
Highly dispersed and disordered nickel–iron layered hydroxides and sulphides: robust and high-activity water oxidation catalysts†
Manjunath
Chatti
 ab,
Alexey M.
Glushenkov
c,
Thomas
Gengenbach
d,
Gregory P.
Knowles
a,
Tiago C.
Mendes
a,
Amanda V.
Ellis
c,
Leone
Spiccia
ab,
Rosalie K.
Hocking
e and
Alexandr N.
Simonov
*ab
ab,
Alexey M.
Glushenkov
c,
Thomas
Gengenbach
d,
Gregory P.
Knowles
a,
Tiago C.
Mendes
a,
Amanda V.
Ellis
c,
Leone
Spiccia
ab,
Rosalie K.
Hocking
e and
Alexandr N.
Simonov
*ab
aSchool of Chemistry, Monash University, Victoria 3800, Australia. E-mail: alexandr.simonov@monash.edu
bARC Centre of Excellence for Electromaterials Science, Monash University, Victoria 3800, Australia
cDepartment of Chemical Engineering, The University of Melbourne, Parkville, Victoria 3010, Australia
dCommonwealth Scientific and Industrial Research Organisation Manufacturing, Clayton, Victoria 3168, Australia
eDepartment of Chemistry and Biotechnology, Swinburne University of Technology, Victoria 3122, Australia
First published on 25th April 2018
Abstract
The present work introduces a rapid low-temperature microwave-assisted synthesis of nickel(iron) layered hydroxides and sulphides that exhibit robust catalytic activity for electrooxidation of alkaline water – the most feasible source of electrons for any renewable fuel synthesis. The procedures require not more than an hour to complete at 120–150 °C with quantitative yields of: (i) few-atomic-layers thick porous sheets of Ni0.75Fe0.25(OH)2+x with surface area ABET = 149 m2 g−1, and (ii) interconnected Ni0.75Fe0.25S2+y particles of few nanometers in size covered with a thin oxide/hydroxide layer having ABET = 87 m2 g−1. These and other morphological and structural features of the materials were inferred from XRD, XPS, Ni- and Fe-edge EXAFS/XANES, TEM/SAED, EDX mapping, SEM, N2 adsorption–desorption, and electrochemical techniques. At lower loadings on the electrode surface (≤0.01 mg cm−2), the specific activity for water (1 M KOH) electrooxidation at 0.3 V overpotential is 210 A g−1 for Ni0.75Fe0.25(OH)2+x, and 384 A g−1 for Ni0.75Fe0.25S2+y, which excels the performance of the best-performing analogues. The enhanced electrocatalytic activity of sulphides over hydroxides is defined by the better electrical conductivity and different nature of the electrochemically active surface species. At higher loadings, the activity of the microwave-synthesised NiFe catalysts is found to be partially limited by agglomeration, though still high enough to enable the water oxidation rate of 10 mA cmgeom−2 at overpotentials of only 0.270 ± 0.005 (flat support) and 0.21 V (foam support) with Ni0.75Fe0.25S2+y. The developed methods offer a new facile strategy for the creation of high-performing multicomponent catalysts.
Introduction
The concept of renewable fuels based on the conversion of solar, hydro or wind energy into electricity and the use of this essentially perpetual electrical power to drive the electrosynthesis of energy-rich compounds from the abundant feedstock, e.g. water or dinitrogen, is considered among the few practical sustainable solutions to support the world energy sector of the future. By definition, a reductive fuel-forming process in this scheme must be coupled to an electron-donating half-reaction, viz. oxidation of an abundant compound that does not impact the environment. In this context, the water oxidation reaction, often referred to as the oxygen evolution reaction (OER: 4OH− ⇄ O2 + 2H2O + 4e−), is currently seen as the only globally viable option and is therefore central to the overall concept of renewable fuels. When the OER is combined with the hydrogen evolution reaction (HER: 2H2O + 2e− ⇄ H2 + 2OH−) on two spatially separated electrodes, electrochemical decomposition of water into molecular hydrogen and oxygen (2H2O ⇄ 2H2 + O2), a process that was discovered more than two centuries ago,1–3 is achieved.Production of dihydrogen via electrolysis of alkaline water is an established technology, which, however, still requires improvements in efficiency to be implemented on a scale that would satisfy the worldwide need.4,5 Both HER and OER require catalytically active electrodes to proceed at sufficiently high rates, and improving relevant catalysts is an obvious path towards better performing electrolysers. In this context, the multi-step OER holds the key, and its sluggish kinetics remains one of the major factors that limit the productivity of the entire process.6,7 Many reasonably efficient catalytic systems based on transition metals including prohibitively scarce Ir and Ru oxides have been reported (Table S1†). Of these, materials where nickel(iron) oxides/oxyhydroxides fulfil the catalytic function remain one of the most efficient OER catalysts for operation at high pH and have been used as anode electrocatalysts for alkaline water splitting long before the recent increase of research interest in the field8–10 (Tables 1 and S1†).
One class of the intensively studied OER catalysts are layered hydroxides of transition metals (Ru, Ir, Co, Ni, Fe, etc.) and their combinations for applications in both electrocatalytic and photocatalytic water splitting.21–29 The best-performing noble metal free systems of this type are nickel–iron layered double hydroxides (LDHs), which were first introduced by Dai and colleagues.13 Later, Müller and co-workers demonstrated that the catalytic activity of the NiFe LDHs synthesised by a pulsed-laser ablation method can be significantly improved by tuning the crystallinity of the catalyst and the nature of interlayer anions.12,30 Further investigations identified that the performance of these materials is limited by their low intrinsic electrical conductivity,13,16 which opened up an opportunity to improve the activity further. For example, Hu et al. exfoliated sheets of NiFe LDHs to overcome the charge-transfer limitations seen in agglomerated multi-layered stacks and thereby achieved enhanced catalytic activity.11 Another solution to the conductivity issue, which was implemented in the initial report13 and was further exploited by other groups, is immobilisation of LDHs, in the form of dispersed sheets, on a high-surface-area substrate.13,15,31,32 Although very impressive electrocatalytic performance has been achieved using carbon-based supports for NiFe-LDH catalysts (Table 1), these materials do not demonstrate satisfactory stability and slowly degrade.13,14 This is in the first place due to the thermodynamic instability of carbon at very positive potentials.33,34 Interestingly, persistent deterioration in the performance of unsupported nickel–iron hydroxides has also been reported.11,13,35 Possible reasons include trivial exfoliation of materials from the surface of glassy carbon electrodes used in these studies, which inevitably undergo oxidative degradation during long-term tests, or electrochemically induced changes to the catalysts.35 Conversely, the use of high surface area copper36 or nickel35 to immobilise NiFe LDHs was reported to partially alleviate both instability and conductivity problems.
| Catalyst (loading/mg cm−2, electrode) | Synthesis method | A BET/m2 g−1 | η 10/Vc | i cat /A g−1 | j BET /mA cm−2 | Ref. | ||
|---|---|---|---|---|---|---|---|---|
| 0.25 V | 0.30 V | 0.25 V | 0.30 V | |||||
| a All data were reported corrected for ohmic losses, except for those in ref. 15. b Chalcogenides are transformed into corresponding (oxy)hydroxides under the OER conditions, either at the surface or completely. c Overpotential required to achieve 10 mA cm−2 (normalised to the geometric surface area) derived from quasi-steady-state data (if available) or voltammograms. d Current density normalised to the catalyst mass (i) or BET surface area (j) at η = 0.25 and 0.30 V. e Glassy carbon. f Not available. g Estimated from Cdl provided in relevant publication and the ratio of Cdl to BET external surface area obtained herein. h Reduced graphene oxide. i Value in m2 cmgeom−2. j External surface area. k High temperature. | ||||||||
| Ni0.75Fe0.25-LDH (0.07, GCe) | Hydrothermal, exfoliation | n.a.f | 0.310 | 7 | 114 | n.a. | n.a. | 11 |
| Ni0.78Fe0.22-LDH (0.44, HOPG) | Pulsed laser ablation | 221 | 0.280 | 7.2 | 72 | 0.003 | 0.03 | 12 |
| Ni0.83Fe0.17-LDH (0.20, GC) | Multistep solvothermal | n.a. | 0.320 | 10 | 35 | n.a. | n.a. | 13 |
| Ni0.83Fe0.17-LDH/CNT (0.25, carbon paper) | Multistep solvothermal | n.a. | 0.230 | 80 | n.a. | n.a. | n.a. | 13 |
| Ni0.7Fe0.3-LDH (0.25, GC) | Precipitation, exfoliation | n.a. (325)g | 0.320 | 8 | 25 | n.a. (0.002)g | n.a. (0.007)g | 14 |
| Ni0.7Fe0.3-LDH/rGOh (0.25, GC) | Precipitation, exfoliation | n.a. (520)g | 0.210 | 200 | n.a. | n.a. (0.02)g | n.a | 14 |
| Ni0.5Fe0.5-LDH (1, Nifoam) | Solvothermal | 0.24i | 0.240 | 12 | 32 | 0.005 | 0.01 | 15 |
| Ni0.75Fe0.25(OH)2+x | Microwave | 149 (91)j | This work | |||||
| (0.01, GC) | 0.391 | 8.4 | 210 | 0.0092 | 0.23 | |||
| (0.07, GC) | 0.336 | 7.6 | 71 | 0.0083 | 0.078 | |||
| (0.21, GC) | 0.282 | 14 | 74 | 0.015 | 0.081 | |||
| Ni0.8Fe0.2S2 (0.18, GC) | Hydrothermal, HTk sulphidation | n.a. | 0.230 | 100 | 222 | n.a. | n.a. (1.6)g | 16 |
| Ni0.5Fe0.5Sx/graphene (0.25, GC) | Coprecipitation, hydrothermal | n.a. (180)g | 0.286 | 20 | 72 | n.a. (0.01)g | n.a. (0.04)g | 17 |
| Ni0.7Fe0.3S2 (3, Nifoam) | Hydrothermal, HT sulphidation | 11 | 0.198 | 20 | 40 | 0.17 | 0.37 | 18 |
| Fe–Ni3S2 (n.a., FeNifoam) | Solvothermal | 0.02i | 0.282 | n.a. | n.a. | 0.03 | 0.1 | 19 |
| NixFe1−xSe2 (4.1, Nifoam) | Solvothermal | 109 | 0.195 | 58 | n.a. | 0.06 | n.a. | 20 |
| Ni0.75Fe0.25S2+y | Microwave | 87 | This work | |||||
| (0.01, GC) | 0.333 | 104 | 384 | 0.12 | 0.44 | |||
| (0.14, GC) | 0.275 | 28 | 165 | 0.032 | 0.19 | |||
| (0.28, GC) | 0.270 | 16 | 98 | 0.018 | 0.11 | |||
| ±0.005 | ±2 | ±10 | ±0.003 | ±0.02 | ||||
Another class of OER catalysts that are now receiving increased attention are nickel, iron and other transition metal chalcogenides (Table 1), many of which were initially devised as electrocatalysts for hydrogen evolution.37 Under the harsh conditions of a functioning water-splitting anode, the catalytically active species are still metal (oxy)hydroxides generated via electrooxidation. This transformation has been found to propagate deep into the bulk for selenide catalysts,20 but is typically limited to the surface layer for sulphides,16 which are of particular interest herein. The high electrical conductivity of the nickel–iron sulphide core sustains efficient charge transfer towards a thin layer of a true catalyst on the surface of the particles and thereby enables very high catalytic activity for water oxidation. Electronic effects of underlying sulphides on the surface (oxy)hydroxide layer might also contribute to the improvements in catalytic performance, though a robust universal evidence for this is yet to be obtained.
One technological limitation that applies to both NiFe-LDH and sulphides are laborious and resource-intense synthesis procedures required to produce these materials. Synthesis of the most OER-active NiFe-LDH catalysts reported in the literature involved long-term solvothermal and other treatments, typically on the timescale of days and with the requirements of very careful control over the reaction environment.13,31,38 Thus prepared NiFe-LDHs were further used as precursors for nanostructured sulphides that were obtained via either a high-temperature gas-phase process16,18,39 or a slow solution-phase solvothermal reaction.17,40 An alternative, potentially cheaper and easier to scale method is a microwave-assisted solution-phase synthesis, which has not been applied before to produce NiFe-based catalysts for water electrooxidation and is the subject of the present work.
Synthesis of inorganic nanostructured materials in solutions under microwave irradiation is a fast-growing area of research.41 Interaction of microwaves with reaction mixtures, mainly via solvents, allows for rapid and homogeneous heating, which provides shorter reaction times, enhanced throughput and decreased energy consumption as compared to other methods, such as solvothermal.42 In some cases, the localised hotspot heating ability of microwaves enables formation of nanostructured materials with unique morphology and improved catalytic and other physicochemical properties.43 Herein, a simple microwave-assisted solution-phase synthetic route is introduced to produce nickel and nickel–iron hydroxides and disulphides, and the electrocatalytic properties of the resulting materials for water oxidation are scrutinised.
Experimental
Materials
All reagents were of analytical grade and were used as received from suppliers (Sigma Aldrich, Alfa Aesar, and Merck) without further purification. Ni foam (>99.99%; thickness 1.6 mm; surface density 346 g m−2; porosity ≥95%, 80–110 pores per inch) was purchased from Marketech International. Ultrapure water (Synergy Milli-Q; 18.2 MΩ cm at 24 ± 1 °C) was used throughout all experiments.Synthesis procedures
All microwave-assisted syntheses were undertaken in 30 mL Pyrex vials using a CEM Discover SP microwave system with Activent technology. The microwave power was automatically adjusted to maintain the required temperature by operating the system in a standard mode.Synthesis of Ni0.9Fe0.1 LDHs was initially performed under hydrothermal conditions by following the experimental protocol reported by Long et al.40 Briefly, NiCl2 (1.45 mL, 1 M) Fe(NO3)3·9H2O (0.145 mL, 1 M), urea (5.6 mL, 0.5 M) and trisodium citrate (2 mL, 0.01 M) aqueous solutions were added to water (5 mL) and heated at 150 °C in a sealed 25 mL Teflon vessel for 24 h. The produced material was washed with water and ethanol three times and dried in an oven at 70 °C overnight.
Microwave-assisted syntheses of layered hydroxides were performed using water as a solvent. Reaction mixtures for the synthesis of Ni(OH)2 and Ni0.75Fe0.25(OH)2+x layered hydroxides were prepared by dissolving Ni(CH3COO)2·4H2O (1.0 and 0.75 mmol, respectively) with Fe(NO3)3·9H2O (0 and 0.25 mmol, respectively) in water (10 mL), adding urea (4 mmol) and sonicating (Elmasonic S300H bath with an operating power of 1500 W) for 5 minutes at ambient temperature. The obtained clear precursor solutions were transferred into microwave vessels that were sealed with septum caps. Reactions were conducted at a constant temperature of 120 or 180 °C for 1 h and then the solutions were allowed to cool to ambient temperature naturally. The obtained materials were collected by centrifugation (8000 rpm; 20 min), washed with ethanol 5 times and dried in an oven at 70 °C overnight.
Solvothermal sulphidation was attempted using the procedure reported by Long et al.40 NiFe LDH precursor (80 mg) was dispersed into ethanol (40 mL), followed by the addition of thioacetamide (0.1125 g). The mixture was then transferred into a 50 mL Teflon-lined stainless steel autoclave and was subsequently heated at 120 °C for 6 h. The product was collected by centrifugation and washed with ethanol at least 3 times.
Microwave-assisted sulphidation of layered hydroxides was undertaken using absolute ethanol as a solvent and thioacetamide as a source of sulphur. In a typical synthesis, thioacetamide (64 mg) was dissolved in absolute ethanol (15 mL), an LDH precursor (50 mg) was added, and the mixture was sonicated for 30 min. The microwave-assisted synthesis was completed as described above at 120, 150 or 180 °C for 30 min. The obtained products were centrifuged (8000 rpm, 20 min), washed three times with ethanol and dried at 70 °C overnight.
Selected microwave-synthesised Ni0.75Fe0.25S2 samples were additionally annealed in a tube furnace in high purity N2 at 350, 450 or 600 °C. The set temperature was reached with a sequential increment of 5° min−1 and maintained for 2 h. The samples were exposed to air after naturally cooling down to ambient temperature.
Physical characterisation
Electrochemical experiments
All electrochemical measurements were performed using a Bio-Logic VSP electrochemical workstation in a three-electrode configuration. Either 1 M KOH (assumed pH = 13.6) or 0.5 M H2SO4 (measured pH = 0.3) was employed as the electrolyte solution, which was saturated with high purity Ar for experiments that involved measurements at potentials more negative than 1.0 V vs. the reversible hydrogen electrode (RHE). Measurements at 60 °C were undertaken with the cell fully immersed into a thermostatted water bath (Thermoline Scientific), and the temperature of the solution in the working-electrode compartment was monitored using a conventional high-precision thermometer.Working electrodes were prepared using either glassy carbon (GC; ϕ 3 mm, BAS) or nickel foam (Nifoam; Marketech International) substrates. Prior to use, glassy carbon electrodes were polished to mirror finish using 1.0, 0.3 and 0.05 μm alumina powders (Alfa Aesar), thoroughly washed with water followed by sonication in a water–ethanol mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 vol) for 30 min. Nifoam electrodes were cleaned by sonicating in concentrated HCl (ca. 32 wt%) for 10 min, copiously washing with water and sonicating in acetone, water, and absolute ethanol for 15 min in each solvent. Catalyst inks were prepared by dispersing 1–5 mg of a material in 1 mL of water–isopropanol (3:1 vol) mixture containing 30 μL of 5 wt% Nafion (ethanol solution) by sonication for 30 min. To obtain dispersions with lower concentrations, the 1 mg mL−1 ink was diluted with the same solvent system and sonicated again for 30 min. Freshly dispersed catalyst inks were drop-cast in small aliquots onto clean and dry GC (loading 0.004–0.35 mg cm−2) or Nifoam (loading 1–3 mg cm−2) substrates and dried in an oven at 70 °C for 2 hours prior to testing.
:3 vol) for 30 min. Nifoam electrodes were cleaned by sonicating in concentrated HCl (ca. 32 wt%) for 10 min, copiously washing with water and sonicating in acetone, water, and absolute ethanol for 15 min in each solvent. Catalyst inks were prepared by dispersing 1–5 mg of a material in 1 mL of water–isopropanol (3:1 vol) mixture containing 30 μL of 5 wt% Nafion (ethanol solution) by sonication for 30 min. To obtain dispersions with lower concentrations, the 1 mg mL−1 ink was diluted with the same solvent system and sonicated again for 30 min. Freshly dispersed catalyst inks were drop-cast in small aliquots onto clean and dry GC (loading 0.004–0.35 mg cm−2) or Nifoam (loading 1–3 mg cm−2) substrates and dried in an oven at 70 °C for 2 hours prior to testing.
High surface area Pt wire (A&E Metals) was used as an auxiliary electrode, which was confined in a separate compartment separated with a low-porosity (P4) glass frit. An Ag|AgCl|KCl(sat.) (CHI) reference electrode was also confined in a glass tube with a P4 glass frit, the tip of which was positioned within a few mm of the working electrode surface. Potentials were recalculated to the RHE scale using the relationship, E/V vs. RHE = E/V vs. Ag|AgCl|KCl(sat.) + 0.059 pH + 0.197 V. Uncompensated resistance (Ru) was derived from the electrochemical impedance spectra (frequency range 200 KHz to 100 mHz; amplitude 0.01 V) recorded at potentials where no significant faradaic processes occur. Typical Ru values were within the range 13–18 Ω and below 1 Ω for the experiments with GC and Nifoam electrodes, respectively. Where specifically mentioned, the reported potentials were manually post-corrected for the ohmic losses by subtracting the I(t)Ru product.
Results and discussion
Synthesis
Thermally induced hydrolysis of urea was used by Costantino et al.45 as a convenient and efficient method to produce mixed hydroxide materials during the hydrothermal reaction. Later, Liu and co-workers46 extended this technique by involving trisodium citrate as a chelating agent for the synthesis of highly crystalline NiFe-LDH. The method was further refined and optimised by different groups and used for the solvothermal synthesis of various types of NiFe-LDH electrocatalysts for water splitting.11,13,18,40 Herein, the formation of Ni0.9Fe0.1 hydroxides in the presence of urea and citrate was demonstrated to occur under microwave conditions at 180 °C (Fig. S1†), within a much shorter reaction time of 1 h compared to 24 h at 150 °C typically required for the solvothermal approach (Table S2†). However, when drop-cast onto a flat electrode support, the films of these microwave-synthesised materials displayed poor mechanical properties and were prone to cracking and exfoliation. In contrast, Ni0.9Fe0.1 LDHs that were synthesised hydrothermally following the protocol from the work of Long et al.40 could be easily deposited as stable layers. On this basis, our initial attempts to produce sulphides were undertaken using the latter material as a precursor (Table S2†). Surprisingly, we were unable to reproduce the results from ref. 40 for the solvothermal conversion of Ni0.9Fe0.1 LDH into the corresponding sulphides using thioacetamide as a source of sulphur (Fig. S1†). This observation emphasises the very high sensitivity of the solvothermal synthetic strategy to even subtle differences in the reaction environment, which we aimed to avoid by strictly following the procedures reported by Long et al.40 Conversely, complete sulphidation was reproducibly achieved herein after only 30 min of microwave treatment of hydrothermally synthesised Ni0.9Fe0.1 hydroxides with thioacetamide in ethanol at 150 °C (Fig. S1†), but again the material formed could not be deposited in the form of stable and homogeneous layers onto an electrode.Eventually, we synthesised Ni and Ni0.75Fe0.25 hydroxides via microwave irradiation of citrate-free aqueous solutions and nickel(II) acetate as the precursor (Fig. 1a; Table S2†). The synthesis takes 1 h and requires a temperature of only 120 °C. The powders obtained form uniform and robust layers upon drop-casting. Subsequent 30 min microwave treatment of these materials with thioacetamide in ethanol at 120–180 °C produces corresponding sulphides (Fig. 1b; Table S2†). The product yields were at least 95%, based on changes in the solution UV-vis absorption during hydroxide synthesis (Fig. S2†) and XRD data (Fig. 1). The overall synthetic procedure is summarised in Scheme 1.
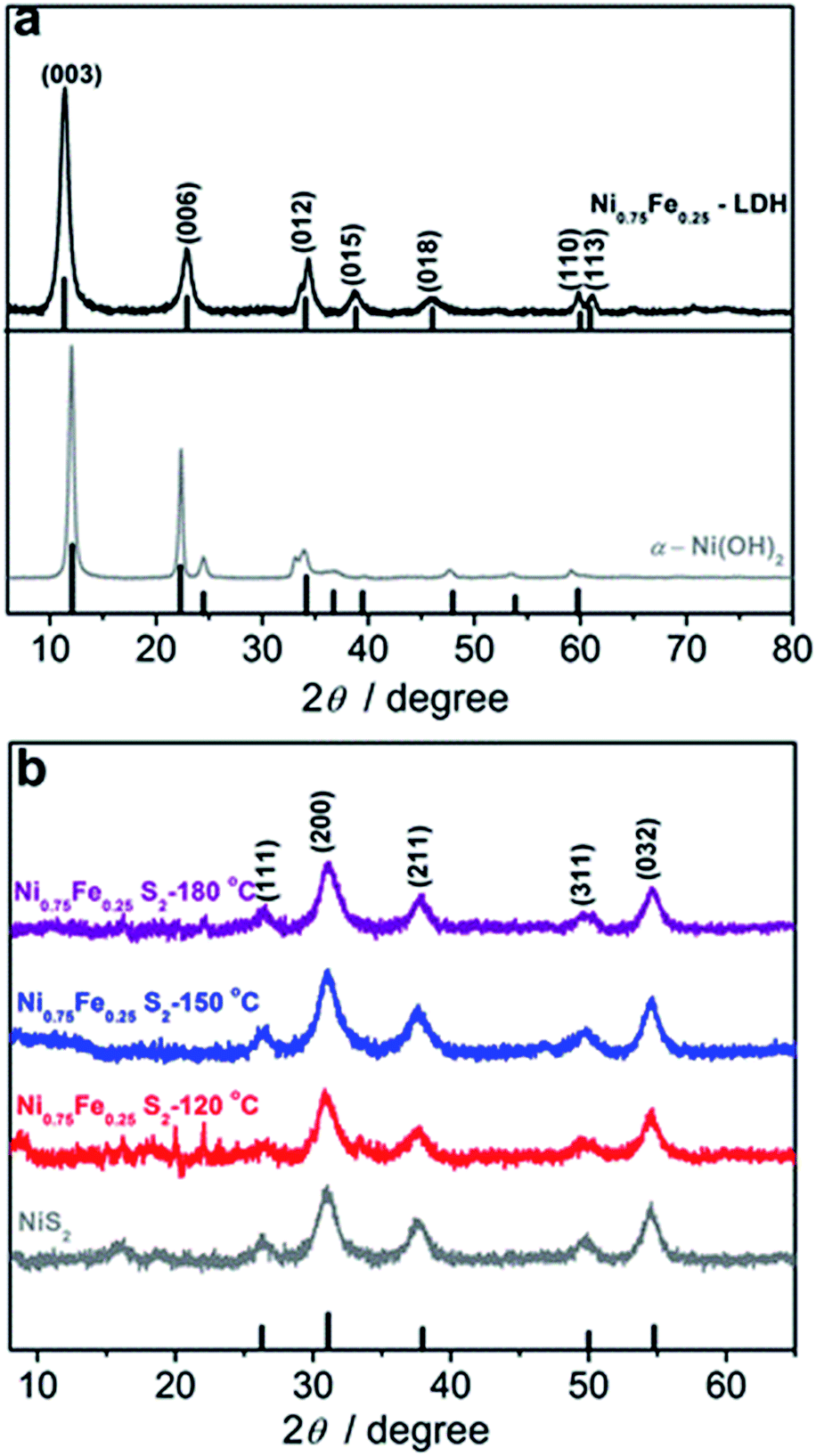 | ||
| Fig. 1 X-ray diffraction patterns of microwave-synthesised (a, grey) Ni layered hydroxide, (a, black) Ni0.75Fe0.25 layered hydroxide, (b, grey) NiS2 and (b, red, blue and purple) Ni0.75Fe0.25S2. The temperature during synthesis was maintained at 120 °C for hydroxides, 150 °C for NiS2, and 120 (red), 150 (blue) and 180 °C (purple) for Ni0.75Fe0.25S2. Vertical lines indicate the tabulated positions and relative intensities for (a, bottom) α-Ni(OH)2 (PDF#38-0715), (a, top) Ni0.75Fe0.25 LDH (JCPDS# 38-0715), and (b) NiS2 (PDF#11-0099). | ||
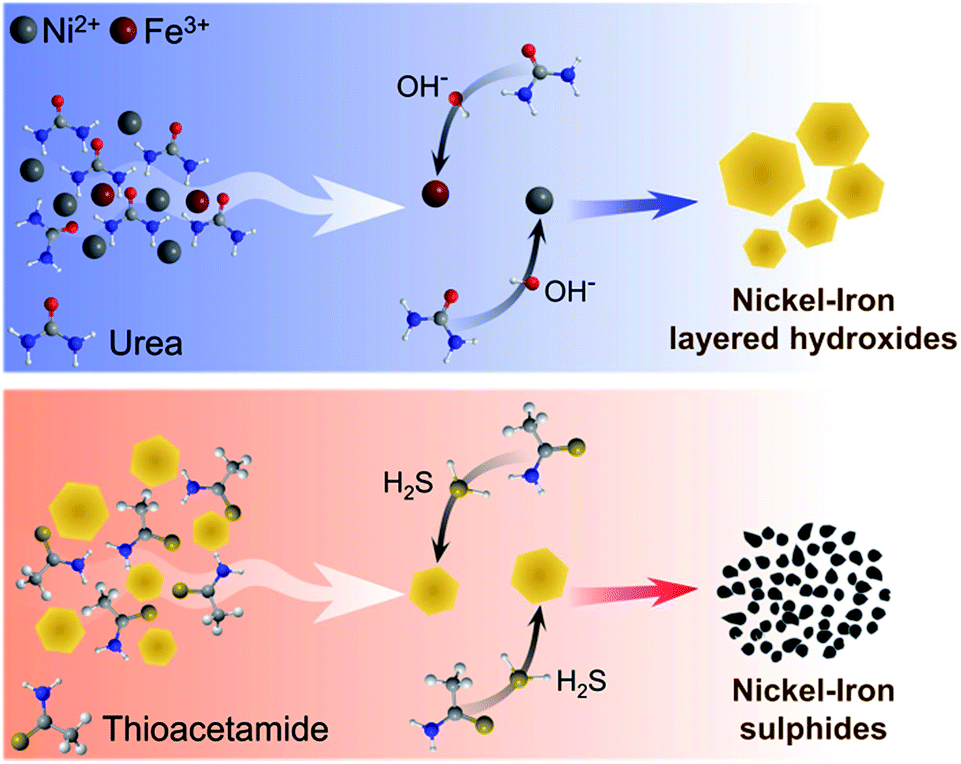 | ||
| Scheme 1 A plausible mechanism of the microwave-assisted formation of nickel(iron) layered hydroxides and sulphides. | ||
Characterisation of hydroxide materials
The juxtaposition of X-ray diffractograms obtained for the microwave-synthesised Ni and Ni0.75Fe0.25 hydroxides and relevant tabulated data confirms the formation of target hexagonal phases (Fig. 1a). The absence of detectable peaks corresponding to β-Ni(OH)2 in the XRD pattern for Ni0.75Fe0.25 hydroxide suggests that iron and nickel occupy the same sites in the crystal lattice of the isomorphous structure.13 The high relative intensity of the (003) and (006) diffraction peaks for the produced α-Ni(OH)2 and Ni0.75Fe0.25(OH)2+x materials as compared to tabulated diffraction data indicates increased periodicity in these crystallographic directions, which is typical for LDHs. On the other hand, the intensity of these peaks found herein is significantly lower than that reported for nickel–iron analogues produced using different methods,11,13,40 with the exception of essentially completely exfoliated materials.11 This observation suggests that microwave synthesis facilitates exfoliation of hydroxide sheets, as corroborated by microscopic data presented later in the text. Mean crystallite sizes (dXRD) derived from the width at half maximum of (003) and (006) diffraction peaks were 17–22 nm for α-Ni(OH)2, and ∼9 nm for Ni0.75Fe0.25(OH)2+x.Analysis of the Ni and Fe-edge X-ray absorption near-edge structure (XANES) data obtained for Ni0.75Fe0.25-LDH and comparison with reference spectra reveal that NiII and FeIII are the main oxidation states of nickel and iron in this material (Fig. S3a†). Extended X-ray absorption fine structure (EXAFS) data are consistent with the layered hydroxide structure (Fig. S3b†), in agreement with the XRD results. X-ray photoelectron spectroscopy further corroborates the formation of Ni0.75Fe0.25-LDH (Fig. 2). The Ni:Fe ratio derived from XPS was close to 3:1, i.e. consistent with the bulk composition. The high-resolution XP spectra suggest that NiII is the main nickel state (Fig. 2a), iron is mainly present as FeIII (Fig. 2b), while the O 1s spectrum is mostly due to hydroxide species (Fig. S4†).47 The XPS observations are consistent with the XAS and XRD data and thereby indicate the conservation of long-range order in the examined material.
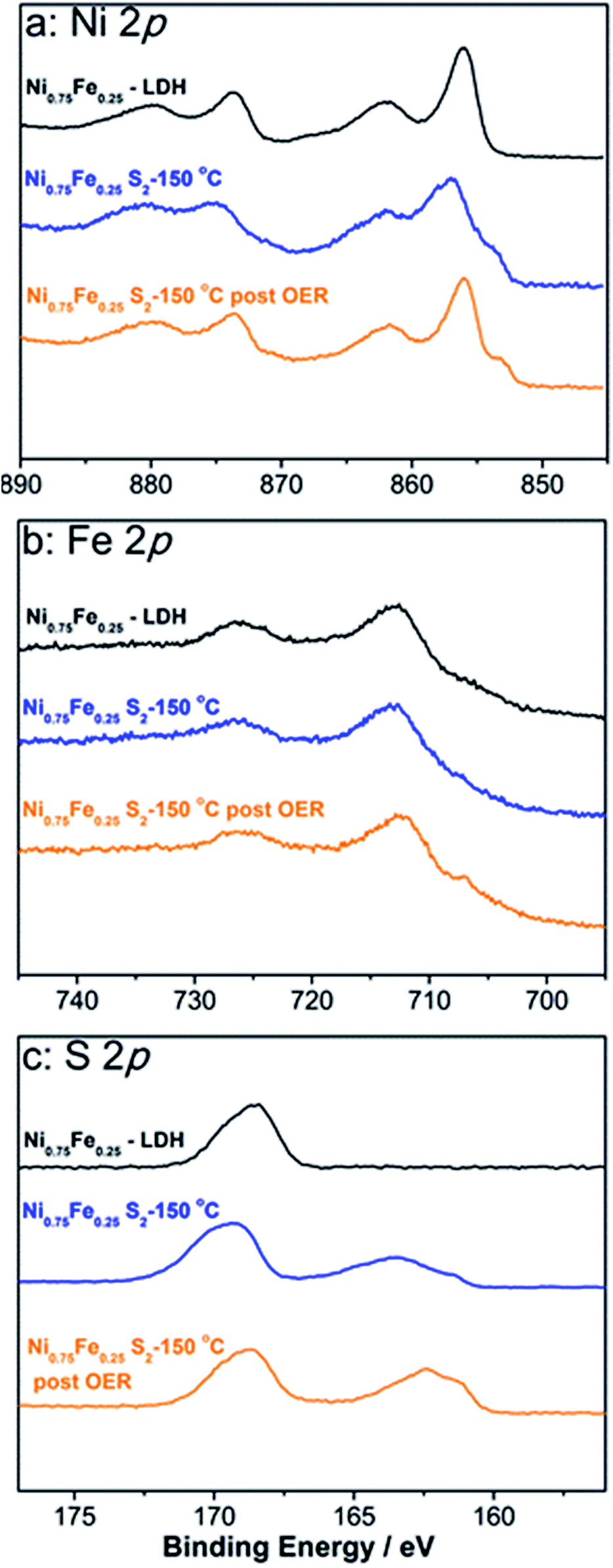 | ||
| Fig. 2 High-resolution (a) Ni 2p, (b) Fe 2p, and (c) S 2p spectra for microwave-synthesised Ni0.75Fe0.25(OH)2+x (black) and Ni0.75Fe0.25S2 (synthesised at 150 °C) that was freshly prepared (blue) or used to catalyse the OER at 1.5 V for 12 h at 25 °C (orange). Peaks at 168.5–168 eV in S 2p spectra originate from a sulphate contamination of unknown origin. | ||
Transmission electron microscopy reveals the thin sheet morphology of microwave-synthesised α-Ni(OH)2 (Fig. S5†) and Ni0.75Fe0.25(OH)2+x (Fig. 3). The hydroxide sheets with lateral dimensions ranging from ten to hundreds of nanometers are self-organised into flower-like architectures (Fig. S5a,†3a and b). The thickness of the Ni0.75Fe0.25(OH)2+x sheets, estimated from edge-on orientation images, is typically in the range of 1–10 nm. The selected area electron diffraction (SAED) pattern collected from “flower” aggregates reveals the polycrystalline nature of the microwave-synthesised hydroxides (Fig. 3c and S5a†). Bright-field images taken on individual sheets show inhomogeneous contrast and evidence the presence of polycrystalline structures in both bimetallic (Fig. 3d) and α-Ni(OH)2 samples (Fig. S5b and c†). High-resolution imaging of individual Ni0.75Fe0.25(OH)2+x sheets reveals a significant concentration of pores (Fig. 3e) that most probably contribute to the high surface area of this material. It can also be seen that extended single-crystalline areas exist within the nanosheets and such areas can envelop the pores present in the structure (Fig. 3f).
 | ||
| Fig. 3 TEM characterisation of microwave-synthesised Ni0.75Fe0.25-LDH: (a, b) bright-field images of flower-like architectures composed of hydroxide sheets; (c) selected area electron diffraction pattern indexed according to JCPDS# 38-0715; (d) bright-field image and (e) high-resolution image of an individual sheet. Panel (f) shows an enlarged section of a high-resolution image from panel e. | ||
Analysis of the textural characteristics of Ni0.75Fe0.25(OH)2+x was undertaken by measuring N2 adsorption–desorption isotherms at 77 K and processing the data using the Brunauer–Emmett–Teller (BET) model (Fig. S6†). Under the assumptions of this model, the material contains a significant number of micropores, which theoretically might represent the interlayer space between sheets. The BET total and observed external surface areas are 149 ± 2 and 91 m2 g−1, respectively.
Characterisation of sulphides
XRD patterns of the materials obtained upon microwave treatment of α-Ni(OH)2 and Ni0.75Fe0.25(OH)2+x with thioacetamide are consistent with the pyrite NiS2 phase (Fig. 1b). Thus, nickel and iron ions randomly occupy the cationic positions in the crystal lattice of produced Ni0.75Fe0.25S2+y,17 similar to the case of Ni0.75Fe0.25(OH)2+x.The Ni0.75Fe0.25S2+y sample synthesised at 120 °C exhibited weak diffraction peaks corresponding to the parent LDH, whereas XRD results attest to essentially complete sulphidation at 150 and 180 °C. Nickel–iron sulphides produced via the microwave-assisted method exhibited mean crystallite sizes of 5–6 nm, which were essentially independent of the temperature maintained during the synthesis and were smaller than that for the hydroxide precursor. Similar disordering occurred upon sulphidation of α-Ni(OH)2, which produced NiS2 with dXRD = 7 nm.
Further insights into the structure of the microwave-synthesised bimetallic sulphides were derived from XAS analysis. Fe K-edge XANES data for Ni0.75Fe0.25S2+y suggest the presence of mixed iron hydroxide/sulphide phases with a predominant FeIII oxidation state (Fig. S7†). At the Ni K-edge, the XANES energy shift is consistent with that for NiS2 (pyrite) attesting to a sulphide-bound redox state (II) as predominant for nickel (Fig. 4a). The presence of a hydroxide component is again evident from these data. However, the level of structural disorder of the hydroxide phase(s) in Ni0.75Fe0.25S2+y is very high, which presumably renders it undetectable by X-ray diffraction. The EXAFS data for Ni0.75Fe0.25S2+y are consistent with those for NiS2 (pyrite), but are substantially dampened (Fig. 4b). The EXAFS at both nickel and iron edges can be fitted with a simple three parameter model derived from the known crystal structure of nickel pyrite (Fig. S8, Table S3†). The fact that nearly the same fit can be obtained from both metal edges implies that iron atoms are occupying nickel sites in this material rather than, for example, decorating the surface of the material in a different state. Thus, the XAS data are consistent with the observations from XRD (Fig. 1b) confirming that the microwave-synthesised material contains Ni0.75Fe0.25S2+y with a pyrite-like structure.
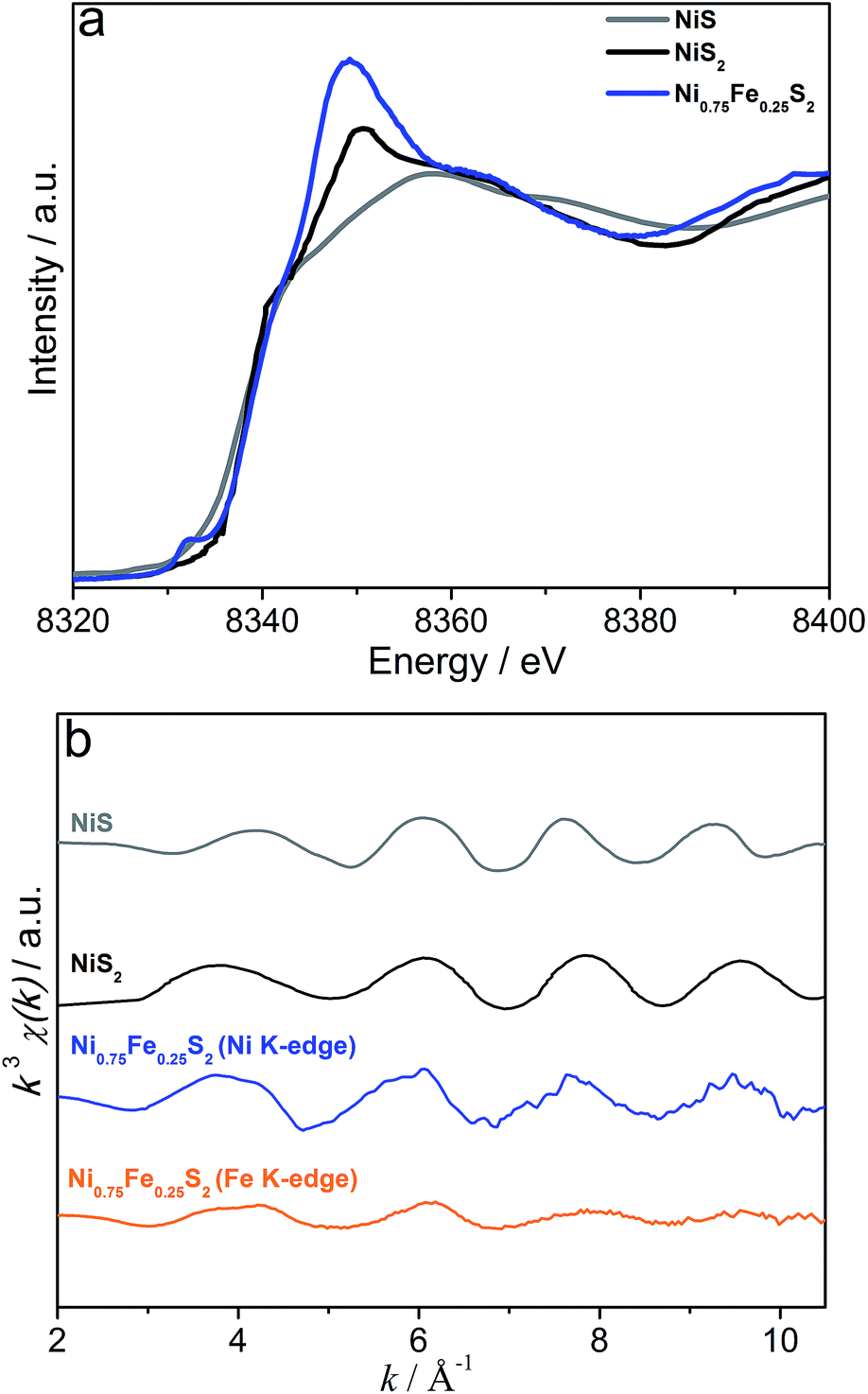 | ||
| Fig. 4 (a) Ni K-edge XANES (blue) and (b) Ni (blue) and Fe (orange) K-edge EXAFS spectra of Ni0.75Fe0.25S2+y (microwave-synthesised at 150 °C). Grey and black traces show reference data for NiS and NiS2 (pyrite), respectively. | ||
Outcomes of the XPS analysis of Ni0.75Fe0.25S2+y (Fig. 2) were consistent with both XRD and XAS, showing a Ni:Fe atomic ratio close to 3:1 and confirming that the predominant oxidation states of nickel and iron are II and III, respectively. Sulphide species were also unambiguously detected as a peak at 163 eV in the S 2p spectrum (Fig. 2c), though the concentration of sulphides derived from XPS (Ni:Fe:S− ≈ 3:1:2) was significantly lower than the stoichiometrically expected value. This is interpreted in terms of the formation of an (oxy)hydroxide surface layer and is further supported by the Ni 2p spectrum, where signals are mainly due to hydroxide species and only a shoulder at binding energies below 854 eV is ascribed to nickel sulphide (Fig. 2a). However, this hydroxide shell is very thin, as suggested by the unambiguous detection of the sulphide species in the S 2p spectrum.
TEM analysis of the microwave-synthesised NiS2 and Ni0.75Fe0.25S2+y shows that these materials represent aggregates of interconnected nanocrystalline grains (Fig. S5d, e† and 5). The SAED patterns are consistent with the XRD data and exhibit polycrystalline rings corresponding to a pyrite phase (Fig. 5b). Bright field imaging of Ni0.75Fe0.25S2+y at higher magnification reveals the presence of very small grains with the sizes of about 1 nm and below, which are visualised as small diffracting dark spots (Fig. 5d). The morphology of Ni0.75Fe0.25S2+y is consistent with a comparatively high surface area of approximately 87 m2 g−1 derived from the N2 adsorption/desorption isotherms (Fig. S6b†). Finally, energy-dispersive X-ray (EDX) mapping conducted for the Ni0.75Fe0.25S2+y material under scanning electron microscopic conditions confirmed homogeneous spatial distributions of nickel, iron, and sulphur within the microwave-synthesised particles (Fig. 5e). The Ni:Fe elemental ratio derived from the EDX mapping analysis was 3:1.
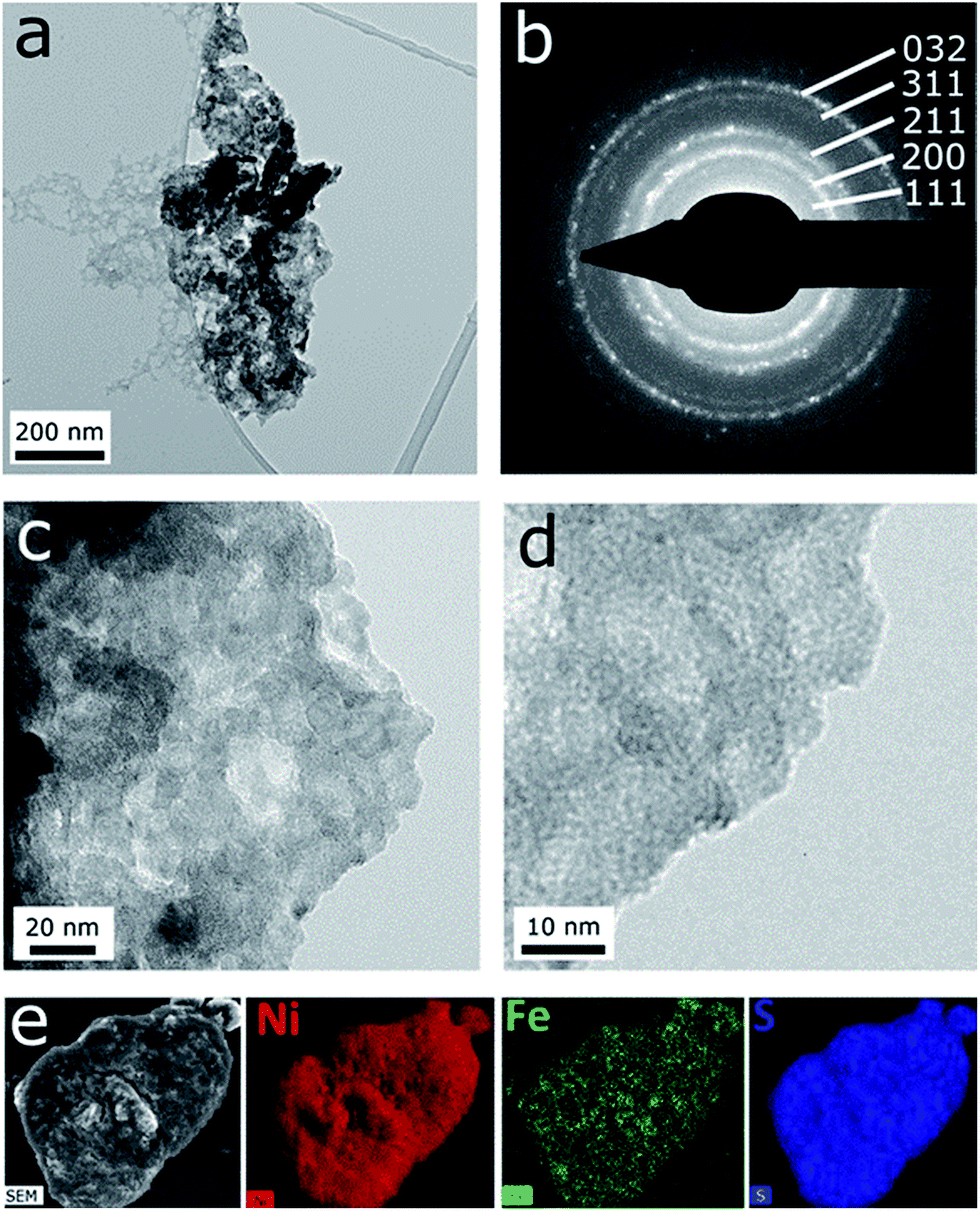 | ||
| Fig. 5 Microscopic characterisation of Ni0.75Fe0.25S2 synthesised under microwave conditions at 150 °C: (a, c, d) bright-field TEM images of polycrystalline aggregates, (b) selected area electron diffraction pattern, and (e) SEM image and energy-dispersive X-ray nickel, iron and sulphur elemental maps. | ||
To improve the crystallinity, several Ni0.75Fe0.25S2+y samples obtained under microwave conditions at 150 °C were additionally calcined in a N2 atmosphere for 2 h. Calcination at 350 °C enhanced dXRD for the double-metal pyrite phase up to 25 nm and did not induce significant qualitative changes to the XRD pattern, although the evolution of new faint diffraction peaks was noted (Fig. S9†). However, segregation of iron to the surface resulting in a Ni:Fe atomic ratio of approximately 2:1 was revealed by XPS upon such treatment (Fig. S10†). Increasing the annealing temperature to 450 °C enhanced the intensity of new diffraction peaks and significantly affected relative intensities of the pyrite diffraction signals, while heating at 600 °C caused degradation of the material with the formation of a complex mixture of unknown nature (Fig. S10†). The surface of the material treated at 600 °C was enriched with iron (Ni:Fe = 0.8:1) and contained a very small amount of sulphur, as determined by XPS (Fig. S10†).
In summary, physical characterisation of the examined microwave-synthesised materials is consistent with the formation of Ni(Fe) layered hydroxides and sulphides. TEM characterisation reveals that the employed synthesis conditions promote exfoliation of polycrystalline porous Ni0.75Fe0.25(OH)2+x sheets. Sulphidation of this LDH produces a network of highly disordered Ni0.75Fe0.25S2+y nanoparticles, whose surface is covered with metal hydroxides.
Electrocatalytic activity
Numerous reports on the use of nickel(iron) based materials as anode and cathode water-splitting catalysts operating at different pH values instigated us to explore the electrocatalytic performance of the microwave-synthesised hydroxides and sulphides towards both hydrogen and oxygen evolution reactions in acidic and alkaline solutions. Given the chemical instability of nickel(iron) hydroxides at very low pH, only sulphides were investigated in 0.5 M H2SO4. On the anodic side, oxidation current densities of the order of mA cm−2 were achieved at potentials more positive than 2.2 V on the RHE scale (Fig. S11a†), which unsurprisingly attests to the unsatisfactory catalytic performance of Ni0.75Fe0.25S2 for the OER under these harsh conditions.Voltammetric measurements to test the HER catalytic activity of NiS2 and Ni0.75Fe0.25S2 in 0.5 M H2SO4 were undertaken with the potential of the working electrode always kept below 0 V vs. RHE to suppress possible dissolution. Reasonably high reductive current densities reproducibly manifesting in the first voltammetric cycles at potentials more negative than ca. −0.2 V vs. RHE were not observed in subsequent potential sweeps (Fig. S11†), again indicating poor stability of these catalysts in acidic solutions (however, visual inspection of the electrodes did not reveal any changes to the catalyst layers, i.e. the materials did not undergo significant dissolution). These findings contradict recent reports on the high and stable HER catalytic activity of nickel and nickel–iron sulphides under similar conditions.39,40 This discrepancy might be because of the disordered and highly dispersed nature of the microwave-synthesised materials (Fig. 1, 4 and 5). To verify this hypothesis, Ni0.75Fe0.25S2+y with improved crystallinity (after calcination in N2 at 350 °C) was tested for its activity in the HER, but the same unsatisfactory behaviour and very low activity were found again (Fig. S11a†). The aforementioned observations hold true for both NiS2 and Ni0.75Fe0.25S2+y, irrespective of temperatures maintained during synthesis and annealing procedures (Fig. S11†). Presumably, the inability to improve the performance of the microwave-synthesised bimetallic sulphides by calcination was at least in part due to changes in the surface Ni:Fe ratio induced by such treatment.
In contrast to the results obtained in 0.5 M H2SO4, robust electrocatalytic performance for H2 evolution is demonstrated by the microwave-synthesised hydroxides and sulphides in 1 M KOH (Fig. S12†). Cyclic voltammograms recorded in N2-saturated and intensively stirred solutions demonstrate the superior catalytic performance of Ni0.75Fe0.25S2+y and NiS2 as compared to their hydroxide counterparts. Interestingly, no significant influence of the presence of iron and the synthesis temperature on the HER performance of sulphides was identified under alkaline conditions. The best H2 evolution performance among the examined materials was demonstrated by Ni0.75Fe0.25S2+y produced at 150 °C. Flat electrodes modified with this catalyst at a loading of Γ = 0.28 mg cm−2 provided the reductive current density of 10 mA cmgeom−2 at −0.38 V vs. RHE. The specific activity of this catalyst at −0.3 V vs. RHE was estimated as 8 A g−1. Notwithstanding that this value is most probably the lower limit of the true intrinsic catalytic activity of the material (vide infra), it can be concluded that the microwave-synthesised nickel(iron) sulphides are not highly active catalysts for H2 evolution.48
Further experiments focused on the electrocatalytic oxidation of water in the presence of 1 M KOH, where microwave-synthesised nickel–iron systems demonstrated much more impressive performance and sulphides again significantly outperformed their hydroxide counterparts (Fig. 6). The quasi-steady-state nature of the electrocatalytic parts of the presented voltammetric curves was confirmed by measurements undertaken at varying potential scan rates and chronoamperometry at selected potentials (Fig. S13†). The activity indicator “overpotential at the OER current density of 10 mA cmgeom−2” (η10) that is widely used in the literature does not allow for direct comparisons of intrinsic catalytic activity as η10 depends on Γ and the electrode geometry/structure. In essence, this parameter is most useful when high-loading, high-surface-area electrodes constructed for application in end devices are compared. We provide relevant η10 data, but discussions below are mainly based on the OER current densities at defined overpotentials normalised to the mass (i) and BET external surface area (j) of catalysts (Table 1).
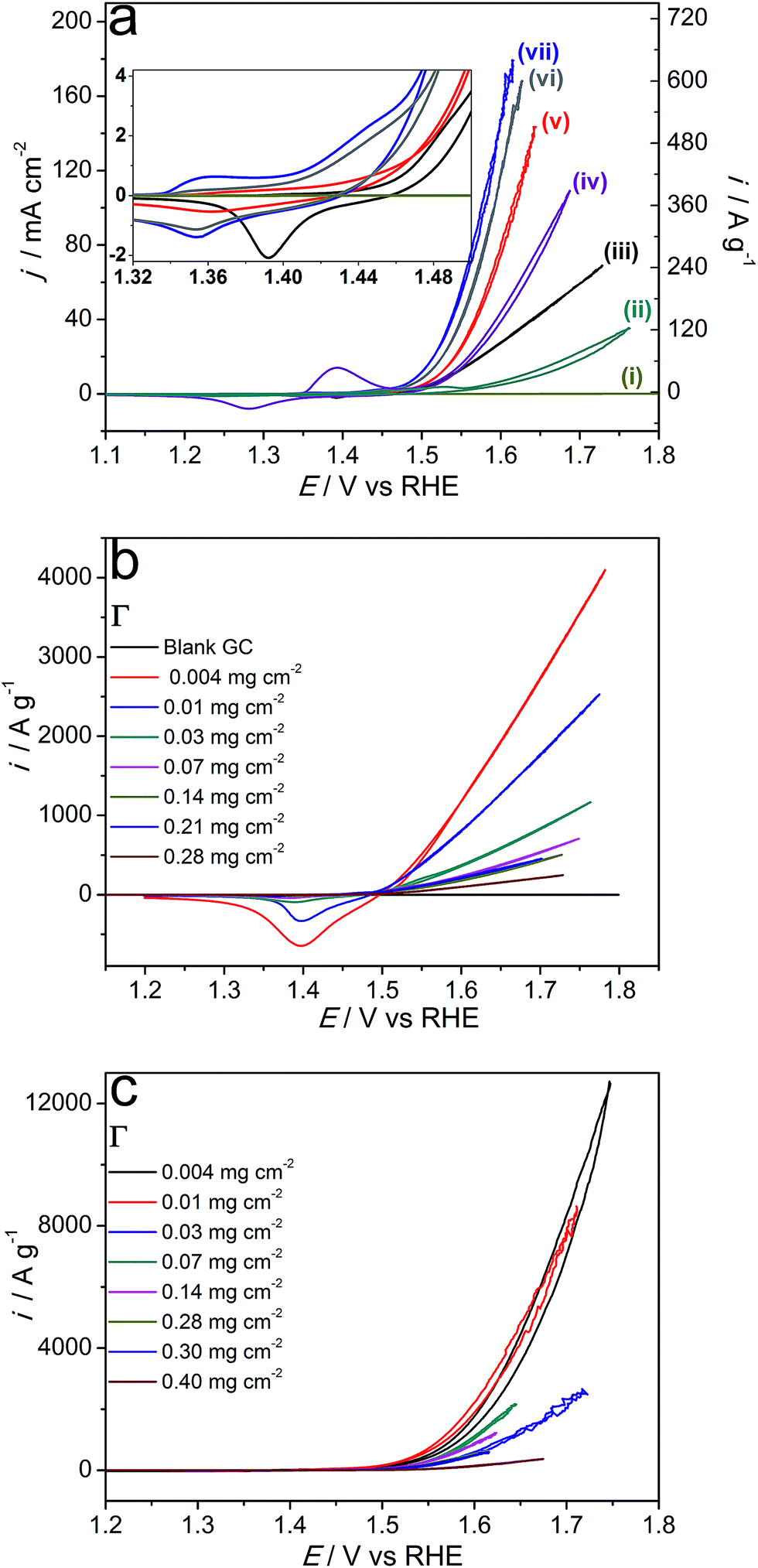 | ||
| Fig. 6 (a) Cyclic voltammograms (scan rate 0.005 V s−1) for the oxidation of stirred 1 M KOH with a glassy carbon electrode that was (i) unmodified, or coated with 0.28 mg cm−2 of microwave-synthesised (ii) α-Ni(OH)2, (iii) Ni0.75Fe0.25(OH)2+x, (iv) NiS2 or Ni0.75Fe0.25S2+y produced at (v) 120, (vi) 180 or (vii) 150 °C. The inset shows magnified plots of redox transformations of the bimetallic materials preceding the water oxidation wave. Panels (b) and (c) show effects of catalyst loading (Γ/mg cm−2) on the voltammetric behaviour and catalytic activity of Ni0.75Fe0.25(OH)2+x and Ni0.75Fe0.25S2+y (synthesised at 150 °C), respectively. Currents are normalised to the electrode surface area or catalyst mass. All data were post-corrected for the IRu drop (Ru = 13–15 Ω). | ||
Although the OER Tafel slopes derived from the voltammetric data were similar for all examined bimetallic sulphides within the 0.034–0.044 V dec−1 range (η range 0.22–0.25 V; data not shown), the sulphidation temperature noticeably influenced the specific electrocatalytic performance of Ni0.75Fe0.25S2+y. Specifically, the material synthesised at 150 °C outperformed the samples prepared at 120 and 180 °C (Fig. 6a). Comparison of the intensities of redox transformations prior to the onset of the OER electrocatalytic wave suggests that this improvement is most probably due to the greater accessibility of the Ni(Fe) electroactive sites for the electrodes functionalised with sulphides obtained at 150 °C (inset to Fig. 6a).
Conversely, similar considerations cannot be invoked to rationalise the superiority of Ni0.75Fe0.25S2+y over Ni0.75Fe0.25(OH)2+x, which provide comparable surface concentrations of metal-based active sites (cf. cathodic sweeps in the inset to Fig. 6a). The specific electrochemical double-layer capacitances are also similar for these two types of materials (Fig. S14†), as are the external BET surface areas (Table 1). Therefore, the higher electrocatalytic activity of Ni0.75Fe0.25S2+y can be attributed to their excellent electrical conductivity as compared to LDHs. Indeed, nickel–iron sulphides synthesised at 150 °C provided sheet resistance of only 33 mΩ square−1 when deposited as thin layers on a glass substrate, while the conductivity of similarly produced Ni0.75Fe0.25(OH)2+x coatings could not be reliably measured by a four-point probe. Besides, voltammetric OER curves registered with Ni0.75Fe0.25(OH)2+x demonstrated an ohmic behaviour (linear dependence of current on potential) at current densities above 10 mA cmgeom−2. However, another equally or even more important factor that can contribute to differences in the specific catalytic activities of the microwave-synthesised sulphides and hydroxides is dissimilarity in the nature of electrochemically active Ni(Fe) species present on the surface of these materials that manifests in cyclic voltammetric data (inset to Fig. 6a).
The processes within the 1.32–1.50 V vs. RHE potential range can be attributed to the Ni2+/3+ redox transformations.49–51 The voltammetric behavior of Ni0.75Fe0.25(OH)2+x within this range is consistent with the previously reported data for nickel–iron hydroxides with the same Ni:Fe ratio.52 For Ni0.75Fe0.25S2+y, similar redox transformations, though at slightly more negative potentials can also be identified. However, at least one different type of redox active species, presumably nickel-based (oxidation process at ca. 1.35 V), is also present on the surface of the sulphide-based electrocatalyst. These observations are particularly interesting in the light of identical surface concentrations of nickel and iron derived from XPS, and attest to significant differences in the electrochemical/electrocatalytic behaviour of (oxy)hydroxides formed upon electrooxidation of the bimetallic sulphide surface and LDHs. This is further confirmed by comparisons of the surface-area normalised catalytic activities of Ni0.75Fe0.25(OH)2+x and Ni0.75Fe0.25S2+y (Table 1).
To enable reliable quantification of the intrinsic OER catalytic activity, effects of the catalyst loading on the voltammetric behaviour of synthesised materials were examined (Fig. 6b and c, S15†). Mass-normalised electrocatalytic activity derived from these data gradually increases with a decrease in Γ, which identifies that the performance of the catalysts is limited by agglomeration. Indeed, SEM analysis of drop-cast Ni0.75Fe0.25OH2+x and Ni0.75Fe0.25S2+y reveals the formation of comparatively crude aggregates of these materials on a flat support surface (Fig. S16†). Variation of the loading of Ni0.75Fe0.25OH2+x also induced notable changes in the voltammetric behaviour of this catalyst. In particular, decreasing Γ pronouncedly enhanced the mass-normalised intensity of the process for reduction of the nickel(iron) species manifesting at ca. 1.4 V vs. RHE in the cathodic sweeps of the voltammograms (Fig. 6b). This observation indicates that agglomeration is suppressed when a smaller amount of Ni0.75Fe0.25OH2+x is deposited and more exfoliated hydroxide sheets, as those visualised by TEM (Fig. 3), become exposed to the solution. Correlation of this phenomenon with changes in the electrocatalytic performance (Fig. 6b, Table 1) suggests that basal planes of the microwave-synthesised nickel–iron layered hydroxides significantly contribute to the OER activity. Interestingly, the specific activity of Ni0.75Fe0.25OH2+x did not stabilise even at very low Γ of 0.004–0.01 mg cm−2 (Fig. 6b), which identifies the electrocatalytic performance of this material presented herein as a lower limit. Determination of the true intrinsic OER activity was found to be challenging due to unsatisfactory reproducibility of the results obtained at catalyst loadings below 0.004 mg cm−2. In contrast, the water oxidation current densities for Ni0.75Fe0.25S2+y converged to similar values at Γ = 0.004–0.01 mg cm−2 (Fig. 6c). In this case, the dependence of the specific activity on loading is attributed to the agglomeration effect only.
The stability of the microwave-synthesised materials was examined during long-term potentiostatic and galvanostatic experiments. Ni0.75Fe0.25S2 synthesised at 150 °C was found to be capable of sustaining water electrooxidation for many hours at ambient temperature with negligible loss in the initial current density (Fig. 7a). Interestingly, XPS analysis of the sulphide sample after this experiment revealed only a minor decrease in the concentration of sulphide species in the near-surface layers, which were still detectable after 12 h of operation (Fig. 2c). TEM analysis of the similarly treated sample did not show any notable changes in the microstructure and morphology (Fig. S17†). These observations indicate that a thin active layer of (oxy)hydroxide may impede the degradation of the catalyst and thus maintain its high activity. Robust operation of the catalyst during the HER under ambient conditions is also noted (Fig. 7a).
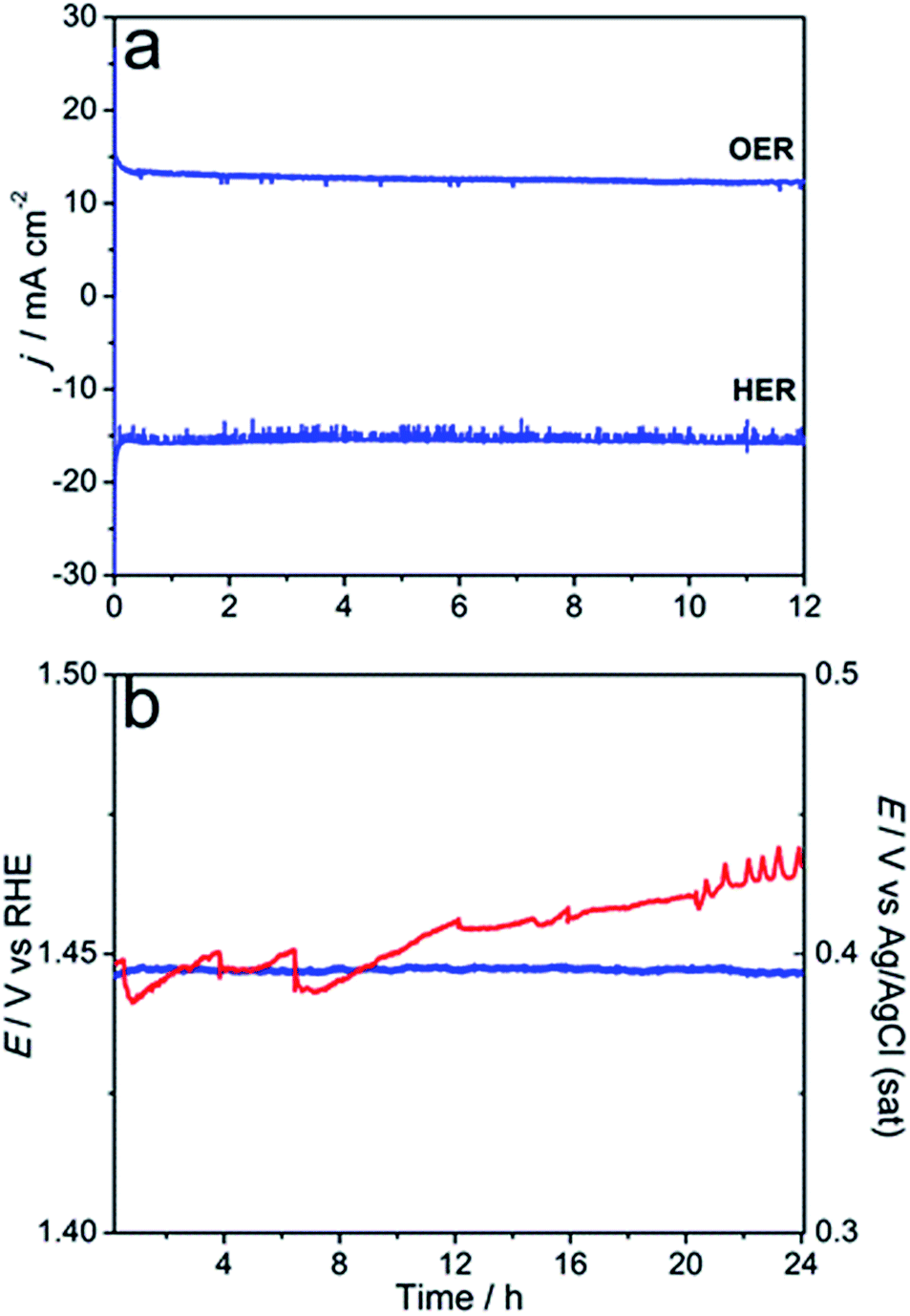 | ||
| Fig. 7 Long-term oxidation and reduction of water (1 M KOH) catalysed by Ni0.75Fe0.25S2+y (synthesised under microwave irradiation at 150 °C): (a) potentiostatic experiments for the OER (1.5 V vs. RHE) and HER (−0.4 V vs. RHE) at 25 °C using a flat glassy carbon support and 0.28 mg cm−2 catalyst loading; (b) galvanostatic experiments for the OER at 10 mA cm−2 at 25 (blue; left-hand-side ordinate axis) and 60 °C (red; right-hand-side ordinate axis) using a nickel foam support and 3 mg cmgeom−2 catalyst loading. Solutions were intensively stirred during experiments. | ||
Further assessment of the stability of Ni0.75Fe0.25S2+y was undertaken using a porous Ni foam support modified with the microwave-synthesised material at Γ = 3 mg cmgeom−2 (optimised on the basis of the voltammetric data; Fig. S18†). Galvanostatic test at ambient temperature did not show any degradation over the period of 24 h, whereof the water oxidation current density of 10 mA cm−2 was stably maintained at η10 = 0.21 V (Fig. 7b). Further, the durability of Ni0.75Fe0.25S2/Nifoam electrodes was examined at 60 °C. Under these harsher conditions, ca. 0.03 V increase in the overpotential needed to sustain the OER at the rate of 10 mA cm−2 was observed over 24 h of the test, but the performance seems to stabilise at this point. Overall, the results summarised in Fig. 6 and 7 attest to the capacity of the microwave-synthesised nickel–iron sulphides to operate as efficient catalysts for water electrooxidation on a long timescale, notwithstanding the very high level of structural disorder in these materials.
Table 1 juxtaposes the OER activities of the microwave-synthesised Ni0.75Fe0.25S2 and Ni0.75Fe0.25(OH)2+x catalysts with those of the best-performing (to the best of our knowledge) analogues reported in the literature. Microwave-synthesised Ni0.75Fe0.25(OH)2+x exhibit similar mass-normalised activity to that of the most active unsupported catalysts of this class, including [Ni0.78Fe0.22(OH)2](NO3)y(OH)x−y LDHs synthesised by the pulsed-laser ablation technique12 and completely exfoliated layered hydroxides with the Ni:Fe ratio as in our work.11 However, catalyst synthesis in ref. 11 and 12 either required advanced equipment or involved days of hydrothermal reaction followed by 24 h of exfoliation through mechanical stirring under inert conditions. Similarly performing Ni0.75Fe0.25(OH)2+x material reported herein was obtained after only 1 h of the microwave reaction at 120 °C.
Unfortunately, very limited data on the surface area of the NiFe LDH materials are available in the literature. This precludes comparisons in terms of the surface area-normalised water oxidation current densities, which are useful for establishing the differences/similarities in the nature of true electrocatalytically active species. Our attempt to use the specific double-layer capacitance provided in one of the publications on Ni0.7Fe0.3-LDH14 to estimate ABET for this catalyst produced j values that were significantly lower than that for the microwave-synthesised Ni0.75Fe0.25(OH)2+x (Table 1). However, the specific surface area estimated via this approach for Ni0.7Fe0.3-LDH from ref. 14 is unrealistically high.
Hitherto, the best-known nickel–iron sulphide-based catalyst for water electrooxidation was reported by Golberg et al.16 In fact, the only catalyst that outperforms this system in terms of mass-weighed activity are gelled FeCoW mixed metal oxyhydroxides53 (Table S1†). At typically employed loadings (0.1–0.3 mg cm−2), microwave-synthesised Ni0.75Fe0.25S2+y (150 °C) yield in performance to the material developed in ref. 16 at 0.25 V, although the activities become comparable at the OER overvoltage of 0.30 V (Table 1). At low Γ, the intrinsic activity of the Ni0.75Fe0.25S2+y catalyst introduced herein is very high, and in fact, the highest reported for any NiFe-based water oxidation catalyst to date (Table 1). Interestingly, the specific capacitance of the Ni0.75Fe0.25 oxyhydroxide/disulphide catalyst from ref. 16 (ca. 1.3 F g−1) is approximately 6-fold lower than that of Ni0.75Fe0.25S2+y obtained under microwave conditions at 150 °C (ca. 7.7 F g−1; Fig. S14c†), which can be tentatively extrapolated to a comparatively low ABET and outstandingly high jBET for the catalyst synthesised by Golberg and colleagues (Table 1). At the same time, comparison of the intensities of reductive peaks in cyclic voltammograms for Ni0.75Fe0.25S2+y synthesised herein at 150 °C (Fig. 6a, inset) and Ni0.75Fe0.25 oxyhydroxide/disulphide from ref. 16 suggests that the latter exhibits at least 7-fold higher mass-weighed concentration of electroactive Ni(Fe) species. It can be hypothesised that these two factors compensate for each other and result in comparable intrinsic mass-weighed electrocatalytic activities for two different types of nickel–iron sulphide catalysts. However, analysis of the data in Table 1 also suggests that deposition of microwave-synthesised Ni0.75Fe0.25S2+y on a high surface area support while preventing significant agglomeration might produce a catalyst that will outperform the material developed in ref. 16 irrespective of the loading on the electrode surface.
Finally, it is additionally emphasised that the microwave synthesis procedures developed herein are very simple as compared to alternative methods summarised in Table 1, but enable fabrication of similar or better-performing catalysts. This provides important technological advantages. Equally important is that the electrocatalytic performance of the microwave-synthesised materials is perfectly reproducible (Fig. S19†), further demonstrating the reliability of this method.
Conclusions
In summary, the present study introduces an effective and economical microwave-assisted solution-phase strategy for the synthesis of polycrystalline nickel(iron) layered hydroxides and sulphides. As-synthesised materials are highly dispersed and exhibit a significant degree of structural disorder, which enables a very high yet robust catalytic activity for water electrooxidation in alkaline solutions. Investigation of the performance-loading dependencies for the microwave-synthesised catalysts allowed for the determination of their intrinsic electrocatalytic activities, which are higher than those of analogues reported in the literature. When Ni0.75Fe0.25(OH)2+x and Ni0.75Fe0.25S2+y are immobilised on electrodes at high loadings, their catalytic activity is partially suppressed by agglomeration. Nevertheless, these electrodes still exhibit the state-of-the-art water oxidation performance at the level of the best catalysts currently known. In particular, nickel foam functionalised with Ni0.75Fe0.25S2+y provides the water electrooxidation rate of 10 mA cmgeom−2 at an overpotential of 0.21 V at 25 °C. Developing a method to deposit this bimetallic sulphide onto a high surface area corrosion-resistant support while preventing significant agglomeration is expected to provide even better water oxidation performance. To the best of our knowledge, the present study is the first report on the microwave-assisted synthesis of nickel(iron) layered hydroxide and sulphide water oxidation catalysts. Apart from being rapid and facile, this synthetic method also opens up many possibilities for structural/morphological tuning and doping of electromaterials for further improvements in catalytic and other properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the use of facilities within the Monash X-ray Platform (funded by Australian Research Council grant LE130100072) and Monash Centre for Electron Microscopy (Monash University), the Australian Synchrotron for providing access to the XAS beam-line (project ID M12592), Dr B. Johannessen, Dr P. Kappen and Dr C. Glover for the support in XAS experiments, and Dr K. Nairn for proofreading the manuscript. Transmission electron microscopy was carried out at the Bio21 Advanced Microscopy Facility (the University of Melbourne). Funding of this work by the Australian Research Council through the ARC Centre of Excellence for Electromaterials Science (CE140100012) is gratefully appreciated.References
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef PubMed.
- K. Shen, X. Chen, J. Chen and Y. Li, ACS Catal., 2016, 6, 5887–5903 CrossRef.
- R. M. Navarro, M. A. Peña and J. L. G. Fierro, Chem. Rev., 2007, 107, 3952–3991 CrossRef PubMed.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef PubMed.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef PubMed.
- V. Vij, S. Sultan, A. M. Harzandi, A. Meena, J. N. Tiwari, W.-G. Lee, T. Yoon and K. S. Kim, ACS Catal., 2017, 7, 7196–7225 CrossRef.
- J. Landon, E. Demeter, N. İnoğlu, C. Keturakis, I. E. Wachs, R. Vasić, A. I. Frenkel and J. R. Kitchin, ACS Catal., 2012, 2, 1793–1801 CrossRef.
- O. Diaz-Morales, I. Ledezma-Yanez, M. T. M. Koper and F. Calle-Vallejo, ACS Catal., 2015, 5, 5380–5387 CrossRef.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 Search PubMed.
- B. M. Hunter, J. D. Blakemore, M. Deimund, H. B. Gray, J. R. Winkler and A. M. Müller, J. Am. Chem. Soc., 2014, 136, 13118–13121 CrossRef PubMed.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef PubMed.
- W. Ma, R. Ma, C. Wang, J. Liang, X. Liu, K. Zhou and T. Sasaki, ACS Nano, 2015, 9, 1977–1984 CrossRef PubMed.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef PubMed.
- M. Zhou, Q. Weng, X. Zhang, X. Wang, Y. Xue, X. Zeng, Y. Bando and D. Golberg, J. Mater. Chem. A, 2017, 5, 4335–4342 Search PubMed.
- B.-Q. Li, S.-Y. Zhang, C. Tang, X. Cui and Q. Zhang, Small, 2017, 13, 1700610 CrossRef PubMed.
- J. Yu, G. Cheng and W. Luo, J. Mater. Chem. A, 2017, 5, 15838–15844 Search PubMed.
- C.-Z. Yuan, Z.-T. Sun, Y.-F. Jiang, Z.-K. Yang, N. Jiang, Z.-W. Zhao, U. Y. Qazi, W.-H. Zhang and A.-W. Xu, Small, 2017, 13, 1604161 CrossRef PubMed.
- X. Xu, F. Song and X. Hu, Nat. Commun., 2016, 7, 12324 CrossRef PubMed.
- S.-M. Xu, T. Pan, Y.-B. Dou, H. Yan, S.-T. Zhang, F.-Y. Ning, W.-Y. Shi and M. Wei, J. Phys. Chem. C, 2015, 119, 18823–18834 Search PubMed.
- Y. Tang, R. Wang, Y. Yang, D. Yan and X. Xiang, ACS Appl. Mater. Interfaces, 2016, 8, 19446–19455 Search PubMed.
- Y. Zhao, B. Li, Q. Wang, W. Gao, C. J. Wang, M. Wei, D. G. Evans, X. Duan and D. O'Hare, Chem. Sci., 2014, 5, 951–958 RSC.
- J. Guo, C. Mao, R. Zhang, M. Shao, M. Wei and P. Feng, J. Mater. Chem. A, 2017, 5, 11016–11025 Search PubMed.
- S. J. Kim, Y. Lee, D. K. Lee, J. W. Lee and J. K. Kang, J. Mater. Chem. A, 2014, 2, 4136–4139 Search PubMed.
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef PubMed.
- L. Fagiolari, A. Scafuri, F. Costantino, R. Vivani, M. Nocchetti and A. Macchioni, ChemPlusChem, 2016, 81, 1060–1063 CrossRef.
- F. Basile, G. Fornasari, M. Gazzano and A. Vaccari, Appl. Clay Sci., 2000, 16, 185–200 CrossRef.
- G. Chen, T. Wang, J. Zhang, P. Liu, H. Sun, X. Zhuang, M. Chen and X. Feng, Adv. Mater., 2018, 30, 1706279 CrossRef PubMed.
- B. M. Hunter, W. Hieringer, J. R. Winkler, H. B. Gray and A. M. Muller, Energy Environ. Sci., 2016, 9, 1734–1743 Search PubMed.
- X. Long, J. Li, S. Xiao, K. Yan, Z. Wang, H. Chen and S. Yang, Angew. Chem., Int. Ed., 2014, 53, 7584–7588 CrossRef PubMed.
- C. Xiao, Y. Li, X. Lu and C. Zhao, Adv. Funct. Mater., 2016, 26, 3515–3523 CrossRef.
- O. V. Cherstiouk, A. N. Simonov, N. S. Moseva, S. V. Cherepanova, P. A. Simonov, V. I. Zaikovskii and E. R. Savinova, Electrochim. Acta, 2010, 55, 8453–8460 CrossRef.
- Y. Yi, G. Weinberg, M. Prenzel, M. Greiner, S. Heumann, S. Becker and R. Schlögl, Catal. Today, 2017, 295, 32–40 CrossRef.
- C. Andronescu, S. Barwe, E. Ventosa, J. Masa, E. Vasile, B. Konkena, S. Möller and W. Schuhmann, Angew. Chem., Int. Ed., 2017, 56, 11258–11262 CrossRef PubMed.
- L. Yu, H. Zhou, J. Sun, F. Qin, F. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2017, 10, 1820–1827 Search PubMed.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097 CrossRef.
- Y. Hou, M. R. Lohe, J. Zhang, S. Liu, X. Zhuang and X. Feng, Energy Environ. Sci., 2016, 9, 478–483 Search PubMed.
- J. Yan, H. Wu, P. li, H. Chen, R. Jiang and S. Liu, J. Mater. Chem. A, 2017, 5, 10173–10181 Search PubMed.
- X. Long, G. Li, Z. Wang, H. Zhu, T. Zhang, S. Xiao, W. Guo and S. Yang, J. Am. Chem. Soc., 2015, 137, 11900–11903 CrossRef PubMed.
- J. Virkutyte and R. S. Varma, Chem. Sci., 2011, 2, 837–846 RSC.
- R. B. N. Baig and R. S. Varma, Chem. Soc. Rev., 2012, 41, 1559–1584 RSC.
- M. Chatti, T. Gengenbach, R. King, L. Spiccia and A. N. Simonov, Chem. Mater., 2017, 29, 3092–3099 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef PubMed.
- U. Costantino, F. Marmottini, M. Nocchetti and R. Vivani, Eur. J. Inorg. Chem., 1998, 1998, 1439–1446 CrossRef.
- Y. Han, Z.-H. Liu, Z. Yang, Z. Wang, X. Tang, T. Wang, L. Fan and K. Ooi, Chem. Mater., 2008, 20, 360–363 CrossRef.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef PubMed.
- L. Chen, X. Dong, Y. Wang and Y. Xia, Nat. Commun., 2016, 7, 11741 CrossRef PubMed.
- A. Seghiouer, J. Chevalet, A. Barhoun and F. Lantelme, J. Electroanal. Chem., 1998, 442, 113–123 CrossRef.
- B. H. R. Suryanto, T. Fang, S. Cheong, R. D. Tilley and C. Zhao, J. Mater. Chem. A, 2018, 6, 4686–4694 Search PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef PubMed.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. García de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00129d |
| This journal is © The Royal Society of Chemistry 2018 |