
Nitrogen-doped porous carbons derived from a natural polysaccharide for multiple energy storage devices†
Yongpeng
Cui
,
Huanlei
Wang
 *,
Xiaonan
Xu
,
Yan
Lv
,
Jing
Shi
,
Wei
Liu
,
Shougang
Chen
and
Xin
Wang
*,
Xiaonan
Xu
,
Yan
Lv
,
Jing
Shi
,
Wei
Liu
,
Shougang
Chen
and
Xin
Wang
Institute of Materials Science and Engineering, Ocean University of China, Qingdao 266100, China. E-mail: huanleiwang@gmail.com; huanleiwang@ouc.edu.cn
First published on 9th November 2017
Abstract
Designing advanced carbon electrodes is considered as one of the most promising directions for energy storage. Herein, we report a facile approach to produce porous carbon nanomaterials. The carbon nanomaterials were prepared via KOH activation using natural polysaccharide-sodium alginate as the precursor with the subsequent introduction of additional nitrogen heteroatoms achieved by further reaction with urea. The optimal electrodes with a high specific surface area (up to 3313 m2 g−1), interconnected porosity, and rich nitrogen (∼7.2 wt%) and oxygen (∼7.4 wt%) doping can achieve an excellent electrochemical performance in supercapacitors and lithium ion batteries. When these materials are employed as supercapacitor electrodes, they achieved an outstanding specific capacitance of 267 F g−1 at 1 A g−1 and an extremely high rate performance with 76.8% capacitance retention ratio in an alkaline electrolyte. In addition, a high capacitance of 197 F g−1 at 0.5 A g−1 with a high capacitance retention ratio of 52.9% at 100 A g−1 can be achieved in an ionic liquid electrolyte. When tested as lithium ion battery anodes, an extraordinarily high specific capacity of 1455 mA h g−1 and a stable energy storage performance up to 500 cycles were observed. The present study highlights that high-performance carbon electrodes can be produced by using sustainable precursor and can be used in multiple energy storage systems.
Introduction
The present global energy shortage problem is of great concern, and energy storage and conversion is an important aspect to be considered in order to enable the sustainable development of our economy and society.1–4 Emerging high-performance electrochemical storage and conversion devices such as supercapacitors and lithium-ion batteries have been at the forefront of research being conducted by both industry and academia over the past few decades.5–7 Supercapacitors are primarily used in high power electronic devices i.e., devices exhibiting high power densities, short charge/discharge times, and long-life cycle stabilities,8–10 while lithium-ion batteries are primarily installed in high energy density storage devices,11 which are essential for our daily life and production because of our dependence on numerous mobile electronic devices and hybrid electric vehicles.12,13 However, the widespread application of these electrochemical devices presents multiple technical limitations; for example, the supercapacitors require to achieve higher energy densities while the cycling stability and power density of the lithium-ion batteries require improvement.5,13Carbon materials are predominantly used as electrode materials in energy storage applications due to their controllable morphologies, well-developed pore structures, and excellent electrochemical performances.9,14–16 Activated carbons,17,18 carbide derived carbons,19,20 template carbons,21,22 porous carbons,23,24 carbon nanotubes and graphene25,26 have been widely investigated for their potential applications as electrode materials. Among these, porous carbon has been considered as a promising candidate for supercapacitors and lithium ion batteries owing to its controllable pore structure, high surface area, and excellent chemical stability.27,28 Recently, natural polysaccharides (e.g. chitosan,29,30 agarose,31 starch,32 alginate,33 and cellulose34) have been used as a promising precursor for carbon production because of their availability, low cost and eco-friendliness. Alginate, which is composed of β-D-mannuronate (M) and α-L-guluronate (G) monomers, is a naturally occurring nontoxic polysaccharide isolated from brown algae.35 Industrial alginate production is approximately 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 metric tons annually, and alginate and its salts have been widely used for preparing carbon-based electrodes.36 It is anticipated that sodium alginate would eventually become an ideal precursor for carbon production, since it is easily extracted from marine biomass and can form a 3D hierarchical porous framework during the carbonization process. Generally, the surface area and porosity of the carbon nanomaterials play an important role in obtaining a high electrochemical performance. The main challenge for the synthesis of porous carbons is to appropriately design the porous structures. To overcome this challenge, the ‘activation technique’ has been considered as an efficient way to tune the porosity.
000 metric tons annually, and alginate and its salts have been widely used for preparing carbon-based electrodes.36 It is anticipated that sodium alginate would eventually become an ideal precursor for carbon production, since it is easily extracted from marine biomass and can form a 3D hierarchical porous framework during the carbonization process. Generally, the surface area and porosity of the carbon nanomaterials play an important role in obtaining a high electrochemical performance. The main challenge for the synthesis of porous carbons is to appropriately design the porous structures. To overcome this challenge, the ‘activation technique’ has been considered as an efficient way to tune the porosity.
To further improve the electrochemical performance, the surface wettability and electrical conductivity of the electrode materials can be modified via doping with heteroatoms (e.g. O, N, S, P, and B).15,37–40 Among these, nitrogen-doped carbon materials have been investigated extensively. As an electron donor, the nitrogen doping can not only improve the electrolyte wettability of the electrode materials but also promote the formation of pseudocapacitance by surface redox reactions.37,41,42 Herein, we adopted a facile synthesis route for a large-scale production of nitrogen-doped carbon nanomaterials using sodium alginate as the carbon precursor, urea as the nitrogen source, and potassium hydroxide as the activating agent. The nitrogen-doped nanomaterials exhibit high specific surface areas (up to 3313 m2 g−1), interconnected hierarchical porosities, and abundant nitrogen (∼7.2–30.5 wt%) and oxygen (∼4.7–8.5 wt%) dopants. When tested as electrode materials for supercapacitors and lithium ion batteries, these carbon nanomaterials were able to achieve fairly high specific capacities, excellent rate capabilities, and long cycling stabilities, demonstrating that they are potential electrodes for advanced energy storage and conversion devices.
Experimental
Material synthesis
Sodium alginate (2 g) and urea (2 g) were thoroughly dissolved in distilled water along with the addition of potassium hydroxide (0–4 g). The mixture was stirred at room temperature for 12 h, followed by freeze drying to form a sponge-like precursor. The sponge-like precursor was then heated to the target temperature (800 °C) for 1 h under a constant nitrogen stream with a heating rate of 3 °C min−1. Finally, the carbon materials were isolated after washing with 3 M HCl and sufficient deionized water, filtration, and drying the residue at 80 °C. The nitrogen-doped sodium alginate-derived carbons are denoted as N,SAC-n, where n represents the mass ratio of potassium hydroxide to sodium alginate. For comparison, the sodium alginate-derived carbon (labelled as SAC) is also prepared without adding urea and KOH.Material characterization
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were performed to characterize the morphology and microstructure using a Hitachi S4800 (at 10 kV) and Tecnai G2 F20 (at 200 kV), respectively. Powder X-ray diffraction (XRD) patterns and Raman spectra were collected on a Bruker D8 Advance diffractometer (Cu-Kα radiation, 2θ = 10–80°) and a microscopic confocal Raman spectrometer (Lab RAM HR800), respectively, to analyse the carbon structure. X-ray photoelectron spectroscopy (XPS) analysis was used to determine the near-surface chemical composition using a Thermo ESCALAB 250 XI spectrometer. Elemental analysis was carried out by a conventional combustion method (CHN, Vario EL cube). N2 sorption isotherms were measured at 77 K to confirm the surface area and porosity on a Micromeritics 3 Flex™ surface characterization analyzer. To evaluate the packing density of the carbon, a given amount of carbon was pressed in a mold at a fixed pressure of 10 MPa, and the measurements were repeated 3 times with an error less than 3%.Electrochemical evaluation
All the electrochemical evaluations, including cyclic voltammetry (CV), galvanostatic charge–discharge, and electrochemical impedance spectroscopy (EIS), were performed using a Gamry Interface 1000 electrochemical work station and a Land battery measurement system (CT2001A) at room temperature. A slurry containing 80 wt% as-obtained carbon materials, 10 wt% Super P, and 10 wt% poly (vinylidenedifluoride) in N-methyl-2-pyrrolidinone was used for the electrode preparation.In KOH electrolyte, the supercapacitive performance was evaluated by the three-electrode system. The slurry was coated on nickel foam to obtain a working electrode, while platinum foil and Hg/HgO were used as the counter and reference electrodes, respectively. In ionic liquid electrolyte, the super-capacitive performance was evaluated using the two-electrode system, for which the slurry was coated onto a stainless steel disc for obtaining the working electrode and two symmetric electrodes separated by a porous polymeric separator were used to fabricate CR2032 coin cells inside an Ar-filled glove box (<0.1 ppm of both oxygen and water). 1-Ethyl-3-methylimidazolium tetrauoroborate (EMIM BF4) was used as the electrolyte.
For the three-electrode system, the specific capacitance of the working electrode (C, F g−1) was calculated by the following formula based on the galvanostatic charge–discharge test:
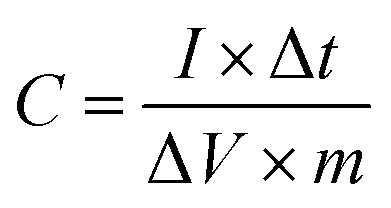
where I (A) is the discharge current, Δt (s) is the full discharge time, ΔV (V) is the discharge voltage excluding the IR drop, and M (g) is the mass of active carbon material on both electrodes. The specific energy density (E, W h kg−1) and power density (P, W kg−1) were calculated using the following relationships:
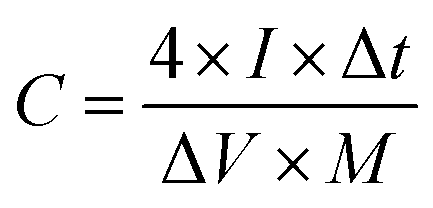
where ΔV is the cell voltage after ohmic drop (V), and t is the discharge time (s).
Electrochemical characterizations of the lithium-ion batteries were also performed using CR2032 coin cells. The working electrode was prepared by coating the slurry on a Cu foil substrate. 1 M LiPF6 in ethylene carbonate–dimethyl carbonate was used as the electrolyte, polyethene was used as the separator, and a Li metal foil was used as the counter electrode. The CR2032 coin cells were assembled inside an Ar-filled glove box (<0.1 ppm of both oxygen and water).
Results and discussion
Physicochemical properties of nitrogen-doped carbons
Scheme 1 illustrates the synthesis process employed for nitrogen-doped sodium alginate-derived carbons. In the first step, sodium alginate, urea, and/or potassium hydroxide were mixed together in an aqueous solution; urea plays a role in supplying the nitrogen species and KOH acts as activating agent to generate additional porosity. Then, a sponge-like precursor was obtained by a simple freeze-drying process. Finally, nitrogen-doped porous carbons were achieved after one-step carbonization/activation process in nitrogen atmosphere. During KOH activation, the KOH is melted at above 400 °C, which involves the main reaction of 6KOH + 2C = 2K + 3H2 + 2K2CO3, the decomposition of K2CO3, and the reactions between potassium species and carbon. SEM and TEM analyses were carried out to examine the morphology and structure of the nitrogen-doped carbons. From the SEM image (Fig. S1a†), the SAC sample prepared without the addition of urea and KOH exhibits an interconnected 3D morphology consisting of macropores and carbon walls. However, the morphology of the N,SAC-0 sample changed with the interconnected structure split into fractures due to the addition of urea (Fig. S1b†). With the assistance of KOH activation, the thickness of the carbon walls reduced (Fig. S1c and d†). In particular, the N,SAC-2 sample exhibited a sheet-like morphology owing to the carbon consumption, which occurs during KOH activation. From the TEM images (Fig. 1), it can be observed that the SAC demonstrates an interconnected porous structure, while N,SAC-0 exhibits thick sheet-like carbon walls after nitrogen doping. With the increase in KOH loading, a highly transparent sheet-like morphology can be observed. The high-resolution TEM images (the insets of Fig. 1) clearly show a highly porous and graphitic structure. It is evident that the highly defective structure can offer extra active sites for charge storage.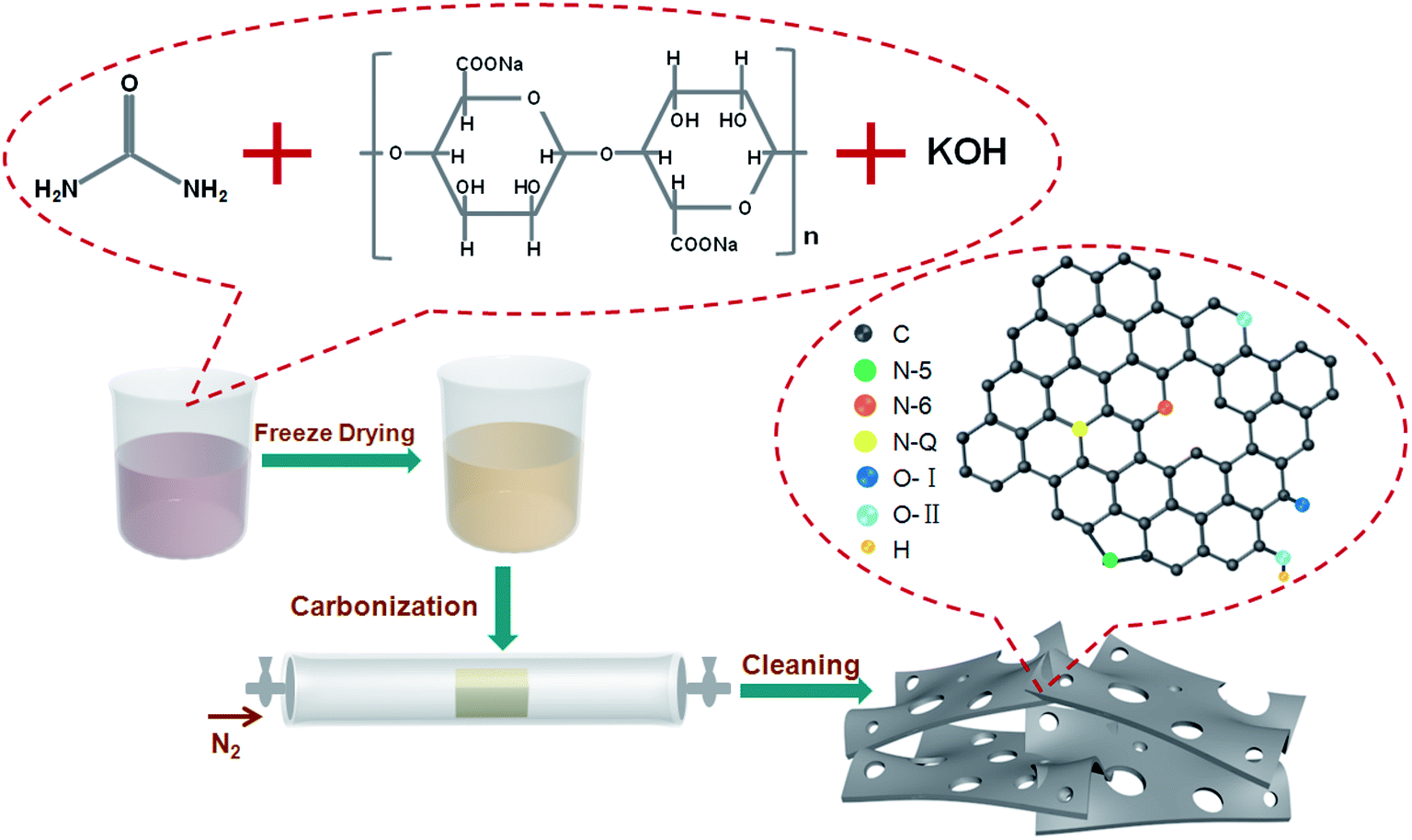 | ||
| Scheme 1 Schematic illustration of the synthesis process for nitrogen-doped sodium alginate derived carbons. | ||
 | ||
| Fig. 1 TEM micrographs for sodium alginate derived carbons: (a) SAC, (b) N,SAC-0, (c) N,SAC-1, and (d) N,SAC-2. | ||
In order to illustrate the porosity of the as-prepared carbon nanomaterials, the nitrogen adsorption–desorption studies were performed. As shown in Fig. 2a, a large quantity of adsorbed nitrogen was observed in the low-pressure range (P/Po < 0.01), indicating the existence of a large number of micropores. Moreover, the broadening of the ‘knee’ in the relatively low pressure range (P/Po < 0.4) and the observed hysteresis loop in the medium–high pressure range (P/Po > 0.4) suggest the presence of abundant mesopores. The textural parameters are listed in Table 1. The surface area of SAC sample is as high as 1069 m2 g−1. The presence of sodium derivatives and the release of CO2, CO, and H2O during the pyrolysis of sodium alginate can promote the development of porosity. For the N,SAC-0 sample, the surface area (1034 m2 g−1) is similar to that of SAC, indicating that the nitrogen species can infiltrate the carbon framework without altering the specific surface area. However, the KOH activated samples have a considerably larger surface area. The surface areas of N,SAC-1 and N,SAC-2 samples are 2699 and 3313 m2 g−1, respectively. Simultaneously, the change in the total pore volume is consistent with that of the specific surface area; the total pore volume increased from 0.65 cm3 g−1 for SAC to 1.69 cm3 g−1 for N,SAC-2. Fig. 2b shows the pore size distributions calculated from the adsorption isotherms. All samples display hierarchical porosity and possess a high proportion of macro/mesopores (38.64–48.72%). As compared to conventional microporous activated carbons, the high macro/mesopore proportion is favorable for rapid diffusion of ions, and excellent rate capability can be expected.43,44
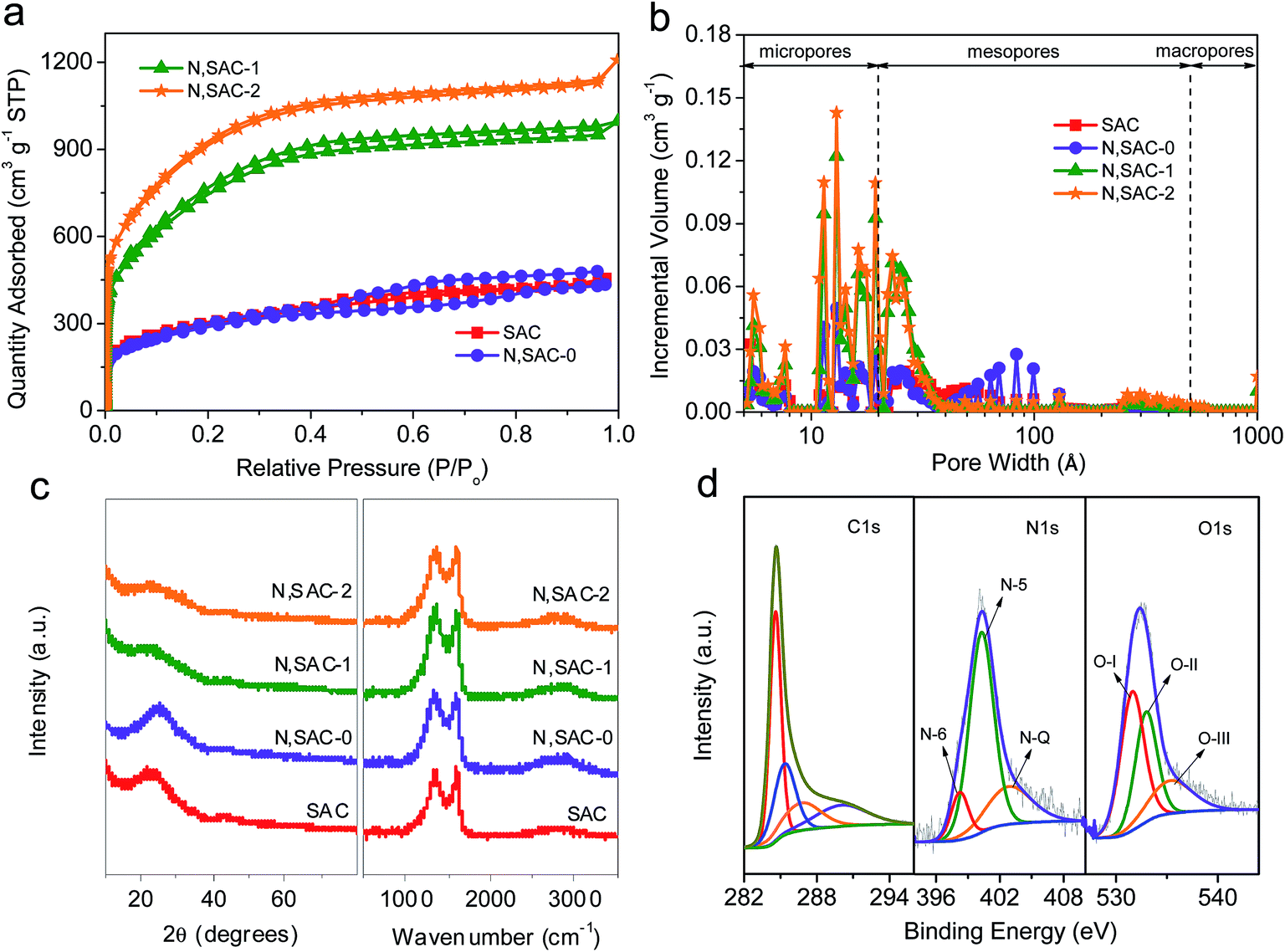 | ||
| Fig. 2 (a) The nitrogen adsorption–desorption isotherms of the as-obtained samples. (b) The pore size distributions calculated from the adsorption isotherms using density functional theory method. (c) The XRD patterns and the Raman spectra of all the samples. (d) The high-resolution XPS C 1s, N 1s and O 1s spectra of N,SAC-2. | ||
| Sample | S BET (m2 g−1) | V t (cm3 g−1) | ρ (g cm−3) | Pore vol (%) | I D/IG | C EMIMBF (F g−1) | C KOH (F g−1) | C KOH (μF cm−2) | C Li (mA h g−1) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| V <1 nm | V 1–2 nm | V >2 nm | |||||||||
| a Specific surface area was calculated by the Brunauer–Emmett–Teller (BET) method. b The total pore volume was determined by the density functional theory (DFT) method. c ρ represents the packing density. d The capacitance at current density of 0.5 A g−1 in EMIMBF4 electrolyte. e The capacitance at current density of 1 A g−1 in 2 M KOH aqueous solution. f The surface area-normalized capacitance was obtained at 1 A g−1 in 2 M KOH aqueous solution. g The discharge capacity at current density rolls back to 0.1 A g−1 for the 5th cycle as anode material for LIB. | |||||||||||
| SAC | 1069 | 0.65 | 0.50 | 17.15 | 34.54 | 48.32 | 2.94 | 61 | 168 | 15.7 | 531 |
| N,SAC-0 | 1034 | 0.62 | 0.54 | 16.22 | 36.06 | 47.73 | 3.33 | 102 | 192 | 18.5 | 649 |
| N,SAC-1 | 2699 | 1.40 | 0.39 | 11.43 | 45.91 | 42.66 | 3.33 | 149 | 251 | 9.3 | 918 |
| N,SAC-2 | 3313 | 1.69 | 0.35 | 13.05 | 48.31 | 38.64 | 3.45 | 197 | 267 | 8.1 | 954 |
The structural differences of the as-prepared carbon samples were investigated by XRD and Raman analyses (Fig. 2c). Two weak and broad diffraction peaks at around 2θ = 23–25° and 43–44° corresponding to the (002) and (101) planes of graphite, respectively, imply that the carbons are disordered. The Raman spectra exhibit a G-band at ∼1600 cm−1 (associated with graphitic carbon) and a D-band at 1338–1357 cm−1 (associated with defects) (Fig. S2†).45 The high ID/IG ratio implies a high degree of disorder, in agreement with the XRD analysis. The N,SAC samples have a higher ID/IG ratio (3.33–3.45) than that of the SAC sample (2.94). This can be understood from the fact that the N-doping and KOH activation can provide more structural defects, leading to a higher degree of disorder.18
In order to study the differences in the chemical composition and the character of the nitrogen/oxygen-containing functional groups, XPS analysis was carried out. The XPS spectra reveal some significant differences between the samples (Fig. S3†). The N-doped samples display evident characteristic N-peaks at around 400 eV. With the addition of urea, the carbons are rich in nitrogen with an atomic ratio in the range of 7.2–30.5 wt%. With the increase in KOH loading, the content of nitrogen significantly decreases, indicating that nitrogen-based functional groups can be easily removed during KOH activation. Even for N,SAC-2, a high nitrogen content of 7.2 wt% was maintained. Fig. 2d and S4–S6† show the high-resolution XPS spectra of all samples. The high-resolution XPS C 1s spectra exhibit four peaks at 284.6, 285.1 ± 0.2, 286.6 ± 0.2, and 290.0 ± 0.2 eV in the carbon frameworks, which represent the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–O/CN, CO/C–N, and –COOR groups, respectively.18,46 From the N 1s spectra, three characteristic peaks centered at 398.2 ± 0.2, 400.1 ± 0.2, and 402.7 ± 0.2 eV, corresponding to pyridinic N (N-6), porrolic N (N-5), and quaternary N (N-Q), respectively, were observed.47–49 From the O 1s spectra we can infer that there are three types of oxygen-containing functional groups, corresponding to CO (O–I, 531.2 ± 0.2 eV), C–OH/C–O–C (O-II, 532.6 ± 0.2 eV), and COOH (O-III, 535.3 ± 0.2 eV).44,50 According to previous reports, the heteroatoms in the carbon frameworks can be involved in surface redox reactions, which can largely improve the overall capacity of the carbon materials.3,51 In order to further understand the role of the functional groups in electrochemical performance of the electrodes, it is essential to clarify the types of heteroatoms in the carbon matrix. The locations of the N-containing and O-containing functional groups within carbon frameworks are described in Scheme 1. Table S1† lists the relative concentrations of N-moieties and O-moieties. For the N-containing functional groups, the amide groups in urea easily react with the phenol and/or carboxylic groups in sodium alginate to form a pyridine–nitrogen doping in the carbons, leading to the high proportion of N-5 group, observed in the N,SAC-0 sample. A notable change is the decrease of N-6 with the increase in KOH loading, which indicates that the N-6 group is preferentially eliminated due to KOH etching.52 The N-5 and N-Q groups can act as active sites to allow rapid electron transfer and enhance the capacitance, contributing towards the pseudocapacitance.9,15,53 For oxygen, the O-I content is slightly increased, while the O-II content is slightly decreased with an increase in KOH loading. It is widely accepted that the O-I and O-II groups play a dominant role in providing the pseudocapacitance. For further investigating the overall nitrogen content in the carbons, elemental analysis was performed using a conventional combustion method. It is worth noting that the nitrogen content is lower than that calculated by XPS analysis (Table 2), indicating that an abundance of nitrogen functional groups are present on the surface of the carbon. Therefore, the overall capacity values of carbon materials can be improved by surface redox reactions through the numerous N-containing and O-containing functional groups.9,15,53–55
C, C–O/CN, CO/C–N, and –COOR groups, respectively.18,46 From the N 1s spectra, three characteristic peaks centered at 398.2 ± 0.2, 400.1 ± 0.2, and 402.7 ± 0.2 eV, corresponding to pyridinic N (N-6), porrolic N (N-5), and quaternary N (N-Q), respectively, were observed.47–49 From the O 1s spectra we can infer that there are three types of oxygen-containing functional groups, corresponding to CO (O–I, 531.2 ± 0.2 eV), C–OH/C–O–C (O-II, 532.6 ± 0.2 eV), and COOH (O-III, 535.3 ± 0.2 eV).44,50 According to previous reports, the heteroatoms in the carbon frameworks can be involved in surface redox reactions, which can largely improve the overall capacity of the carbon materials.3,51 In order to further understand the role of the functional groups in electrochemical performance of the electrodes, it is essential to clarify the types of heteroatoms in the carbon matrix. The locations of the N-containing and O-containing functional groups within carbon frameworks are described in Scheme 1. Table S1† lists the relative concentrations of N-moieties and O-moieties. For the N-containing functional groups, the amide groups in urea easily react with the phenol and/or carboxylic groups in sodium alginate to form a pyridine–nitrogen doping in the carbons, leading to the high proportion of N-5 group, observed in the N,SAC-0 sample. A notable change is the decrease of N-6 with the increase in KOH loading, which indicates that the N-6 group is preferentially eliminated due to KOH etching.52 The N-5 and N-Q groups can act as active sites to allow rapid electron transfer and enhance the capacitance, contributing towards the pseudocapacitance.9,15,53 For oxygen, the O-I content is slightly increased, while the O-II content is slightly decreased with an increase in KOH loading. It is widely accepted that the O-I and O-II groups play a dominant role in providing the pseudocapacitance. For further investigating the overall nitrogen content in the carbons, elemental analysis was performed using a conventional combustion method. It is worth noting that the nitrogen content is lower than that calculated by XPS analysis (Table 2), indicating that an abundance of nitrogen functional groups are present on the surface of the carbon. Therefore, the overall capacity values of carbon materials can be improved by surface redox reactions through the numerous N-containing and O-containing functional groups.9,15,53–55
| Sample | Elemental analysis (wt%) | XPS composition (wt%) | |||||
|---|---|---|---|---|---|---|---|
| C | H | N | Oa | C | O | N | |
| a The content of oxygen calculated from elemental analysis as a difference to 100%, because the ash is neglected. | |||||||
| SAC | 86.6 | 2.5 | — | 10.9 | 95.3 | 4.7 | — |
| N,SAC-0 | 66.0 | 3.7 | 18.0 | 12.3 | 63.1 | 6.4 | 30.5 |
| N,SAC-1 | 72.0 | 3.7 | 9.8 | 14.5 | 78.7 | 8.5 | 12.8 |
| N,SAC-2 | 79.1 | 3.4 | 4.5 | 13.0 | 85.4 | 7.4 | 7.2 |
Electrochemical behavior for supercapacitors
The sodium alginate-derived carbon nanomaterials with high surface area, heteroatom doping, and hierarchical porosity consisting of micropores and mesopores is expected to achieve excellent electrochemical performance for supercapacitors. To demonstrate this, we first evaluated the supercapacitive performance of the as-prepared carbon materials in 2 M KOH electrolyte using a three-electrode configuration. Fig. 3a compares the CV curves of all the as-prepared carbon materials at a scan rate of 10 mV s−1. All samples were expected to exhibit curves with quasi-rectangular shapes due to the large surface area of the materials, indicating excellent electrochemical behavior of the carbons. However, for the nitrogen-doped N,SACs, reversible redox ‘humps’ can be observed in the CV curves, suggesting the presence of pseudocapacitance. It is worth noting that the integrated area beneath the CV curves for the N,SAC samples are significantly larger than that of the SAC sample, validating that the electrical double-layer capacitance can be enhanced by increasing the surface area of the electrodes and the pseudocapacitance of the carbon materials could be improved by introducing nitrogen/oxygen functional groups. The CV curves of all the electrodes maintained a quasi-rectangular shape even at 200 mV s−1, indicating smooth ion diffusion within the pore channels (Fig. 3b and S7†). The N,SAC-2 specimen displayed the largest integrated area beneath its CV curves at every scan rate, demonstrating that N,SAC-2 has the highest specific capacitance.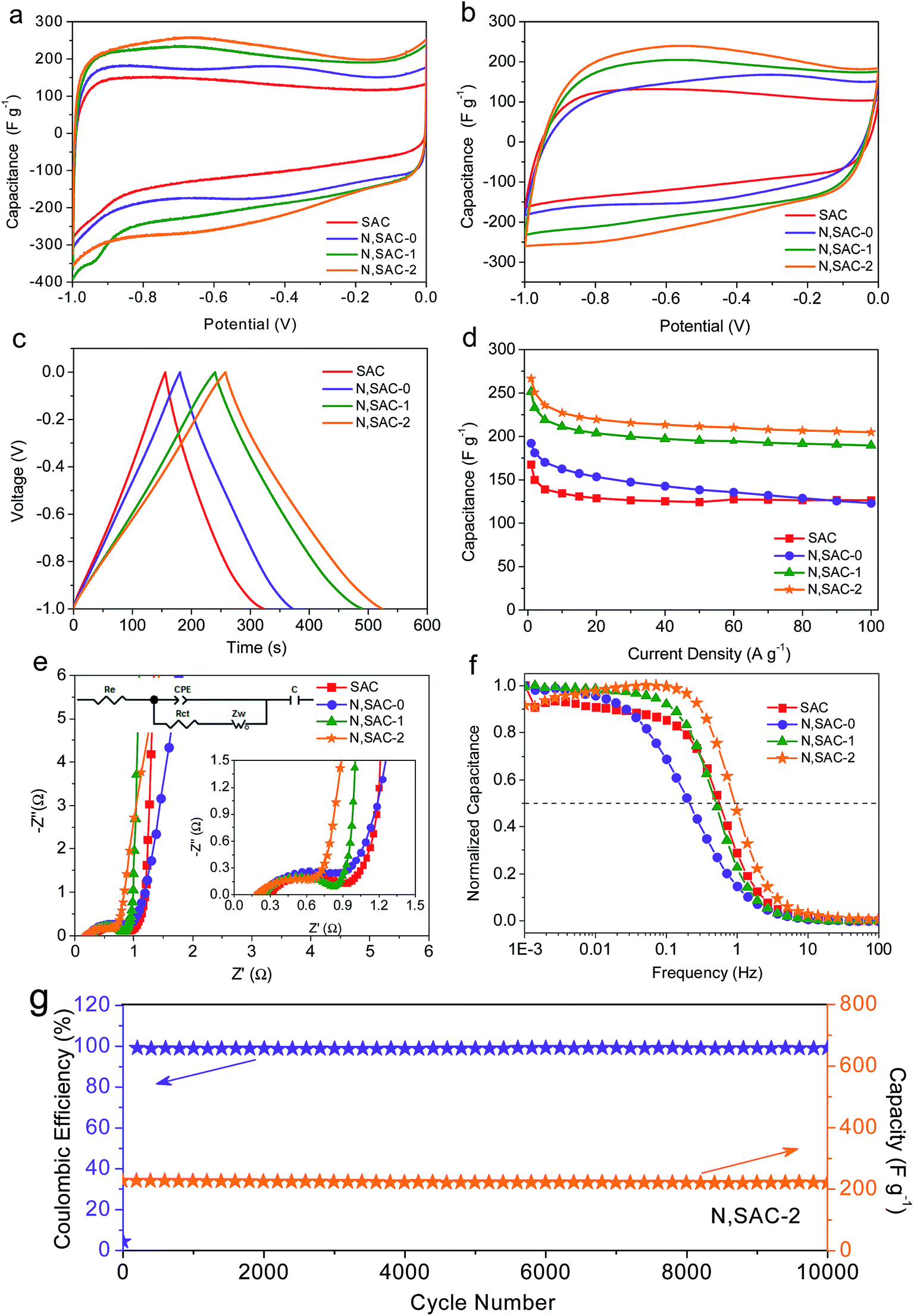 | ||
| Fig. 3 Electrochemical properties of all the samples tested for supercapacitors in 2 M KOH electrolyte. CV curves at (a) 10 mV s−1 and (b) 200 mV s−1. (c) Galvanostatic charge–discharge profiles at 1 A g−1. (d) Specific capacitances at different current densities. (e) Nyquist plots of all the electrodes. (f) Frequency response of all electrodes. (g) Cyclic stability of the N,SAC-2 at a current density of 10 A g−1 for 10000 cycles. | ||
Fig. 3c shows the galvanostatic charge–discharge profiles of the as-prepared carbon electrodes at 1 A g−1. All samples exhibit symmetrical and slightly distorted curves, manifesting their excellent coulombic efficiency as well as the coexistence of electric double-layer capacitance and pseudocapacitance. Consistent with the CV analysis, the N,SAC-2 sample exhibits the highest specific capacitance (as high as 267 F g−1 at 1 A g−1), which is comparable to that of most of the previously reported carbons (Table S2†). Even at 100 A g−1, the N,SAC-2 maintains a high specific capacitance of 205 F g−1, which offers an excellent rate performance with a high capacitance retention ratio of 76.8% (Fig. 3d). The relatively high specific surface area and hierarchical porous structure of the N,SAC-2 sample offers passageway for ion-transport and storage. Moreover, both SAC and N,SAC-0 samples have similar surface areas and pore size distributions; however, N,SAC-0 exhibits a higher specific capacitance at 1 A g−1. The explanation for this can be found in the nitrogen doping in N,SAC-0 sample. It is worth noting that N,SAC-0 displays faster capacitance fading (capacitance retention ratio: 64.1%), with the increase in current density than that of SAC (capacitance retention ratio: 75.4%), indicating that the pseudocapacitance fades more quickly at a higher rate. As for the SAC sample, the surface area-normalized capacitance at 1 A g−1 in a 2 M KOH aqueous solution is 15.7 μF cm−2, while that for the N,SAC-0 sample is as high as 18.5 μF cm−2. The numerous N-containing and O-containing functional groups provide the defects and active sites to facilitate pseudocapacitance.15,56,57 With an increase in KOH-loading, the surface area significantly increases with a decrease in the number of functional groups, resulting in a smaller surface area-normalized capacitance for N,SAC-1 and N,SAC-2 (9.3 μF cm−2 for N,SAC-1 and 8.1 μF cm−2 for N,SAC-2).
The results of the electrochemical impedance spectroscopy measurements for the as-obtained electrodes in the 2 M KOH electrolyte are shown Fig. 3e and f. The electrolyte resistance (Re), calculated based on the intercept of the Nyquist plots with the real axis at high frequency, is in the range of 0.20–0.30 Ω, indicating good conductivity in alkaline electrolyte.58,59 The semicircle in the medium–high frequency range indicates charge transfer resistance (Rct, 0.52–0.79 Ω) at the electrode/electrolyte interface. The N,SAC-2 sample exhibits the smallest charge transfer resistance owing to the highly developed porosity. The almost vertical line in the low frequency range represents ideal capacitive behavior.60,61 Moreover, the operating frequency (fo) at which the capacitance is 50% of its maximum value and the characteristic relaxation time (τo) can further indicate the rapid ion-transfer ability.51,62 For example, the N,SAC-2 sample exhibits the best frequency response with values of fo = 0.99 Hz and τo = 1.01 s, demonstrating superior ion transport properties. It is worth noting that the N,SAC-0 sample displays the lowest operating frequency (0.22 Hz), implying that the ‘tangling’ of functional groups can hinder ion transport. In order to validate the cycle stability, the cycle life test was performed for N,SAC-2 at a current density of 10 A g−1 (Fig. 3g). It is interesting to note that only a 3% capacitance decrease is observed after 10000 cycles. Moreover, the coulomb efficiency is close to 100% during cycling, demonstrating very good cycle stabilities for practical applications.
It is well-known that achieving both high energy and power density is of great significance for actual applications. In order to realize this, we further evaluated the symmetric supercapacitors for all obtained samples in ionic liquid electrolyte. Fig. 4a and b shows the CV curves with scan rates of 10 and 200 mV s−1. It can be observed that the CV curves at 10 mV s−1 exhibit small redox humps for nitrogen-doped samples, indicating that the functional groups can react with ionic liquids to provide pseudocapacitance. The rectangular shape can be maintained even at 200 mV s−1 or higher, thus providing further evidence for the excellent ion transport behavior (Fig. S8†). The CV curves of N,SAC-2 samples are less distorted, demonstrating that a high surface area with developed porosity is favorable for rapid ion transport. Fig. 4c and S9† shows the galvanostatic charge–discharge curves of all samples measured at 1, 10, 20, 50, and 100 A g−1. The GCD curves of N,SACs exhibit nearly linear shapes with evident humps, suggesting the coexistence of electric double-layer capacitance and pseudocapacitance. Fig. S10† presents the relationship between voltage drop and current density. The high IR drop at higher current can be attributed to the higher ion transport resistance. It can be observed that the N,SAC-0 sample exhibits the largest IR drop. The explanation for this can be found in the high amount of nitrogen-containing groups for N,SAC-0, and the presence of tangled functional groups, which can lead to a high resistance for ion transport. The specific capacitances for SAC, N,SAC-0, N,SAC-1, and N,SAC-2 are 61, 102, 149, and 197 F g−1, respectively, at 0.5 A g−1. Similarly, heteroatom doping and increased surface area can significantly affect the specific capacitance. The higher capacitance of N,SAC-0 than that of the SAC sample can be ascribed to the nitrogen doping in the N,SAC-0 sample due to their similar surface area. Moreover, on comparing SAC, N,SAC-1, and N,SAC-2 samples, we can observe that the capacitance is proportional to the surface area, indicating that surface area plays an important role in determining the specific capacitance. In particular, the capacitance retention ratio of N,SAC-2 can reach up to 52.9% at room temperature and 100 A g−1 due to the well-developed porosity (Fig. 4d). The N,SAC-2 sample can maintain 86.7% of the initial capacitance at 10 A g−1 after 10000 cycles (Fig. 4e), and the decay in the capacitance can be ascribed to the presence of the nitrogen and oxygen containing functional groups.
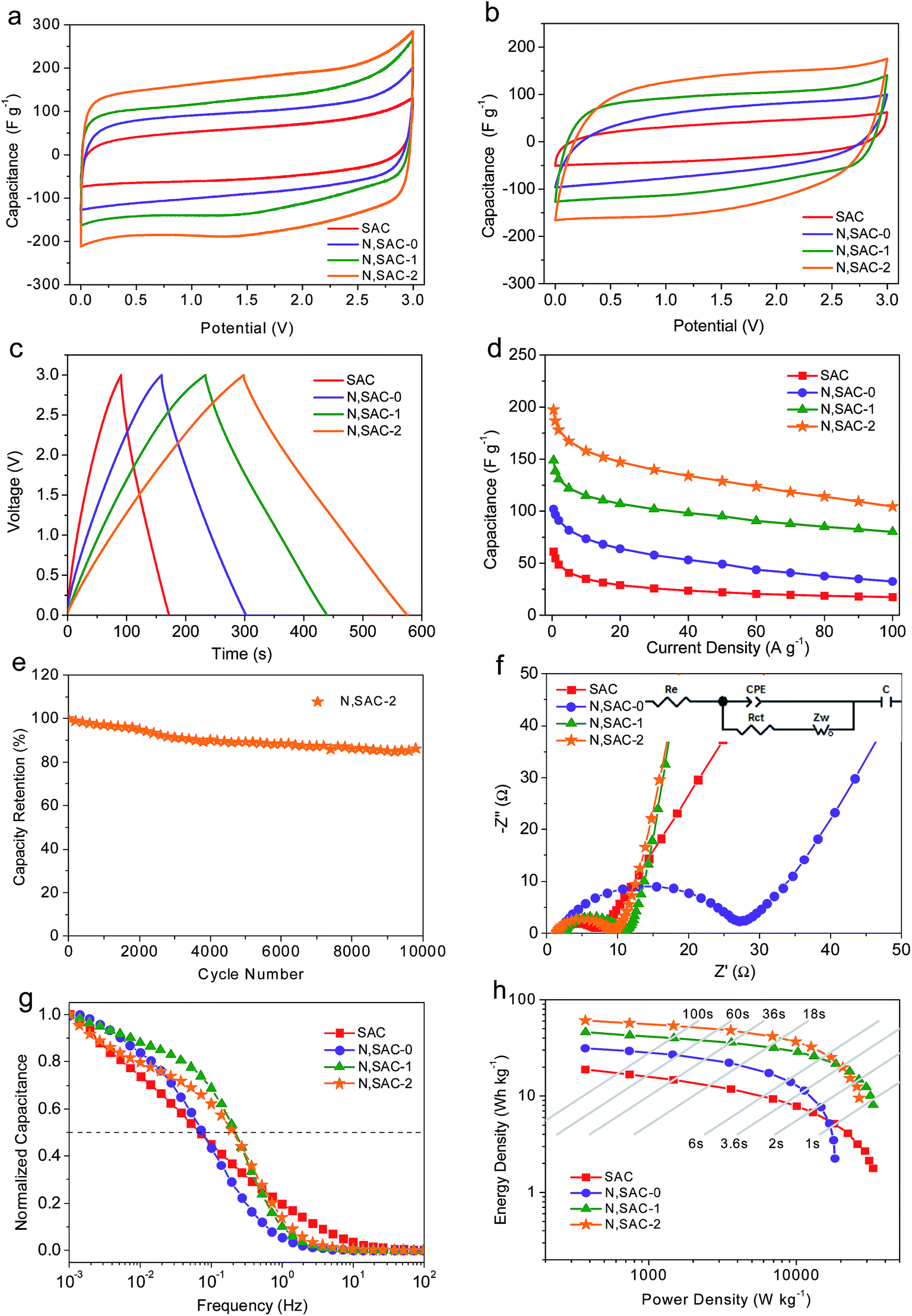 | ||
| Fig. 4 Electrochemical properties of all the samples tested for supercapacitors in ionic liquid electrolyte. CV curves at (a) 10 mV s−1 and (b) 200 mV s−1. (c) Galvanostatic charge–discharge profiles at 1 A g−1. (d) Specific capacitances at different current densities. (e) Cyclic stability of the N,SAC-2 at a current density of 10 A g−1 for 10000 cycles. (f) Nyquist plots of all the electrodes. (g) Frequency response of all electrodes. (h) Ragone plots of symmetric supercapacitors. | ||
Fig. 4f and g show the EIS measurements of all the electrodes in an ionic liquid electrolyte. The equivalent series resistance is around 1.4–2.5 Ω, indicating good conductivity in the EMIMBF4 electrolyte. Similarly, the semicircle in the medium–high frequency range indicates charge transfer resistance (4.7–22.3 Ω) at the electrode/electrolyte interface. The N,SAC-0 sample exhibits the largest charge-transfer resistance, further confirming that the presence of abundant functional groups can inhibit ion diffusion. The almost vertical line in the low frequency range represents the ideal capacitive behavior. Moreover, the N,SAC-2 sample exhibits the best frequency response with values of fo = 0.22 Hz and τo = 4.5 s, demonstrating superior ion transport properties.
Fig. 4h displays the Ragone plots of all the carbon samples based on the total mass of the active materials in the ionic liquid. The N,SAC-2 based supercapacitors yield an active mass normalized energy density of 61 W h kg−1 at a power density of 373 W kg−1. Even at a high power of 20 kW kg−1, an energy density of 21 W h kg−1 was obtained, which is superior to or at least comparable to previously reported carbon-based symmetric supercapacitors in ionic liquid electrolytes (Table S2†), such as porous carbon nanosheets (25.4 W h kg−1 at 15 kW kg−1),63 hemp-based derived carbons (18–19 W h kg−1 at 19 kW kg−1),64 and 3D nitrogen-doped porous carbon (19.8 W h kg−1 at 6.22 kW kg−1).65 When the device was totally charged/discharged in less than 1s, a high energy density of 10 W h kg−1 was maintained for N,SAC-1, demonstrating the outstanding rate capability. For most commercial applications, the volumetric value is of equal importance to the gravimetric value. Based on the mass density (0.35–0.54 g cm−3, Table 1), the energy density can reach up to 26 W h L−1.
Electrochemical behavior for lithium-ion battery anodes
The lithium-ion battery performance of the sodium alginate-derived carbon nanomaterials was investigated using coin-type half cells in a 1 M LiPF6 electrolyte. Fig. 5a and S11† present the CV curves of the as-obtained carbons for the first, second, and fifth cycles at a scan rate of 0.1 mV s−1 with a voltage range of 0.01–3.0 V. An evident cathodic peak appeared over the potential range of 0.01–1.0 V in the first CV scan and 0.01–0.5 V during the following scan. This peak is more pronounced in the first cycle scan, due to electrolyte decomposition, the formation of a solid electrolyte interphase (SEI) layer, and the trapping of lithium ions in the carbons.15,53,66 For the subsequent cycles, the CV curves were nearly similar to each other, indicating the high electrochemical reversibility of electrodes. The smooth “box-like” curves of the CV at high potentials of 0.5–3.0 V imply typical capacitive lithium storage behavior due to the physisorption of lithium ions and chemisorption on the graphitic defects and functional groups.67 It is worth to note that the “box-like” curves of the N,SACs are significantly broader than that of the SAC sample, demonstrating enhanced lithium storage capacity associated with heteroatom doping and high surface area with developed porosity. | ||
| Fig. 5 Electrochemical properties of the all the samples tested for lithium-ion battery anodes. (a) CV curves and (b) galvanostatic charge–discharge profiles for N,SAC-2. (c) Specific capacitances at different current densities. (d) The cycling test of the N,SAC-2 electrode at a current density of 2 A g−1 for 500 cycles. | ||
Fig. 5b and S12† show the galvanostatic charge–discharge curves of the as-obtained carbon electrode within the voltage range of 0.01–3.0 V vs. Li+/Li. When the potential shifts downward, the capacity can be realized by the electric double layer, the surface redox reaction of lithium ions with functional groups, the chemisorption at the defective graphitic layers, and the lithium intercalation between the graphitic layers.68 The initial reversible charge capacity of the SAC sample is 774 mA h g−1, while the N,SAC-0 sample exhibits a larger charge capacity (893 mA h g−1) at 0.1 A g−1. This can be explained by the heteroatom doping, indicating enhanced lithium storage capacity for N,SAC-0. As expected, the initial reversible charge capacity of the N,SAC-1 and N,SAC-2 increased to 1334 and 1455 mA h g−1, respectively, due to the larger specific surface area, which can facilitate a larger amount of Li ions to adsorb on the surface of carbon and insert in the graphitic layers. This capacity is about 3 times higher than the theoretical capacity of graphite (372 mA h g−1), indicating the excellent lithium ion storage performance. The initial coulombic efficiency of the carbon electrodes is in the range of 51.4–55.4% at 0.1 A g−1, which is higher than that of N-doped carbon nanosheets (49.2%),9 nitrogen-doped carbon nanofiber aerogels (47%),69 and N-rich mesoporous carbon (29–54%).70 The large irreversible capacity loss is primarily attributed to the electrolyte decomposition and the formation of the dense SEI film.15 This phenomenon is usually observed for high surface area carbons, and can hinder their practical application as anodes for lithium ion batteries.
Furthermore, the rate capability of the sodium alginate-derived carbon nanomaterials is described under different current densities ranging from 0.1 A g−1 to 10 A g−1 for 10 cycles at each rate (Fig. 5c). At a current density of 0.5 A g−1 for the fifth cycle, the charge capacity of the N,SAC-2 sample is 613 mA h g−1. Even at ultrahigh current densities of 5 and 10 A g−1, the charge capacity of N,SAC-2 was maintained at 223 and 173 mA h g−1, respectively. When the current density was switched back to 0.1 A g−1, the specific capacity of N,SAC-2 recovered to 950 mA h g−1. It is worth noting that N,SAC-1 and N,SAC-2 exhibit similar rate performances, which can be ascribed to the combined effects of surface area and nitrogen doping. The higher surface area of N,SAC-2 can be favorable for improving the capacity, while the lower nitrogen content in N,SAC-2 can lead to the decreased capacity. The excellent cycling stability of the N,SAC-2 electrode was also evaluated at a current density of 2 A g−1 for 500 cycles. As shown in Fig. 5d, the capacity is maintained at ∼300 mA h g−1 after 500 cycles with no sign of degradation, which indicates that this carbon electrode can be used as a potential anode for practical applications. Moreover, the coulombic efficiency is close to 100% during cycling. The excellent rate capability, high lithium storage capacity and superior cycling stability of the sodium alginate-derived carbon electrodes again indicate that they can be used as promising anode materials for lithium ion batteries as compared to the previously reported carbon electrodes (Table S3†). In order to gain a better understanding of the electrochemical behavior, the EIS for the N,SAC-2 electrode was measured before and after 500 cycles (Fig. S13†). The Nyquist plot showed a relatively large semicircle at medium–high frequencies before cycling, which represents the charge-transfer resistance (Rct). It is worth noting the emergence of a second inconspicuous semicircle in the medium–high frequency region after cycling, which represents a new resistance (Rf) due to the SEI formation and Li+ ion transport through the SEI layer.46 The value of Rct + Rf is 10.4 Ω after cycling, calculated using the equivalent circuit model, which is lower than the Rct (42.2 Ω) of the as-prepared electrode. The above results indicate a high Li+ ion transfer speed at the SEI/electrolyte interface, which is favorable for providing excellent cycling stability. Moreover, the value of the equivalent series resistance Re increases from 1.7 Ω before cycling to 6.1 Ω after 500 cycles owing to the SEI growth and electrode material disintegration.
To summarize, the above results suggest that nitrogen-doped sodium alginate-derived carbons can be used as advanced electrodes for both supercapacitors and lithium ion batteries. The high surface area, interconnected hierarchical porosity, and partially graphitized and heteroatom-doped carbon materials can lead to large, reversible, and stable electrochemical energy storage. First, the high surface area with interconnected hierarchical porosity provides sufficient electrode–electrolyte interface area and abundant transport channels for charge accumulation and ion diffusion. Second, the heteroatom doping in the carbon structure introduces active sites for extra charge accumulation and is involved in surface redox reactions for additional pseudocapacitance. Benefiting from the above-mentioned features and their synergistic effects, nitrogen-doped sodium alginate-derived carbon materials can be favorable for electrodes with excellent electrochemical properties.
Conclusions
We have demonstrated a facile approach for the synthesis of nitrogen-doped carbon materials with a high specific surface area, interconnected hierarchical porosity, and heteroatom doping. A one-step carbonization/activation process was developed using a natural polysaccharide (sodium alginate) as the carbon source, urea as the nitrogen source, and potassium hydroxide as the activating agent. These nitrogen-doped carbon electrodes achieved excellent electrochemical performance when incorporating in supercapacitors and lithium ion batteries. The specific capacitance for supercapacitors is as high as 267 F g−1 in KOH electrolyte with a high capacitance retention ratio of 76.8% at 100 A g−1. The symmetric supercapacitors can deliver an energy density of 61 W h kg−1 in an ionic liquid electrolyte. As electrode materials for lithium ion batteries, a reversible lithium storage capacity of 1455 mA h g−1 was obtained. Benefiting from simple synthetic method and low-cost availability of the precursors, nitrogen-doped sodium alginate-derived carbon materials can be produced on a large scale for next-generation energy storage and renewable delivery devices with high energy, high power capacity and long life spans.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21471139, and 51402272), Seed Fund from Ocean University of China, and Fundamental Research Funds for the Central Universities (No. 841562011).References
- C. Liu, F. Li, L. P. Ma and H. M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed
.
- K. Xie and B. Wei, Adv. Mater., 2014, 26, 3592–3617 CrossRef CAS PubMed
- J. Yan, Q. Wang, T. Wei and Z. Fan, Adv. Energy Mater., 2014, 4, 1300816 CrossRef
- J. B. Goodenough, Energy Environ. Sci., 2014, 7, 14–18 CAS
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed
- Y. Wang, Y. Song and Y. Xia, Chem. Soc. Rev., 2016, 45, 5925–5950 RSC
- J. Yang, H. Wu, M. Zhu, W. Ren, Y. Lin, H. Chen and F. Pan, Nano Energy, 2017, 33, 453–461 CrossRef CAS
- L. Kou, T. Huang, B. Zheng, Y. Han, X. Zhao, K. Gopalsamy, H. Sun and C. Gao, Nat. Commun., 2014, 5, 3754 CAS
- J. H. Hou, C. B. Cao, F. Idrees and X. L. Ma, ACS Nano, 2015, 9, 2556–2564 CrossRef CAS PubMed
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed
- P. Roy and S. K. Srivastava, J. Mater. Chem. A, 2015, 3, 2454–2484 CAS
- H. D. Yoo, E. Markevich, G. Salitra, D. Sharon and D. Aurbach, Mater. Today, 2014, 17, 110–121 CrossRef CAS
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC
- W. H. Yu, H. L. Wang, S. Liu, N. Mao, X. Liu, J. Shi, W. Liu, S. G. Chen and X. Wang, J. Mater. Chem. A, 2016, 4, 5973–5983 CAS
- F. Beguin, V. Presser, A. Balducci and E. Frackowiak, Adv. Mater., 2014, 26, 2219–2251 CrossRef CAS PubMed
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250–1280 CAS
- N. Mao, H. Wang, Y. Sui, Y. Cui, J. Pokrzywinski, J. Shi, W. Liu, S. Chen, X. Wang and D. Mitlin, Nano Res., 2017, 10, 1767–1783 CrossRef CAS
- B. Dyatkin, O. Gogotsi, B. Malinovskiy, Y. Zozulya, P. Simon and Y. Gogotsi, J. Power Sources, 2016, 306, 32–41 CrossRef CAS
- M. Oschatz, L. Borchardt, M. Thommes, K. A. Cychosz, I. Senkovska, N. Klein, R. Frind, M. Leistner, V. Presser, Y. Gogotsi and S. Kaskel, Angew. Chem., Int. Ed., 2012, 51, 7577–7580 CrossRef CAS PubMed
- T. Q. Lin, I. W. Chen, F. X. Liu, C. Y. Yang, H. Bi, F. F. Xu and F. Q. Huang, Science, 2015, 350, 1508–1513 CrossRef CAS PubMed
- S. Xu, Y. Lv, X. Zeng and D. Cao, Chem. Eng. J., 2017, 323, 502–511 CrossRef CAS
- D. Zhu, Y. Wang, W. Lu, H. Zhang, Z. Song, D. Luo, L. Gan, M. Liu and D. Sun, Carbon, 2017, 111, 667–674 CrossRef CAS
- Z. Xu, X. Zhuang, C. Yang, J. Cao, Z. Yao, Y. Tang, J. Jiang, D. Wu and X. Feng, Adv. Mater., 2016, 28, 1981–1987 CrossRef CAS PubMed
- Y. Yang, C. Han, B. Jiang, J. Iocozzia, C. He, D. Shi, T. Jiang and Z. Lin, Mater. Sci. Eng., R, 2016, 102, 1–72 CrossRef
- L. Wen, F. Li and H. M. Cheng, Adv. Mater., 2016, 28, 4306–4337 CrossRef CAS PubMed
- Y. Jin, K. Tian, L. Wei, X. Zhang and X. Guo, J. Mater. Chem. A, 2016, 4, 15968–15979 CAS
- X. Yang, C. Wei and G. Zhang, J. Mater. Sci., 2016, 51, 5565–5571 CrossRef CAS
- P. Hao, Z. Zhao, Y. Leng, J. Tian, Y. Sang, R. I. Boughton, C. P. Wong, H. Liu and B. Yang, Nano Energy, 2015, 15, 9–23 CrossRef CAS
- L. Chai, Q. Qu, L. Zhang, M. Shen, L. Zhang and H. Zheng, Electrochim. Acta, 2013, 105, 378–383 CrossRef CAS
- P. Feng, W. Wang, K. Wang, S. Cheng and K. Jiang, J. Mater. Chem. A, 2017, 5, 10261–10268 CAS
- A. Varzi and S. Passerini, J. Power Sources, 2015, 300, 216–222 CrossRef CAS
- I. Kovalenko, B. Zdyrko, A. Magasinski, B. Hertzberg, Z. Milicev, R. Burtovyy, I. Luzinov and G. Yushin, Science, 2011, 334, 75–79 CrossRef CAS PubMed
- Y. Cui, H. Wang, N. Mao, W. Yu, J. Shi, M. Huang, W. Liu, S. Chen and X. Wang, J. Power Sources, 2017, 361, 182–194 CrossRef CAS
- D. Li, C. Lv, L. Liu, Y. Xia, X. She, S. Guo and D. Yang, ACS Cent. Sci., 2015, 1, 261–269 CrossRef CAS PubMed
- S. N. Pawar and K. J. Edgar, Biomaterials, 2012, 33, 3279–3305 CrossRef CAS PubMed
- J. Zhang, J. Zhou, D. Wang, L. Hou and F. Gao, Electrochim. Acta, 2016, 191, 933–939 CrossRef CAS
- M. Seredych and T. J. Bandosz, J. Mater. Chem. A, 2013, 1, 11717–11727 CAS
- J. Yi, Y. Qing, C. Wu, Y. Zeng, Y. Wu, X. Lu and Y. Tong, J. Power Sources, 2017, 351, 130–137 CrossRef CAS
- L. Zhang, G. Xia, Z. Guo, X. Li, D. Sun and X. Yu, Int. J. Hydrogen Energy, 2016, 41, 14252–14260 CrossRef CAS
- X. Yan, Y. Liu, X. Fan, X. Jia, Y. Yu and X. Yang, J. Power Sources, 2014, 248, 745–751 CrossRef CAS
- J. Han, G. Xu, B. Ding, J. Pan, H. Dou and D. R. MacFarlane, J. Mater. Chem. A, 2014, 2, 5352–5357 CAS
- J. Niu, R. Shao, J. Liang, M. Dou, Z. Li, Y. Huang and F. Wang, Nano Energy, 2017, 36, 322–330 CrossRef CAS
- J. Ding, H. Wang, Z. Li, K. Cui, D. Karpuzov, X. Tan, A. Kohandehghan and D. Mitlin, Energy Environ. Sci., 2015, 8, 941–955 CAS
- W.-Y. Tsai, R. Lin, S. Murali, L. Li Zhang, J. K. McDonough, R. S. Ruoff, P.-L. Taberna, Y. Gogotsi and P. Simon, Nano Energy, 2013, 2, 403–411 CrossRef CAS
- J. Xu, M. Wang, N. P. Wickramaratne, M. Jaroniec, S. Dou and L. Dai, Adv. Mater., 2015, 27, 2042–2048 CrossRef CAS PubMed
- Y. Deng, Y. Xie, K. Zou and X. Ji, J. Mater. Chem. A, 2016, 4, 1144–1173 CAS
- C. Xuan, Z. Wu, W. Lei, J. Wang, J. Guo and D. Wang, ChemCatChem, 2017, 9, 809–815 CrossRef CAS
- Y. Yang, D.-M. Tang, C. Zhang, Y. Zhang, Q. Liang, S. Chen, Q. Weng, M. Zhou, Y. Xue, J. Liu, J. Wu, Q. H. Cui, C. Lian, G. Hou, F. Yuan, Y. Bando, D. Golberg and X. Wang, Energy Environ. Sci., 2017, 10, 979–986 CAS
- H. Liu, H. Song, X. Chen, S. Zhang, J. Zhou and Z. Ma, J. Power Sources, 2015, 285, 303–309 CrossRef CAS
- Y. Li, G. Wang, T. Wei, Z. Fan and P. Yan, Nano Energy, 2016, 19, 165–175 CrossRef CAS
- Z. Li, Z. Xu, H. Wang, J. Ding, B. Zahiri, C. M. B. Holt, X. Tan and D. Mitlin, Energy Environ. Sci., 2014, 7, 1708 CAS
- Z. Li, Z. Xu, X. Tan, H. Wang, C. M. B. Holt, T. Stephenson, B. C. Olsen and D. Mitlin, Energy Environ. Sci., 2013, 6, 871 CAS
- C. Hu, L. Wang, Y. Zhao, M. Ye, Q. Chen, Z. Feng and L. Qu, Nanoscale, 2014, 6, 8002–8009 RSC
- W. H. Shin, H. M. Jeong, B. G. Kim, J. K. Kang and J. W. Choi, Nano Lett., 2012, 12, 2283–2288 CrossRef CAS PubMed
- H. Hou, X. Qiu, W. Wei, Y. Zhang and X. Ji, Adv. Energy Mater., 2017, 1602898 CrossRef
- X. Du, Z. Zhang, W. Liu and Y. Deng, Nano Energy, 2017, 35, 299–320 CrossRef CAS
- J. Zhang and X. S. Zhao, ChemSusChem, 2012, 5, 818–841 CrossRef CAS PubMed
- Z. Wu, W. Ren, L. Xu, F. Li and H. Cheng, ACS Nano, 2011, 5, 5463–5471 CrossRef CAS PubMed
- J. Hou, C. Cao, X. Ma, F. Idrees, B. Xu, X. Hao and W. Lin, Sci. Rep., 2014, 4, 7260 CrossRef CAS PubMed
- B. E. Wilson, S. He, K. Buffington, S. Rudisill, W. H. Smyrl and A. Stein, J. Power Sources, 2015, 298, 193–202 CrossRef CAS
- D. Zhou, H. Wang, N. Mao, Y. Chen, Y. Zhou, T. Yin, H. Xie, W. Liu, S. Chen and X. Wang, Microporous Mesoporous Mater., 2017, 241, 202–209 CrossRef CAS
- C. Chen, D. Yu, G. Zhao, B. Du, W. Tang, L. Sun, Y. Sun, F. Besenbacher and M. Yu, Nano Energy, 2016, 27, 377–389 CrossRef CAS
- H. Wang, Z. Xu, A. Kohandehghan, Z. Li, K. Cui, X. Tan, T. J. Stephenson, C. K. King'ondu, C. M. B. Holt, B. C. Olsen, J. K. Tak, D. Harfield, A. O. Anyia and D. Mitlin, ACS Nano, 2013, 7, 5131–5141 CrossRef CAS PubMed
- J. Pu, C. Li, L. Tang, T. Li, L. Ling, K. Zhang, Y. Xu, Q. Li and Y. Yao, Carbon, 2015, 94, 650–660 CrossRef CAS
- F. Zheng, Y. Yang and Q. Chen, Nat. Commun., 2014, 5, 5261 CrossRef CAS PubMed
- J. Ji, J. Liu, L. Lai, X. Zhao, Y. Zhen, J. Lin, Y. Zhu, H. Ji, L. L. Zhang and R. S. Ruoff, ACS Nano, 2015, 9, 8609–8616 CrossRef CAS PubMed
- Z. Weng, F. Li, D. W. Wang, L. Wen and H. M. Cheng, Angew. Chem., Int. Ed., 2013, 52, 3722–3725 CrossRef CAS PubMed
- G. Ye, X. Zhu, S. Chen, D. Li, Y. Yin, Y. Lu, S. Komarneni and D. Yang, J. Mater. Chem. A, 2017, 5, 8247–8254 CAS
- Y. Mao, H. Duan, B. Xu, L. Zhang, Y. Hu, C. Zhao, Z. Wang, L. Chen and Y. Yang, Energy Environ. Sci., 2012, 5, 7950 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7se00443e |
| This journal is © The Royal Society of Chemistry 2018 |