
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic study of styrene aziridination by iron(IV) nitrides†
Douglas W.
Crandell
*ab,
Salvador B.
Muñoz
III
a,
Jeremy M.
Smith
 *a and
Mu-Hyun
Baik
*cd
*a and
Mu-Hyun
Baik
*cd
aDepartment of Chemistry, Indiana University, 800 E. Kirkwood Avenue, Bloomington, IN 47405, USA. E-mail: smith962@indiana.edu
bDepartment of Chemistry, Saint Louis University, St. Louis, MO 63103, USA. E-mail: doug.crandell@slu.edu
cCenter for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon 34141, Republic of Korea
dDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 34141, Republic of Korea. E-mail: mbaik2805@kaist.ac.kr
First published on 10th September 2018
Abstract
A combined experimental and computational investigation was undertaken to investigate the mechanism of aziridination of styrene by the tris(carbene)borate iron(IV) nitride complex, PhB(tBuIm)3Fe![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N. While mechanistic investigations suggest that aziridination occurs via a reversible, stepwise pathway, it was not possible to confirm the mechanism using only experimental techniques. Density functional theory calculations support a stepwise radical addition mechanism, but suggest that a low-lying triplet (S = 1) state provides the lowest energy path for C–N bond formation (24.6 kcal mol−1) and not the singlet ground (S = 0) state. A second spin flip may take place in order to facilitate ring closure and the formation of the quintet (S = 2) aziridino product. A Hammett analysis shows that electron-withdrawing groups increase the rate of reaction σp (ρ = 1.2 ± 0.2). This finding is supported by the computational results, which show that the rate-determining step drops from 24.6 kcal mol−1 to 18.3 kcal mol−1 when (p-NO2C6H4)CH
N. While mechanistic investigations suggest that aziridination occurs via a reversible, stepwise pathway, it was not possible to confirm the mechanism using only experimental techniques. Density functional theory calculations support a stepwise radical addition mechanism, but suggest that a low-lying triplet (S = 1) state provides the lowest energy path for C–N bond formation (24.6 kcal mol−1) and not the singlet ground (S = 0) state. A second spin flip may take place in order to facilitate ring closure and the formation of the quintet (S = 2) aziridino product. A Hammett analysis shows that electron-withdrawing groups increase the rate of reaction σp (ρ = 1.2 ± 0.2). This finding is supported by the computational results, which show that the rate-determining step drops from 24.6 kcal mol−1 to 18.3 kcal mol−1 when (p-NO2C6H4)CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 is used and slightly increases to 25.5 kcal mol−1 using (p-NMe2C6H4)CHCH2 as the substrate.
CH2 is used and slightly increases to 25.5 kcal mol−1 using (p-NMe2C6H4)CHCH2 as the substrate.
Introduction
Aziridines are three-membered heterocycles with properties that are highly advantageous for chemical synthesis. Their large ring strain of ∼27 kcal mol−1 leads to ring-opening and ring-expansion1,2 reactions in which a wide range of functional groups can be regio- and stereoselectively installed. Their utility is not limited to ring-opening reactions, for example N-protected aziridines undergo formal [3 + 2] cycloaddition reactions with dipolarphiles to furnish complex heterocycles.1,3 For these reasons, aziridines are important intermediates in natural product synthesis, e.g. the kainoids, (−)-mesembrine, (−)-platynesine, sphingosines, actinomycin, (±)-epicapreomycidine, and feldamycin.1,4,5 In addition to this synthetic utility, the aziridine functionality is also present in a small number of naturally occurring molecules with antibiotic and antitumor properties e.g. azinomycins, mitomycins, FR-900482, ficellomycin, miraziridine, maduropeptin, and azicemicins.5,6The metal-catalyzed transfer of nitrenes (NR) to alkenes is an appealing and concise synthetic route to aziridines that has attracted substantial efforts towards the development of efficient and versatile reaction protocols,7,8 most notably the copper-catalyzed asymmetric aziridination of alkenes by PhINTs, which leads to the formation of N-tosylatedaziridines.9,10 The utility of the copper-catalyzed methodology has been demonstrated in its application to the total synthesis of (+)-agelastatin A.11 Despite this success, the nitrene transfer strategy suffers from a number of severe limitations, many of which are related to the nitrene source, which typically requires an electron withdrawing group (e.g. N-sulfonyl) for successful alkene aziridination. While new catalysts and/or alternate nitrene sources have had some success in addressing these problems, allowing for direct access to the more desirable N–R or N–H substituted aziridines,12–18 these solutions typically require expensive/toxic transition metals and/or costly/hazardous nitrene sources.
We have previously reported that the tris(carbene)borate iron(IV) nitride complex PhB(tBuIm)3FeN191 reacts under thermal conditions with a range of styrenes to yield the corresponding high spin (S = 2) iron(II) aziridino complexes PhB(tBuIm)3Fe–N(CH2CH(C6H4R)) by a two-electron nitrogen atom reaction (Scheme 1).20 This is a rare instance of nitrogen atom transfer from a nitride ligand to an alkene substrate.21,22 Transition metal nitride complexes are generally unreactive towards hydrocarbons.23–28 The aziridino ligand can be released from iron in a subsequent transformation, providing the corresponding N–H aziridine in quantitative yield. Although not catalytic, an appealing aspect of this reaction sequence is that it demonstrates an alternative strategy to the commonly used nitrene transfer methodology for accessing synthetically useful N–H substituted aziridines under mild conditions. Moreover, since metal nitrides are accessible from N2,29 understanding the mechanism of this aziridination reaction may provide insight into methods for functionalizing hydrocarbons using N2 as the nitrogen atom source.
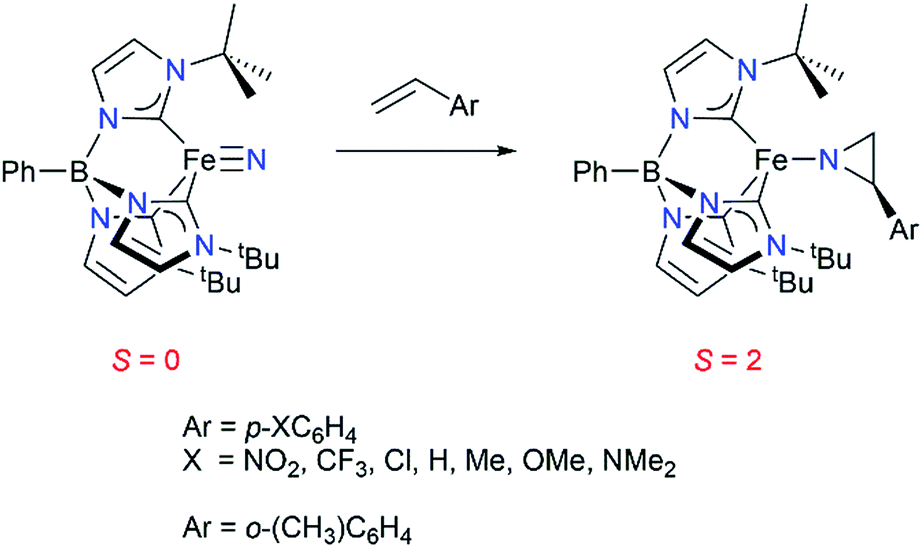 | ||
| Scheme 1 | ||
This unusual reactivity of the tris(carbene)borate iron nitride complexes has prompted us to undertake a combined experimental and theoretical study into the mechanism of aziridine ring formation. This two-electron, nitrogen atom transfer reaction is related to alkene aziridination by transition metal imido complexes. Experimental30–33 and computational34 mechanistic investigations of alkene aziridination by iron imidos35 have generally implicated stepwise mechanisms involving radical intermediates,36 suggesting that the aziridination reactivity of 1 may occur by a similar mechanism. However, it is not clear how the greater charge of the nitride ligand and the greater covalency of the iron nitride bond will influence the energetics of the stepwise radical pathway. Styrene aziridination is also conceptually related to alkene epoxidation via oxygen atom transfer from metal oxo intermediates.37–40 Stepwise mechanisms have also been proposed for alkene epoxidation by high valent iron oxo intermediates in both heme41–43 and nonheme44–49 environments, including pathways involving radical50,51 and cationic intermediates.52,53 The accessibility of these mechanisms may be dictated by spin state changes, where computational studies have suggested the involvement of more than one spin surface (i.e., multistate reactivity)54 in the epoxidation of alkenes by cytochrome P450.55–58 Elucidating the mechanism of aziridination by 1 is expected to provide important insights into the further development of nitrogen atom transfer reactions, particularly for alkene aziridination, where similar multistate reactivity may also be important. Indeed, we note that nitrene transfer reactions of a copper complex supported by redox-active iminosemiquinone ligands have also been proposed to involve multistate reactivity.59 Additionally, multistate reactivity is noted in the posited mechanism for the cyclopropanation reaction of iron alkylidenes with alkenes.60
In this paper we report a combined experimental and computational investigation into the styrene aziridination mechanism. Experimental investigations strongly suggest a stepwise pathway, which is confirmed by the computational investigation. The latter studies also provide insight into the effect of spin state considerations on the mechanism of aziridination. These studies are important for the further development of the nitrogen atom transfer strategy for hydrocarbon functionalization.
Experimental
General considerations
All manipulations were performed under a nitrogen atmosphere by standard Schlenk techniques or in an M. Braun Labmaster glovebox. Glassware was dried at 150 °C overnight. Diethyl ether, n-pentane, tetrahydrofuran, and toluene, were purified by the Glass Contour solvent purification system. Deuterated benzene was first dried with CaH2, then over Na/benzophenone, and then vacuum transfer into a storage container. Before use, an aliquot of each solvent was tested with a drop of sodium benzophenone ketyl in THF solution. All reagents were purchased from commercial vendors and used as received. Complex 1 was prepared according to a literature procedure.191H NMR data were recorded on a Varian Unity 400 MHz or a Varian Inova 500 MHz spectrometer at 25 °C.Mechanistic investigations
A J-Young NMR tube was charged with 1 (10 mg, 0.016 mmol, 1 equiv.) and 1 equivalent of the corresponding styrene and C6D6 (1.5 mL). The 1H NMR spectrum of the reaction mixture was obtained immediately after styrene. The reaction solution was heated in an oil bath at 65 °C. The 1H NMR spectrum of the reaction was collected at regular time intervals using a thirty second relaxation delay. Reaction plots were generated by integrating the resonances of the reactants and products for each time measurement, with the integrations normalized to residual tetrahydrofuran present in the reaction. The average of two resonances for each species was used to determine the molar concentration with an estimated error of less than 5%.Computational details
All calculations were performed using density functional theory60,61 as implemented in the Jaguar 8.1 suite of ab initio quantum chemistry programs.62 Geometry optimizations using Grimme's D3 dispersion corrections63 were performed with the B3LYP functional64–68 using the 6-31G** basis set with Fe represented using the Los Alamos LACVP basis set that includes relativistic core potentials.69–71 More accurate single point energies were computed from the optimized geometries along with Dunning's correlation-consistent triple-ζ basis set, cc-pVTZ(-f) that includes a double set of polarization functions.72 Fe was represented using a modified version of LACVP, designated as LACV3P, in which the exponents were decontracted to match the effective core potential with triple-ζ quality. Additional single point energy calculations were done using the M06 functional.73 Vibrational frequencies were computed at the B3LYP/6-31G** level of theory to derive zero point energy and vibrational entropy corrections from unscaled frequencies. Entropy here refers specifically to the vibrational/rotational/translational entropy of the solute(s), as the continuum model includes the entropy of the solvent. All intermediates were confirmed as minima on the potential energy surface having zero imaginary frequencies. Transition states were confirmed to possess only one imaginary frequency. Solvation energies were evaluated using a self-consistent reaction field (SCRF) approach based on accurate numerical solutions of the linearized Poisson–Boltzmann equation.74–77Solvation calculations were carried out on the optimized gas-phase geometries using a dielectric constant of ε = 2.284 for benzene. As with all continuum models, the solvation energies are subject to empirical parameterization of the atomic radii that are used to generate the solute surface. We employ the standard set of optimized radii for H (1.150 Å), B (2.042 Å), C (1.900 Å), N (1.600 Å), O (1.600 Å), and Fe (1.456 Å). AF states were modeled using Noodleman's broken symmetry (BS) formalism without spin projection.78–80 The change in solution phase free energy ΔG(sol) was calculated as follows:
| ΔG(sol) = ΔG(gas) + ΔΔGsolv |
| ΔG(gas) = ΔH(gas) − TΔS(gas) |
| ΔH(gas) = ΔE(SCF) + ΔZPE |
Results and discussion
Based on the proposals for oxygen atom transfer from the ferryl intermediate of cytochrome P450,81,82 at least three mechanisms for alkene aziridination by the iron nitride complex can be envisioned, as summarized in Scheme 2: (1) a concerted two-electron reaction between the iron nitride and alkene directly provides the aziridino product; (2) a cationic stepwise mechanism provides a carbocationic intermediate that subsequently undergoes ring closure; and (3) a stepwise radical mechanism in which an iron(III) imido and an organoradical intermediate undergoes ring closure. These three mechanisms are expected to have different stereochemical outcomes. Specifically, the concerted mechanism is expected to be stereospecific, with the geometry of the alkene translated to the relative stereochemistry of the aziridino product, while the stepwise mechanisms are expected to be non-stereospecific, unless ring closure is fast relative to C–C bond rotation. A fourth possible mechanism, which involves outer sphere electron transfer to provide an iron(III) nitride and styrene radical cation is unlikely based on the very low Fe(IV)/Fe(III) potential (Ered < −2.5 V vs. Fc+/Fc).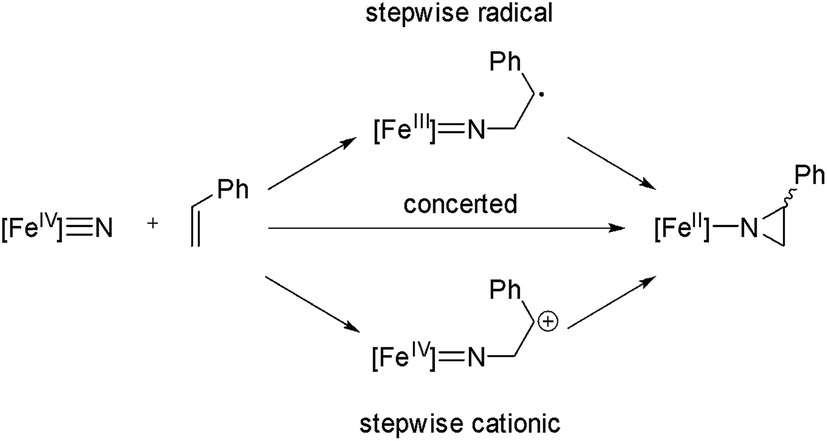 | ||
| Scheme 2 | ||
Mechanistic investigations
Interestingly, thermolysis of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 1 and styrene does not result in complete consumption of the reagents, but instead leads to an equilibrium mixture of the reactants and the corresponding aziridino product 2 (Fig. 1). This result suggests that the aziridination reaction is reversible, allowing us to design an experiment to distinguish the concerted and stepwise mechanisms. Specifically, it is expected that stepwise mechanisms will equilibrate a non-equilibrium mixture of cis- and trans-styrenes, whereas a concerted mechanism is not expected to change the initial isomer ratio. Since steric effects limit the reactivity of 1 to primary styrenes, this hypothesis was tested using a mono-β-deuterated styrene. Consistent with our hypothesis, in addition to providing the expected aziridino product, thermolysis of 1 with a 70:30 mixture of trans- and cis-4-dimethylamino-β-deuterostyrenes results in styrene isomerization (Fig. 2). Styrene isomerization continues after the equilibrium ratio of 1 and the corresponding aziridino product is established, ultimately providing an equimolar mixture of the two stereoisomers. Interestingly, the relative rate of isomerization is subject to an electronic effect, as the isomerization of 4-chloro-β-deuterostyrene is complete within minutes at room temperature, without appreciable formation of the aziridino product. Indeed, 1 is a catalyst for the isomerization 4-chloro-β-deuterostyrene, with equilibrium ratio achieved over the course of a few hours at 65 °C in the presence of 10% 1.
:1 mixture of 1 and styrene does not result in complete consumption of the reagents, but instead leads to an equilibrium mixture of the reactants and the corresponding aziridino product 2 (Fig. 1). This result suggests that the aziridination reaction is reversible, allowing us to design an experiment to distinguish the concerted and stepwise mechanisms. Specifically, it is expected that stepwise mechanisms will equilibrate a non-equilibrium mixture of cis- and trans-styrenes, whereas a concerted mechanism is not expected to change the initial isomer ratio. Since steric effects limit the reactivity of 1 to primary styrenes, this hypothesis was tested using a mono-β-deuterated styrene. Consistent with our hypothesis, in addition to providing the expected aziridino product, thermolysis of 1 with a 70:30 mixture of trans- and cis-4-dimethylamino-β-deuterostyrenes results in styrene isomerization (Fig. 2). Styrene isomerization continues after the equilibrium ratio of 1 and the corresponding aziridino product is established, ultimately providing an equimolar mixture of the two stereoisomers. Interestingly, the relative rate of isomerization is subject to an electronic effect, as the isomerization of 4-chloro-β-deuterostyrene is complete within minutes at room temperature, without appreciable formation of the aziridino product. Indeed, 1 is a catalyst for the isomerization 4-chloro-β-deuterostyrene, with equilibrium ratio achieved over the course of a few hours at 65 °C in the presence of 10% 1.
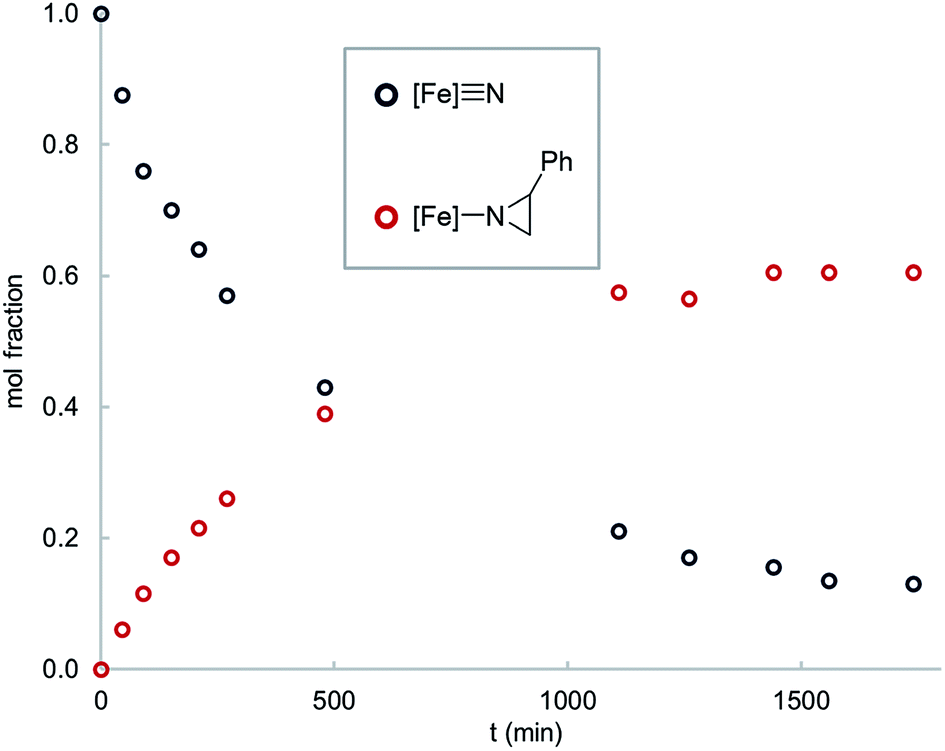 | ||
| Fig. 1 Time course for the reaction of 1 (0.016 mM) with styrene (0.016 mM) in C6D6 solvent at 65 °C. | ||
 | ||
| Fig. 2 Reaction time course for 1 (0.016 mM) with a mixture of trans- and cis-4-dimethylamino-β-deuterostyrenes (0.016 mM) in C6D6 solvent at 65 °C. | ||
These results are most consistent with a stepwise pathway, assuming that the rate of ring closure is fast. However, they do not provide strong evidence into the nature of the intermediate in the stepwise mechanism, i.e. radical or cationic. We do not observe a dramatic difference in the relative reaction rate in different solvents, suggesting that radical intermediates are most likely. However, we are unable to provide definitive experimental evidence in favor of a radical intermediate as attempts at radical trapping or inhibition (e.g. with TEMPO or 9,10-dihydroanthracene) did not have a notable effect on the reaction.
A series of para-substituted styrenes was used to investigate the impact of the electronic effects on the aziridination reaction. Qualitatively, we observe that electron-withdrawing substituents increase the rate of reaction, and very electron poor styrenes, e.g. CH2CH(p-CF3C6H4) react at room temperature. More quantitative insight into the electronic effect on reaction rate was obtained from the relative rates of aziridination as determined by the method of initial rates. The resulting Hammett plot shows that electron-withdrawing substituents increase the rate of reaction (Fig. 3), similarly to nitrogen atom transfer from iron(IV) nitrides to triarylphosphines.83 There is an excellent correlation with the Hammett parameter σp (ρ = 1.2 ± 0.2) suggesting that there is negative charge development in the transition state.
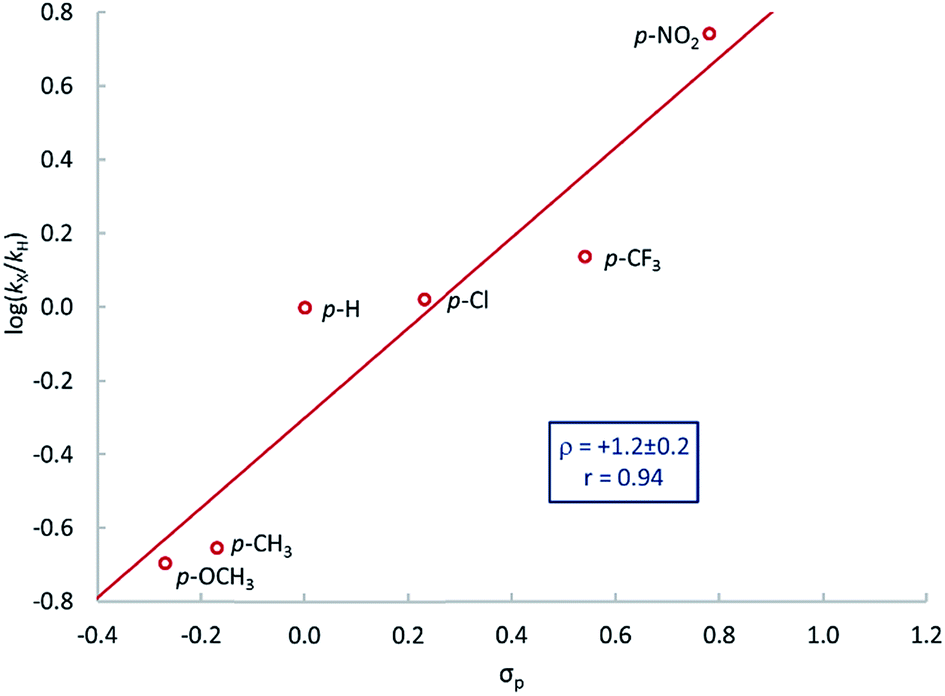 | ||
| Fig. 3 Hammett plot for the reaction between 1 and CH2CH(p-XC6H4) in C6D6 solvent at 65 °C. | ||
In summary, the experimental investigations strongly suggest a reversible, stepwise pathway for styrene aziridination. The lack of a solvent effect as well as the increase in reaction rate with electron-withdrawing styrene substituents is reminiscent of a radical addition reaction, where the nitride has nucleophilic character. However, since the experimental results do not provide strong evidence for radical intermediate, the nature of the mechanism cannot be unambiguously determined.
Computer models
Given the experimental uncertainty regarding the mechanism of styrene aziridination by 1, we have undertaken a computational investigation to better elucidate the reaction pathway. In addition to the full system, we have also performed calculations on a simplified tris(carbene)borate ligand where the alkyl groups bound to the N-heterocyclic carbenes are truncated to H atoms. The results using the simplified model are consistent with those using the full system and in some cases the small model system was used in lieu of the bulkier model for computational simplicity. The calculations suggest that the steric bulk of the tris(carbene)borate has only a minor impact on the reaction energies and all calculated results using the small model system are consistent with those of the full model.The ground state for 1 is the singlet (S = 0), which is calculated to lie 2.6 kcal mol−1 below the triplet (S = 1) state. Calculations using the M06 functional suggest that this energy gap is slightly larger at 5.3 kcal mol−1, but both functionals are in agreement with regards to predicted spin states for all important species and overall mechanistic conclusions of the computational investigation. The quintet state is found significantly higher in energy at 19.0 kcal mol−1 relative to the singlet and the d-orbital splitting for the singlet state is shown in Fig. 4 and is mechanistically irrelevant. Thus, we can conclude that the singlet and triplet states are most significant to understanding the reactivity of 1. An open-shell singlet configuration for 1 was also considered, but we were unable to locate such despite significant efforts. The orbitals shapes for the triplet are similar, but 136α is occupied. The orbital energies for the frontier d-orbitals for the triplet are presented alongside those of the singlet. The C3v geometry of the low-spin complex leads to an approximate 2 + 1 + 2 orbital splitting with the two lowest degenerate frontier orbitals being doubly occupied.84 The LUMO is the Fe-dz2 orbital, which is significantly stabilized by small σ*-based interactions with the carbene fragments displaying significant amounts of admixed character from the iron s and pz orbitals that reduce the net overlap with the nitrogen pz orbital,13,14 as illustrated in Fig. 4. This stabilization results in the triplet state for 1 being found only slightly above the singlet ground state energetically.
 | ||
| Fig. 4 (a) Isodensity surface plots (isodensity value = 0.07 au) of five Fe 3d-based molecular orbitals for simplified tris(carbene)borate model with (b) qualitative frontier orbital diagram for singlet (S = 0) and triplet (S = 1) states. | ||
The computed structures of the singlet and triplet states of 1 are shown in Fig. 5. Selected structural parameters from the crystal structure of 1 are given in the ESI† for comparison and are in generally good agreement with those of the calculated structure for the closed-shell singlet state.13,48 The triplet state exhibits several geometric distortions that result from populating the Fe-dz2 orbital. The Fe-nitride bond lengthens from 1.498 Å to 1.558 Å and the angle defined by B–Fe–N distorts significantly from linearity (179°) to 156° to decrease the unfavorable overlap of the now occupied antibonding orbital. In the quintet state, the nitride ligand distorts even more to an angle of 148°. All of the Fe–carbene carbon bonds also are lengthened slightly as there are still some σ-type interactions with the Fe-dz2 orbital. One Fe–C distance, however, is substantially longer than the other two Fe–C bond distances as promotion of an electron into the Fe-dz2 orbital results in an asymmetric occupation of the previously degenerate occupied metal-based orbitals leading to a first-order Jahn–Teller-like distortion.
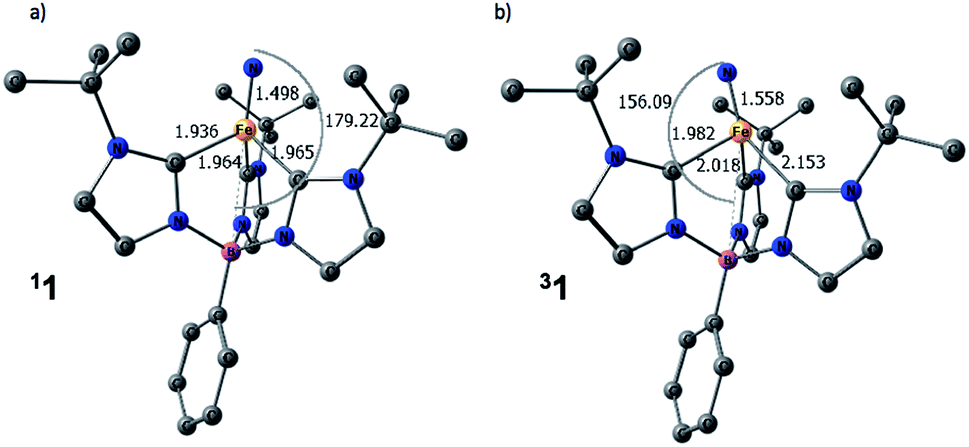 | ||
| Fig. 5 Calculated structures of (a) singlet and (b) triplet spin states for 1. Hydrogen atoms are omitted for clarity. [Unit: Å] | ||
The electronic structures for the different spin states of 1 provide insights into possible reaction pathways for styrene aziridination. On the ground state S = 0 surface, a stepwise mechanism involving an open-shell singlet (OSS) has a kinetic barrier for C–N bond formation of 27.4 kcal mol−1. While the experimental investigations are most consistent with a stepwise mechanism for styrene aziridination, we computationally probed the feasibility of a concerted addition of styrene. This mechanism involves two-electron transfer from the styrene π-orbital to the Fe-dz2 σ*-orbital of the ground state closed-shell singlet 1. However, we were unable to locate a transition state for the concerted mechanism. Interestingly, we were also able to locate a transition state for stepwise C–N bond formation on the closed shell singlet surface, but the barrier was found to be significantly higher in energy at 31.5 kcal mol−1 with respect to the singlet ground state of 1. This closed-shell TS, where the nitride ligand forms a bond to only one of the sp2 carbons of styrene, would lead to the formation of a carbocationic intermediate. Such an intermediate would also be consistent with the experimental stereochemical observations, however, we could not find a stable intermediate in our calculations on the closed shell surface. The ring closure reaction between this putative carbocation and the incipient nitrene nitrogen atom is be expected to be rapid, making this closed-shell attack of the styrene on the nitride a good approximation for the barrier in the truly concerted mechanism.
On the other hand, the low-energy triplet state that is only 2.6 kcal mol−1 above the singlet ground state potentially provides access to a radical type mechanism in which single electron transfer from the styrene π-orbital to the Fe-dz2 σ*-orbital leads to a biradical intermediate. While experimental attempts at radical trapping did not have a significant effect on the outcome of the aziridination reaction, this does not necessarily preclude the possibility of radical intermediates. For instance, the biradical intermediate may be too short-lived to trap intermolecularly, but sufficiently long-lived to allow for C–C bond rotation prior to transfer of the second electron concomitant with aziridine ring closure.
In good agreement with experimental results, our calculations on the full reaction coordinate also suggest that the reaction of Fe(IV)-nitride with styrene occurs in a stepwise manner, where the nitride ligand initially forms a bond to one of the alkenyl carbons of the styrene substrate. The computed reaction profile for the stepwise addition is shown in Fig. 6. The initial C–N bond formation step 1-TS, which is also expected to be rate-determining, is most favorable on the triplet potential energy surface, with a barrier of 24.6 kcal mol−1. This is consistent with the experimental results, where heat is required to drive the reaction and no intermediates are observed. Furthermore, the low barrier in the reverse direction (15.9 kcal mol−1) is consistent with the cis/trans isomerization of the styrene substrate that is observed experimentally. Unsurprisingly, the quintet surface, which starts 19.0 kcal mol−1 above the singlet ground state, has a very high barrier for styrene addition of 33.8 kcal mol−1.
 | ||
| Fig. 6 Reaction profile for aziridination of styrene by an Fe(IV)-nitride, showing closed shell singlet (S = 0), triplet (S = 1) and quintet (S = 2) spin surfaces. Only stepwise mechanisms are shown as concerted pathways were found to be inaccessible energetically. | ||
The electrophilic nitride fragment of the triplet is preferentially attacked by the β-carbon of the incoming styrene substrate as the phenyl group will help to better stabilize the radical resulting from one-electron transfer to the metal center. A study using (Z)-prop-1-en-1-ylbenzene as the substrate in a small model system (R groups on the NHC donors truncated to H) reveals that the energetic stabilization realized by attacking the β- rather than the α-carbon is about 11 kcal mol−1 at the transition state (see the ESI† for more details).
Scheme 3 depicts the electron density polarization and corresponding change in orbital occupation that occurs on the triplet surface during the course of the aziridination reaction prior to the intersystem crossing to the quintet product. In complex 1, there is a small amount of α-spin density that bleeds out onto the nitride fragment of the triplet (Mulliken spin population of 0.169). Following the formation of the C–N bond, however, there is a significant accumulation of spin density on the nitrogen atom in intermediate 31a, which is the most stable biradical intermediate found at 8.7 kcal mol−1 above the singlet ground state reactant 11. The corresponding quintet and open-shell singlet intermediates, 51a and 11a, are 1.6 and 4.1 kcal mol−1 higher in energy, respectively. During C–N bond formation an α-electron is transferred from the alkene π-orbital to Fe. The Mulliken spin population consequently increases from 1.81 in 31 to 3.21 in 31a leaving an excess of β-electron density of 0.67 β on the former α-carbon of the styrene. The remainder of the excess β-spin (−0.42) has been distributed on the nitrene nitrogen atom. At the transition state for C–N bond formation, 31-TS, the C–N bond distance is 1.944 Å and a majority of the α-electron density has been transferred to Fe, which has a spin population of 2.63. Surprisingly, there is already a significant amount of β-spin present on the nitride fragment (0.49 β) with a population of only 0.26 β on the α-carbon of the styrene. This β-spin on the nitrene nitrogen reacts in a radical coupling mechanism with the slight excess of α-spin of 0.15 on the β-carbon of the incoming styrene to form the C–N bond. The structures of 31-TS and intermediate 31a are shown in Fig. 7. The CC bond distance has lengthened to 1.384 Å in 31-TS from 1.338 Å in free styrene and further increases to a distance of 1.498 Å in 31a. Simultaneously, the Fe–N bond distance lengthens from 1.558 Å in 31 to 1.634 Å at the transition state and finally to 1.748 Å in the biradical intermediate, consistent with a decrease in the bond order of the Fe–N bond from three to two due to population of an Fe–N π*-orbital.
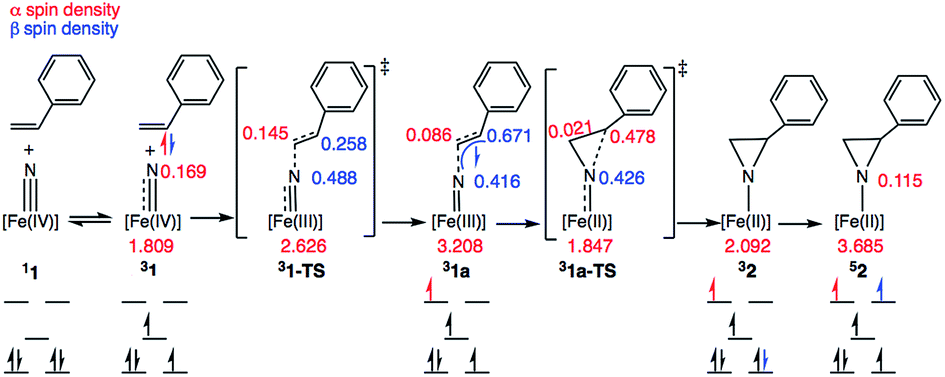 | ||
| Scheme 3 | ||
 | ||
| Fig. 7 Optimized structures with selected bond distances in Å of 31-TS and 31a. Black values indicate bond lengths in Å, red values indicate α Mulliken spin densities, and blue values are excess β spin densities. | ||
After formation of intermediate 31a, the second electron transfer takes place through the movement of a β-electron from the styrene π-orbital to iron, reducing the metal from Fe(III) to Fe(II). This electron transfer leads to an excess of α-electron density on the α-carbon of styrene, which subsequently couples with the excess β-electron density on the nitrene nitrogen atom in a facile radical recombination reaction. This closes the three-membered ring with a barrier of just 12.18 kcal mol−1 on the triplet potential energy surface. An analysis of the Mulliken spin densities at the transition state 31a-TS reveals that the spin density on Fe drops from 3.208 in 31a to 1.847 at the TS, consistent with an S = 1 Fe(II) ion. The spin density on the nitrene nitrogen atom at the TS (0.43) is essentially the same as in the biradical intermediate (0.42). The spin population on the α-carbon of the styrene also changes from having excess β-electron density (0.67) to surplus α-electron density (0.48). Thus, a full β-electron has been transferred from the styrene substrate to iron. However, because the β-spin is distributed across both the nitrene nitrogen and the α-carbon of styrene in 31a, the electron transfer makes styrene α-electron density available to couple with the nitrene β-spin, thereby facilitating ring closure. In contrast to the low barrier on the triplet surface, the barrier to ring closure is calculated to be 23.2 kcal mol−1 on the open-shell singlet surface, which is unsurprising given that generation of the aziridino product is 2.0 kcal mol−1 uphill with respect to the singlet ground state of 1. Consistent with experimental results, the final aziridino complex is calculated to be most stable as a quintet, found to be 16.5 kcal mol−1 over the corresponding triplet species.13,27,57–59 We were unable to locate a true transition state for aziridine ring closure on the quintet surface. This is likely due to the fact that the spin densities of 0.42 on the nitrene nitrogen and 0.77 on the styrene α-carbon in the biradical intermediate 51a facilitate a very rapid recombination event that forms the aziridine. Using the small model system described above, a linear synchronous transit scan reveals that the formation of the second C–N bond formation is likely barrierless on the electronic potential energy surface (see the ESI† for more details). The intersystem crossing, which is likely helped by the metal center, may occur prior to the ring closing step, or may only occur after product formation once a large thermodynamic driving force for the quintet state is established. Our model calculations are therefore suggestive of multistate reactivity, where radical coupling between styrene and the nitride fragment proceeds more efficiently on the triplet potential energy surface compared to the ground state singlet surface. The triplet intermediate may be long-lived enough to permit C–C bond rotation before another spin flip to the quintet state enables facile ring closure.
As quantified by the Hammett study mentioned above, electron-withdrawing p-substituents increase the rate of styrene aziridination. As revealed by the calculated barriers for the rate-determining C–N bond formation step (Table 1), the computations replicate this trend for the p-substituted styrenes, p-NO2 and p-NMe2, as well as the previously discussed, unsubstituted, p-H styrene. Importantly, analysis of the complete mechanism for all three substrates with the small model system (see above) reveals that the triplet potential energy surface is the most favorable for each substrate, allowing us to only model the rate-determining step 31-TS for p-NO2 and p-NMe2 for the full system. The computational models reveal that the lowest barrier to reaction with p-NO2 styrene is just 18.3 kcal mol−1, which is readily overcome even under ambient conditions, consistent with the experimental observation that this substrate reacts at room temperature. The unsubstituted substrate and p-NMe2 substituted analog show much more similar kinetic barriers at 24.6 and 25.6 kcal mol−1, respectively, with the barrier to C–N bond formation with p-NMe2 calculated to be slightly higher, consistent with experiment.
| Styrene | 31-TS (kcal mol−1) | Bond length | Mulliken spin density | |||||
|---|---|---|---|---|---|---|---|---|
| C–N (Å) | Fe–N (Å) | C1–C2 (Å) | Fe | N | C1 | C2 | ||
| p-NO2 | 18.27 | 2.005 | 1.573 | 1.386 | 1.781α | 0.044β | 0.060β | 0.182α |
| p-H | 24.61 | 1.944 | 1.634 | 1.384 | 2.626α | 0.488β | 0.145α | 0.258β |
| p-NMe2 | 25.55 | 1.957 | 1.641 | 1.384 | 2.715α | 0.523β | 0.172α | 0.321β |
Interestingly, the calculations reveal that the reaction of 1 with p-NO2 styrene has a significantly different electronic structure at the transition state, whereas the other two substrates resemble each other much more closely (Table 1). Notably, the TS for p-NO2 occurs very early with respect to electron transfer. The C–N bond distance at the transition state 31-TS is substantially longer p-NO2 (2.005 Å) than for p-H (1.944 Å) and p-NMe2 (1.957 Å). Additionally, the iron nitride bond is essentially unactivated in the p-NO2 case with a length of 1.573 Å compared 1.558 Å for 31 and 1.498 Å for the ground state species 11. In contrast, p-H and p-NMe2 display much longer bond lengths of 1.634 and 1.641 Å, respectively. Whereas 31-TS for the p-H and p-NMe2 substrates occurs much later with respect to the first electron transfer and demonstrate significant radical character on the nitride fragment and both carbon atoms of the alkene, the spin densities for p-NO2 are largely unchanged with respect to 31, suggesting limited electron transfer between the nitride fragment and styrene at the transition state when the substrate bears and electron-withdrawing group. Since the electron-withdrawing nitro group would typically destabilize radical formation, its presence encourages the reaction to proceed through a transition state that closely resembles the initial reactants in electronic structure. The minimal amount of electronic and structural reorganization required at the transition state for the p-NO2 substrate likely helps facilitate a faster rate of reaction compared to the unsubstituted and p-NMe2 substrates where larger structural changes accompany the development of significant radical cation character at the transition state.
Conclusions
A combined experimental and computational investigation into the aziridination of styrenes by the Fe(IV) nitride complex 1 reveals that the reaction proceeds as an asynchronous addition of the styrene to the nitride moiety. While we were able to locate a pathway leading to aziridine formation on the same spin state surface as the singlet ground state for 1, the most energetically favorable mechanism actually occurs on the triplet surface. This multistate character therefore results in the rate determining step being on a different spin surface than the reactant ground state. The triplet state imparts slight nucleophilic nitridyl character to the nitride fragment, thereby promoting the stepwise radical addition of the styrene. It is likely that the styrene substrate also plays a role in stabilizing this radical character during the reaction. The accessibility of a spin state surface with a lower barrier towards C–N bond formation is therefore critical for facilitating styrene aziridination.This combined experimental and computational investigation provides the first evidence for multistate reactivity by a transition metal nitride complex. Despite the differences in bonding with the related oxo and imido ligands (e.g. ligand charge, covalency), complex 1 is able to access a different spin state surface that facilitates the aziridination reactivity. It is worth comparing the mechanism of alkene aziridination by 1, which involves nitrogen atom transfer, with that determined for high valent iron porphyrin tosylimido complexes, which involve nitrene transfer. In the latter case, multistate reactivity requires access to lower spin states,33 whereas access to a higher spin state is required to facilitate styrene aziridination by 1. This difference in spin state effects, which stems from the different ground state electronic structures, provides a rationale for the differential electronic selectivity of 1 and iron porphyrin complexes. Thus, while electron poor styrenes that better stabilize the high spin transition state react more rapidly for 1, in the case of iron porphyrins aziridination is postulated to be faster with more electron rich styrenes.33
The mechanistic insights from this study also suggest strategies for rationally tuning the aziridination reaction. For example, ligand design strategies that increase the nitridyl radical character (e.g. by decreasing the relative energy of the dz2-derived orbital) may increase the reactivity of 1 towards alkene substrates that are less stabilizing of the radical character. Substrates bearing electron-withdrawing groups require minimal electronic reorganization at the rate-determining transition state, leading to faster reaction rates. Moreover, the stepwise nature of the reaction mechanism also suggests a strategy for stereospecific aziridination, where chiral substituents on the tris(carbene)borate ligands are expected to impart stereocontrol on the ring closing step. Studies towards exploiting these novel insights are underway in our laboratories.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SBM and JMS acknowledge support from IU and DOE-BES (DE-FG02-08ER15996). We thank the Institute for Basic Science in Korea for financial support (IBS-R10-D1).Notes and references
- A. Padwa, Aziridines and Azirines: Monocyclic, in Comprehensive Heterocyclic Chemistry III, ed. C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, pp. 1–104 Search PubMed.
- J. T. Njardarson, Catalytic Ring Expansion Adventures, Synlett, 2013, 24, 787–803 CrossRef.
- A. L. Cardoso and T. M. V. D. Pinho e Melo, Aziridines in Formal [3 + 2] Cycloadditions: Synthesis of Five-Membered Heterocycles, Eur. J. Org. Chem., 2012, 2012, 6479–6501 CrossRef.
- H. Pellissier, Recent Developments in Asymmetric Aziridination, Tetrahedron, 2010, 66, 1509–1555 CrossRef.
- C. Botuha, F. Chemla, F. Ferreira and A. Pérez-Luna, Aziridines in Natural Product Synthesis, in Heterocycles in Natural Product Synthesis, ed. K. C. Majumdar and S. K. Chattopadhyay, 2011 Search PubMed.
- C. J. Thibodeaux, W.-C. Chang and H.-W. Liu, Enzymatic Chemistry of Cyclopropane, Epoxide, and Aziridine Biosynthesis, Chem. Rev., 2012, 112, 1681–1709 CrossRef PubMed.
- I. D. G. Watson, L. Yu and A. K. Yudin, Advances in Nitrogen Transfer Reactions Involving Aziridines, Acc. Chem. Res., 2006, 39, 194–206 CrossRef PubMed.
- T. G. Driver, Recent Advances in Transition Metal-Catalyzed N-atom Transfer Reactions of Azides, Org. Biomol. Chem., 2010, 8, 3831–3846 RSC.
- D. A. Evans, M. M. Faul, M. T. Bilodeau, B. A. Anderson and D. M. Barnes, Bis(oxazoline)–Copper Complexes as Chiral Catalysts for the Enantioselective Aziridination of Olefins, J. Am. Chem. Soc., 1993, 115, 5328–5329 CrossRef.
- Z. Li, K. R. Conser and E. N. Jacobsen, Asymmetric Alkene Aziridination with Readily Available Chiral Diimine-based Catalysts, J. Am. Chem. Soc., 1993, 115, 5326–5327 CrossRef.
- B. M. Trost and G. Dong, New Class of Nucleophiles for Palladium-Catalyzed Asymmetric Allylic Alkylation. Total Synthesis of Agelastatin A, J. Am. Chem. Soc., 2006, 128, 6054–6055 CrossRef PubMed.
- S. Cenini, S. Tollari, A. Penoni and C. Cereda, Catalytic Amination of Unsaturated Hydrocarbons: Reactions of p-nitrophenylazide with Alkenes Catalysed by Metallo-porphyrins, J. Mol. Catal. A, 1999, 137, 135–146 CrossRef.
- S. A. Cramer and D. M. Jenkins, Synthesis of Aziridines from Alkenes and Aryl Azides with a Reusable Macrocyclic Tetracarbene Iron Catalyst, J. Am. Chem. Soc., 2011, 133, 19342–19345 CrossRef PubMed.
- D. M. Jenkins, Atom-Economical C2 + N1 Aziridination: Progress towards Catalytic Intermolecular Reactions Using Alkenes and Aryl Azides, Synlett, 2012, 23, 1267–1270 CrossRef.
- S. A. Cramer, R. Hernández Sánchez, D. F. Brakhage and D. M. Jenkins, Probing the Role of an FeIV Tetrazene in Catalytic Aziridination, Chem. Commun., 2014, 50, 13967–13970 RSC.
- J. L. Jat, M. P. Paudyal, H. Gao, Q.-L. Xu, M. Yousufuddin, D. Devarajan, D. H. Ess, L. Kürti and J. R. Falck, Direct Stereospecific Synthesis of Unprotected N–H and N–Me Aziridines from Olefins, Science, 2014, 343, 61–65 CrossRef PubMed.
- Z. Ma, Z. Zhou and L. Kürti, Direct and Stereospecific Synthesis of N–H and N-Alkyl Aziridines from Unactivated Olefins Using Hydroxylamine-O-Sulfonic Acids, Angew. Chem., Int. Ed., 2017, 56, 9886–9890 CrossRef PubMed.
- C. L. Keller, J. L. Kern, B. D. Terry, S. Roy and D. M. Jenkins, Catalytic Aziridination with Alcoholic Substrates via a Chromium Tetracarbene Catalyst, Chem. Commun., 2018, 54, 1429–1432 RSC.
- J. J. Scepaniak, M. D. Fulton, R. P. Bontchev, E. N. Duesler, M. L. Kirk and J. M. Smith, Structural and Spectroscopic Characterization of an Electrophilic Iron Nitrido Complex, J. Am. Chem. Soc., 2008, 130, 10515–10517 CrossRef PubMed.
- S. B. Muñoz, W.-T. Lee, D. A. Dickie, J. J. Scepaniak, D. Subedi, M. Pink, M. D. Johnson and J. M. Smith, Styrene Aziridination by Iron(IV) Nitrides, Angew. Chem., Int. Ed., 2015, 54, 10600–10603 CrossRef PubMed.
- S. N. Brown, Insertion of a Metal Nitride into Carbon–Carbon Double Bonds, J. Am. Chem. Soc., 1999, 121, 9752–9753 CrossRef.
- A. G. Maestri, S. D. Taylor, S. M. Schuck and S. N. Brown, Cleavage of Conjugated Alkenes by Cationic Osmium Nitrides: Scope of the Reaction and Dynamics of the Azaallenium Products, Organometallics, 2004, 23, 1932–1946 CrossRef.
- J. M. Smith, Reactive Transition Metal Nitride Complexes, in Progress in Inorganic Chemistry, ed. K. D. Karlin, Wiley, 2014 Search PubMed.
- A. G. Maestri, K. S. Cherry, J. J. Toboni and S. N. Brown, [4 + 1] Cycloadditions of Cyclohexadienes with Osmium Nitrides, J. Am. Chem. Soc., 2001, 123, 7459–7460 CrossRef PubMed.
- W. L. Man, W. W. Y. Lam, H. K. Kwong, S. M. Yiu and T. C. Lau, Ligand-Accelerated Activation of Strong C–H Bonds of Alkanes by a (Salen)ruthenium(VI)–Nitrido Complex, Angew. Chem., Int. Ed., 2012, 51, 9101–9104 CrossRef PubMed.
- W. L. Man, J. Xie, P. K. Lo, W. Y. Lam William, S. M. Yiu, K. C. Lau and T. C. Lau, Functionalization of Alkynes by a (Salen)ruthenium(VI) Nitrido Complex, Angew. Chem., Int. Ed., 2014, 53, 8463–8466 CrossRef PubMed.
- J. Xie, W.-L. Man, C.-Y. Wong, X. Chang, C.-M. Che and T.-C. Lau, Four-Electron Oxidation of Phenols to p-Benzoquinone Imines by a (Salen)ruthenium(VI) Nitrido Complex, J. Am. Chem. Soc., 2016, 138, 5817–5820 CrossRef PubMed.
- W.-H. Chiu, W.-M. Cheung, M.-C. Chong, H. H.-Y. Sung, I. D. Williams and W.-H. Leung, Tetranuclear Ruthenium Cyclopentadienyl and Pentamethylcyclopentadienyl Complexes Containing Bridging Nitrido Ligands, Organometallics, 2018, 37, 1034–1039 CrossRef.
- (a) C. E. Laplaza and C. C. Cummins, Science, 1995, 268, 861–863 CrossRef PubMed; (b) Q. Liao, A. Cavaille, N. Saffon-Merceron and N. Mezailles, Angew. Chem., Int. Ed., 2016, 55, 11212–11216 CrossRef PubMed; (c) T. J. Hebden, R. R. Schrock, M. K. Takase and P. Mueller, Chem. Commun., 2012, 48, 1851–1853 RSC; (d) I. Klopsch, M. Finger, C. Wuertele, B. Milde, D. B. Werz and S. Schneider, J. Am. Chem. Soc., 2014, 136, 6881–6883 CrossRef PubMed; (e) K. Arashiba, A. Eizawa, H. Tanaka, K. Nakajima, K. Yoshizawa and Y. Nishibayashi, Bull. Chem. Soc. Jpn., 2017, 90, 1111–1118 CrossRef; (f) B. M. Lindley, R. S. van Alten, M. Finger, F. Schendzielorz, C. Wuertele, A. J. M. Miller, I. Siewert and S. Schneider, J. Am. Chem. Soc., 2018, 140, 7922–7935 CrossRef PubMed.
- M. Nakanishi, A.-F. Salit and C. Bolm, Iron-Catalyzed Aziridination Reactions, Adv. Synth. Catal., 2008, 350, 1835–1840 CrossRef.
- A. C. Mayer, A.-F. Salit and C. Bolm, Iron-Catalysed Aziridination Reactions Promoted by an Ionic Liquid, Chem. Commun., 2008, 5975–5977 RSC.
- S. Liang and M. P. Jensen, Half-Sandwich Scorpionates as Nitrene Transfer Catalysts, Organometallics, 2012, 31, 8055–8058 CrossRef.
- E. T. Hennessy, R. Y. Liu, D. A. Iovan, R. A. Duncan and T. A. Betley, Iron-mediated Intermolecular N-group Transfer Chemistry with Olefinic Substrates, Chem. Sci., 2014, 5, 1526–1532 RSC.
-
(a) Y. Moreau, H. Chen, E. Derat, H. Hirao, C. Bolm and S. Shaik, NR Transfer Reactivity of Azo-Compound I of P450. How Does the Nitrogen Substituent Tune the Reactivity of the Species toward C–H and CC Activation?, J. Phys. Chem. B, 2007, 111, 10288–10299 CrossRef PubMed;
(b) R. Patra, G. Coin, L. Castro, P. Dubourdeaux, M. Clémancey, J. Pécaut, C. Lebrun, P. Maldivi and J.-M. Latour, Rational Design of Fe Catalysts for Olefin Aziridination through DFT-Based Mechanistic Analysis, Catal. Sci. Technol., 2017, 7, 4388–4400 RSC.
- C. Damiano, D. Intrieri and E. Gallo, Aziridination of Alkenes Promoted by Iron or Ruthenium Complexes, Inorg. Chim. Acta, 2018, 470, 51–67 CrossRef.
- K. L. Klotz, L. M. Slominski, M. E. Riemer, J. A. Phillips and J. A. Halfen, Mechanism of the Iron-Mediated Alkene Aziridination Reaction: Experimental and Computational Investigations, Inorg. Chem., 2009, 48, 801–803 CrossRef PubMed.
- C.-M. Che and J.-S. Huang, Metalloporphyrin-based Oxidation Systems: From Biomimetic Reactions to Application in Organic Synthesis, Chem. Commun., 2009, 3996–4015 RSC.
- K. Schröder, K. Junge, B. Bitterlich and M. Beller, Fe-Catalyzed Oxidation Reactions of Olefins, Alkanes, and Alcohols: Involvement of Oxo- and Peroxo Complexes, Top. Organomet. Chem., 2011, 33, 83–109 CrossRef.
- H. Srour, P. Le Maux, S. Chevance and G. Simonneaux, Metal-Catalyzed Asymmetric Sulfoxidation, Epoxidation and Hydroxylation by Hydrogen Peroxide, Coord. Chem. Rev., 2013, 257, 3030–3050 CrossRef.
- X. Huang and J. T. Groves, Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins, Chem. Rev., 2018, 118, 2491–2553 CrossRef PubMed.
- J. T. Groves, T. E. Nemo and R. S. Myers, Hydroxylation and Epoxidation Catalyzed by Iron–Porphine Complexes. Oxygen Transfer from Iodosylbenzene, J. Am. Chem. Soc., 1979, 101, 1032–1033 CrossRef.
- J. T. Groves, R. C. Haushalter, M. Nakamura, T. E. Nemo and B. J. Evans, High-valent Iron–Porphyrin Complexes Related to Peroxidase and Cytochrome P-450, J. Am. Chem. Soc., 1981, 103, 2884–2886 CrossRef.
- H. Fujii, Electronic Structure and Reactivity of High-valent Oxo Iron Porphyrins, Coord. Chem. Rev., 2002, 226, 51–60 CrossRef.
- M. H. Lim, J.-U. Rohde, A. Stubna, M. R. Bukowski, M. Costas, R. Y. N. Ho, E. Münck, W. Nam and L. Que, AnFeIVO Complex of a Tetradentate Tripodal Nonheme Ligand, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3665 CrossRef PubMed.
- J. Bautz, P. Comba, C. opezdeLaorden, M. Menzel and G. Rajaraman, Biomimetic High-Valent Non-Heme Iron Oxidants for the cis-Dihydroxylation and Epoxidation of Olefins, Angew. Chem., Int. Ed., 2007, 46, 8067–8070 CrossRef PubMed.
- J. Annaraj, S. Kim, M. S. Seo, Y.-M. Lee, Y. Kim, S.-J. Kim, Y. S. Choi, H. G. Jang and W. Nam, An Iron(II) Complex with a N3S2 Thioether Ligand in the Generation of an Iron(IV)-oxo Complex and its Reactivity in Olefin Epoxidation, Inorg. Chim. Acta, 2009, 362, 1031–1034 CrossRef.
- A. Thibon, J.-F. Bartoli, S. Bourcier and F. Banse, Mononuclear Iron Complexes Relevant to Nonheme Iron Oxygenases. Synthesis, Characterizations and Reactivity of Fe-Oxo and Fe-Peroxo Intermediates, Dalton Trans., 2009, 9587–9594 RSC.
- W. Ye, R. J. Staples and E. V. Rybak-Akimova, Oxygen Atom Transfer Mediated by an Iron(IV)/Iron(II) Macrocyclic Complex Containing Pyridine and Tertiary Amine Donors, J. Inorg. Biochem., 2012, 115, 1–12 CrossRef PubMed.
- W. Nam, High-Valent Iron(IV)–Oxo Complexes of Heme and Non-Heme Ligands in Oxygenation Reactions, Acc. Chem. Res., 2007, 40, 522–531 CrossRef PubMed.
- W. J. Song, Y. O. Ryu, R. Song and W. Nam, Oxoiron(IV) Porphyrin π-Cation Radical Complexes with a Chameleon Behavior in Cytochrome P450 Model Reactions, J. Bio. Inorg. Chem., 2005, 10, 294–304 CrossRef PubMed.
- K. K. Singh, M. K. Tiwari, B. B. Dhar, K. Vanka and S. Sen Gupta, Mechanism of Oxygen Atom Transfer from FeV(O) to Olefins at Room Temperature, Inorg. Chem., 2015, 54, 6112–6121 CrossRef PubMed.
- J. P. Collman, T. Kodadek and J. I. Brauman, Oxygenation of Styrene by Cytochrome P-450 Model Systems. A Mechanistic Study, J. Am. Chem. Soc., 1986, 108, 2588–2594 CrossRef.
- A. J. Castellino and T. C. Bruice, Intermediates in the Epoxidation of Alkenes by Cytochrome P-450 Models. 1. cis-Stilbene as a Mechanistic Probe, J. Am. Chem. Soc., 1988, 110, 158–162 CrossRef.
- D. Schröder, S. Shaik and H. Schwarz, Two-State Reactivity as a New Concept in Organometallic Chemistry, Acc. Chem. Res., 2000, 33, 139–145 CrossRef.
- S. P. de Visser, F. Ogliaro, N. Harris and S. Shaik, Multi-State Epoxidation of Ethene by Cytochrome P450: A Quantum Chemical Study, J. Am. Chem. Soc., 2001, 123, 3037–3047 CrossRef PubMed.
- S. Shaik, S. P. de Visser, F. Ogliaro, H. Schwarz and D. Schröder, Two-State Reactivity Mechanisms of Hydroxylation and Epoxidation by Cytochrome P-450 Revealed by Theory, Curr.Opin. Chem. Biol., 2002, 6, 556–567 CrossRef PubMed.
- D. Kumar, B. Karamzadeh, G. N. Sastry and S. P. de Visser, What Factors Influence the Rate Constant of Substrate Epoxidation by Compound I of Cytochrome P450 and Analogous Iron(IV)-Oxo Oxidants?, J. Am. Chem. Soc., 2010, 132, 7656–7667 CrossRef PubMed.
- D. Kumar, R. Latifi, S. Kumar, E. V. Rybak-Akimova, M. A. Sainna and S. P. de Visser, Rationalization of the Barrier Height for p-Z-styrene Epoxidation by Iron(IV)-Oxo Porphyrin Cation Radicals with Variable Axial Ligands, Inorg. Chem., 2013, 52, 7968–7979 CrossRef PubMed.
- Y. Ren, K. Cheaib, J. Jacquet, H. Vezin, L. Fensterbank, M. Orio, S. Blanchard and M. Desage-El Murr, Copper-Catalyzed Aziridination with Redox-Active Ligands: Molecular Spin Catalysis, Chem.–Eur. J., 2018, 24, 5086–5090 CrossRef PubMed.
- J. Liu, L. Hu, L. Wang, H. Chen and L. Deng, An Iron(II) Ylide Complex as a Masked Open-Shell Iron Alkylidene Species in Its Alkylidene-Transfer Reactions with Alkenes, J. Am. Chem. Soc., 2017, 139, 3876–3888 CrossRef PubMed.
- R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- T. Ziegler, Approximate Density Functional Theory as a Practical Tool in Molecular Energetics and Dynamics, Chem. Rev., 1991, 91, 651–667 CrossRef.
- Jaguar 8.1, Schrödinger, Inc., New York, NY, 2014 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. D. Becke, Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef.
- A. D. Becke, Density-Functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density, Phys. Rev. B, 1988, 37, 785–789 CrossRef.
- J. C. Slater, Quantum Theory of Molecules and Solids: The Self-Consistent Field for Molecules and Solids, McGraw-Hill, New York, 1974, vol. 4 Search PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin-Density Calculations – A Critical Analysis, Can. J. Phys., 1980, 58, 1200–1211 CrossRef.
- P. J. Hay and W. R. Wadt, Ab initio Effective Core Potentials for Molecular Calculations - Potentials for the Transition-Metal Atoms Sc to Hg, J. Chem. Phys., 1985, 82, 270–283 CrossRef.
- W. R. Wadt and P. J. Hay, Ab initio Effective Core Potentials for Molecular Calculations - Potentials for Main Group Elements Na to Bi, J. Chem. Phys., 1985, 82, 284–298 CrossRef.
- P. J. Hay and W. R. Wadt, Ab initio Effective Core Potentials for Molecular Calculations - Potentials for K to Au Including the Outermost Core Orbitals, J. Chem. Phys., 1985, 82, 299–310 CrossRef.
- T. H. Dunning Jr, Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- Y. Zhao and D. G. Truhlar, The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- B. Marten, K. Kim, C. Cortis, R. A. Friesner, R. B. Murphy, M. N. Ringnalda, D. Sitkoff and B. Honig, New Model for Calculation of Solvation Free Energies: Correction of Self-Consistent Reaction Field Continuum Dielectric Theory for Short-Range Hydrogen-Bonding Effects, J. Phys. Chem., 1996, 100, 11775–11788 CrossRef.
- M. Friedrichs, R. H. Zhou, S. R. Edinger and R. A. Friesner, Poisson-Boltzmann Analytical Gradients for Molecular Modeling Calculations, J. Phys. Chem. B, 1999, 103, 3057–3061 CrossRef.
- S. R. Edinger, C. Cortis, P. S. Shenkin and R. A. Friesner, Solvation Free Energies of Peptides: Comparison of Approximate Continuum Solvation Models With Accurate Solution of the Poisson-Boltzmann Equation, J. Phys. Chem. B, 1997, 101, 1190–1197 CrossRef.
- A. A. Rashin and B. Honig, Reevaluation of the Born Model of Ion Hydration, J. Phys. Chem., 1985, 89, 5588–5593 CrossRef.
- L. Noodleman, Valence Bond Description of Antiferromagnetic Coupling in Transition Metal Dimers, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef.
- L. Noodleman and E. R. Davidson, Ligand Spin Polarization and Antiferromagnetic Coupling in Transition Metal Dimers, Chem. Phys., 1986, 109, 131–143 CrossRef.
- L. Noodleman, T. Lovell, W.-G. Han, J. Li and F. Himo, Quantum Chemical Studies of Intermediates and Reaction Pathways in Selected Enzymes and Catalytic Synthetic Systems, Chem. Rev., 2004, 104, 459–508 CrossRef PubMed.
- F. P. Guengerich and T. L. Macdonald, Chemical Mechanisms of Catalysis by Cytochromes P-450: A Unified View, Acc. Chem. Res., 1984, 17, 9–16 CrossRef.
- P. R. Ortiz de Montellano, Substrate Oxidation by Cytochrome P450 Enzymes, in Cytochrome P450: Structure, Mechanism, and Biochemistry, ed. P. R. Ortiz de Montellano, Springer International Publishing, Cham, 2015, pp. 111–176 Search PubMed.
- L. Bucinsky, M. Breza, W.-T. Lee, A. K. Hickey, D. A. Dickie, I. Nieto, J. A. DeGayner, T. D. Harris, K. Meyer, J. Krzystek, A. Ozarowski, J. Nehrkorn, A. Schnegg, K. Holldack, R. H. Herber, J. Telser and J. M. Smith, Spectroscopic and Computational Studies of Spin States of Iron(IV) Nitrido and Imido Complexes, Inorg. Chem., 2017, 56, 4751–4768 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional computational details, coordinates of the optimized structures. See DOI: 10.1039/c8sc03677b |
| This journal is © The Royal Society of Chemistry 2018 |