings partly occupied by aqua ligands is interesting. The edge of the triangle is ca. 7.6 Å with an in-circle diameter of ca. 5.4 Å (
653.5 Å3/64029.6 Å3 per cell 96152.0 Å3 and 106649.0 Å3, respectively, corresponding to 56.8%/60.0% of the total crystal volume. It is plausible that the different estimated solvent-accessible voids between the two isostructural cage clusters are due to the presence of different solvent molecules as well as the different collecting temperatures of the crystallographic data. That the hollow cages have two opposite triangle windows between two metallomacrocyclic rings partly occupied by aqua ligands is interesting. The edge of the triangle is ca. 7.6 Å with an in-circle diameter of ca. 5.4 Å (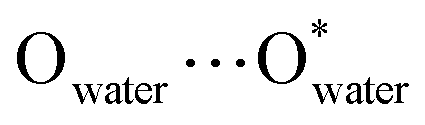 , Fig. S3, ESI†). Furthermore, irregular pockets between three cages are also observed with dimensions ranging from 4.0 to 11.7 Å (
, Fig. S3, ESI†). Furthermore, irregular pockets between three cages are also observed with dimensions ranging from 4.0 to 11.7 Å (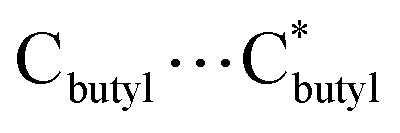 , Fig. S6, ESI†). By packing the cages along the c axis, one can find irregular channels with permanent pores (pore A, within cages) and packing pores (pore B, between adjacent cages) in ABBA mode (Fig. 2, S7 and S8, ESI†). The porous structure is expected to be useful for the uptake of gas molecules upon activation by desolvation.
, Fig. S6, ESI†). By packing the cages along the c axis, one can find irregular channels with permanent pores (pore A, within cages) and packing pores (pore B, between adjacent cages) in ABBA mode (Fig. 2, S7 and S8, ESI†). The porous structure is expected to be useful for the uptake of gas molecules upon activation by desolvation.
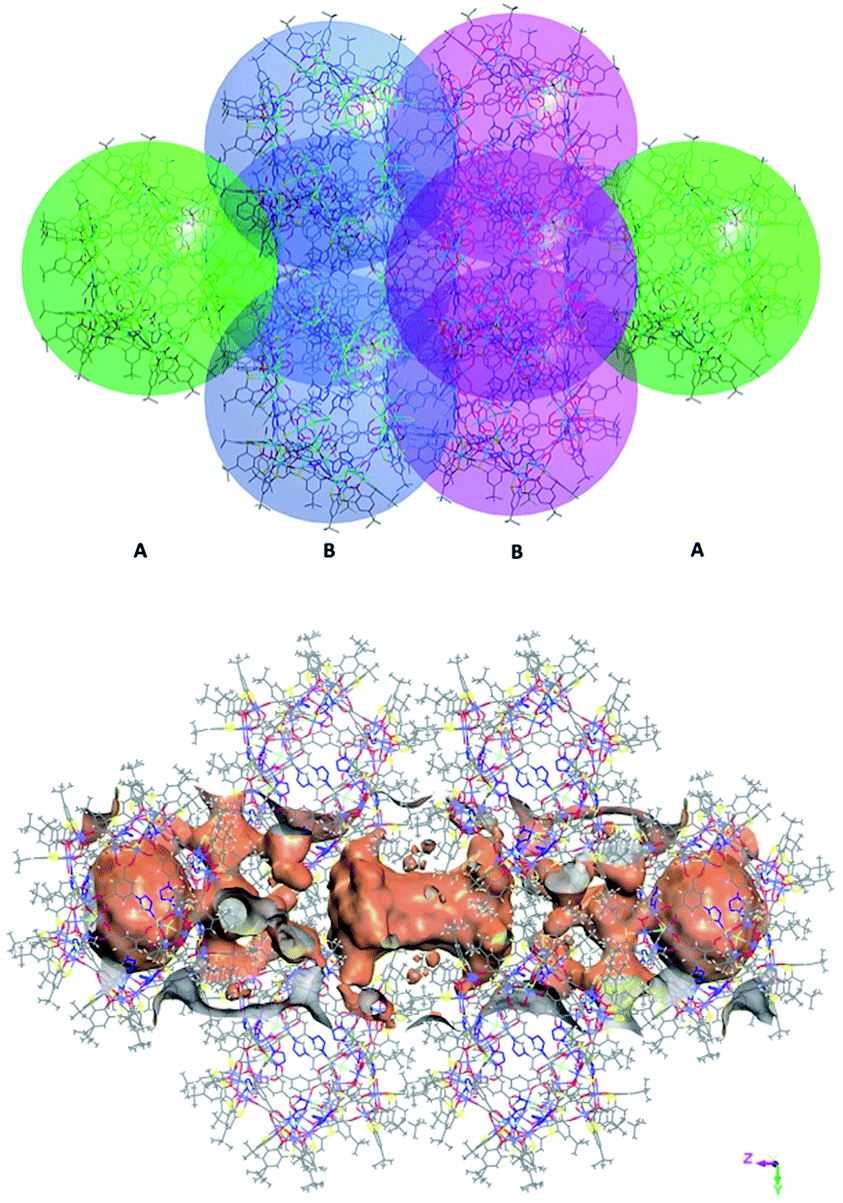 | ||
| Fig. 2 View of packing mode of the cages (up) and surface plot of the pores formed by arrangement in/between the cages (bottom). | ||
Stability and porosity
Both PXRD and thermogravimetric analysis indicated that LSHU01 and LSHU02 lost lattice solvent molecules between room temperature and 80 °C under vacuum (Fig. S9–S11, ESI†). Negligible weight loss in the temperature range of 80–150 °C for LSHU01 suggests complete removal of organic solvents and maintenance of structural integrity (Fig. 3 and S11–S13, ESI†). Weight loss in the range of 150–250 °C can be attributed to loss of the coordinated water molecules and Cl atoms (calc: 3.3%, found: 3.1%).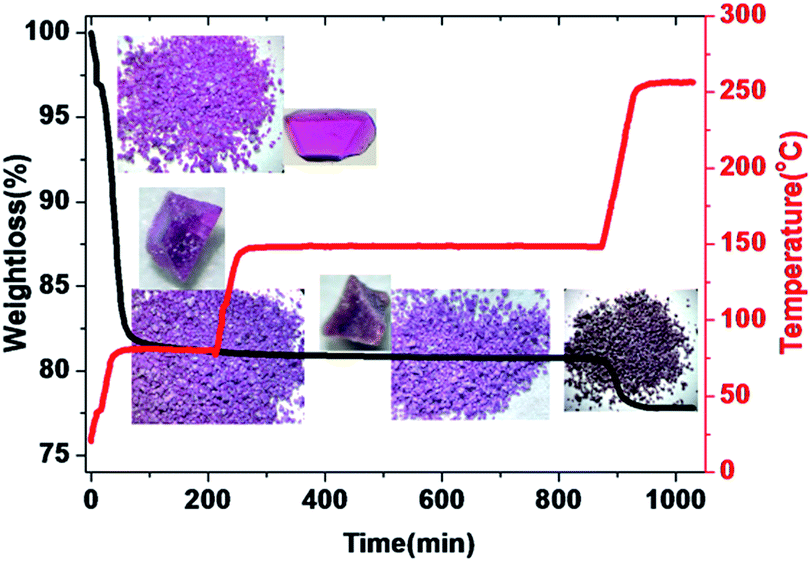 | ||
| Fig. 3 Temperature programmed desorption (TPD) of LSHU01 (for example, heating rate 3 °C min−1 in a vacuum and maintained at 80 °C for 3 hours, 150 °C for 10 hours and 250 °C for 1 hour). Inset: crystal pictures and morphology corresponding to different states. The samples were placed at ambient temperature for several hours before the TPD experiment. | ||
In order to assess the porosity of the M48 cages, argon adsorption measurements were performed at 87 K using LSHU01′ and LSHU01′′ (LSHU01 activated at 80 °C and 150 °C, respectively), for example LSHU01 and LSHU02 are isostructural and the cage clusters showed similar outside dimensions and inner cavities (Fig. S14, ESI†). The gas sorption showed a non-reversible type-I isotherm with large hysteresis upon desorption of LSHU01′. Such a behavior is probably due to the presence of the voids of the cage cluster and the extrinsic voids between cages as evidenced by the broad pore size distributions (PSDs) (0.50–1.10 nm with two peaks at ca. 0.51 nm and 0.65 nm, respectively, Fig. S14, inset†). A typical reversible type-I isotherm was obtained using LSHU01′′; its PSD is similar to that of LSHU01′ but the decrease of surface areas at 0.65 nm suggested different microstructures of these two activated samples. The observation of the PSD at ca. 0.51 nm that can be ascribed to the cage windows for both LSHU01′ and LSHU01′′ suggested the undamaged framework of cage clusters after higher temperature evacuation. For LSHU01′, the Langmuir and Brunauer–Emmett–Teller (BET) surface areas are estimated to be 803.09 m2 g−1 and 582.02 m2 g−1, while for LSHU01′′ the corresponding values are 607.95 m2 g−1 and 438.55 m2 g−1, respectively. The decreases in micropore areas (from 411.94 to 360.88 m2 g−1, 12.39%), external surface areas (from 170.08 to 77.67 m2 g−1, 54.33%), and micropore volume (from 0.1776 to 0.1600 cm3 g−1, 9.91%) for LSHU01′′ with respect to LSHU01′ are attributed to the decrease of free space between cages caused by the more compact packing of cages evacuated at higher temperature, consistent with Horvath–Kawazoe (HK) and Barrett–Joyner–Halenda (BJH) absorption PSDs (Fig. S15 and S16, ESI†).
Gas absorption and separation
The CH4, C2H4, C2H6, C3H8, and CO2 uptake experiments were conducted at 295 K in the pressure range of 0–100 kPa using LSHU01′ and LSHU01′′ (Fig. 4 right and Fig. S17–S23, ESI†). For CH4, the absorption isotherms are almost identical at about 0.49 mmol g−1 for LSHU01′ and LSHU01′′ at 1 bar. The adsorption capacity of LSHU01′ at 1 bar for C2H4, C2H6, C3H8, and CO2 is 2.32, 2.49, 3.75, and 1.77 mmol g−1, respectively, all being higher than the corresponding values (2.17, 2.33, 3.31, and 1.67 mmol g−1) obtained with the use of LSHU01′′ (Fig. S23, ESI†). The calculated isosteric heat (Qst) of adsorption at zero loading is 26.27 and 39.50 kJ mol−1 for CH4, 20.53 and 25.79 kJ mol−1 for C2H4, 23.51 and 32.15 kJ mol−1 for C2H6, and 38.16 and 47.76 kJ mol−1 for C3H6 by virial equation and dual-site Langmuir Freundlich (DSLF) equation fitting, respectively.52–55 The values remained essentially unchanged for the loading of CH4, C2H4, C2H6, and CO2 using the virial method. The values by the DSLF method are slightly higher in the lower gas uptake region but approached the values obtained using the virial method at 1 bar. The obtained Qst plots for C3H8 using virial and DSLF equations are almost identical and can be used to describe the whole absorption isotherms (Fig. S20, ESI†). The difference in the calculated Qst values using the virial equation and the DSLF method indicated the dynamic absorption behavior of the discrete Co48 cages.55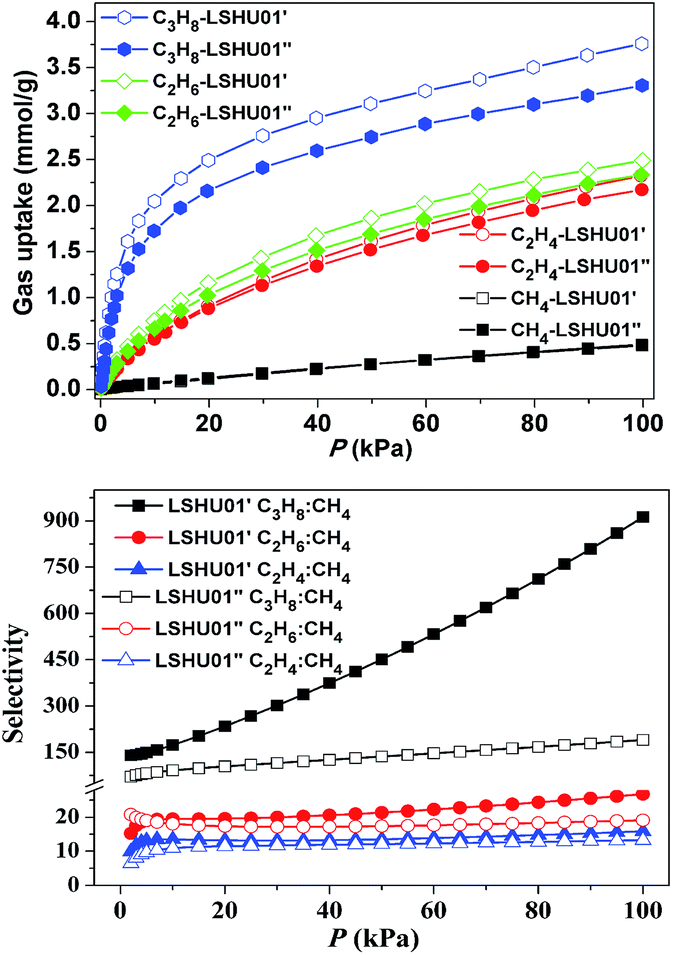 | ||
| Fig. 4 CH4, C2H4, C2H6 and C3H8 adsorption isotherms at 295 K (up) and gas mixture adsorption selectivity predicted by IAST at 295 K for LSHU01′ and LSHU01′′ (P: 1–100 kPa, gas ratios: 50:50, bottom). | ||
For binary mixtures composed of equimolar C2-hydrocarbons or CO2 and CH4, the selectivity is below 30 for both LSHU01′ and LSHU01′′ (Fig. 4 right), which is comparable to many systems previously reported.56–60 However, the initial selectivity for the C3H8/CH4 mixture was 140.1 and 70.5 and reached 912.6 and 189.8 at 295 K and 1 bar for LSHU01′ and LSHU01′′, respectively (Fig. S22 and S23, ESI†). It is highlighted that the selectivity for C3H8/CH4 under ambient conditions of LSHU01′ is about 30 fold that of the closed Ni40 cage30 and the best performance among metal–organic polymers (Table S3, ESI†). The extremely high selectivity values for C3H8/CH4 strongly suggest that it is feasible to separate the pair in a vacuum swing adsorption process using the activated Co48 sample as an adsorbent.
Frequency response
To understand why LSHU01′ showed higher selectivity than LSHU01′′ in the adsorption of C3H8 over CH4 at 295 K, frequency response (FR) spectra of C3H8 and CH4 were obtained. Both in-phase (IP) and out-of-phase (OP) FR signals of C3H8 can be detected using both LSHU01′ and LSHU01′′ (Fig. 5), but not for CH4 (Fig. S24, ESI†). The OP curves of C3H8 spectra, well fitted by the Yasuda sorption theoretical model,61,62 correspond to three adsorption processes (P1–P3) for LSHU01′ and LSHU01′′, which can be ascribed to the absorption in the molecular cages, between cages, and on the interface of the matrix from low frequency to high frequency, respectively. The reduction of the response intensity of IP and OP for LSHU01′′versusLSHU01′ indicates the lower absorption of C3H8 molecules, which is consistent with the decrease in BET surface areas and the C3H8-absorption experiments. Furthermore, the fitted OP response peaks P1–P3 are shifted to lower frequency (from 0.03 to 0.02 Hz for P1, from 0.22 to 0.16 Hz for P2, and from 1.74 to 0.97 Hz for P3) for LSHU01′′ with respect to LSHU01′, pointing to the lower absorption rate of C3H8 molecules.55,63 The observations detailed above together indicate that the free space between the cages and matrix interface plays an important role in the uptake and separation of C3H8 over CH4 under ambient conditions. It is also concluded that the absorption properties are sensitively dependent on the pore size, the moderate surface areas in accordance with gas kinetic diameters, and/or the polarizability of the gas molecules.64–66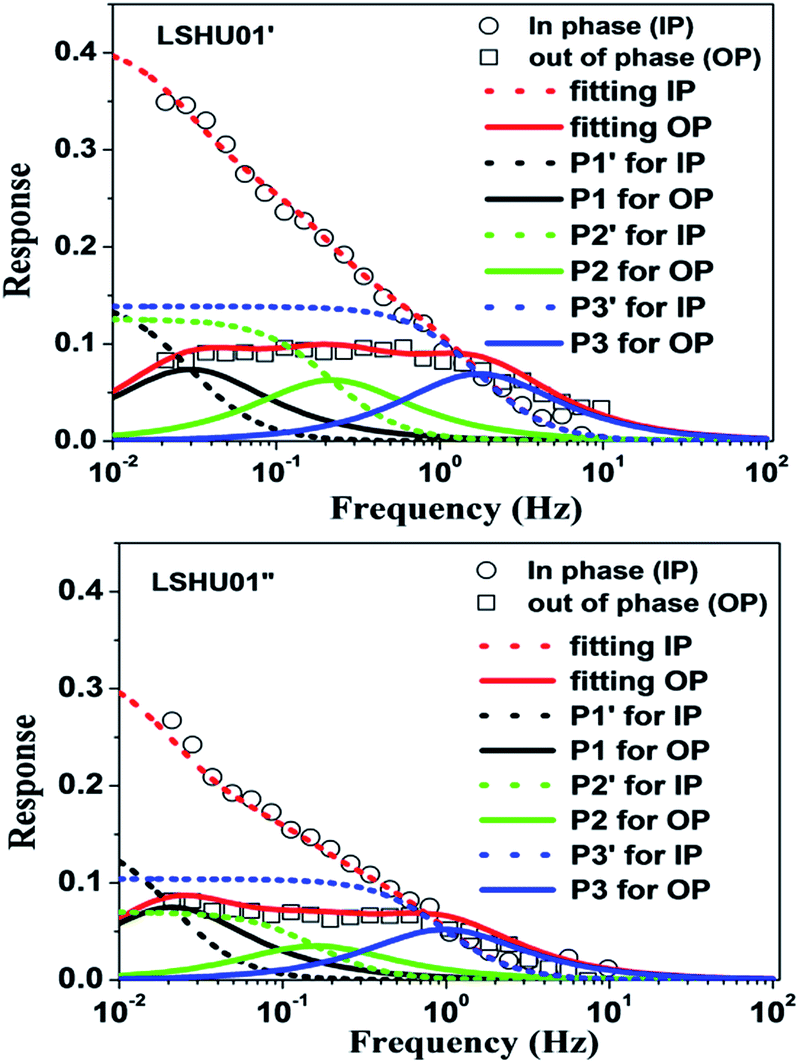 | ||
| Fig. 5 Frequency response (FR) spectra of C3H8 for LSHU01′ (up) and LSHU01′′ (bottom) at 0.133 kPa, 0–10 Hz. | ||
Conclusions
In summary, we have obtained and structurally characterized two record-setting high-nuclearity M48 (M = Co, Ni) cage clusters. It has been shown that higher-nuclearity Co and Ni cages can be built by using M4-TC4A SBUs and asymmetric ligands. The merohedral icosahedron-type structures with large pores and windows allow us to investigate gas sorption properties of the crystalline materials. The activated Co48 crystals were proved to be a competitive candidate for separating C3H8 from CH4. The activation temperature for Co48 crystals has a profound effect on the extrinsic voids between the cage clusters which are believed to be primarily responsible for the observed absorption properties and selectivity. Efforts aimed at the synthesis of even higher-nuclearity cage clusters are ongoing.Experimental section
Materials and measurements
p-tert-Butylthiacalix[4]arene (H4TC4A)67 and 5-(1H-tetrazol-1-yl)isophthalic acid (H2L)68 were synthesized according to a literature method, respectively. Other reagents were purchased from commercial sources and used as received. TGA was performed on a Perkin Elmer Pyris1 TGA thermo-gravimetric analyzer. FT-IR spectra using KBr pellets were taken on a Perkin Elmer Spectrum GX spectrometer. UV-vis spectra were recorded on an Agilent Cary 5000 spectrometer. MALDI-TOF mass data were collected on a Bruker Autoflex III Smartbeam MALDI-TOF mass spectrometer. CHNS elemental analysis was performed on an EAI CE-440 instrument. Magnetic susceptibility measurements were performed on a Quantum Design MPMS XL-5 SQUID system in the temperature range of 2–300 K, and diamagnetic corrections for the sample and sample holder were applied. X-ray photoelectron spectroscopic (XPS) measurements were carried out with an ESCALAB 250Xi using a monochromatic Al Kα X-ray source (1486.6 eV).Synthesis of LSHU01 and LSHU02
H2L was added to a suspension of MCl2·6H2O (M = Co, Ni) (0.95 g, 0.4 mmol) and H4TC4A (0.09 g, 0.125 mmol) in a m:n (v/v) CHCl3–CH3OH mixed solvent (total 10 mL, m = 8, n = 2 for Co; m = 5, n = 5 for Ni) with stirring for 10 min and then transferred into a 20 mL Teflon-lined autoclave which was kept at 130 °C for 3 days and then slowly cooled to room temperature at about 4 °C h−1. Red/green crystals were isolated by filtration and then washed with m:n CHCl3–CH3OH and dried in a vacuum at room temperature. Yield (0.094 g and 0.043 g): ca. 55% and 26% with respect to H4TC4A for LSHU01 and LSHU02, respectively. Elemental analysis: calculated (%) for [M48(C40H44S4O4)12(C9H4N4O4)18Cl12(H2O)6]: M = Co, C 44.76, H 3.82, N 6.25, S 9.53; M = Ni, C 44.79, H 3.82, N 6.25, S 9.54; found: M = Co, C 44.60, H 3.96, N 6.15, S 9.38, found: M = Ni, C 44.60, H 3.96, N 6.15, S 9.38 (after being dried in a vacuum at 80 °C).
Single crystal X-ray diffraction
The intensity data were recorded on a Bruker D8 QUEST system with Mo-Kα radiation (λ = 0.71073 Å). The crystal structures were solved by means of direct methods and refined by employing full-matrix least squares on F2 (SHELXTL-2014).69 Even the low temperature data set obtained at about 100 K for the compound LSHU01 reveals severely disordered solvents within the lattice interstices that are difficult to interpret thus complicating efforts to give precise estimates of the molecular formula. The diffraction data were treated by the “SQUEEZE” method as implemented in PLATON (see ESI† for details).70 All non-hydrogen atoms were refined anisotropically, and hydrogen atoms of the organic ligands were generated theoretically onto the specific atoms and refined isotropically with fixed thermal factors. Since the crystals do not diffract very well at high angles due to the structural disorder, the determined 2θ is 22.0141° and 20.1027° for LSHU01 and LSHU02, respectively. The R factors in the final structural refinement are also relatively large but typical of such systems.27,29,45 Refinement parameters and crystallographic data, selected bond distances and BVS calculations for LSHU01 and LSHU02 are shown in Tables S1 and S2 in the ESI,† respectively. CCDC 1526121 and 1526122 contain the supplementary crystallographic data of LSHU01 and LSHU02 for this paper, respectively.Gas adsorption experiments
Ultra-high-purity grade gas (>99.99) was used throughout the adsorption experiments. The measured sorption isotherms have been recorded at least two times to confirm the reproducibility within experimental errors. About 150 mg of methanol solvent-exchanged Co48 samples were activated at 80 °C and 150 °C for 10 hours according to the TPD experiment, respectively. Low-pressure gas sorption experiments are carried out on Micromeritics ASAP 2020M automatic volumetric instruments for Ar and on an Intelligent Gravimetric Analyser (IGA-003 Hiden Analytical Ltd., Warrington, UK.) for CO2, CH4, C2H4, C2H6 and C3H8, respectively. Buoyancy corrections for the samples were applied for gravimetric measurements. Desorption was achieved by placing the samples under a dynamic vacuum at 80 °C or 150 °C for three hours, respectively. Ar isotherms were measured using a liquid argon bath (87 K). Other gas isotherms, e.g. for CO2, CH4, C2H4, C2H6 and C3H8, were measured at 273 K and 295 K, respectively. The specific surface areas are determined using the Brunauer–Emmett–Teller and Langmuir equations from the Ar sorption data. The pore size distribution was obtained from the DFT, HK, and BJH models in the Micromeritics ASAP 2020 software package based on the Ar sorption at 87 K.Frequency response measurements
Frequency response measurements were carried out on a high-accuracy differential Baratron pressure transducer (MKS 698A11TRC). The frequencies were controlled using an on-line computer, which was also used for the acquisition of the pressure data from the Baratron. An accurate amount of sample (ca. 30 mg) was scattered in a plug of glass wool and degassed under a high vacuum (<10−3 Pa) at 80 °C or 150 °C for 6 h. Prior to the measurement, the sorbate vapor from the supply side of the vapor reservoir was admitted to the sorption chamber and equilibrated over the sample at a certain pressure.63Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the financial support from the National Natural Science Foundation of China (No. 21301170), Educational Bureau of Liaoning Province for the Fundamental Research of Key Lab (No. L2017LZD002), the Talent Scientific Research/Scientific Research Cultivation Fund of LSHU (No. 2015XJJ-009/2016PY-003) and the Shenzhen Nobel Prize Scientists Laboratory Project (C17213101). We thank Dr Yanzhen Zheng of Xi'an Jiaotong University for the measurements of magnetic susceptibility and valuable discussions.Notes and references
- D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 351–360 CrossRef PubMed.
- T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247–262 RSC.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650–1659 CrossRef PubMed.
- Q. F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144–1147 CrossRef PubMed.
- C. Papatriantafyllopoulou, E. E. Moushi, G. Christou and A. J. Tasiopoulos, Chem. Soc. Rev., 2016, 45, 1597–1628 RSC.
- G. E. Kostakis, S. P. Perlepes, V. A. Blatov, D. M. Proserpio and A. K. Powell, Coord. Chem. Rev., 2012, 256, 1246–1278 CrossRef.
- A. Escuer, J. Esteban, S. P. Perlepes and T. C Stamatatos, Coord. Chem. Rev., 2014, 275, 87–129 CrossRef.
- N. Ahmad, A. H. Chughtai, H. A. Younus and F. Verpoort, Coord. Chem. Rev., 2014, 280, 1–27 CrossRef.
- M. Manoli, S. Alexandrou, L. Pham, G. Lorusso, W. Wernsdorfer, M. Evangelisti, G. Christou and A. J. Tasiopoulos, Angew. Chem., Int. Ed., 2016, 55, 679–684 CrossRef PubMed.
- L. Vilà-Nadal and L. Cronin, Nat. Rev. Mater., 2017, 2, 17054 CrossRef and references there in..
- A. J. Tasiopoulos, A. Vinslava, W. Wernsdorfer, K. A. Abboud and G. Christou, Angew. Chem., Int. Ed., 2004, 43, 2117–2121 CrossRef PubMed.
- T. Liu, Y. J. Zhang, Z. M. Wang and S. Gao, J. Am. Chem. Soc., 2008, 130, 10500–10501 CrossRef PubMed.
- T. S. M. Abedin, L. K. Thompson, D. O. Miller and E. Krupicka, Chem. Commun., 2003, 708–709 RSC.
- M. Murugesu, R. Clérac, C. E. Anson and A. K. Powell, Chem. Commun., 2004, 1598–1599 RSC.
- Y. L. Bai, X. L. Bao, S. R. Zhu, J. H. Fang, M. Shao and H. Y. Shi, Eur. J. Inorg. Chem., 2014, 1275–1278 CrossRef.
- A. L. Dearden, S. Parsons and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2001, 40, 151–154 CrossRef PubMed.
- Z. Y. Zhang, K. Y. Zheng, T. L. Xia, L. J. Xu and R. Cao, Chem.–Eur. J., 2016, 22, 17576–17580 CrossRef PubMed.
- P. Alborés and E. Rentschler, Angew. Chem., Int. Ed., 2009, 48, 9366–9370 CrossRef PubMed.
- C. Zhang, R. S. Patil and J. L. Atwood, Adv. Inorg. Chem., 2018, 71, 247–276 CrossRef.
- J. L. Atwood, E. K. Brechin, S. J. Dalgarno, R. Inglis, L. F. Jones, A. Mossine, M. J. Paterson, N. P. Power and S. J. Teat, Chem. Commun., 2010, 46, 3484–3486 RSC.
- C. Zhang, R. S. Patil, C. L. Barnes and J. L. Atwood, Cryst. Growth Des., 2017, 17, 4541–4543 CrossRef.
- A. S. Rathnayake, K. A. Feaster, J. White, C. L Barnes, S. J. Teat and J. L. Atwood, Cryst. Growth Des., 2016, 16, 3562–3564 CrossRef.
- C. Zhang, R. S. Patil, C. Liu, C. L. Barne and J. L. Atwood, J. Am. Chem. Soc., 2017, 139, 2920–2923 CrossRef PubMed.
- Y. F. Bi, S. C. Du and W. P. Liao, Coord. Chem. Rev., 2014, 76, 61–72 CrossRef.
- R. Kumar, Y. O. Lee, V. Bhalla, M. Kumar and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4824–4870 RSC.
- A. Ovsyannikov, S. Solovieva, I. Antipin and S. Ferlay, Coord. Chem. Rev., 2017, 352, 151–186 CrossRef.
- Y. F. Bi, X. T. Wang, W. P. Liao, X. F. Wang, X. W. Wang, H. J. Zhang and S. Gao, J. Am. Chem. Soc., 2009, 131, 11650–11651 CrossRef PubMed.
- A. Gehin, S. Ferlay, J. M. Harrowfield, D. Fenske, N. Kyritsakas and M. W. Hosseini, Inorg. Chem., 2012, 51, 5481–5486 CrossRef PubMed.
- Y. F. Bi, S. T. Wang, M. Liu, S. C. Du and W. P. Liao, Chem. Commun., 2013, 49, 6785–6787 RSC.
- X. X. Hang, B. Liu, X. F. Zhu, S. T. Wang, H. T. Han, W. P. Liao, Y. L. Liu and C. H. Hu, J. Am. Chem. Soc., 2016, 138, 2969–2972 CrossRef PubMed.
- Y. P. Qiao, L. Zhang, J. Li, W. Lin and Z. Q. Wang, Angew. Chem., Int. Ed., 2016, 55, 12778–12782 CrossRef PubMed.
- Y. Fang, Z. F. Xiao, J. L. Li, C. Lollar, L. J. Liu, X. Z. Lian, S. Yuan, S. Banerjee, P. Zhang and H. C. Zhou, Angew. Chem., Int. Ed., 2018, 57, 5283–5287 CrossRef PubMed.
- Y. Fang, J. L. Li, T. Togo, F. Y. Jin, Z. F. Xiao, L. J. Liu, H. Drake, X. Z. Lian and H. C. Zhou, Chem, 2018, 4, 555–563 Search PubMed.
- C. X. Tan, J. J. Jiao, Z. J. Li, Y. Liu, X. Han and Y. Cui, Angew. Chem., Int. Ed., 2018, 57, 2085–2090 CrossRef PubMed.
- S. T. Wang, X. H. Gao, X. X. Hang, X. F. Zhu, H. T. Han, W. P. Liao and W. Chen, J. Am. Chem. Soc., 2016, 138, 16236–16239 CrossRef PubMed.
- S. T. Wang, X. H. Gao, X. X. Hang, X. F. Zhu, H. T. Han, X. K. Li, W. P. Liao and W. Chen, J. Am. Chem. Soc., 2018, 140, 6271–6277 CrossRef PubMed.
- M. Liu, S. C. Du, Y. F. Bi and W. P. Liao, Inorg. Chem. Commun., 2014, 41, 96–99 CrossRef.
- K. C. Xiong, F. L. Jiang, Y. L. Gai, D. Q. Yuan, L. Chen, M. Y. Wu, K. Z. Su and M. C. Hong, Chem. Sci., 2012, 3, 2321–2325 RSC.
- F. R. Dai, U. Sambasivam, A. J. Hammerstrom and Z. Q. Wang, J. Am. Chem. Soc., 2014, 136, 7480–7491 CrossRef PubMed.
- M. Liu, W. P. Liao, C. H. Hu, S. C. Du and H. J. Zhang, Angew. Chem., Int. Ed., 2012, 51, 1585–1588 CrossRef PubMed.
- F. R. Dai and Z. Q. Wang, J. Am. Chem. Soc., 2012, 134, 8002–8005 CrossRef PubMed.
- S. C. Du, C. H. Hu, J. C. Xiao, H. Q. Tan and W. P. Liao, Chem. Commun., 2012, 48, 9177–9179 RSC.
- H. Q. Tan, S. C. Du, Y. F. Bi and W. P. Liao, Inorg. Chem., 2014, 53, 7083–7085 CrossRef PubMed.
- S. C. Du, T. Q. Yu, W. P. Liao and C. H. Hu, Dalton Trans., 2015, 44, 14394–14402 RSC.
- Y. F. Bi, S. C. Du and W. P. Liao, Chem. Commun., 2011, 47, 4724–4726 RSC.
- S. M. Zhang, Z. Chang, T. L. Hu and X. H. Bu, Inorg. Chem., 2010, 49, 11581–11586 CrossRef PubMed.
- R. A. Bilbeisi, T. K. Ronson and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 9027–9030 CrossRef PubMed.
- A. Burke, D. Ellis, B. T. Giles, B. E. Hodson, S. A. Macgregor, G. M. Rosair and A. J. Welch, Angew. Chem., Int. Ed., 2003, 42, 225–228 CrossRef PubMed.
- R. B. King, Inorg. Chim. Acta, 1992, 198–200, 841–861 CrossRef.
- Y. Wang, M. Moses-DeBusk, L. Stevens, J. K. Hu, P. Zavalij, K. Bowen, B. I. Dunlap, E. R. Glaser and B. Eichhorn, J. Am. Chem. Soc., 2017, 139, 619–622 CrossRef PubMed.
- S. Pasquale, S. Sattin, E. C. Escudero-Adán, M. Martínez-Belmonte and J. de Mendoza, Nat. Commun., 2012, 3, 785–791 CrossRef PubMed.
- J. L. C. Rowsell and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 1304–1315 CrossRef PubMed.
- L. T. Du, Z. Y. Lu, K. Y. Zheng, J. Y. Wang, X. Zheng, Y. Pan, X. Z. You and J. F. Bai, J. Am. Chem. Soc., 2013, 135, 562–565 CrossRef PubMed.
- T. Runčevski, M. T. Kapelewski, R. M. Torres-Gavosto, J. D. Tarver, C. M. Brown and J. R. Long, Chem. Commun., 2016, 52, 8251–8254 RSC.
- D. T. Geng, M. Zhang, X. X. Hang, W. J. Xie, Y. C. Qin, Q. Li, Y. F. Bi and Z. P. Zheng, Dalton Trans., 2018, 47, 9008–9013 RSC.
- J. R. Li, R. J. Kuppler and H. C Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- Y. B. He, R. Krishna and B. L. Chen, Energy Environ. Sci., 2012, 5, 9107–9120 RSC.
- Y. B. He, Z. J. Zhang, S. C. Xiang, F. R. Fronczek, R. Krishna and B. L. Chen, Chem.–Eur. J., 2012, 18, 613–619 CrossRef PubMed.
- B. Li, H. L. Wang and B. L. Chen, Chem.–Asian J., 2014, 9, 1474–1498 CrossRef PubMed.
- K. Adil, Y. Belmabkhout, R. S. Pillai, A. Cadiau, P. M. Bhatt, A. H. Assen, G. Maurin and M. Eddaoudi, Chem. Soc. Rev., 2017, 46, 3402–3430 RSC.
- Y. Yasuda, J. Phys. Chem., 1976, 80, 1867–1869 CrossRef.
- G. Onyestyák, J. Valyon and L. V. C. Rees, Stud. Surf. Sci. Catal., 2005, 158, 1027–1034 CrossRef.
- L. J. Song and L. V. C. Rees, Microporous Mesoporous Mater., 2000, 35, 301–314 CrossRef.
- W. Sapp, B. Gifford, Z. Q. Wang and D. S. Kilin, RSC Adv., 2017, 7, 11180–11190 RSC.
- P. Murphy, S. J. Dalgarno and M. J. Paterson, J. Phys. Chem. A, 2016, 120, 824–839 CrossRef PubMed.
- P. K. Thallapally, S. J. Dalgarno and J. L. Atwood, J. Am. Chem. Soc., 2006, 128, 15060–15061 CrossRef PubMed.
- N. Iki, C. Kabuto, T. Fukushima, H. Kumagai, H. Takeya, S. Miyanari, T. Miyashi and S. Miyano, Tetrahedron, 2000, 56, 1437–1443 CrossRef.
- S. V. Voitekhovich, A. N. Vorobév, P. N. Gaponik and O. A. Ivashkevich, Chem. Heterocycl. Compd., 2005, 41, 999–1004 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Scheme, magnetic susceptibility measurements, MALDI-TOF mass spectra, PXRD, TGA, FT-IR spectra, UV-vis-NIR spectra, additional figures and tables. CCDC 1526121 and 1526122. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03193b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |