
Electrocatalytic reduction of CO2 to produce higher alcohols†
Shamsa
Munir
abc,
Amir Rahimi
Varzeghani
ab and
Sarp
Kaya
 *abd
*abd
aMaterial Science and Engineering, Koç University, Istanbul, Turkey. E-mail: sarpkaya@ku.edu.tr
bKoç University TÜPRAŞ Energy Center, Istanbul, Turkey
cChemistry Department, Women University Swabi, Swabi, Pakistan
dChemistry Department, Koç University, Istanbul, Turkey
First published on 22nd August 2018
Abstract
Electrodeposited and thermally oxidized copper surfaces have been documented in recent years to produce simple alcohols. In this work, we endeavored to study the electrochemical reduction of CO2 at different electrodes prepared via the electrodeposition method, namely, Cu–Cu2O, Cu–Cu2O–ZnO, and Cu–ZnO. In addition, thermally oxidized Cu (Cu-TO) was also investigated. C1, C2, and C3 species were produced on Cu–Cu2O–ZnO, Cu–Cu2O, and Cu–ZnO. The highest faradaic efficiency (FE of 97.4%) of the liquid products (methanol, formate, n-propanol, acetone) was evidenced on Cu–ZnO. The formation of C3 species with high FE on the Cu–ZnO electrode is attributed to the fast C–C–C coupling at the Cu–Zn interface. On thermally oxidized Cu, the total FE of the liquid products (methanol, formate, ethanol, acetate, n-propanol) was found to be 58.51%, which is considerably closer to the already reported values for these electrodes. Moreover, the Cu–Cu2O–ZnO electrode revealed selectivity toward methanol production. Detailed morphological and elemental analyses of the electrode, performed using XPS, Raman spectroscopy, and FESEM, as well as activity measurements to obtain an insight into the mechanistic pathways, reveal that C–C coupling is favored on Cu0 sites rather than Cu2O. Moreover, methanol formation seems to proceed via O coordination of CO2 to Cu–Cu2O surface having (100) facets, whereas C coordination is favored on Cu-TO with (111) exposed faces, resulting in Cu0 sites. The localized formation of ZnO nanoflowers was observed on Cu–ZnO electrodes after the electrochemical reduction of CO2, which is attributed to the mechanistic pathway involving chemical steps, leading to the formation of C3 species.
1. Introduction
The electrochemical conversion of CO2 to liquid fuels is one of the most promising incentives to utilize atmospheric CO2 whose concentration is predicted to increase to ∼570 ppm by the end of the century.1 CO2 reduction chemistry is fairly rich and various gas- and liquid-phase products could be obtained. For instance, the production of formic acid with high faradaic efficiencies (FEs) using Cu- or Cu-oxide-based electrodes has been reported.2–4 Alternative systems such as Pd-multiwalled carbon nanotubes5 have also been utilized for the electrocatalytic conversion of CO2 to produce formic and acetic acids with FEs of 34.5 and 52.3%, respectively. However, the conversion of CO2 to alcohols is of special interest. The production of methanol, from otherwise wasted CO2, has considerable significance since a large quantity of this chemical is manufactured worldwide.6 In addition, the high operating temperature and pressure required for the synthesis procedures demand large energy investments.7 Therefore, mild and environmentally friendly methods of CO2 conversion to methanol have become more significant. Similarly, the selective production of ethanol using CO2 reduction is one of the bigger incentives to develop efficient and cost-effective electrode materials for this reaction.Methanol production using oxidized Cu is reported in one of the earlier studies by Frese et al.; 240% FE for methanol using 0.5 M KHCO3 at −1.9 V (SCE) has been evidenced.8 However, the onset potential of methanol formation was around −0.4 V (SCE).8 Since FEs are determined based on six electron transfers for methanol, values greater than 100% show the involvement of both electrochemical and chemical steps in the reaction mechanism.8,9 Since then, several studies have documented the formation of methanol with appreciable amounts using Cu-oxide catalysts.10 The property of copper oxide surfaces to produce liquid fuels have been investigated using core–shell nanoparticles11 and composites materials.12–14 Methanol has been reported with 42.7% FE on Cu/CuO core–shell catalysts.15 The selective production of methanol with a 95% FE is documented by using hybrid CuO/Cu2O semiconductor nanorod arrays6,16 in a photoelectrochemical setup. The photoelectrocatalytic efficiency is determined by the potential gradient applied simultaneously with light irradiation.17,18
The production of higher alcohols, such as ethanol and propanol, is challenging, and only few researchers have confirmed their formation with appreciable FEs.19–23 A recent investigation by Kanan et al. has demonstrated that nanocrystalline Cu prepared from Cu2O can produce multi-carbon oxygenates including ethylene, ethanol, and n-propanol with high total FE of 57%.24 A combined FE of 46% is reported for ethylene and ethanol in alkaline electrolytes using a gas diffusion electrode (GDE) covered with Cu nanoparticles. A comparison of several studies under different conditions reveals that higher FEs for C2 products are obtained under alkaline conditions.22
The present work is aimed toward a comparative study of electrodes with different compositions and morphologies. Four different electrodes were prepared and investigated for the electrocatalytic reduction of CO2. Detailed elemental and morphological analyses of electrodes were performed to propose mechanistic pathways for C1, C2, and C3 compounds. Cu2O–ZnO composite electrodes were also tested to produce liquid fuels. Interestingly, we observed the highest FE of C3 compounds on electrodeposited ZnO on Cu metal rather than Cu-oxide-based electrodes.
2. Experimental
2.1. Materials and chemicals
A Cu-plate with a thickness of 0.25 mm (99.98% trace metal basis) was employed to prepare a cathode for the electrochemical reduction of CO2. CuSO4·5H2O (>98.0%), DL lactic acid (90.0%), and NaOH (>97.0%), purchased from Sigma Aldrich, were used to prepare the electrodeposition solution of Cu2O. ZnSO4 (>99.0%) was used to prepare the electrodeposition solution of ZnO. KHCO3 (99.95% trace metal basis) was utilized as an electrolyte for CO2 reduction. A two-compartment cell with a Nafion® NRE-212 (thickness: 0.002 in.) separator and a three-electrode assembly was employed. A Pt and Ag/AgCl electrode served as the counter- and reference electrodes, respectively. All the solutions were prepared in deionized water.2.2. Characterization
All the fabricated electrodes were characterized using FESEM-EDS, (Zeiss Ultra Plus), XRD (Bruker D8 Advance), Raman spectroscopy (Reinshaw Invia), and XPS (Thermo K alpha). The potentiostatic measurements were carried out using a Bio-Logic potentiostat (VSP 300). The product analysis of the liquid samples was done using a 600 MHz Agilent Premium Compact NMR employing the following parameters. Wet 1D solvent: D2O; observe: H1; decoupler C13 32 scans; relaxation delay: 1.5 s; acquisition time: 4 s; and spectral width: 9615.4 Hz.2.3. Fabrication of electrodes
Four different types of electrodes were prepared on a 1 × 1 cm2 Cu plate using electrodeposition and thermal oxidation methods. At first, the electrodeposition of Cu2O on Cu was performed from the deposition solution of 0.4 M CuSO4 and 3.0 M lactic acid. The pH of the solution was adjusted to 11.0 by adding NaOH pellets. Electrodeposition was carried out in a single-compartment cell at −0.45 V for 15 min using a counter-electrode made of Pt. A reddish-brown layer of Cu2O was formed over the Cu plate. These electrodes were named as Cu–Cu2O.Cu2O and ZnO electrodes were fabricated by first depositing Cu2O in the same manner as mentioned above. ZnO was then electrodeposited on Cu2O from 0.1 M aqueous solution of ZnSO4 by applying a constant potential of −1.3 V for 5 min. These electrodes are represented as Cu–Cu2O–ZnO in the forthcoming sections. ZnO was also deposited directly onto Cu to observe the effect of ZnO on the electrochemical reduction of CO2. The last set of electrodes was prepared by the thermal oxidation of 1 × 1 cm2 copper foil in a furnace at 500 °C for 1 h, resulting in the formation of a black CuO layer on copper (Cu–CuO). However, the black CuO layer was removed, exposing reddish Cu2O beneath it (Cu-TO).
2.4. Electrochemical reduction experiments
Electrochemical reduction of CO2 was performed in a specially built two-compartment cell, each filled with 75 ml 0.1 M KHCO3.25,26 The working and Ag/AgCl reference electrodes were placed in one of the compartments, whereas a Pt counter-electrode was placed separately in the other compartment. Before each measurement, CO2 was bubbled into the cathodic compartment for 15 min to saturate the electrolyte. At first, electrochemical potential, ranging from −1.0 to −1.8 V, was applied using Cu–Cu2O as the cathode. Liquid samples were taken out from the electrochemical cell after 30 min for NMR analysis. Afterward, experiments were performed at −1.7 V for all the fabricated electrodes, including Cu–Cu2O owing to the formation of higher alcohols at this voltage. The quantification of the gaseous product was not performed in this work.3. Results and discussion
The fabricated electrodes, namely, Cu–Cu2O, Cu–Cu2O–ZnO, Cu–ZnO, and Cu-TO, were used to test the electrochemical reduction (ER) of CO2 in 0.1 M KHCO3. All the electrodes were analyzed by various techniques, including FESEM, XRD, Raman spectroscopy, and XPS before and after the ER experiments.Electrochemical experiments were first performed using Cu–Cu2O as the cathode. In these experiments, a potential of −1.0 to −1.8 V vs. Ag/AgCl was applied for 30 min each, and the samples were collected to observe the effect of the applied potential on product distribution. This electrode will be referred to as Cu–Cu2O DV in the subsequent sections, where DV stands for different voltages. The current density (j)–time (t) plots at various applied voltages are shown in Fig. 1a. The current density increased rapidly in the first 15 min, corresponding to the reduction of Cu2O, as reported in the previous studies.27 After this initial increase, the current density became constant, yielding a value of −0.15 mA cm−2 at −1.0 V. The applied potential was then increased to −1.2 V and the reaction was continued for 30 min, while CO2 was bubbled in the cathodic compartment.
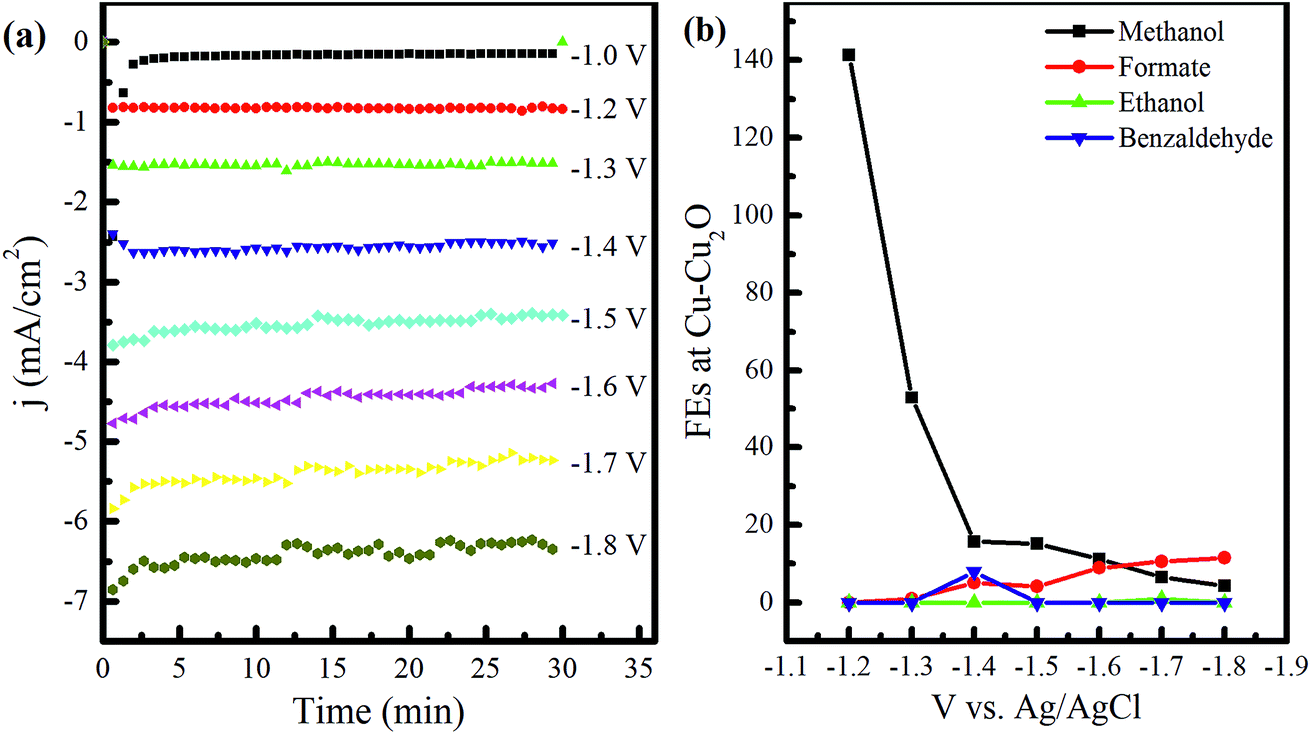 | ||
| Fig. 1 (a) j–t plots of electrochemical reduction of CO2 in 0.1 M KHCO3 at the applied potential of −1.0 to −1.8 V using Cu–Cu2O electrodes. (b) FEs of the major liquid products as a function of applied potential. | ||
The experiments were performed in the same way by increasing the cathodic potential, by a unit of 0.1 until −1.8 V, and the samples for quantitative product analysis were taken after half an hour each time. The stability of current over the experimental time exhibits the electrode's stability or a marginal deactivation due to impurities.28 In addition, the noise at higher applied voltages is attributed to the formation of bubbles at the electrode surface, resulting in current fluctuations. IR drop could be another cause of these fluctuations as it becomes difficult to maintain constant potential due to large bubbles.
The NMR analysis of the samples unveiled the formation of different compounds owing to the applied voltage (see Fig. S1 in the ESI†). The FEs of the observed products at various voltages are shown in Fig. 1b. Only methanol was witnessed at −1.0 to −1.2 V. Formate appeared at −1.3 V and its amount as well as FE kept on increasing with the voltage. However, the decrease in the total FE of the liquid products at this stage might indicate the presence of CH4, which can be produced at more negative potentials, as reported in the literature.29,30 Benzaldehyde formation was noticed at −1.4 V corresponding to the NMR peaks at 10.1 and a collection of peaks between 7.5–7.8 ppm31 with the FE of 7.9%. Acetone was also detected at this voltage, yielding a single peak at 2.2 ppm. Ethanol formation was evident at −1.7 V, indicating CH3 and CH2 peaks at 1.05 and 3.53 ppm, respectively, with the OH peak embedded in the water peak.28,32 However, the FE of ethanol was found to be 0.96% only (see Table S1 in the ESI†). Ethanol peaks vanished at −1.8 V after 30 min of further electrochemical reduction in the same electrolyte, indicating its decomposition (see Fig. S1 in the ESI†). The FE of methanol was found to be 141% at −1.2 V, revealing the involvement of chemical steps in the electrochemical reduction mechanism. A value of 240% has been reported previously by Frese et al.8 FE greater than 100% can be observed if the number of electrons for the electrocatalytic reduction of CO2 is less than 6. According to the theoretical analysis performed by Frese, the FE of 240% indicated the involvement of 3 faradaic electrons. The chemical steps proposed were the reduction of CO2 (at the expense of Cu surface) forming COads and Oads (that is equivalent to 2e− reduction to CO) and the reaction of CO with H2O to form HCOads and OHads. From this, we can predict that the number of faradaic electrons in our case is 4. Since Cu has great affinity toward oxygen, we anticipate the chemical adsorption of CO2 to form COads and Oads as a chemical step involved in methanol synthesis, since it provides a low energy pathway as compared to direct electron transfer.8
After an initial experiment (employing a voltage range from −1.0 to −1.8 V), we selected −1.7 V to perform additional electrochemical reduction experiments using other electrodes, owing to the production of C2 species at this voltage. The electrochemical CO2 reduction was carried out for 7 h in the same two-compartment cell with fresh 0.1 M KHCO3 solution as an electrolyte, and the samples were drawn after 1, 3, 5, and 7 h for NMR analyses. The NMR analysis of the samples using Cu–Cu2O and Cu–Cu2O–ZnO at −1.7 V is shown in Fig. S2.† Evidently, only methanol can be seen in both the cases after 1 h, with FEs of 60.75 and 67.22% with regard to Cu–Cu2O and Cu–Cu2O–ZnO electrodes, respectively (also see Table S2 in the ESI†). The formation of methanol using Cu-oxide-based electrodes has already been observed;15 however, there are conflicting reports demonstrating no methanol formation.24 This discrepancy can be attributed to the electrode fabrication process and experimental conditions. Moreover, it is evident that the concentration of methanol is relatively stable with the passage of time, which can be attributed to its conversion into methane or other gaseous products, thereby maintaining it at an almost constant level in the solution.33,34 Alternatively, it could be related to the modification of the active phase for methanol formation at early stages of the reduction reaction.
Formate appears after 3 h of ER on Cu–Cu2O, whereas methanol remains the only product on Cu–Cu2O–ZnO throughout the experiment. No ethanol peak was observed on Cu–Cu2O. When the only difference is that −1.7 V was directly applied rather than increasing the voltage in steps, the absence of ethanol suggests that it is not only the applied voltage responsible for its production but also the electrochemical treatment of the electrodes. Increasing the voltage in steps also leads to different FEs. For Cu–Cu2O, the FE of methanol was found to be 6.56% as the voltage was gradually increased to −1.7 V (Fig. 1b), whereas the direct application of the same voltage yielded much higher FEs (Fig. 2). Further characterization of the electrodes revealed different morphologies of the two electrodes after electrochemical experiments, which might be responsible for the absence of ethanol and differences in the FEs. This will be explained in the forthcoming section.
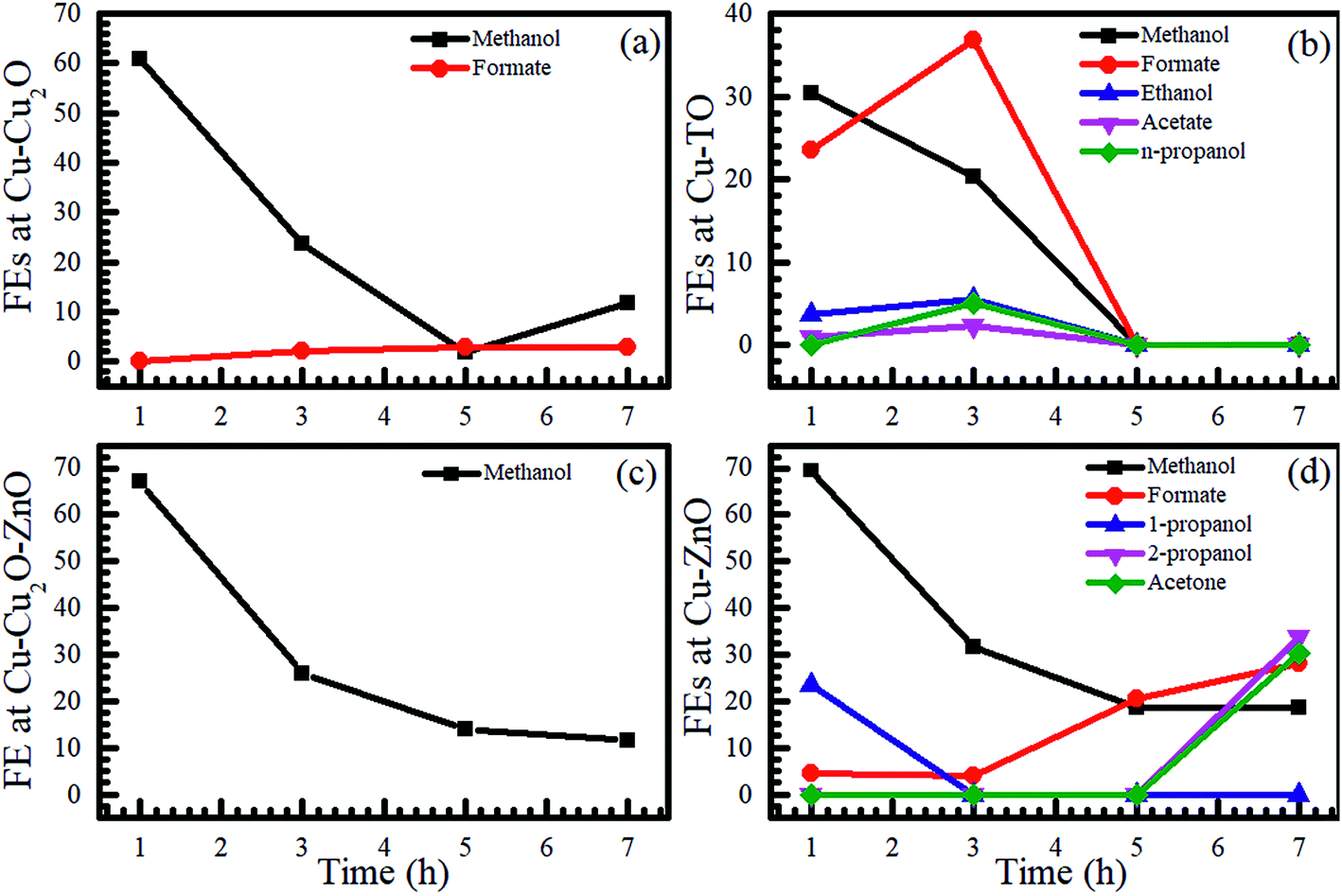 | ||
| Fig. 2 Time evolutions of the FEs of various products formed using (a) Cu–Cu2O, (b) Cu-TO, (c) Cu–Cu2O–ZnO, and (d) Cu–ZnO electrodes at −1.7 V in 0.1 M KHCO3. | ||
To investigate the influence of the substrate-ZnO interface on product distribution, Cu–ZnO electrodes were also employed for the electrochemical reduction of CO2 at −1.7 V for 7 h and samples were taken after 1, 3, 5, and 7 h. Fairly interestingly, we observed the formation of n-propanol/2-propanol and acetone along with formate and methanol (Fig. 2 and S3†). The FE of 2-propanol after 7 h was 33.65%, whereas that of acetone was 30.13% (also see Table S2 in the ESI†). These results reveal that the morphology, composition, and interface have a fundamental role in determining the product distribution of CO2 reduction reaction. The formation of n-propanol was reported recently using graphene/ZnO/Cu2O electrocatalyst35 with FE of 30%, indicating that it is not unlikely to obtain C3 compounds with high FE using these catalysts.
We also investigated the effect of thermally prepared Cu-TO electrodes on the electrochemical reduction of CO2 and compared them with Cu–Cu2O electrodes prepared via the electrodeposition method. It was found that Cu-TO produced C2 and C3 compounds, i.e., ethanol, acetate, and n-propanol, in addition to methanol and formate. The total FE of the liquid products was found to be 58.51% after 1 h, approximately equal to the total FE observed at Cu–Cu2O (60.75%). However, the total FE of the liquid products decreased to 25.68% on Cu–Cu2O after 3 h, while that of Cu-TO increased to 64.94%, revealing the promising potential of thermally oxidized Cu electrodes to produce liquid fuels as compared to the electrodeposited one. The formation of ethanol and acetate has been reported previously on thermally oxidized copper surfaces,24 where the formation of C2 compounds is attributed to the grain boundaries, which can support the surface structures that are not stable enough for individual nanocrystals.24
It should be noted that none of the C2 species was produced on Cu–ZnO, but C3 compounds were formed with good FE, yielding faster C–C–C coupling on the latter electrode. We believe that the Cu–ZnO interface plays a fundamental role in the formation of C3 species.
Summarizing the NMR results, we observe that the highest FE of the liquid products was observed on Cu–ZnO, including C1 (methanol, formate) and C3 species (propanol, acetone). In addition, only C1 products were witnessed on Cu–Cu2O and Cu–Cu2O–ZnO at −1.7 V, where the latter electrode was found to be the most selective for methanol. C2 species (ethanol and acetate) were observed only on Cu-TO and Cu–Cu2O DV. Further, n-propanol was also formed on Cu-TO after 3 h. In the forthcoming section, we will investigate the composition and morphology of the electrodes before and after electrochemical reduction of CO2 to get an insight into the mechanistic pathways leading to these products.
3.1. Characterization of electrodes
To explore the factors governing the formation and selectivity of C1, C2, and C3 compounds, electrodes were characterized with X-ray photoelectron spectroscopy, Raman spectroscopy, and X-ray diffraction. FESEM images were also taken before and after the electrochemical reduction of CO2 to obtain a morphological insight.Cu 2p XPS of Cu–Cu2O electrodes before and after electrochemical reduction is shown in Fig. 3a. The Cu 2p XPS spectrum taken from as-prepared Cu–Cu2O shows main peaks and satellite features that are characteristic to Cu2O. A substantial decline in the intensity of Cu 2p peaks can be observed in the electrodes after ER at −1.7 V for 7 h. The emergence of high binding energy at 934 eV and the satellite features indicate the formation of CuO after the electrochemical reduction experiment. This might be the result of the interaction mechanism of CO with the electrodes via oxygen atoms, which can later lead to the formation of methanol and other alcohols, leaving behind oxidized copper. Moreover, we also observed the blackening of the electrodes at certain parts after electrochemical reduction, which probably corresponds to the formation of CuO. The oxidation of Cu surface (because of CO2 interaction) and its stability have been addressed previously by Frese et al., revealing that the oxide layer on Cu is observable as the rate of cathodic decomposition is generally slow.8 It should be noted that any air oxidation of the electrode usually results in the formation of a reddish Cu(I) oxide layer and not Cu(II) oxide. Moreover, the blackish Cu(II) oxide in our case was formed during the electrochemical reaction, as observed from the color changes during the experiment.
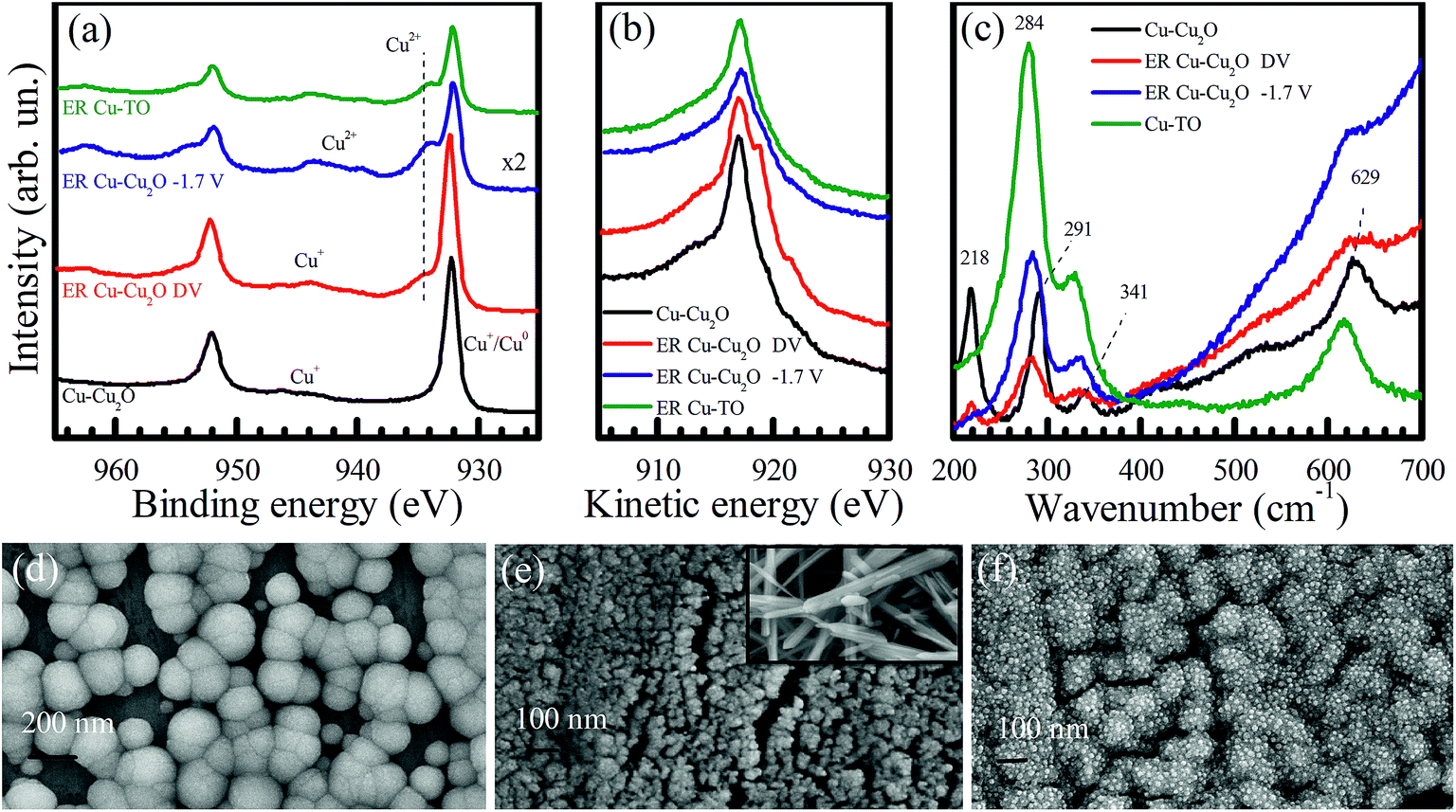 | ||
| Fig. 3 (a) Cu 2p XPS, (b) Cu LMM Auger electron, and (c) Raman spectra of Cu–Cu2O before ER (black), ER Cu–Cu2O–DV after gradually reaching to −1.7 V (red), ER Cu–Cu2O at −1.7 V (blue), and ER Cu-TO (green). FESEM images of Cu–Cu2O (d) before ER, (e) after ER of 7 h at −1.7 V (inset shows black region of the electrode), and (f) after ER at different voltages. The images are taken at 150 kX magnification. | ||
The Cu–Cu2O electrode, which is subjected to increasing voltage from −1.0 to −1.8 V for 30 min each (labelled as Cu–Cu2O–DV), yields a slightly increased intensity of Cu 2p peaks, employing the formation of metallic copper. However, it should be noted that the 2p1/2 and 2p3/2 peaks of Cu0 and Cu+ overlap with each other; therefore, it is difficult to distinguish between them.11 The XPS of the Cu-TO electrode after the reaction also shows more electrochemically reduced copper as compared to Cu–Cu2O, owing to the lower Cu2+ to main peak ratio. Both Cu–Cu2O–DV and Cu-TO also exhibit the 2p3/2 peak at 934 eV, which is characteristic of Cu2+, suggesting that CuO is formed in these electrodes, too (like Cu–Cu2O). A complete XPS analysis of these electrodes evidenced an increase in the intensity of O 1s peaks (particularly in ER involving Cu–Cu2O), and therefore, confirms the proposition of CO2 interaction to the substrate via oxygen atoms, leaving behind surface-bound oxygen in the end (Fig. S4†).
Cu LMM Auger spectra is more illustrative in terms of identifying Cu0 and Cu+ species. As shown in Fig. 3b, a gradual cathodic potential increase would induce spectral changes that are attributable to the formation of Cu0 on the Cu–Cu2O electrode. The more pronounced peaks at 918.8 and 921.6 eV indicate reduction.36 Hence, both CuO and Cu0 existed in our electrochemically reduced electrodes. Cu LMM Auger spectra taken from the Cu–Cu2O electrode after the direct application of −1.7 V and Cu-TO electrode refer primarily to CuO structures on the electrode surfaces.
To further investigate the formation of CuO on the electrode surfaces, we obtained their Raman spectra before and after the electrochemical reduction experiments (Fig. 3c). Four major peaks positioned at 218, 291, 341, and 629 cm−1 are observed in the Cu–Cu2O electrode. The peaks at 284, 334, and 620 cm−1 correspond to CuO after the electrochemical reduction of CO2.37–39 CuO peaks were confirmed by taking the Raman spectrum of the black region (CuO) of the thermally prepared electrode. The peaks of this region matched well with the peaks observed in the electrochemically reduced electrodes, thereby validating our XPS results. Moreover, a decreased intensity of the peak at 218 cm−1 also indicates the disappearance of Cu2O from the electrode surface.
Keeping in view the findings of the XPS and Raman spectroscopy, we discuss the morphology of the electrodes using FESEM. A high magnification image of Cu–Cu2O before ER shows spherical nanoparticles of Cu2O of the size of around 200 nm (Fig. 3d). Most of the particles have exposed (100) faces having four sides, while some of them are pentagonal. After reaction for 7 h, these nanospheres are transformed into highly dispersed, much smaller nanoparticles (Fig. 3e). The inset of Fig. 3e shows the FESEM image of the blackened region on the Cu–Cu2O electrode after ER, confirming the presence of CuO nanorods.
Interestingly, Cu–Cu2O DV electrode has different morphology (Fig. 3f). It reveals that Cu2O nanospheres are not completely disintegrated and display small embedded particles of around 16 nm. It is anticipated that these are metallic Cu0 nanoparticles, since their high content is evidenced from the XPS and Raman spectra.
Fig. 4 shows the FESEM of Cu-TO electrodes before and after subjecting them to −1.7 V. The Cu-TO electrodes contain a mix of CuO (black) and Cu2O (reddish), despite the fact that most of the black CuO fell off before electrochemical reduction. The black region (CuO) under FESEM was found to comprise needle-like structures of various lengths.32,40 Underneath these needle-like structures, Cu2O nanoparticles are present.41Fig. 4b shows the portion of the electrode with no black spots and is composed of particles of around 200 nm.24 ER involving Cu-TO indicates that the particle size is reduced to half, i.e., 100 nm. Most of the nanoparticles are octahedral and edge- or corner-truncated octahedra with (111) exposed planes.42–44 The needle-like structures, probably corresponding to CuO, were also observed at some parts of the electrode. Recalling that we observed the formation of C2 compounds (ethanol and acetate) only on Cu–Cu2O DV and Cu-TO, it is important to address similar features of these electrodes, which might be responsible for C–C coupling. For example, an elemental analysis from the XPS data indicated a lesser amount of CuO on Cu-TO and relatively smaller amount Cu–Cu2O on DV as compared to ER involving Cu–Cu2O at 1.7 V (see Table S3 in the ESI†).
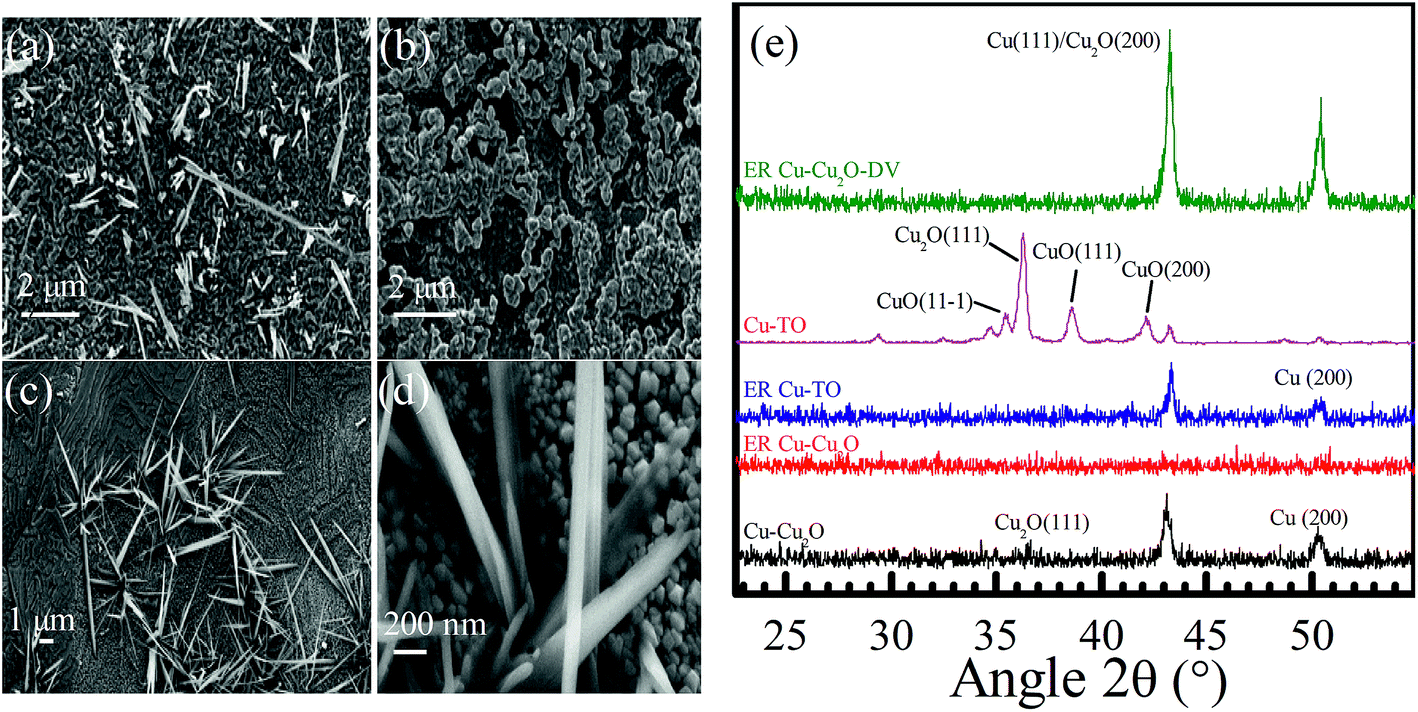 | ||
| Fig. 4 Cu-TO before ER (a) at 10 kX, (b) at 20 kX after ER of 4 h at −1.7 V, (c) at 10 kX, and (d) at 100 kX. (e) XRD spectra of Cu–Cu2O, ER–Cu–Cu2O, ER Cu–Cu2O DV, and ER Cu-TO. | ||
Grazing incidence XRD was performed to obtain an insight into the crystal structure of the electrodes before and after initiating the electrochemical reduction of CO2. The peaks observed at 36.6, 36.2, 38.6, 42.2, 43.1, and 50.3° are attributed to Cu2O(111), Cu2O(111), CuO(111), CuO(200), Cu2O(200), and Cu(200), respectively (Fig. 4e).42,45–47 After the electrochemical reduction of CO2 at −1.7 V, the intensities of all the peaks are completely diminished, which can be interpreted as the conversion of crystalline Cu2O to an amorphous form and partial reductions due to surface reactions. Thermally oxidized electrodes, contrary to ER involving Cu–Cu2O, shows the disappearance of the Cu2O(111) peak after reaction, but Cu(111) and Cu(200) are preserved, indicating the enrichment of Cu0. Similarly, the ER involving Cu–Cu2O DV displayed the escalation of (111) and (200) peaks, yielding even higher metallic Cu content in these electrodes after electrochemical reduction, approving our assessments from XPS and Raman spectroscopy. Recalling that only Cu-TO and Cu–Cu2O DV were capable of producing C2 species, we can infer that C–C coupling is favored on Cu0 rather than Cu2O.
In other words, most of the Cu2O was converted into metallic Cu0. It seems as if metallic Cu grains were somehow responsible for the formation of C2 species. This proposition was further confirmed from the XRD pattern of these electrodes and will be demonstrated in the forthcoming discussion.
XPS spectra were also taken for Cu–Cu2O–ZnO and Cu–ZnO electrodes before and after the electrochemical reduction of CO2 at −1.7 V. The Zn 2p3/2 region is shown in Fig. 5a. The Zn 2p3/2 peaks located at 1022 for Cu–Cu2O–ZnO electrodes are characteristic of ZnO, which are reduced in intensity and shifted toward a lower binding energy of 1021.4 eV after ER, indicating changes in the valence of Zn to more reduced values. As compared to Cu–Cu2O–ZnO, the Zn 2p3/2 XPS peak of Cu–ZnO (before ER) shows a considerably shifted Zn-2p3/2 peak (1021.8 eV), which might be due to the metallic Cu substrate instead of Cu2O, indicating the presence of Zn0 along with Zn2+.48 In addition, complete reduction to metallic Zn is not observed after ER, which is contrary to ER involving Cu–Cu2O–ZnO electrodes. Recalling the formation of C3 compounds on Cu–ZnO, we anticipate that the Cu–Zn interface has a fundamental role in faster C–C–C coupling.
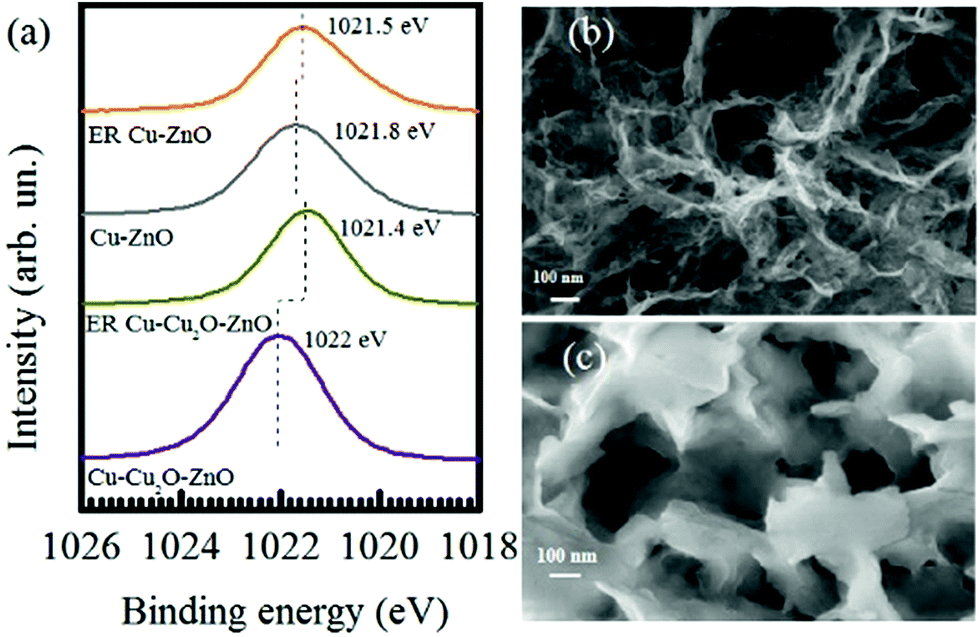 | ||
| Fig. 5 (a) Zn-2p3/2 XPS spectra of Cu–Cu2O–ZnO and Cu–ZnO electrodes before and after the electrochemical reduction of CO2 and FESEM images of Cu–Cu2O–ZnO (b) before ER and (c) after ER of 7 h at −1.7 V. The images are taken at 150 kX magnification. | ||
The FESEM of Cu–Cu2O–ZnO (Fig. 5b and c) electrodes exhibits a sheet-like structure of ZnO before electrochemical reduction, which are transformed into thicker grooved nanosheets after reaction.49–51
The structure of Cu–ZnO before and after electrochemical reduction ER is of prime importance (shown in Fig. 6). Different morphologies have been observed on ER involving Cu–ZnO electrodes. For example, we observed highly organized nanoflowers in some areas. The EDX of this portion depicts the presence of Zn and O only (see Fig. S5 in the ESI†). According to the previous studies, the formation of ZnO nanoflowers proceeds via the precipitation of Zn(OH)2, which is dissolved to a considerable extent in water in the form of Zn(OH)42−.52–54 ZnO nanoflowers are developed from Zn(OH)42− when its concentration is increased in the solution above a critical value.54 The formation of ZnO nanoflowers during the electrochemical reduction of CO2 can be favored due to the basic pH (8.4) of the electrolyte, resulting in the initial formation of Zn(OH)2. However, we did not observe ZnO nanoflowers on the as-prepared Cu–ZnO electrodes as the pH was less than 7 under electrodeposition conditions. Fig. 6d shows another image of the Cu–ZnO electrode after ER, which yields the growth of nanoflowers at an intermediate stage.55 It should be noted that nanoflowers were localized to some areas. The EDX of other regions indicate the presence of metallic Zn with almost vanished O (see Fig S5 in the ESI†). The localized formation of nanoflowers can be a consequence of C3 species forming at the electrode surface.
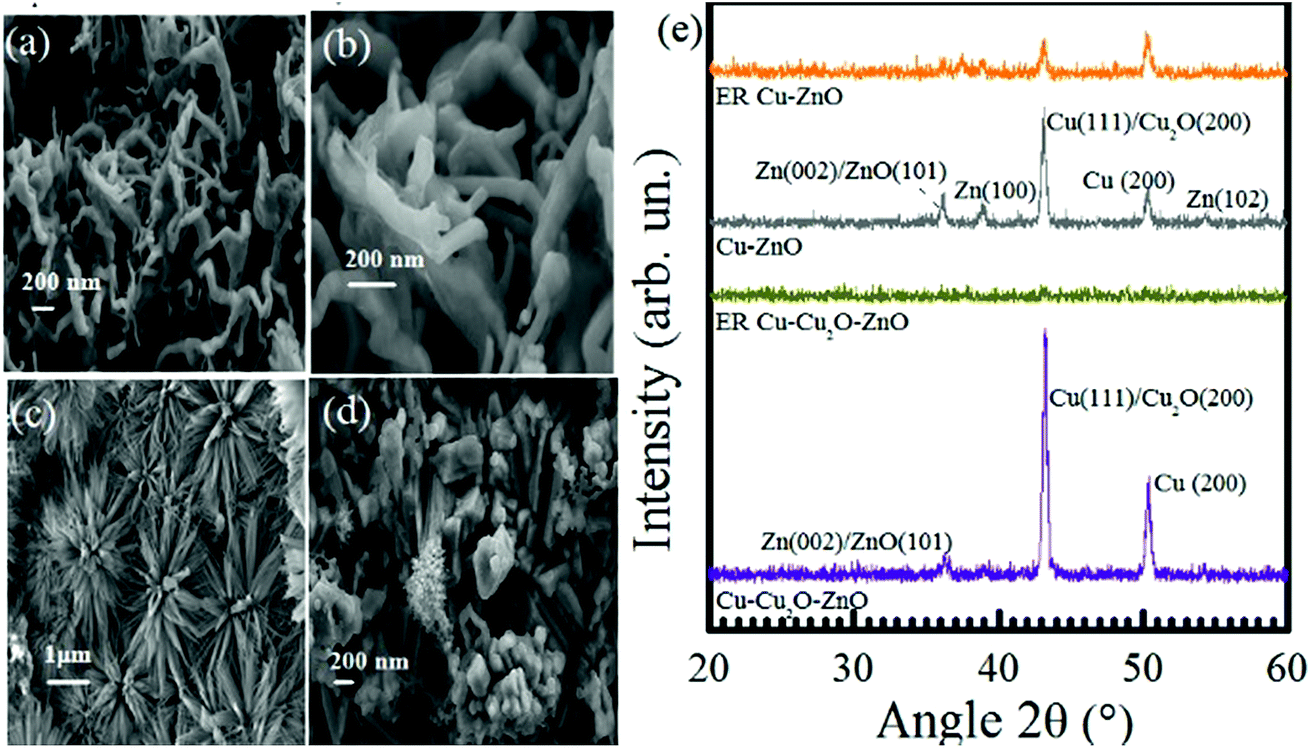 | ||
| Fig. 6 FESEM images of Cu–ZnO, (a and b) before ER at 50 kX and 150 kX, respectively (c and d) after ER of 7 h at −1.7 V at 30 kX and 50 kX, respectively. (e) XRD spectra of Cu–Cu2O–ZnO, ER-Cu-Cu2O–ZnO, Cu–ZnO, and ER Cu–ZnO. | ||
The XRD pattern of Cu–ZnO and Cu–Cu2O–ZnO before and after subjecting to −1.7 V for 7 h is shown in Fig. 6e. Four peaks are located for the Cu–ZnO electrode, out of which three correspond to Zn(102), Zn(100), and Zn(002) or ZnO(101), while two represent Cu(200) and Cu(111) or Cu2O(200) planes.56–58 After the ER of CO2, Cu–Cu2O–ZnO electrodes display complete conversion to the amorphous phase like that of Cu–Cu2O, while the diffraction peaks of Cu–ZnO are decreased in intensity but still visible. Therefore, the XRD pattern of electrodes forming only C1 species (Cu–Cu2O, Cu–Cu2O–ZnO) possess the same XRD features, i.e., conversion from the crystalline to amorphous form is witnessed after the electrochemical reduction of CO2. On the other hand, XRD patterns of the electrodes producing C2 species (Cu-TO, Cu–Cu2O DV) are related to each other, indicating the presence of metallic copper at the end of the reaction.
3.2. Proposed mechanisms
The distribution of liquid products at various electrodes as depicted by H-NMR reveals the formation of C1 compounds (formate and methanol) on all the electrodes used in this study. However, considering CO2 reduction at −1.7 V, C2 compounds (i.e., ethanol and acetate) were observed on Cu-TO and Cu–Cu2O DV, whereas C3 compounds (propanol and acetone) were produced in appreciable amounts at the Cu–ZnO surface.Methanol was a major product, along with a little formate, on Cu–Cu2O electrodes. The mechanism of the formation of C1 products (methanol and formate) has been documented by previous researchers and is thought to proceed via formyl radical, HCOads on Cu(111).11,59 In addition, the conversion of Cu2O to metallic Cu is also documented during the electrochemical reduction of CO2 by various groups. However, our Cu2O nanoparticles are not octahedral, but they rather have (100) exposed faces. In addition, we witnessed the formation of CuO on the electrode surface after the electrochemical reaction,8 as evidenced by the XPS and Raman spectra. This led us to propose a different reduction mechanism involving the interaction of CO2 molecule via O atoms (Scheme 1b). Subsequent electron and proton additions would lead to the formation of methanol and evolution of H2. It can also be speculated that single e−/H+ transfer to the surface methoxy group might lead to the production of methane, leaving behind O. The O left on the Cu surface ultimately results in the formation of CuO. Formate can be formed in the initial steps via the reduction of the adsorbed anion radical, but since it is observed in a very small amount as compared to methanol, the overall result will be the oxidation of Cu to CuO according to the proposed mechanism.
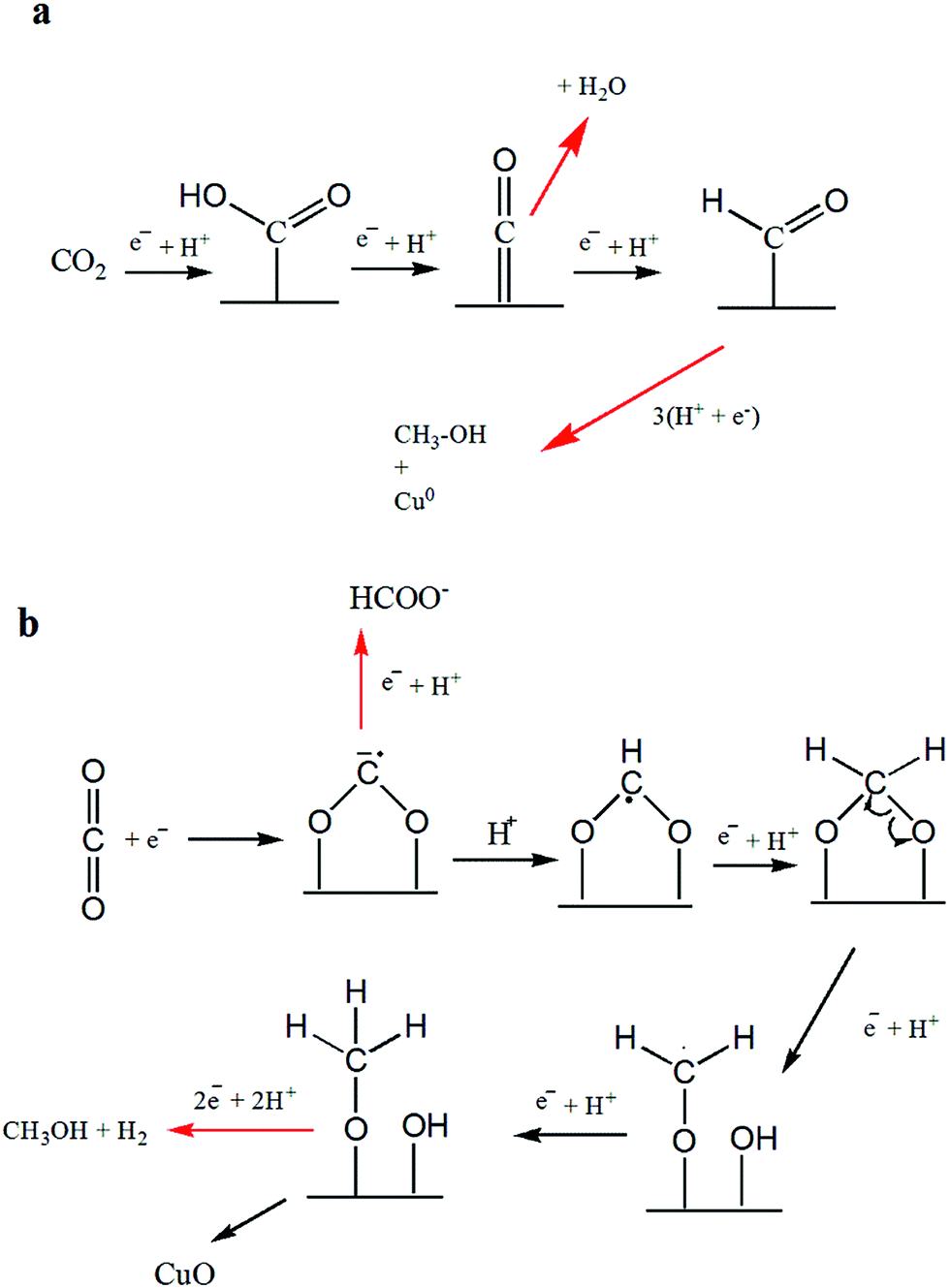 | ||
| Scheme 1 Formation mechanism of C1 products: (a) methanol formation on Cu-TO electrode at −1.7 V and as reported in literature, (b) methanol and formate on Cu–Cu2O leaving behind CuO. | ||
It should be noted that the amount of detected CuO is more on the Cu–Cu2O subjected to −1.7 V, as compared to Cu–Cu2O DV and Cu-TO as explained previously, despite the fact that methanol is produced in all of them. This can be attributed to the structural variations among them. Cu-TO shows more octahedral, and edge- or corner-truncated octahedra with exposed (111) faces, when analyzed under FESEM (Fig. 4d), which are known to favor a different mechanistic pathway for methanol formation as reported in previous studies, and therefore, do not result in surface oxidation responsible for the formation of CuO.59 The methanol formation in this case is accomplished by the attachment of CO2via C atom instead of O, which is thought to be a probable pathway on the (111) surface, ultimately leaving metallic Cu (Scheme 1a). In addition, the higher FE of the formate on Cu-TO might also be a contributing factor for observing a lesser percentage of CuO. Similarly, Cu–Cu2O DV electrode also indicates a lesser percentage of CuO and more Cu0 after electrochemical reduction. Although we are unable to see the exposed faces in this case, but XRD reveals a similar pattern for both Cu-TO and Cu–Cu2O DV electrodes, suggesting similar mechanistic pathways leading to methanol.
NMR shows methanol as the sole product on the Cu–Cu2O–ZnO electrode. In this case, contrary to Cu–Cu2O in which oxidation took place, the XPS results reveal the reduction of ZnO, implying that the interaction of CO2via O atoms is not involved. Hence, a similar mechanism to that of Cu-TO is expected (Scheme 1a).
C2 products, i.e., ethanol and acetate, were produced in small amounts at the surface of Cu-TO. The reduction of CO2 to C2 compounds requires C–C coupling, which is predicted to occur on the (100) surface.59 C–C coupling can take place after two HCO molecules are adsorbed side by side.59 Since adsorbed HCO species are also formed for CO2 reduction to methanol, it is quite possible that they undergo C–C coupling by the addition of 2e−s and produce ethanol or acetate by subsequent steps, as shown in Scheme 2.
 | ||
| Scheme 2 Mechanistic pathway leading to the formation of C2 compounds, ethanol and acetate. | ||
C3 compounds, i.e., propanol and acetone, were produced with good FEs on Cu–ZnO electrodes requiring 18 and 16e−s, respectively. The C3 pathway requires the coupling of three C atoms. The probable mechanism for the electrochemical reduction of CO2 to n-propanol/2-propanol and acetone is shown in Scheme 3. The steps are similar to the mechanism predicted for ethanol until C–C bond formation. At this point, HCOads (formed by the addition of 3e−, 3H+ to CO2) is coupled with C2 species to form a C3 adsorbed intermediate. From here onwards, multiple pathways could lead to the formation of n-propanol/2-propanol and acetone. The electrochemical steps with 18 and 16e−s leads to the formation of n-propanol/2-propanol and acetone, respectively, leaving behind metallic Zn. The second pathway involves a chemical step (equivalent to 1e− transfer) at the end, resulting in the formation of Zn(OH)2 and explains the formation of localized ZnO nanoflowers, as shown by the FESEM images.
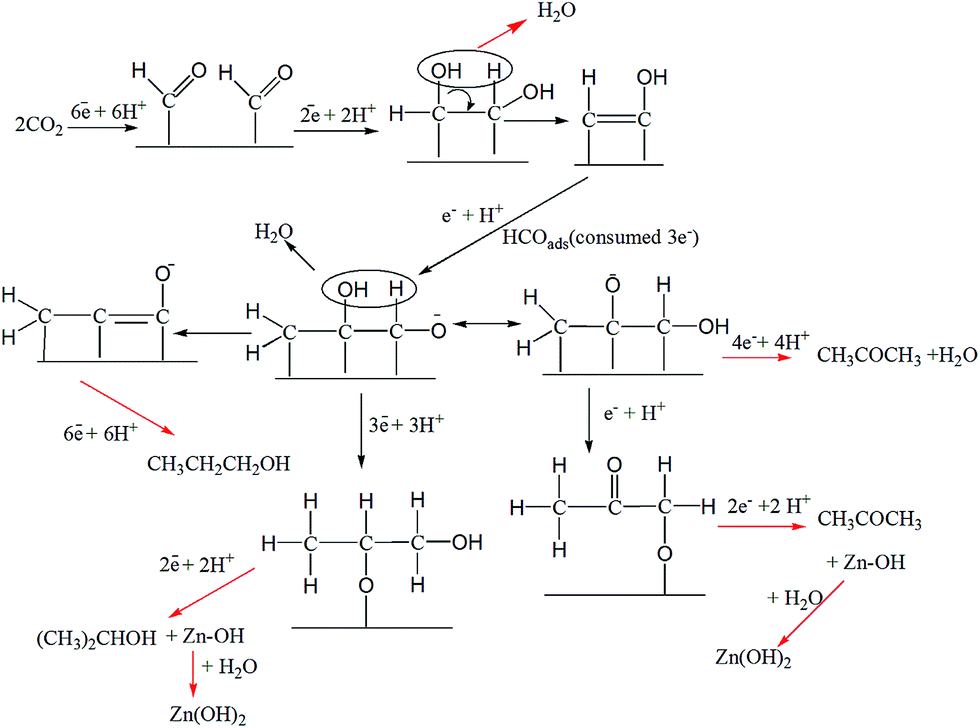 | ||
| Scheme 3 Mechanistic pathways leading to the formation of 2-propanol and acetone on Cu–ZnO. | ||
4. Conclusions
A comparative study of Cu–Cu2O, Cu–Cu2O–ZnO, Cu–ZnO, and Cu-TO electrodes for the electrochemical reduction of CO2 at −1.7 V established the formation of C1 compounds at Cu–Cu2O and Cu–Cu2O–ZnO. Moreover, the composite electrode Cu–Cu2O–ZnO displayed the highest selectivity for methanol production. Electrodeposited Cu–ZnO was found to be the most efficient toward producing higher alcohols, yielding a total of 97.4% FE of liquid products (methanol, formate, propanol, and acetone). We believe that the chemistry of the Cu–Zn interface plays a fundamental role in the production of C3 species. The total FE of 58.5% was witnessed on Cu-TO, which is in good agreement with the literature (57%).24 The elemental and morphological characterizations of the cathode materials reveal that methanol formation proceeds via the O coordination of CO2 to Cu–Cu2O surface having (100) facets, leaving behind CuO at the end, whereas C coordination is favored on Cu-TO with (111) exposed facets, resulting in Cu0 sites. The localized appearance of ZnO nanoflowers on the Cu–ZnO electrode after electrochemical reduction is the result of mechanistic pathways leading to propanol and acetone involving a chemical step equivalent to one electron transfer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Shamsa Munir thanks to the Scientific and Technological Research Council of Turkey (TUBITAK), Sarp Kaya thanks to The Science Academy and Turkish Academy of Sciences (TUBA) for financial support. Authors would like to thank TARLA for the collaborative research support.References
- X. Xiaoding and J. Moulijn, Energy Fuels, 1996, 10, 305–325 CrossRef.
- R. Kas, R. Kortlever, A. Milbrat, M. T. Koper, G. Mul and J. Baltrusaitis, Phys. Chem. Chem. Phys., 2014, 16, 12194–12201 RSC.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef.
- R. Kas, K. K. Hummadi, R. Kortlever, P. de Wit, A. Milbrat, M. W. Luiten-Olieman, N. E. Benes, M. T. Koper and G. Mul, Nat. Commun., 2016, 7, 10748 CrossRef PubMed.
- G. Lu, H. Wang, Z. Bian and X. Liu, Sci. World J., 2013, 2013, 424617 Search PubMed.
- G. Ghadimkhani, N. R. de Tacconi, W. Chanmanee, C. Janaky and K. Rajeshwar, Chem. Commun., 2013, 49, 1297–1299 RSC.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek and J. F. Sanz, Science, 2014, 345, 546–550 CrossRef PubMed.
- K. W. Frese, J. Electrochem. Soc., 1991, 138, 3338–3344 CrossRef.
- M. Le, M. Ren, Z. Zhang, P. T. Sprunger, R. L. Kurtz and J. C. Flake, J. Electrochem. Soc., 2011, 158, E45–E49 CrossRef.
- S. Singh, L. M. Aeshala and A. Verma, International Journal of Innovative Research and Development, 2012, 1, 155–160 Search PubMed.
- Y. Lan, S. Ma, J. Lu and P. J. Kenis, Int. J. Electrochem. Sci., 2014, 9, 7300–7308 Search PubMed.
- R. S. Kumar, S. S. Kumar and M. A. Kulandainathan, Electrochem. Commun., 2012, 25, 70–73 CrossRef.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- W. DA HYE, C. H. Choi, J. Chung and S. I. Woo, Appl. Catal., B, 2014, 158, 217–223 Search PubMed.
- J. Albo, M. Alvarez-Guerra, P. Castaño and A. Irabien, Green Chem., 2015, 17, 2304–2324 RSC.
- K. Rajeshwar, N. R. de Tacconi, G. Ghadimkhani, W. Chanmanee and C. Janáky, ChemPhysChem, 2013, 14, 2251–2259 CrossRef PubMed.
- H. O. Finklea, 1988.
- M. V. B. Zanoni, J. J. Sene and M. A. Anderson, J. Photochem. Photobiol., A, 2003, 157, 55–63 CrossRef.
- S. Ikeda, K. Ito and H. Noda, 2009.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Nat. Commun., 2014, 5 Search PubMed.
- D. Raciti, K. J. Livi and C. Wang, Nano Lett., 2015, 15, 6829–6835 CrossRef PubMed.
- S. Ma, M. Sadakiyo, R. Luo, M. Heima, M. Yamauchi and P. J. Kenis, J. Power Sources, 2016, 301, 219–228 CrossRef.
- V. Yadav and M. Purkait, Energy Fuels, 2015, 29, 6670–6677 CrossRef.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef PubMed.
- J. Bugayong and G. L. Griffin, ECS Trans., 2015, 66, 61–65 CrossRef.
- C.-C. Tsai, J. Bugayong and G. L. Griffin, 2012.
- D. Ren, Y. Deng, A. D. Handoko, C. S. Chen, S. Malkhandi and B. S. Yeo, ACS Catal., 2015, 5, 2814–2821 CrossRef.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- X. Sun, X. Cao and P. Hu, Sci. China: Chem., 2015, 58, 553–564 CrossRef.
- C.-C. Tsai, J. Bugayong and G. L. Griffin, MRS Online Proc. Libr., 2012, 1446 Search PubMed.
- V. Polshettiwar and R. S. Varma, Org. Biomol. Chem., 2009, 7, 37–40 RSC.
- C. S. Chen, J. H. Wan and B. S. Yeo, J. Phys. Chem. C, 2015, 119, 26875–26882 CrossRef.
- T. Mizuno, A. Naitoh and K. Ohta, J. Electroanal. Chem., 1995, 391, 199–201 CrossRef.
- S. Kaneco, M. Yabuuchi, H. Katsumata, T. Suzuki and K. Ohta, Fuel Chem. Div. Prepr, 2002, 47, 71–72 Search PubMed.
- R. Geioushy, M. M. Khaled, K. Alhooshani, A. S. Hakeem and A. Rinaldi, Electrochim. Acta, 2017, 245, 456–462 CrossRef.
- S. Poulston, P. Parlett, P. Stone and M. Bowker, Surf. Interface Anal., 1996, 24, 811–820 CrossRef.
- L. Debbichi, M. Marco de Lucas, J. Pierson and P. Kruger, J. Phys. Chem. C, 2012, 116, 10232–10237 CrossRef.
- C.-H. Tsai, P.-H. Fei and C.-H. Chen, Materials, 2015, 8, 5715–5729 CrossRef PubMed.
- W. Wang, P. Zhang, L. Peng, W. Xie, G. Zhang, Y. Tu and W. Mai, CrystEngComm, 2010, 12, 700–701 RSC.
- M. Ma, K. Djanashvili and W. A. Smith, Phys. Chem. Chem. Phys., 2015, 17, 20861–20867 RSC.
- S. Choopun, N. Hongsith and E. Wongrat, in Nanowires, InTech, 2010 Search PubMed.
- Y.-H. Won and L. A. Stanciu, Sensors, 2012, 12, 13019–13033 CrossRef PubMed.
- M.-C. Kim, S.-J. Kim, S.-B. Han, D.-H. Kwak, E.-T. Hwang, D.-M. Kim, G.-H. Lee, H.-S. Choe and K.-W. Park, J. Mater. Chem. A, 2015, 3, 23003–23010 RSC.
- A. K. Mishra and D. Pradhan, Cryst. Growth Des., 2016, 16, 3688–3698 CrossRef.
- L. Xu, X. Chen, Y. Wu, C. Chen, W. Li, W. Pan and Y. Wang, Nanotechnology, 2006, 17, 1501 CrossRef.
- A. Rittermeier, S. Miao, M. K. Schröter, X. Zhang, M. W. van den Berg, S. Kundu, Y. Wang, S. Schimpf, E. Löffler and R. A. Fischer, Phys. Chem. Chem. Phys., 2009, 11, 8358–8366 RSC.
- C.-H. Chen, T. Yamaguchi, K.-i. Sugawara and K. Koga, J. Phys. Chem. B, 2005, 109, 20669–20672 CrossRef PubMed.
- F.-H. Wang, K.-N. Chen, C.-M. Hsu, M.-C. Liu and C.-F. Yang, Nanomaterials, 2016, 6, 88 CrossRef PubMed.
- N. M. Al-Hada, E. B. Saion, A. H. Shaari, M. A. Kamarudin, M. H. Flaifel, S. H. Ahmad and S. A. Gene, PLoS One, 2014, 9, e103134 CrossRef PubMed.
- K.-H. Kim, B. Kumar, K. Y. Lee, H.-K. Park, J.-H. Lee, H. H. Lee, H. Jun, D. Lee and S.-W. Kim, Sci. Rep., 2013, 3, 2017 CrossRef PubMed.
- M. Wu, H. Ye, F. Zhao and B. Zeng, Sci. Rep., 2017, 7, 39778 CrossRef PubMed.
- R. Wahab, S. Ansari, Y. Kim, H. Seo, G. Kim, G. Khang and H.-S. Shin, Mater. Res. Bull., 2007, 42, 1640–1648 CrossRef.
- S. Hussain, T. Liu, M. Kashif, L. Lin, S. Wu, W. Guo, W. Zeng and U. Hashim, Mater. Sci. Semicond. Process., 2014, 18, 52–58 CrossRef.
- J. Fan, T. Li and H. Heng, Bull. Mater. Sci., 2016, 39, 19–26 CrossRef.
- N. Kıcır, T. Tüken, O. Erken, C. Gumus and Y. Ufuktepe, Appl. Surf. Sci., 2016, 377, 191–199 CrossRef.
- P. S. da Silva, J. M. Maciel, K. Wohnrath, A. Spinelli and J. R. Garcia, 2013.
- O. Lupan, L. Chow, G. Chai and H. Heinrich, Chem. Phys. Lett., 2008, 465, 249–253 CrossRef.
- J.-H. Lin, R. A. Patil, R. S. Devan, Z.-A. Liu, Y.-P. Wang, C.-H. Ho, Y. Liou and Y.-R. Ma, Sci. Rep., 2014, 4, 6967 CrossRef PubMed.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00258d |
| This journal is © The Royal Society of Chemistry 2018 |