
Ionically cross-linked PEDOT:PSS as a multi-functional conductive binder for high-performance lithium–sulfur batteries†
Longlong
Yan‡
ab,
Xiguang
Gao‡
a,
Joseph Palathinkal
Thomas
c,
Jenner
Ngai
a,
Haig
Altounian
a,
Kam Tong
Leung
 c,
Yuezhong
Meng
*b and
Yuning
Li
*a
c,
Yuezhong
Meng
*b and
Yuning
Li
*a
aDepartment of Chemical Engineering, Waterloo Institute for Nanotechnology (WIN), University of Waterloo, 200 University Ave West, Waterloo, Ontario N2L 3G1, Canada. E-mail: yuning.li@uwaterloo.ca; Fax: +1-519-888-4347; Tel: +1-519-888-4567 ext. 31105
bThe Key Laboratory of Low-Carbon Chemistry & Energy Conservation of Guangdong Province, State Key Laboratory of Optoelectronic Materials and Technologies, School of Materials Science and Engineering, Sun Yat-sen University, Guangzhou 510275, P. R. China. E-mail: mengyzh@sysu.edu.cn
cDepartment of Chemistry, University of Waterloo, 200 University Ave West, Waterloo, Ontario N2L 3G1, Canada
First published on 23rd April 2018
Abstract
Despite their very high theoretical specific capacity, lithium–sulfur (Li–S) batteries still face issues such as low sulfur utilization and poor long-term cycling stability due to the low conductivity of sulfur, lithium polysulfide shuttle effect and large volume change during discharge–charge processes. This work uses a novel multi-functional polymer binder, PEDOT:PSS-Mg2+, to address the above issues. First, PEDOT:PSS is a highly conductive polymer, which improves the conductivity of the cathode composite. Second, cross-linking of PEDOT:PSS with Mg2+ forms a robust network that is able to endure the drastic volume change of the cathode during discharge/charge. Third, the abundant oxygen atoms present in PEDOT:PSS strongly interact with lithium polysulfides to suppress the shuttle effect. Li–S batteries with this new binder showed high initial specific capacity of up to 1097 mA h g−1 and high capacity retention of up to 74% after 250 cycles at 0.5C with a sulfur content of 70 wt% in the cathode, which are significant improvements compared with the corresponding Li–S batteries with a conventional PVDF binder. Additionally, preparation of the cathode material with this new binder uses water as the solvent, avoiding the use of toxic organic solvents such as N-methylpyrrolidone (NMP).
Introduction
The very high theoretical specific capacity of the sulfur cathode (1675 mA h g−1) allows lithium–sulfur (Li–S) batteries to reach a theoretical energy density of 2600 W h kg−1, which is 6 times those of lithium ion batteries (LIBs) based on intercalation compounds such as LiCoO2 and LiFePO4.1–5 In addition, sulfur is inexpensive, environmentally benign and abundant, making it one of the most promising candidate cathode materials for next-generation high performance batteries. Despite the above-mentioned advantages, the practical applications of Li–S batteries are still hampered by several technical barriers. The low electrical conductivities of elemental sulfur (5 × 10−30 S cm−1) and its reduced products (intermediate lithium polysulfides and lithium sulfide) significantly lower the capabilities of Li–S batteries, especially under high discharge–charge current densities.6,7 Furthermore, since the intermediate long chain polysulfides (Li2Sxx = 3–8) are soluble in the electrolyte solution, they can move to the anode during discharging and return to the cathode during the subsequent charging, which is commonly called the “shuttle effect”.8–10 Some of the soluble polysulfides that reach the anode are reduced by lithium to form insoluble Li2S2 and Li2S deposits, which cannot come back to the cathode. Both the shuttle effect and the low electrical conductivity of the insoluble Li2S2 and Li2S deposits formed on the anode surface would result in a reduction of sulfur utilization and hence capacity decay of Li–S batteries over time. Moreover, Li–S batteries also suffer from a large volumetric change (∼80%) due to the dramatic density difference between sulfur in the charged state and Li2S in the charged state, which may gradually lead to an irreversible destruction of the cathode structure and a capacity drop. To address the afore-mentioned issues, approaches involving embedding sulfur in carbon-based host materials such as porous hollow carbon,11–13 carbon nanotubes,14,15 nanofibers,16,17 and various types of graphene18–22 have been explored and some improvements have been achieved.A typical Li–S battery cathode consists of four components: sulfur, conductive carbon additive, binder, and current collector. The binder is an important ingredient, which binds sulfur and conductive carbon together with the current collector to maintain the structural integrity of the cathode. Compared with other cathode components, the binder has been paid much less attention for the improvement of Li–S battery performance. Polyvinylidene fluoride (PVDF) and polyethylene glycol (PEO) are the most commonly used binders for Li–S battery cathodes.23 However, these binders are suboptimal for achieving high cycling stability and high capacity of Li–S batteries because (i) they have linear polymer chains and weaker interchain interaction and cannot cushion the large volume change during discharge/charge cycling to retain the structural integrity of the cathode; (ii) they have relatively weak affinity to polysulfides and cannot effectively prevent the shuttling of polysulfides; (iii) they are electrical insulators and make no contribution to the electron transfer. In particular, for high sulfur loading cathode composites (S% ≥70 wt%), which are mandatory for high energy density Li–S batteries,24,25 the combined amount of the conductive component and binder used would be 20 wt% or less. Therefore, a high-performance binder with high binding strength as well as high electrical conductivity would be ideal. Recently, some efforts have been made to develop new binders to replace PVDF and PEO. For example, Li et al.26 used gum arabic (GA) polymer as the binder to improve the capacity and stability. Bhattacharya et al.27 reported a polyamidoamine (PAMAM) dendrimer-based binder, which can encapsulate sulfur more effectively. Chen et al.28 designed a multifunctional cross-linked binder of PEI-HDI with a 3D network structure for stable Li–S batteries. Although these new binders showed better performance than PVDF and PEO, they are electrical insulators and do not contribute to the improvement of the electrical conductivity of the cathode composite. A few electrically conductive binders such as (reduced) graphene oxide-polyacrylic acid (GOPAA),29 PEDOT:PSS,30 PEDOT:PSS-PAA,31 and poly(9,9-dioctylfluorene-co-fluorenone-co-methylbenzoic ester) (PFM)32 have been reported to improve the cathode performance of Li–S batteries. However, Li–S batteries with these conductive binders have not demonstrated both high capacity and high cycling stability for high sulfur loading (S ≥70 wt%) Li–S batteries.
PDEOT:PSS is the most studied and widely used conductive polymer as the electrode for various electronic and electrical devices such as organic thin film transistors, organic photovoltaics, and batteries because it is solution-processable and both electrically (with electrical conductivity of up to 4600 S cm−1 and higher)33,34 and ionically (with the mobility of K+ ions of up to 2.2 × 10−3 cm2 V−1 s−1)35 highly conductive. In addition, it has been reported that the oxygen atoms in PEDOT:PSS can strongly bond polysulfides,36 which would be beneficial for the improvement of the cathode cycling stability. Despite these advantages, previous studies showed that when PEDOT:PSS alone was used as a binder, very limited improvements in the cathode capacity and cycling stability were obtained.30 A combination of PEDOT:PSS and polyacrylic acid (PAA) could improve both the capacity and cycling stability of the cathode.31 It was suggested that PEDOT:PSS facilitated electron transfer and prevented lithium polysulfide dissolution, while PAA improved the swelling properties of the cathode, leading to better lithium ion conduction. An impressive initial specific capacity of 1121 mA h g−1 was achieved, but the specific capacity still dropped rather rapidly to 834 mA h g−1 (74% of the initial capacity) after 80 cycles.
In this work, we used a very convenient approach to cross-linking PEDOT:PSS to improve the structural stability of the PEDOT:PSS network formed in the Li–S battery cathode. Specifically, we used the divalent Mg2+ ion as a cross-linker for PSS in PEDOT:PSS to form a 3-D network binder, PEDOT:PSS-Mg2+, for the Li–S battery cathode. The Li–S batteries using this PEDOT:PSS-Mg2+ binder achieved a high initial capacity of 1097 mA h g−1 and significantly improved cycling stability with capacity retention of 74% after 250 cycles at 0.5C. Moreover, water was used as the solvent for preparing the cathode, making the fabrication process environmentally friendly.
Results and discussion
PEDOT:PSS is a commercially available water-soluble polymer, whose chemical structure is shown in Fig. 1a. A PEDOT:PSS aqueous dispersion comprises PEDOT nanoparticles wrapped by PSS (polystyrenesulfonic acid), which was added when PEDOT:PSS was synthesized, in order to solubilize PEDOT.33,35 PSS is also a dopant, giving away some of its protons to PEDOT to form the PEDOT:PSS complex, which is highly conductive in the solid state. The PEDOT:PSS dispersion, Clevios PH 1000, used in this study can provide films with high electrical conductivity of up to 1000 S cm−1. The PSS:PEDOT weight ratio in this dispersion is 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating that the sulfonic acid –SO2OH groups in PSS is in a large excess. Therefore, we may utilize the extra –SO2OH groups to crosslink the PSS polymer chains. Since PSS molecules strongly interact with PEDOT molecules, cross-linking of PSS could fasten the PEDOT chains to form a robust conductive PEDOT:PSS 3D network. To cross-link the PSS, we chose the divalent cation, Mg2+, as the cross-linker (Fig. 1). To prepare the cathode material, nano-particulate sulfur (NPS) with an average diameter of ∼110 nm (Fig. S1 and S2†) was used as the active cathode material. At room temperature, NPS and a conductive carbon, Super P (SP), were uniformly dispersed in a PEDOT:PSS aqueous solution (1.2 wt%) by ultrasonication and then a small amount of Mg(NO3)2 was added. The mixture was further sonicated to obtain a stable, uniform dispersion with NPS/SP/PEDOT:PSS/Mg(NO3)2 weight ratio of 70:20:9:1. During mixing, Mg(NO3)2 would react with the –SO2OH groups of PSS, producing the –SO2O–Mg–O–SO2– linkage (an immediate increase in viscosity was observed). A trace amount of by-product HNO3 would form, which could be evaporated during drying. Therefore, in the dried film only the Mg2+ ion of Mg(NO3)2 would remain, which takes up only 2 wt% of the binder or merely 0.2 wt% of the total cathode material. Even with such a small amount of Mg2+, the Mg2+/PSS repeat unit molar ratio would be ∼4.3, which would be sufficient to achieve a high cross-linking density. The as-prepared slurry was blade-coated on Al foil and then dried in a vacuum oven at 50 °C for 12 h. The formed NPS/SP/PEDOT:PSS-Mg2+ layer was peeled off and subjected to Soxhlet extraction with CS2, which is a good solvent for elemental sulfur, to evaluate the sulfur retention ability of this new cathode binder. As comparative references, NPS/SP/PVDF and NPS/SP/PEDOT:PSS (without addition of Mg(NO3)2) samples were also prepared and tested. After extensive extraction for 5 h with boiling CS2, the NPS/SP/PEDOT:PSS-Mg2+ sample retained 14% of the initially loaded sulfur (Table S1†). On the other hand, sulfur in the NPS/SP/PVDF or the NPS/SP/PEDOT:PSS sample was completely lost after extraction for 5 h. The improved solvent resistance of the NPS/SP/PEDOT:PSS-Mg2+ sample against sulfur loss indicates that the PEDOT:PSS-Mg2+ binder has a stronger ability to retain elemental sulfur, as a result of the cross-linked binder network. The slurry of NPS + SP + PEDOT:PSS-Mg(NO3)2 or NPS + SP + PVDF cathode material was then coated on a carbon paper (CP) current collector substrate. The coated substrate was dried in a vacuum oven at 50 °C for 12 h, cut into discs, and assembled into CR2025 coin cells to evaluate the electrochemical performance. As aforementioned, with addition of Mg(NO3)2 the viscosity of the NPS + SP + PEDOT:PSS + Mg(NO3)2 slurry increased notably probably due to the electrostatic interaction of Mg2+ cations with the pendent –SO2O− anions of neighboring PSS chains. The appropriately increased viscosity makes this aqueous slurry stable and facilitates its coating on the CP current collector substrate to form a smooth and uniform cathode composite film. In contrast, the slurry of NPS + SP + PEDOT:PSS without the addition of Mg(NO3)2 could not form a stable slurry, rapidly forming precipitates as soon as agitation was stopped. This posed a challenge to coat the slurry on the CP current collector and no properly working battery with the NPS/SP/PEDOT:PSS cathode was obtained in this study.
:1, indicating that the sulfonic acid –SO2OH groups in PSS is in a large excess. Therefore, we may utilize the extra –SO2OH groups to crosslink the PSS polymer chains. Since PSS molecules strongly interact with PEDOT molecules, cross-linking of PSS could fasten the PEDOT chains to form a robust conductive PEDOT:PSS 3D network. To cross-link the PSS, we chose the divalent cation, Mg2+, as the cross-linker (Fig. 1). To prepare the cathode material, nano-particulate sulfur (NPS) with an average diameter of ∼110 nm (Fig. S1 and S2†) was used as the active cathode material. At room temperature, NPS and a conductive carbon, Super P (SP), were uniformly dispersed in a PEDOT:PSS aqueous solution (1.2 wt%) by ultrasonication and then a small amount of Mg(NO3)2 was added. The mixture was further sonicated to obtain a stable, uniform dispersion with NPS/SP/PEDOT:PSS/Mg(NO3)2 weight ratio of 70:20:9:1. During mixing, Mg(NO3)2 would react with the –SO2OH groups of PSS, producing the –SO2O–Mg–O–SO2– linkage (an immediate increase in viscosity was observed). A trace amount of by-product HNO3 would form, which could be evaporated during drying. Therefore, in the dried film only the Mg2+ ion of Mg(NO3)2 would remain, which takes up only 2 wt% of the binder or merely 0.2 wt% of the total cathode material. Even with such a small amount of Mg2+, the Mg2+/PSS repeat unit molar ratio would be ∼4.3, which would be sufficient to achieve a high cross-linking density. The as-prepared slurry was blade-coated on Al foil and then dried in a vacuum oven at 50 °C for 12 h. The formed NPS/SP/PEDOT:PSS-Mg2+ layer was peeled off and subjected to Soxhlet extraction with CS2, which is a good solvent for elemental sulfur, to evaluate the sulfur retention ability of this new cathode binder. As comparative references, NPS/SP/PVDF and NPS/SP/PEDOT:PSS (without addition of Mg(NO3)2) samples were also prepared and tested. After extensive extraction for 5 h with boiling CS2, the NPS/SP/PEDOT:PSS-Mg2+ sample retained 14% of the initially loaded sulfur (Table S1†). On the other hand, sulfur in the NPS/SP/PVDF or the NPS/SP/PEDOT:PSS sample was completely lost after extraction for 5 h. The improved solvent resistance of the NPS/SP/PEDOT:PSS-Mg2+ sample against sulfur loss indicates that the PEDOT:PSS-Mg2+ binder has a stronger ability to retain elemental sulfur, as a result of the cross-linked binder network. The slurry of NPS + SP + PEDOT:PSS-Mg(NO3)2 or NPS + SP + PVDF cathode material was then coated on a carbon paper (CP) current collector substrate. The coated substrate was dried in a vacuum oven at 50 °C for 12 h, cut into discs, and assembled into CR2025 coin cells to evaluate the electrochemical performance. As aforementioned, with addition of Mg(NO3)2 the viscosity of the NPS + SP + PEDOT:PSS + Mg(NO3)2 slurry increased notably probably due to the electrostatic interaction of Mg2+ cations with the pendent –SO2O− anions of neighboring PSS chains. The appropriately increased viscosity makes this aqueous slurry stable and facilitates its coating on the CP current collector substrate to form a smooth and uniform cathode composite film. In contrast, the slurry of NPS + SP + PEDOT:PSS without the addition of Mg(NO3)2 could not form a stable slurry, rapidly forming precipitates as soon as agitation was stopped. This posed a challenge to coat the slurry on the CP current collector and no properly working battery with the NPS/SP/PEDOT:PSS cathode was obtained in this study.
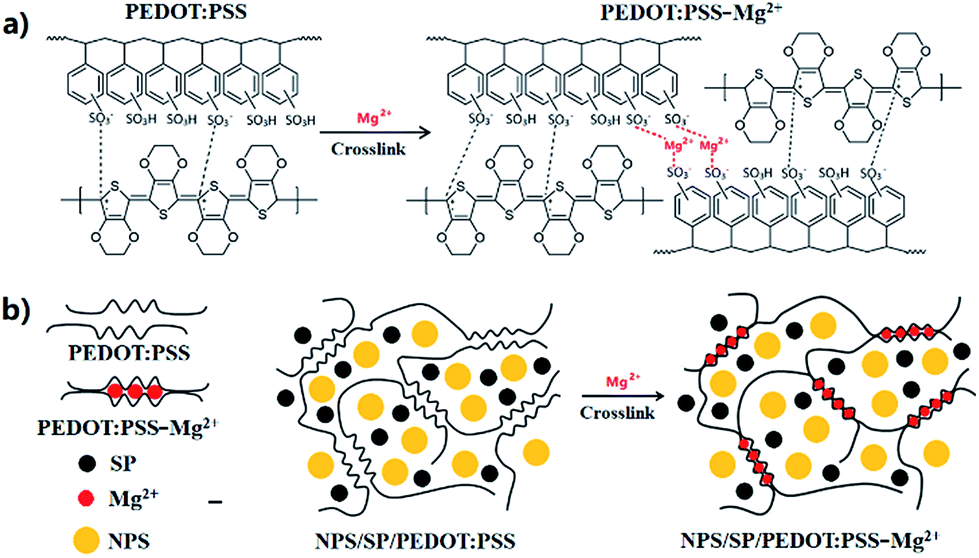 | ||
| Fig. 1 Schematic representation of the PEDOT:PSS-Mg2+ binder structure and NPS/SP/PEDOT:PSS-Mg2+ electrode. The coordination of Mg2+ by the SO3− is the main process in the gel formation and polymer–metal framework stabilization. | ||
To examine whether the Mg2+-crosslinking affects the conductivity of PEDOT:PSS or not, PEDOT:PSS and cross-linked PEDOT:PSS-Mg2+ films (thickness ∼ 80 nm) were prepared and their conductivities were evaluated using a four-point probe method. The average conductivity of PEDOT:PSS films was measured to be 833 S cm−1. After cross-linking by Mg2+, the average conductivity dropped to 668 S cm−1 which corresponds to a ∼20% decrease. Therefore, the cross-linked PEDOT:PSS-Mg2+ films remained highly conductive.
Fig. 2a presents the cathode capacity versus voltage profiles during the second discharge/charge cycle of batteries with NPS/SP/PEDOT:PSS-Mg2+ and NPS/SP/PVDF cathodes. The NPS/SP/PVDF cathode shows a specific capacity of 1079 mA h g−1 at a discharge rate of 0.1C. Its discharge curve exhibits a typical two-step reduction process with the formation of long lithium polysulfide species (Li2Sx, x = ∼3–8) in the first plateau at ∼2.3 V and lithium disulfide (Li2S2) and lithium sulfide (Li2S) in the second plateau at ∼2.1 V.37–42 During the charging process, Li2S was oxidized to form Li2S2 at ∼2.2–2.3 V, Li2S3–8 at ∼2.3 V and then the final product sulfur (S8) at ∼2.4 V. The NPS/SP/PEDOT:PSS-Mg2+ cathode shows a significantly increased specific capacity of 1219 mA h g−1. It also appears that the NPS/SP/PEDOT:PSS-Mg2+ cathode exhibits a higher specific capacity (∼270 mA h g−1) than the NPS/SP/PVDF electrode (∼220 mA h g−1) at the end of the first discharge plateau (Fig. 2a). This result indicates that the reduction reactions to form the soluble polysulfides (Li2S3–8) were facilitated by the PEDOT:PSS-Mg2+ binder, which might be due to the improved electrical conductivity due to the presence of the conductive PEDOT:PSS-Mg2+ binder as well as the stronger interaction of this new binder with soluble polysulfides (to be discussed below). The second discharge plateau at ∼2.1 V to form Li2S2 and Li2S is also much longer for the NPS/SP/PEDOT:PSS-Mg2+ cathode, indicating that a larger amount of soluble polysulfides were involved in this reduction step. This may be contributed by (1) improved charge transfer by the presence of this conductive binder, (2) the suppressed diffusion of soluble polysulfides (Li2Sx, where x > 2) due to the 3D network structure and the strong polysulfide-adsorbing ability of the PEDOT:PSS-Mg2+ binder. It is noteworthy that a smaller voltage hysteresis (ΔE) was observed for the NPS/SP/PEDOT:PSS-Mg2+ cathode, which also suggests that introduction of this conductive binder into the Li–S cathode can promote the reduction of the intermediate polysulfides to Li2S2/Li2S.7
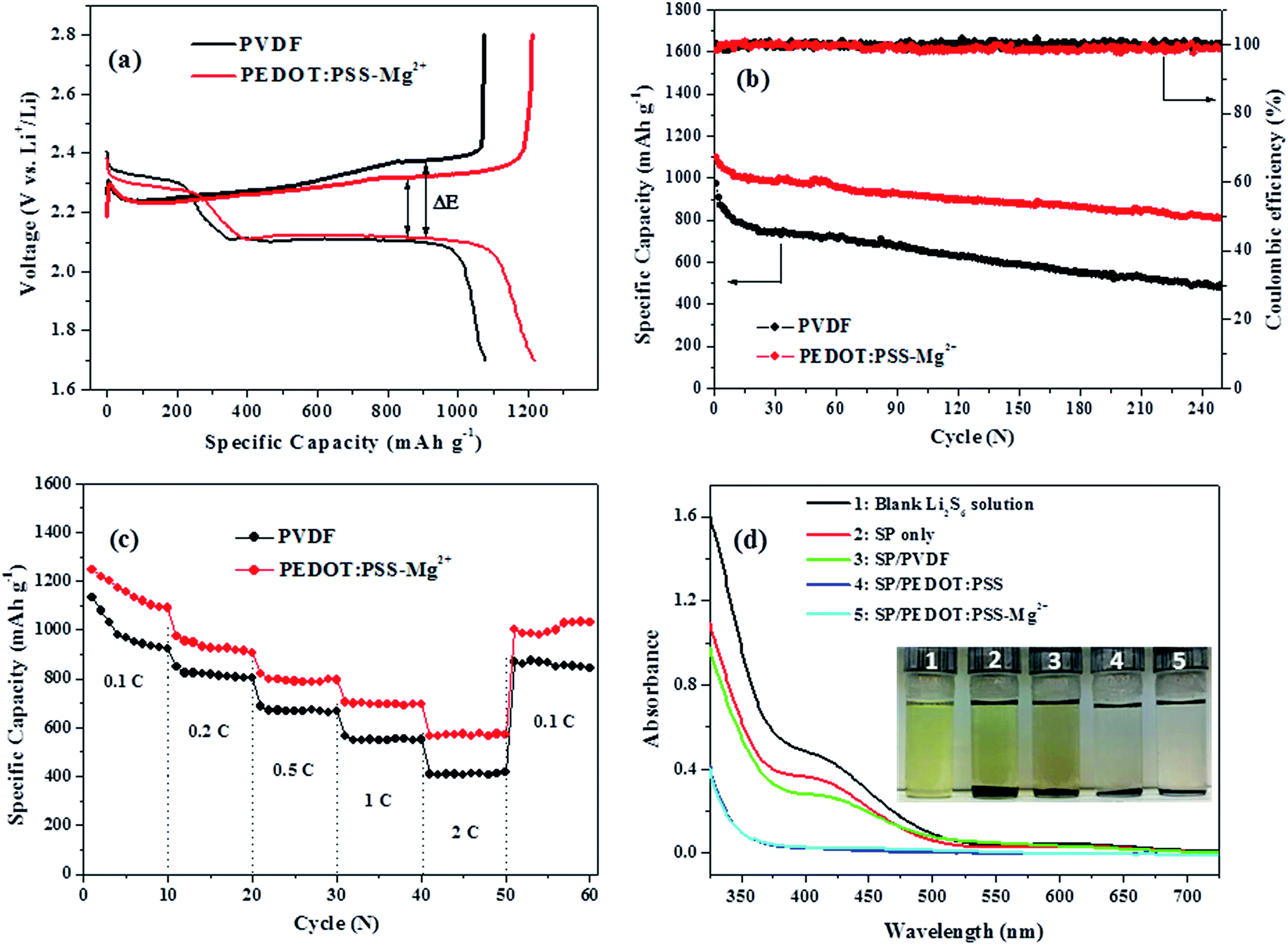 | ||
| Fig. 2 Discharge–charge performance of NPS/SP/PVDF and NPS/SP/PEDOT:PSS-Mg2+ electrodes: (a) typical discharge–charge voltage vs. capacity profile; (b) cycling performance at 0.5C; (c) rate performance from 0.1C to 2C. (d) UV-vis absorption spectra and photos of the Li2S6 solutions in 1,3-dioxolane (DOL)/1,2-dimethoxyethane (DME) (v/v, 1/1) before (blank) and after addition of SP, SP/PVDF, SP/PEDOT:PSS, and SP/PEDOT:PSS-Mg2+. | ||
The cycling stabilities of cathodes with the PVDF binder and PEDOT:PSS-Mg2+ binder at a discharge–charge current rate of 0.5C are shown in Fig. 2b. The cathode with the PVDF binder shows an initial specific capacity of 973 mA h g−1 and a rapid decrease in capacity to 795 mA h g−1 after only 10 cycles. The capacity further dropped to 495 mA h g−1 after 250 cycles, corresponding to a capacity retention of 51%. On the other hand, the cathode with the PEDOT:PSS-Mg2+ binder shows a significantly improved initial specific capacity of 1097 mA h g−1, which is 13% higher than that of the cathode with the PVDF binder. The discharge capacity is maintained at a high value of 807 mA h g−1 after 250 cycles, corresponding to a capacity retention of 74%. As discussed previously, the higher sulfur utilization and better capacity retention of the cathode using the PEDOT:PSS-Mg2+ binder compared to the PVDF binder are most likely due to the highly conductive, cross-linked 3-D network structure and strong polysulfide-adsorbing ability of the PEDOT:PSS-Mg2+ binder. Moreover, the coulombic efficiency of the NPS/SP/PEDOT:PSS-Mg2+ electrode is maintained at >98% after 250 cycles. The capacities at various C-rates of 0.1C, 0.2C, 0.5C, 1C and 2C of the NPS/SP/PEDOT:PSS-Mg2+ and NPS/SP/PVDF cathodes were measured and the results are shown in Fig. 2c. A highly reversible capacity of 1033 mA h g−1 after 60 cycles was obtained when the C-rate was set back to 0.1C for the cathode with the PEDOT:PSS-Mg2+ binder. Highly reversible capacities of ∼790 mA h g−1 at 1C and 576 mA h g−1 at 2C were also achieved. As a comparison, the electrode with the PVDF binder exhibits a capacity of 846 mA h g−1 after 60 cycles when the C-rate was set back to 0.1C. At higher C-rates, the capacities decreased to ∼550 mA h g−1 at 1C and ∼410 mA h g−1 at 2C. Clearly the battery based on the NPS/SP/PEDOT:PSS-Mg2+ cathode demonstrated higher specific capacities at all the tested current densities, which results from the improved conductivity and structural stability of the cathode composite due to the use of the PEDOT:PSS-Mg2+ binder.
Furthermore, the lithium polysulfide-adsorbing ability of the PEDOT:PSS-Mg2+ binder was studied by using UV-vis spectroscopy. Li2S6 was chosen as a representative long chain lithium polysulfide, which was synthesized by the reaction of sulfur with Li2S in a stoichiometric ratio in a mixture solvent of 1,3-dioxolane (DOL)/1,2-dimethoxyethane (DME) (v/v, 1/1).43 Specifically, an SP/PEDOT:PSS-Mg2+ composite with a carbon/polymer weight ratio of 2:1 was added to the Li2S6 solution and kept for 24 h at room temperature before the clear supernatant was taken out for the UV-vis absorption measurement. For comparison, SP, SP/PVDF composite, and SP/PEDOT:PSS composite were also tested similarly. As shown in Fig. 2d, the blank Li2S6 solution showed a shoulder peak at ∼400 nm, which is in agreement with that reported in the literature.43–45 The solution mixed with SP showed a lighter color and its UV-vis spectrum also displayed a decrease in the absorption intensity, indicating that SP has some effect on the adsorption of Li2S6. For the sample mixed with SP/PVDF, a further slight drop in the absorption intensity was observed. On the other hand, the solutions mixed with SP/PEDOT:PSS and SP/PEDOT:PSS-Mg2+ showed much lighter colors and their UV-vis spectra exhibited a complete disappearance of absorption at ∼400 nm, indicating that almost all the Li2S6 species were adsorbed. These results strongly support that the SP/PEDOT:PSS and SP/PEDOT:PSS-Mg2+ composites have excellent Li2S6 (and probably other lithium polysulfides) adsorption ability, which agrees with the theoretical calculation results that the oxygen atoms in PEDOT can strongly bind lithium polysulfides.46 The results also confirmed that the presence of the crosslinking Mg2+ ions in SP/PEDOT:PSS-Mg2+ does not negatively affect its ability for adsorbing the lithium polysulfides compared to SP/PEDOT:PSS. Therefore, the above UV-vis absorption results further demonstrated that the observed better cycling stability of the NPS/SP/PEDOT:PSS-Mg2+ cathode compared to the NPS/SP/PVDF cathode is attributed in part to the stronger lithium polysulfide adsorption ability of the PEDOT:PSS-Mg2+ binder than PVDF.
To further examine the contributions of the PEDOT:PSS-Mg2+ binder to the enhancement of the battery performance, electrochemical impedance spectroscopy (EIS) measurements were performed on the fresh and cycled batteries with PEDOT:PSS-Mg2+ and PVDF binders in the charged state (Fig. 3). The Nyquist plot of each fresh battery exhibits a semicircle in the high frequency region and an inclined line in the low frequency region (Fig. 3a), which can be deconvoluted into an equivalent Randles circuit47 shown in the inset of Fig. 3a. The intercept of the semicircle with the Zreal (the real part of impedance) axis in the high frequency region corresponds to the resistance of the electrolyte solution (Re). The semicircle is ascribed to the charge-transfer resistance (Rct, the diameter of the semicircle) and related capacitance (CPE, constant phase element) at the cathode. The inclined line in the low frequency region is associated with the Li+ diffusion, namely, the Warburg impedance (Wo). As shown in Table 1, the fresh battery with the NPS/SP/PEDOT:PSS-Mg2+ cathode exhibits a much lower Rct (43.1 Ω) compared to that with the NPS/SP/PVDF cathode (78.0 Ω), indicating that the charge transfer resistance decreased significantly when the conductive PEDOT:PSS-Mg2+ binder was used. Since Rct is related to the electronic conduction of the conductive network of the cathode as well as the faradaic charge transfer at the interface of the conductive network and the electrolyte,48,49 the results strongly indicate that the use of the conductive PEDOT:PSS-Mg2+ binder is very effective to reduce the Rct.
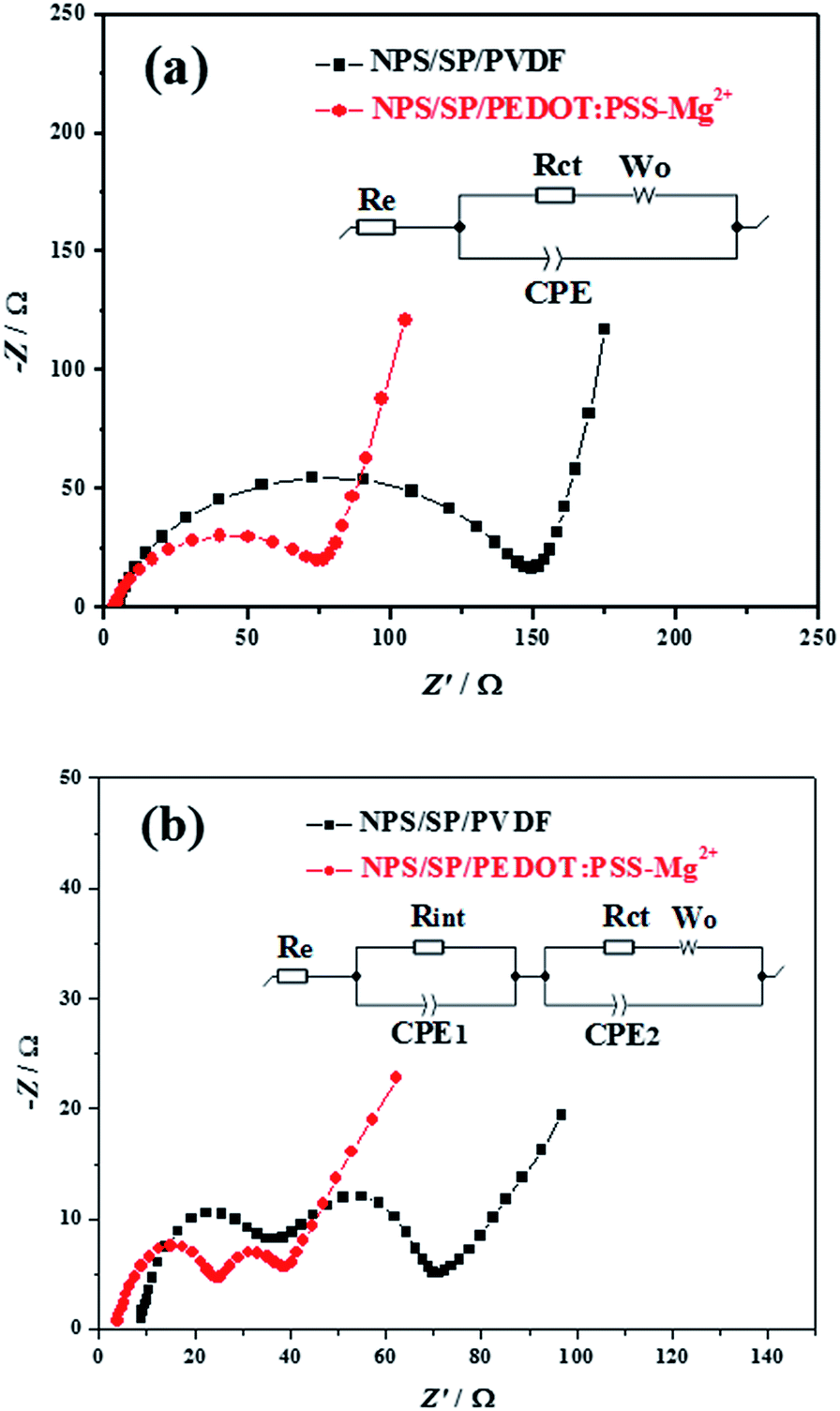 | ||
| Fig. 3 Electrochemical impendence spectroscopic (EIS) data of Li–S batteries with NPS/SP/PVDF and NPS/SP/PEDOT:PSS-Mg2+ electrodes measured in the charge state: (a) fresh cells and (b) after 10 cycles. | ||
| Cathode | Cycle | R e (Ω) | R ct (Ω) | R int (Ω) |
|---|---|---|---|---|
| NPS/SP/PVDF | Fresh cell | 3.6 | 78.0 | — |
| 10th | 8.4 | 24.7 | 17.3 | |
| NPS/SP/PEDOT:PSS-Mg2+ | Fresh cell | 3.3 | 43.1 | — |
| 10th | 3.5 | 12.4 | 13.4 |
After cycling, both batteries showed two semicircles (Fig. 3b), where the semicircle in the high-to-middle frequency region (right) is due to the charge-transfer resistance (Rct) and capacitance (CPE2) of the cathode, while the semicircle in the high frequency region (left) can be ascribed to the interfacial contact resistance (Rint)/capacitance (CPE1) between the electrolyte and the cathode.47,49,50 The semicircle that newly appeared in the high frequency region is mainly due to the formation of an insulating layer (sulfur and some non-oxidized insoluble Li2S2/Li2S after charging) on the surface of the cathode current collector (CP). The absence of this semicircle in the Nyquist plots for the fresh batteries is probably due the fact that part of the CP surface was not covered by the active cathode composite and was able to be in direct contact with the electrolyte solution. The battery with the NPS/SP/PEDOT:PSS-Mg2+ cathode showed a lower Rint of 13.4 Ω than that of the battery with the NPS/SP/PVDF cathode (17.3 Ω), indicating that the PEDOT:PSS-Mg2+ binder in the newly formed layer on the CP surface after cycling can effectively reduce the resistance of this interlayer. Another possible contribution for the appearance of the high-frequency semicircle after cycling might be the deposition of insoluble Li2S2/Li2S on the anode (Li) surface as a result of the diffusion of polysulfides from the cathode, followed by reduction by Li. Therefore, the lower Rint observed for the battery with the NPS/SP/PEDOT:PSS-Mg2+ cathode might also partially result from the stronger ability of the PEDOT:PSS-Mg2+ binder to trap soluble polysulfides within the cathode. For both batteries, the Rct decreased notably, revealing that the surface electrochemical activity is initiated during the discharge–charge processes.47 As observed for their fresh batteries, the Rct of the cycled battery with the NPS/SP/PEDOT:PSS-Mg2+ cathode (12.4 Ω) is still much lower than that of the battery with the NPS/SP/PVDF cathode (24.7 Ω), which again manifests the enhanced charge transfer by the conductive PEDOT:PSS-Mg2+ binder. It is also noticed that the Re of the battery with the NPS/SP/PVDF cathode obviously increased from 3.6 Ω to 8.4 Ω after cycling, which might be caused by the increased viscosity of the electrolyte solution and thus lowered ion mobility due to the presence of unreacted polysulfides in the electrolyte.47 For the battery with the NPS/SP/PEDOT:PSS-Mg2+ cathode, the Re remained almost the same (from 3.3 Ω to 3.5 Ω) after cycling, which can be accounted for by the presence of much less unreacted polysulfides in the electrolyte as supported by the UV-vis absorption spectroscopy experiments (Fig. 3d). Overall, the impedance of the NPS/SP/PEDOT:PSS-Mg2+ cathode is significantly decreased owing to the application of the conductive cross-linked PEDOT:PSS-Mg2+ binder, which supports the improved discharge–charge performance as shown in Fig. 2.
Experimental
Materials and instrumentation
PEDOT:PSS (Clevios PH 1000, 1.2 wt%, with a PEDOT:PSS weight ratio of 1:2.5) was obtained from Heraeus, Germany. Magnesium nitrate (Mg(NO3)2, 99%) was supplied by EM Science, Germany. Carbon paper (CP) (TGP-H-090; 0.28 mm) was obtained from Toray, Japan. Super P (SP; 99%) conductive carbon powder was obtained from Timical, Switzerland. All the chemicals were used without further purification. UV-vis spectra were measured with a Shimadzu UV-2501PC spectrometer. The conductivity measurements of PEDOT:PSS and PEODT:PSS-Mg2+ films on glass substrates were conducted using a four-point probe technique with a Signatone Pro-4 and Keithley 2400 source meter.
Synthesis of NPS
Nano-particulate sulfur (NPS) was synthesized by a previously reported method.51 Aqueous solutions of 80 mM Na2S2O3 (50 mL) and 0.4 M PVP (the concentration was based on the repeat unit of PVP) (50 mL) were mixed at room temperature. Then, concentrated hydrochloric acid (0.4 mL, 37%) was added under stirring. After 2 h, the obtained reaction mixture was centrifuged at 4000 rpm for 15 min to isolate the precipitated particles, which were washed with deionized water 5 times and dried in air and then under vacuum at room temperature to give the NPS product. The sulfur content of the NPS is ∼99% as determined by thermogravimetric analysis (TGA) (Fig. S1†). The average diameter of the NPS is ∼110 nm estimated from the SEM image (Fig. S2†).Preparation of PEDOT:PSS and PEDOT:PSS-Mg2+ films
PEDOT:PSS film was prepared by spin coating PEDOT:PSS aqueous solution (1.2 wt%) on glass substrates and dried at 120 °C in a vacuum oven. PEDOT:PSS-Mg2+ film was prepared by adding a few drops of Mg(NO3)2 solution (11 mM) onto PEDOT:PSS film, waiting for 1 min, then spin-coating, and then drying at 120 °C in a vacuum oven.Preparation of cathodes
NPS (0.21 g) was uniformly dispersed in 1.2 wt% PEDOT:PSS aqueous solution (2.3 mL containing ∼27.6 mg of PEDOT:PSS) in an ultrasonic bath for 30 min. Then SP (0.06 g) was added and the mixture was homogenized in an ultrasonic bath for an additional 40 min. Subsequently, 0.5 M Mg(NO3)2 solution (50 μL containing 3.7 mg of Mg(NO3)2) was added and the mixture was sonicated for another 30 min. The obtained slurry was blade coated on a CP current collector and dried briefly in air before being transferred to a vacuum oven and heated at 50 °C for 12 h to form the NPS/SP/PEDOT:PSS-Mg2+ (NPS/SP/PEDOT:PSS-Mg2+ = 70:20:10 by weight) cathode. The NPS/SP/PVDF cathode using PVDF as the binder was prepared similarly by blade coating a slurry made by mixing NPS, SP, and PVDF at weight ratios of 70:20:10 in N-methylpyrrolidone (NMP). The cathodes were cut into circular disks with a diameter of 12 mm for the battery assembly.
Battery assembly and electrochemical measurements
CR2025 coin cells were assembled in an argon-filled glove box using a CP coated with either the NPS/SP/PEDOT:PSS-Mg2+ or NPS/SP/PVDF composite described above as the cathode. Lithium foil, a Celgard 2500 film, and 1 M LiTFSI (LiN(SO2CF3)2) in a 1:1 volume of DOL/DME solvent with 2 wt% LiNO3 (50 μL per cell) were used as the anode, separator, and electrolyte, respectively. Discharge–charge measurements were carried out in a voltage window of 1.7–2.8 V at different current densities at room temperature with a LAND battery tester. The capacities were calculated based on the mass of sulfur in the cathode materials. Electrochemical impedance spectroscopy (EIS) of fresh and cycled (for 10 cycles) batteries in the charged state was conducted on an SP-300 electrochemical workstation (Bio-Logic Science Instrument).
Li2S6 adsorption testing of SP, SP/PVDF, SP/PEDOT:PSS, and SP/PEDOT:PSS-Mg2+
Li2S6 was synthesized by reacting elemental sulfur with Li2S in a stoichiometric ratio of 5:1 in DOL/DME (v/v, 1/1) in a sealed vessel at 80 °C.43 SP/PVDF, SP/PEDOT:PSS and SP/PEDOT:PSS-Mg2+ composite slurries were prepared as described above for the fabrication of the cathode composites, where the SP/polymer weight ratio was kept at 2:1. The slurries were dried in air and then under vacuum at 50 °C for 12 h. After drying, the composites were ground into fine powders in an agate mortar. Next, SP/PVDF composite (10 mg SP + 5 mg PVDF), SP/PEDOT:PSS (10 mg SP + 5 mg PEDOT:PSS), and SP/PEDOT:PSS-Mg2+ composite (10 mg SP + 5 mg PEDOT:PSS-Mg2+) were added to 3.5 mL of 0.5 mM Li2S6 solution, respectively. The mixtures were well mixed by shaking the vials. After being kept for 24 h at room temperature, the clear supernatants were taken for the UV-vis absorption measurements. As a comparison, the Li2S6 adsorption of a same amount of SP (10 mg) was also tested.
Conclusions
In summary, a multi-functional PEDOT:PSS-Mg2+ binder formed by cross-linking PEDOT:PSS with Mg2+ was developed for the sulfur cathode in Li–S batteries. This new binder has high electrical conductivity, a robust 3-D network structure achieved by the cross-linking of PSS with Mg2+ ions, and a strong binding ability toward lithium polysulfides due to the strong interaction between the oxygen atoms in PEDOT and lithium polysulfides. These functionalities can increase the conduction and charge transfer reactions, cushion the drastic volume change during discharge/charge cycling, and trap the soluble lithium polysulfides in the cathode. The Li–S battery with a cathode using this new binder exhibited an initial capacity of 1097 mA h g−1 and capacity retention of 74% over 250 cycles at 0.5C, which are significant improvements compared with the Li–S battery using a conventional PVDF binder. Moreover, the preparation of the cathode slurry and the subsequent cathode fabrication using the PEDOT:PSS-Mg2+ binder uses water present in the PEDOT:PSS dispersion as the only dispersing solvent, which eliminates the use of any organic solvent, making the fabrication of Li–S batteries more environmentally friendly. Therefore, this study demonstrated that the cross-linked PEDOT:PSS-Mg2+ is a very promising new binder for high-performance Li–S batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council (NSERC) of Canada (Discovery Grants # RGPIN-2016-04366), the Link Project of the National Natural Science Foundation of China and Guangdong Province (Grant No. U1301244), and the National Natural Science Foundation of China (Grant No. 51573215, 21506260).References
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed.
- Y. Yang, G. Zheng and Y. Cui, Chem. Soc. Rev., 2013, 42, 3018 RSC.
- S. Evers and L. F. Nazar, Acc. Chem. Res., 2013, 46, 1135–1143 CrossRef PubMed.
- A. Rosenman, E. Markevich, G. Salitra, D. Aurbach, A. Garsuch and F. F. Chesneau, Adv. Energy Mater., 2015, 5, 1500212 CrossRef.
- H.-J. Peng, J.-Q. Huang, X.-B. Cheng and Q. Zhang, Adv. Energy Mater., 2017, 1700260 CrossRef.
- D. S. Jung, T. H. Hwang, J. H. Lee, H. Y. Koo, R. A. Shakoor, R. Kahraman, Y. N. Jo, M.-S. Park and J. W. Choi, Nano Lett., 2014, 14, 4418–4425 CrossRef PubMed.
- W. Hua, Z. Yang, H. Nie, Z. Li, J. Yang, Z. Guo, C. Ruan, X. Chen and S. Huang, ACS Nano, 2017, 11, 2209–2218 CrossRef PubMed.
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969 CrossRef.
- A. Manthiram, Y. Fu and Y.-S. Su, Acc. Chem. Res., 2013, 46, 1125–1134 CrossRef PubMed.
- Q. Pang, D. Kundu, M. Cuisinier and L. F. Nazar, Nat. Commun., 2014, 5, 4759 CrossRef PubMed.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef PubMed.
- S. Chen, X. Huang, H. Liu, B. Sun, W. Yeoh, K. Li, J. Zhang and G. Wang, Adv. Energy Mater., 2014, 4, 1301761 CrossRef.
- Z. Li, J. Zhang, B. Guan, D. Wang, L.-M. Liu and X. W. Lou, Nat. Commun., 2016, 7, 13065 CrossRef PubMed.
- Y. Zhao, W. Wu, J. Li, Z. Xu and L. Guan, Adv. Mater., 2014, 26, 5113–5118 CrossRef PubMed.
- J. Song, M. L. Gordin, T. Xu, S. Chen, Z. Yu, H. Sohn, J. Lu, Y. Ren, Y. Duan and D. Wang, Angew. Chem., Int. Ed., 2015, 54, 4325–4329 CrossRef PubMed.
- L. Ji, M. Rao, S. Aloni, L. Wang, E. J. Cairns and Y. Zhang, Energy Environ. Sci., 2011, 4, 5053 Search PubMed.
- G. Zheng, Q. Zhang, J. J. Cha, Y. Yang, W. Li, Z. W. Seh and Y. Cui, Nano Lett., 2013, 13, 1265–1270 CrossRef PubMed.
- H. Wang, Y. Yang, Y. Liang, J. T. Robinson, Y. Li, A. Jackson, Y. Cui and H. Dai, Nano Lett., 2011, 11, 2644–2647 CrossRef PubMed.
- G. Zhou, S. Pei, L. Li, D.-W. Wang, S. Wang, K. Huang, L.-C. Yin, F. Li and H.-M. Cheng, Adv. Mater., 2014, 26, 625–631 CrossRef PubMed.
- J. Liu, W. Li, L. Duan, X. Li, L. Ji, Z. Geng, K. Huang, L. Lu, L. Zhou, Z. Liu, W. Chen, L. Liu, S. Feng and Y. Zhang, Nano Lett., 2015, 15, 5137–5142 CrossRef PubMed.
- F. Wu, J. T. Lee, E. Zhao, B. Zhang and G. Yushin, ACS Nano, 2016, 10, 1333–1340 CrossRef PubMed.
- L. Ji, M. Rao, H. Zheng, L. Zhang, Y. Li, W. Duan, J. Guo, E. J. Cairns and Y. Zhang, J. Am. Chem. Soc., 2011, 133, 18522–18525 CrossRef PubMed.
- W. Kang, N. Deng, J. Ju, Q. Li, D. Wu, X. Ma, L. Li, M. Naebe and B. Cheng, Nanoscale, 2016, 8, 16541–16588 RSC.
- B. D. McCloskey, J. Phys. Chem. Lett., 2015, 6, 4581–4588 CrossRef PubMed.
- J. Gao and H. D. Abruña, J. Phys. Chem. Lett., 2014, 5, 882–885 CrossRef PubMed.
- G. Li, M. Ling, Y. Ye, Z. Li, J. Guo, Y. Yao, J. Zhu, Z. Lin and S. Zhang, Adv. Energy Mater., 2015, 5, 1500878 CrossRef.
- P. Bhattacharya, M. I. Nandasiri, D. Lv, A. M. Schwarz, J. T. Darsell, W. A. Henderson, D. A. Tomalia, J. Liu, J.-G. Zhang and J. Xiao, Nano Energy, 2016, 19, 176–186 CrossRef.
- W. Chen, T. Qian, J. Xiong, N. Xu, X. Liu, J. Liu, J. Zhou, X. Shen, T. Yang, Y. Chen and C. Yan, Adv. Mater., 2017, 29, 1605160 CrossRef PubMed.
- G. Xu, Q. Yan, A. Kushima, X. Zhang, J. Pan and J. Li, Nano Energy, 2017, 31, 568–574 CrossRef.
- Z. Wang, Y. Chen, V. Battaglia and G. Liu, J. Mater. Res., 2014, 29, 1027–1033 CrossRef.
- J. Pan, G. Xu, B. Ding, Z. Chang, A. Wang, H. Dou and X. Zhang, RSC Adv., 2016, 6, 40650–40655 RSC.
- G. Ai, Y. Dai, Y. Ye, W. Mao, Z. Wang, H. Zhao, Y. Chen, J. Zhu, Y. Fu, V. Battaglia, J. Guo, V. Srinivasan and G. Liu, Nano Energy, 2015, 16, 28–37 CrossRef.
- L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef.
- B. J. Worfolk, S. C. Andrews, S. Park, J. Reinspach, N. Liu, M. F. Toney, S. C. B. Mannsfeld and Z. Bao, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14138–14143 CrossRef PubMed.
- J. Rivnay, S. Inal, B. A. Collins, M. Sessolo, E. Stavrinidou, X. Strakosas, C. Tassone, D. M. Delongchamp and G. G. Malliaras, Nat. Commun., 2016, 7, 11287 CrossRef PubMed.
- W. Li, Q. Zhang, G. Zheng, Z. W. Seh, H. Yao and Y. Cui, Nano Lett., 2013, 13, 5534–5540 CrossRef PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2011, 11, 172 CrossRef.
- M. Barghamadi, A. Kapoor and C. Wen, J. Electrochem. Soc., 2013, 160, A1256–A1263 CrossRef.
- Z. Lin and C. Liang, J. Mater. Chem. A, 2015, 3, 936–958 Search PubMed.
- A. Kawase, S. Shirai, Y. Yamoto, R. Arakawa and T. Takata, Phys. Chem. Chem. Phys., 2014, 16, 9344–9350 RSC.
- R. Fang, S. Zhao, Z. Sun, D.-W. Wang, H.-M. Cheng and F. Li, Adv. Mater., 2017, 1606823 CrossRef PubMed.
- D. Zheng, D. Liu, J. B. Harris, T. Ding, J. Si, S. Andrew, D. Qu, X.-Q. Yang and D. Qu, ACS Appl. Mater. Interfaces, 2017, 9, 4326–4332 Search PubMed.
- Z. Sun, J. Zhang, L. Yin, G. Hu, R. Fang, H. M. Cheng and F. Li, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed.
- A. Kawase, S. Shirai, Y. Yamoto, R. Arakawa and T. Takata, Phys. Chem. Chem. Phys., 2014, 16, 9344–9350 RSC.
- W. Chen, T. Qian, J. Xiong, N. Xu, X. Liu, J. Liu, J. Zhou, X. Shen, T. Yang, Y. Chen and C. Yan, Adv. Mater., 2017, 29, 1605160 CrossRef PubMed.
- W. Li, Q. Zhang, G. Zheng, Z. W. Seh, H. Yao and Y. Cui, Nano Lett., 2013, 13, 5534–5540 CrossRef PubMed.
- Z. Zhang, L.-L. Kong, S. Liu, G.-R. Li and X.-P. Gao, Adv. Energy Mater., 2017, 7, 1602543 CrossRef.
- F. Y. Fan, W. H. Woodford, Z. Li, N. Baram, K. C. Smith, A. Helal, G. H. McKinley, W. C. Carter and Y.-M. Chiang, Nano Lett., 2014, 14, 2210–2218 CrossRef PubMed.
- Z. Deng, Z. Zhang, Y. Lai, J. Liu, J. Li and Y. Liu, J. Electrochem. Soc., 2013, 160, A553–A558 CrossRef.
- H. Chen, Q. Zou, Z. Liang, H. Liu, Q. Li and Y.-C. Lu, Nat. Commun., 2015, 6, 5877 CrossRef PubMed.
- Z. W. Seh, Q. Zhang, W. Li, G. Zheng, H. Yao and Y. Cui, Chem. Sci., 2013, 4, 3673 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00167g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |