DOI:
10.1039/C8BM01096J
(Paper)
Biomater. Sci., 2019,
7, 104-112
One-month zero-order sustained release and tumor eradication after a single subcutaneous injection of interferon alpha fused with a body-temperature-responsive polypeptide†
Received
7th September 2018
, Accepted 24th November 2018
First published on 27th November 2018
Abstract
Most therapeutic proteins except antibodies necessitate frequent dosing at high concentrations due to their short circulation half-lives, leading to limited therapeutic efficacy, serious adverse side effects and poor patient compliance. Herein we report a strategy of thermoresponsive polypeptide fusion to genetically engineer a super-long-acting interferon alpha fused with a body-temperature-responsive polypeptide. After a single subcutaneous injection in a mouse model, this interferon alpha can in situ form a depot to show a one-month zero-order sustained release, which would enable a once-trimonthly dosing in humans. As a result, it exhibits greatly enhanced tumor accumulation and tumor eradication as well as substantially improved tolerability and biosafety. This strategy provides a promising solution to dramatically enhance the pharmacological performance of therapeutic proteins with short circulation half-lives while reducing the side effects.
1. Introduction
Protein therapeutics has played an increasingly important role in the treatment of various diseases, especially oncology and immune-mediated inflammatory diseases.1,2 However, most of the therapeutic proteins need to be administered frequently at high concentrations due to their short circulation half-lives, resulting in limited therapeutic efficacy and undesirable side effects as well as increased treatment cost and reduced patient compliance.3,4 The poly(ethylene glycol) (PEG) conjugation of proteins, called PEGylation, is one of the most frequently used and effective strategies to extend their circulation half-lives, typically by increasing the hydrodynamic diameter of proteins to reduce renal clearance, improving the in vivo stability, and reducing the immune response.5–9 So far 16 PEGylated proteins have been approved in the world, and much more are in clinical trials.9 However, the PEGylation of proteins has its inherent drawbacks, such as a deficiency in site-selectivity, multiple-step chemical reactions, complicated purification processes, and the lack of a precisely defined polymer length. Alternatively, the genetic fusion of therapeutic proteins with long-circulating proteins such as human serum albumin (HSA) and the fragment crystallizable (Fc) domain of antibodies is another effective approach to extend their circulation half-lives.10 Indeed, 1 HSA and 11 Fc fusion proteins have been approved by the FDA.11 However, HSA fusion usually significantly reduces the activity of the fused therapeutic proteins,12 which may offset the half-life extension.13 Fc fusion also has its own problems, such as immunogenicity, reduced activity and stability, and heterogeneous glycosylation.14 Recently, randomly coiled polypeptides have been genetically fused with therapeutic proteins to improve their in vivo half-lives, called polypeptide fusion, including XTENylation,15 PASylation,16 and ELPylation.17 Nevertheless, the half-life extension by polypeptide fusion is inferior to those by PEGylation, HSA fusion, and Fc fusion. Typically, these half-life extension strategies can prolong the in vivo half-life of therapeutic proteins up to 1–2 days in mouse models, thereby reducing the dosing frequency to once a week. However, it remains a tremendous challenge to further significantly improve the pharmacokinetics using the current half-life extension strategies.
To address this challenge, we combined the half-life extension with thermoresponsive controlled release to develop a strategy of thermoresponsive polypeptide fusion that can overcome the limitation of half-life extension in improving the pharmacokinetics. In this proof-of-concept study, an important therapeutic protein, interferon alpha (IFN), was genetically fused to a thermoresponsive elastin-like polypeptide (ELP) to form a thermoresponsive ELP fused interferon alpha (IFN-ELP). We reasoned that IFN-ELP would phase separate from the solution in situ to form an IFN-ELP depot upon subcutaneous injection if the phase transition temperature (Tt) of IFN-ELP is below local body temperature. We further hypothesized that the IFN-ELP molecules of the depot would gradually release in a soluble form due to the concentration dependence of the Tt and would diffuse into the circulation system over a greatly prolonged period of time. As a result, thermoresponsive ELP fusion would dramatically enhance the pharmacological performance of the drug and reduce the side effects.
2. Experimental section
2.1. Construction, expression and purification of IFN-ELP(V), IFN-ELP(A) and IFN
The DNA sequences encoding IFN-ELP(V) and IFN-ELP(A) were synthesized and inserted in a pET-25b (+) vector using the PRe-RDL method as previously described.18 The sequences of ELP(V) and ELP(A) were composed of 90 duplicates of the pentapeptide sequences Val–Pro–Gly–Val–Gly and Val–Pro–Gly–Ala–Gly, respectively. The constructed plasmids encoding IFN-ELP(V) and IFN-ELP(A) were overexpressed in E. coli and purified by the method of Inverse Transition Cycling (ITC), as described previously with minor modifications.19 IFN was purified using immobilized metal affinity chromatography as described in the published literature.20
2.2. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS)
The molecular weights of protein samples were measured with a 4800 Plus MALDI-TOF/TOF™ Analyzer (Applied Biosystems). The samples were diluted in water and then mixed with the matrix solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v:v). The spectra were calibrated with proteins of known masses.
:1, v:v). The spectra were calibrated with proteins of known masses.
2.3. Dynamic light scattering (DLS)
DLS was carried out with a Zetasizer Nano-zs90 (Malvern) operated at a scattering angle of 90° and a laser wavelength of 633 nm at 4 °C for IFN-ELP(V) and 25 °C for IFN-ELP(A) and IFN. Samples were filtered before detection. Zetasizer software 6.32 was used to analyze the data.
2.4. Circular dichroism (CD)
A Pistar π-180 instrument (Applied Photophysics Ltd) was used to record the CD spectra in the range from 200 nm to 260 nm. Samples were pre-diluted to 0.2 mg mL−1 in H2O.
2.5. Thermoresponsive behaviors
The inverse phase transition behaviors of IFN-ELP(V) and IFN-ELP(A) as a function of temperature were determined by monitoring the absorbance at 350 nm on a SpectraMax M3 Microplate Reader (Molecular Devices). A PBS solution of IFN-ELP was heated at a rate of 1 °C min−1 typically from 4 °C to 80 °C, and then cooled at the same rate to 4 °C to examine the transition reversibility. The transition temperature (Tt) was defined as the temperature at 50% of the maximum turbidity according to the heating profile. To study the Tt as a function of the concentration of IFN-ELP, 200 μL samples at various concentrations were incubated in disposable cuvettes from 4 to 80 °C, and then the absorbance of the sample at 350 nm was monitored every 2 °C.
To investigate the in vitro release of IFN-ELP(V) in the soluble form as a function of time, 5 mg IFN-equivalent IFN-ELP(V) was separated from the solution by centrifugation at 16000g at 37 °C for 10 min. The supernatant was removed and 600 μL PBS was carefully added to submerge the depot. The sample was incubated at 37 °C for 30 days and the IFN concentration in the supernatant was measured at given time points (0.5 d, 1 d, 2 d, 5 d, 10 d, 15 d, 20 d, and 30 d). The release percentage of IFN in the soluble form of IFN-ELP(V) was calculated.
2.6.
In vitro antiproliferative activity
Daudi B cells were seeded at a density of 5000 per well in a 96-well plate (Corning) and serial dilutions (1, 2, 5, 10, 20, 50, 100, 300, 1000, and 10000 pg mL−1) of the IFN, IFN-ELP(A) and IFN-ELP(V) samples in fresh RPMI-1640 medium were added. After 96 h incubation, the activity of the cells was examined by MTT assay using a cell proliferation assay kit (Promega).19 The value of IC50 was calculated and the data were analyzed using the GraphPad Prism 5.0 software.
2.7. Maximum tolerated dose (MTD)
6 week old healthy female SPF BALB/c mice were administered s.c. with IFN-ELP(V) at the doses of 25, 50, 100, and 200 mg IFN-equivalent per kg BW, IFN-ELP(A) at the doses of 10, 15, 25, and 50 mg IFN-equivalent per kg BW or IFN at the doses of 5, 10, 15, and 25 mg IFN per kg BW (n = 3 for each injection). The changes in the mouse body weight and survival were monitored every day for two weeks. The mice that lost over 10% of their pre-treatment body weights were euthanized. The highest dose at which no animal mortality and no more than 10% body weight loss were observed was defined as the MTD. To further study the tolerability of these therapeutic proteins, at 30 days post subcutaneous administration at their MTDs, the mice continued to be subcutaneously injected with them at the MTDs, respectively. The changes in the mouse body weight and survival were monitored every day for two weeks. The mice that lost over 10% of their pre-treatment body weights were euthanized.
2.8. Pharmacokinetics
BALB/c mice with an average body weight of about 20 g were used to evaluate the pharmacokinetics of IFN, IFN-ELP(A) and IFN-ELP(V). The mice were distributed into 3 groups (n = 3 for each group) randomly. IFN-ELP(V), IFN-ELP(A), or IFN was injected intravenously at 30 μg IFN-equivalent per kg BW. Blood samples (100 μL) were drawn from the retro-orbital at given time points (1 min, 5 min, 15 min, 30 min, 1 h, 2 h, 8 h, 24 h, 48 h, and 72 h) after anesthesia with isoflurane, and centrifuged at 4000g for 15 min after standing for 30 min. The IFN concentration was measured by ELISA assay. Simultaneously, plasma from untreated mice (control) was obtained and defined as the background. The data were analysed using Drug analysis System 3.0 to obtain the pharmacokinetic parameters. The mice in each group (n = 3) were subcutaneously injected with IFN-ELP(V), IFN-ELP(A), or IFN at its MTD. At given time points (1 min, 5 min, 15 min, 30 min, 1 h, 2 h, 8 h, 1 d, 2 d, 3 d, 5 d, 7 d, and then once every 3 d), blood samples (100 μL) were drawn from the retro-orbital after anesthesia with isoflurane, centrifuged at 4000g for 15 min after left standing for 30 min. The methods of measuring IFN concentrations were the same as mentioned above. The data were further analyzed using Drug analysis System 3.0 to obtain the pharmacokinetic parameters.
2.9. Biodistribution
The mice with OVCAR-3 tumor burden were randomly distributed into 3 groups (3 mice in each group) and then subcutaneously injected with IFN, IFN-ELP(A) and IFN-ELP(V) at their MTDs, respectively. At 24 h, 72 h and 30 d after the injections, major tissues were harvested and IFN concentrations were measured by ELISA as mentioned above.
2.10. Antitumor efficacy
BALB/c nude mice aged 6 weeks were subcutaneously injected in the right dorsal area with 6 × 106 OVCAR-3 or C8161 cells (0.1 mL). At 30 days and 15 days post the inoculations, the two kinds of tumors were established with an average size of ∼30 mm3, respectively. The animals were randomized into 4 groups (n = 8 to 10 per group for OVCAR-3 tumors and n = 6 to 8 per group for C8161 tumors) and subcutaneously injected with IFN-ELP(V), IFN-ELP(A), and IFN at their MTDs, and saline at the equivalent volume, respectively. The tumor volumes were measured with calipers every three days and calculated using the formula: volume = length × width2 × 0.5. Simultaneously, the mice were weighed. The mice would be considered dead if their tumor volumes were larger than 500 mm3 or the loss of body weight was greater than 15%.
2.11. Biological safety
OVCAR tumor-bearing mice were sacrificed on the 9th day post the subcutaneous injections of IFN, IFN-ELP(A) and IFN-ELP(V) at their MTDs, and their major tissues were collected and subjected to H&E staining for morphology observation using standard procedures. The images were captured with a Nikon Eclipse 90i microscope. At the end of the treatments, blood samples were collected via the retro-orbital for hematology examination. Hematological parameters of blood were detected using a hematology analyzer (SYSMEX). An automatic biochemical analyzer (HITACHI) was used to measure the clinical biochemistry parameters of serum. Blood samples (1 mL) were collected in heparinized-tubes via the retro-orbital of the BALB/c nude mice. The hemolysis ratios were evaluated as previously reported.19
2.12. Statistical analysis
Data were collected as the mean ± standard error of the mean. For the antitumor study, the values were analyzed with a variance (ANOVA) test and then the Tukey HSD test for multiple comparisons between different groups using the SPSS software (SPSS, Chicago IL, USA). The value of P < 0.05 was considered to represent statistical significance. For antiviral effect analyses, a nonparametric Mann–Whitney U test was performed between the two groups. All tests of significance were two-tailed and P < 0.05 was considered statistically significant. Statistical analyses were performed with the GraphPad Prism 5.0 software (Graphpad Software Inc., La Jolla, California).
3. Results and discussion
3.1. Physicochemical characterization
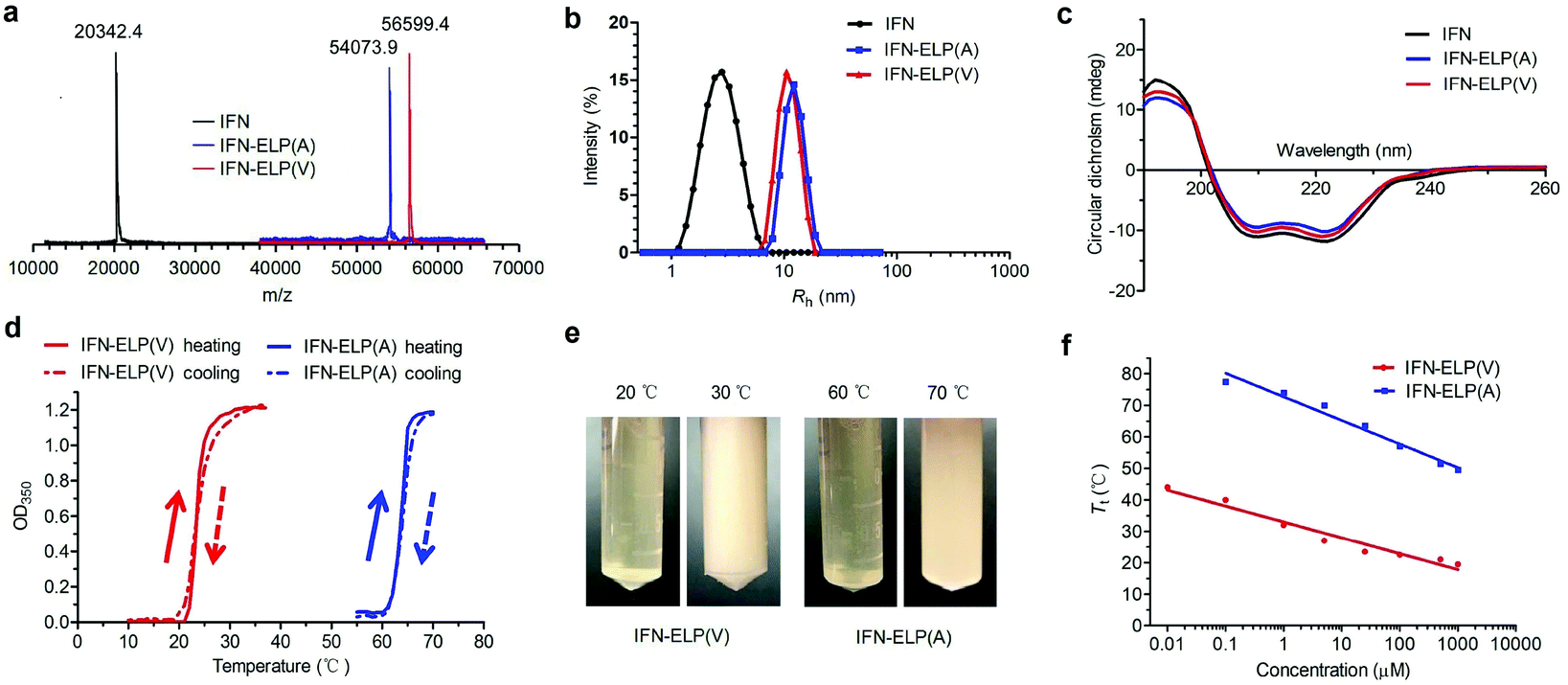
ELPs are polypeptides comprising Val–Pro–Gly–Xaa–Gly repeat units, where Xaa can be any amino acid except proline.21 ELPs are thermoresponsive, that is, they are soluble below their Tt, but become insoluble above their Tt, and vice versa. The Tt of ELPs can be tuned by changing the hydrophobicity of the Xaa and the number of Val–Pro–Gly–Xaa–Gly repeat units. ELPs are biodegradable, non-toxic, and non-immunogenic.22,23 Moreover, they can be genetically fused with proteins and peptides for protein and peptide purification and delivery.17,24–27 IFN is a highly pleiotropic cytokine with antiviral, immunoregulatory and antiproliferative functions, which is widely used in the clinic for the treatment of a variety of viral diseases and cancers. The N- or C-terminus of IFN is far from the receptor binding domains identified.28,29 Based on these principles, we chose to genetically fuse IFN with the N-terminus of ELP(V) and ELP(A) in which the Xaa guest residues are valine (V) and alanine (A), respectively, and the number of repeat units in each ELP is 90. The as-formed IFN-ELP(V) and IFN-ELP(A) were overexpressed in Escherichia coli and then purified by inverse transition cycling (Fig. S1†). The molecular weights of IFN, IFN-ELP(V) and IFN-ELP(A) were determined by matrix-assisted laser desorption/ionization time of flight mass spectroscopy (MALDI-TOF-MS) to be 56599.4, 54073.9 and 20342.4 Da, respectively (Fig. 1a), which are identical to their theoretical values of 56601.3, 54076.5 and 20340.1 Da. The hydrodynamic radii of IFN, IFN-ELP(V) and IFN-ELP(A) were determined by dynamic light scattering (DLS) to be 2.9, 11.1, and 12.8 nm, respectively (Fig. 1b), suggesting that the circulation half-life of IFN would be extended by ELP fusion, considering that the sizes of IFN-ELP(V) and IFN-ELP(A) are much larger than the cutoff size (around 5 nm in radius) of renal filtration.30 Additionally, ELP fusion did not interfere with the second structure of IFN, as indicated by the identical doublets at 209/219 nm in circular dichroism (CD) scans (Fig. 1c). Next, we further studied the thermoresponsive behaviors of IFN-ELP(V) and IFN-ELP(A) using turbidity measurements. Apparently, reversible sharp phase transition behaviors were observed for both of them (Fig. 1d and e). The Tt of IFN-ELP(V) was below body temperature at high concentrations, but can be above body temperature at low concentrations (Fig. 1f and Fig. S2†). This result suggests that IFN-ELP(V) would phase separate from the solution in situ to form an IFN-ELP(V) depot upon subcutaneous injection at high concentrations, but would gradually release from the depot and diffuse into the circulation system in a soluble form at low concentrations. Although the Tt of IFN-ELP(A) is also concentration dependent, it is well above body temperature even if the concentration of IFN-ELP(A) is as high as 500 μM, suggesting that IFN-ELP(A) would not form a depot after subcutaneous injection even at high concentrations. The release profile of the IFN in the soluble form of IFN-ELP(V) as a function of time at 37 °C (Fig. S3†) showed that the release percent increased with time, suggesting that IFN-ELP(V) in a soluble form would be gradually released from the depot upon subcutaneous injection.
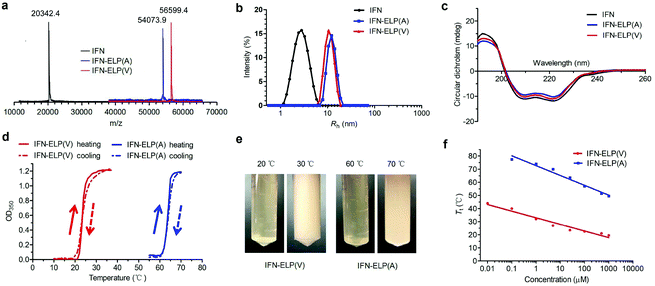 |
| | Fig. 1 Physicochemical characterization of IFN, IFN-ELP(V) and IFN-ELP(A). (a) MALDI-TOF-MS spectra of IFN, IFN-ELP(V) and IFN-ELP(A). (b) DLS analyses of IFN, IFN-ELP(V) and IFN-ELP(A), in which Rh is the hydrodynamic radius. (c) CD spectra of IFN, IFN-ELP(V) and IFN-ELP(A). (d) Turbidity (OD350) of IFN-ELP(V) and IFN-ELP(A) versus temperature at a concentration of 25 μM in PBS. Heating and cooling traces are marked by arrows. (e) Digital images of IFN-ELP(V) and IFN-ELP(A) at temperatures below and above their Tt. (f) The concentration dependence of Tt. | |
3.2.
In vitro antiproliferative and antiviral activities
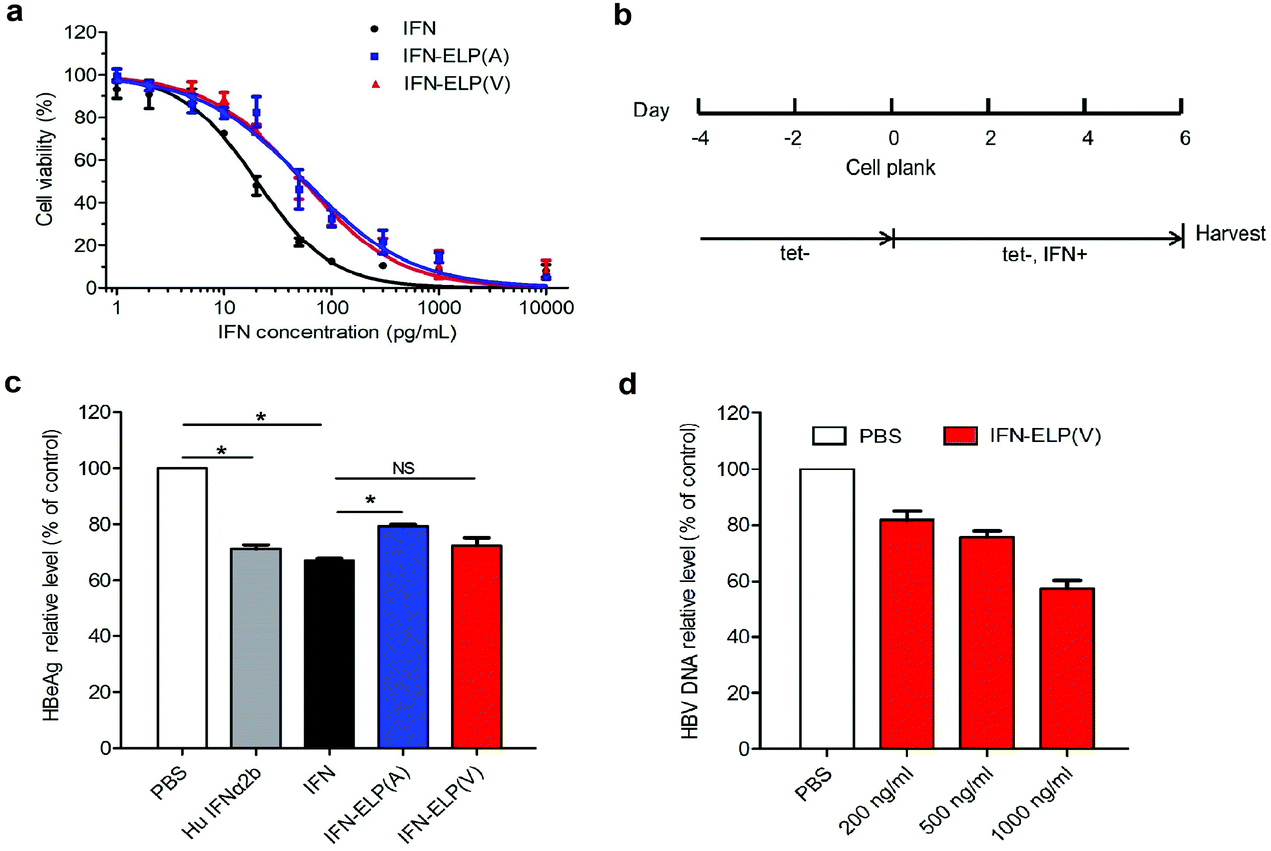
The in vitro antiproliferative activity for IFN-ELP(V) and IFN-ELP(A) was examined using Daudi B cells which are highly sensitive to IFN.31 The half maximal inhibitory concentrations of IFN-ELP(V) (IC50 = 54.6 pg mL−1) and IFN-ELP(A) (IC50 = 56.9 pg mL−1) are nearly 3-fold that of IFN (IC50 = 20.2 pg mL−1) (Fig. 2a). These data indicate that the activity of IFN was reduced to around 37% by ELP fusion, presumably due to the physical shielding of the tethered ELP. However, the IFN activity retention (37%) for ELP fusion is at least 5.3- and 37-fold higher than those for PEGylation (1%–7%)32 and HSA fusion (1%).12 Furthermore, the antiviral activity of IFN, IFN-ELP(V) and IFN-ELP(A) in inhibiting hepatitis B virus (HBV) replication was tested by using the HepAD38 cell strain33 (Fig. 2b). The levels of HBeAg for IFN-ELP(V) and IFN-ELP(A) treatments, which reflect the virological response, decreased to 72.5% and 79.3% of the control of the PBS treatment, respectively, which are comparable to those for commercial recombinant human IFN-α2b (71.3%) and IFN (67.2%) treatments (Fig. 2c). The dose-dependence of the inhibition of HBV DNA replication was observed for IFN-ELP(V) treatment (Fig. 2d). These data show that ELP fusion did not markedly decrease the antiviral activity of IFN.
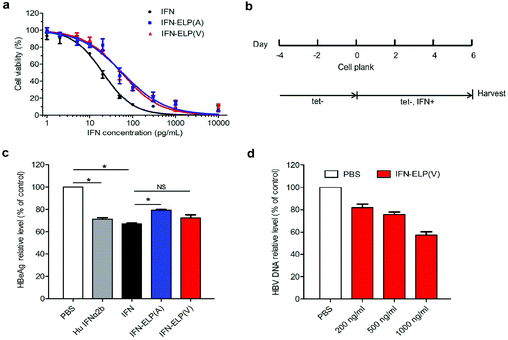 |
| | Fig. 2
In vitro antiproliferative and antiviral activities of IFN, IFN-ELP(V) and IFN-ELP(A). (a) Cytotoxicity of IFN, IFN-ELP(V) and IFN-ELP(A) to Daudi B cells. (b) Schematic illustration of the antiviral experimental procedures. (c) HBeAg relative levels in HepAD38 cell culture medium post treatments (Mann–Whitney U test, *P < 0.05). (d) HBV DNA relative levels in HepAD38 cell culture medium after treatments with different concentrations of IFN-ELP(V). Data are shown as the mean ± standard error of the mean. | |
3.3. Pharmacokinetics and biodistribution
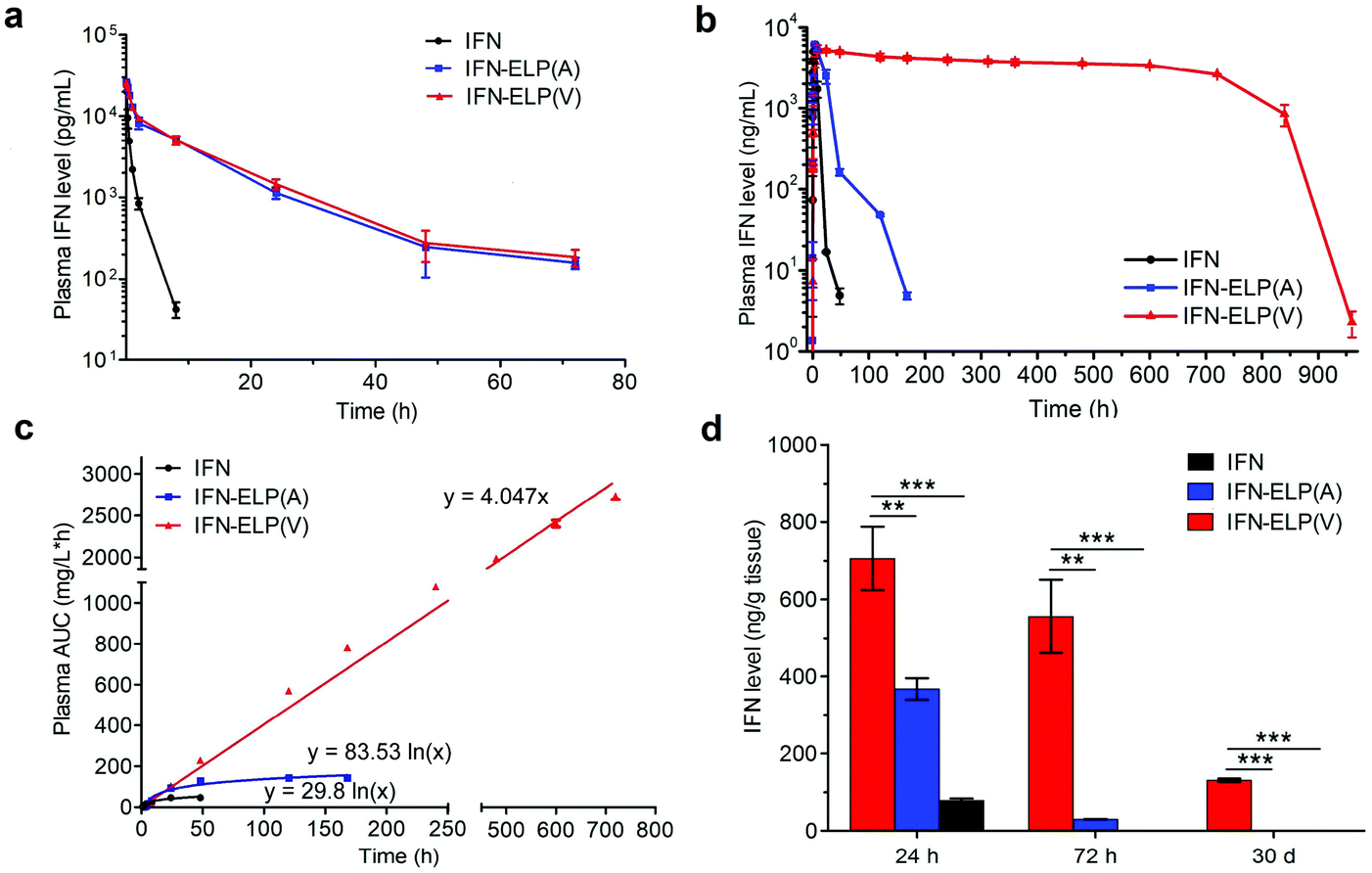
The in vivo pharmacokinetics of IFN, IFN-ELP(V) and IFN-ELP(A) was investigated in a mouse model. Above all, the half-life extension by ELP fusion was confirmed by the intravenous injections of these proteins at a low concentration of 0.1 μM (Fig. 3a). Notably, this concentration is low enough to avoid the phase separation of IFN-ELP(V) in circulation, as suggested in Fig. 1f. The data of Fig. 3a were fitted with a two-compartment model to obtain the pharmacokinetic parameters (Table S1†). The clearance (CL) of IFN (48.7 ± 4.0 mL h−1) is reduced to 4.0 ± 0 mL h−1 for IFN-ELP(V) and IFN-ELP(A). The terminal half-life (t1/2β) of IFN (1.2 ± 0.1 h) is extended to 9.9 ± 0.2 h for IFN-ELP(V) and 9.8 ± 0.4 h for IFN-ELP(A). As a result, the area under the curves (AUCs) of IFN-ELP(V) (150.4 ± 8.7 mg L−1 h−1) and IFN-ELP(A) (147.1 ± 7.5 mg L−1 h−1) was 12.1- and 11.9-fold that of IFN (12.4 ± 1.0 mg L−1 h−1). Unsurprisingly, these data indicate the significantly improved pharmacokinetics by ELP fusion due to the enlarged size and thus reduced renal clearance, as achieved by other half-life extension strategies such as PEGylation, HSA fusion, Fc fusion, and randomly coiled polypeptide fusion.10,11,17 However, the half-life extension by the intravenous injection of soluble IFN-ELP cannot eliminate the peak and valley fluctuations in pharmacokinetics, as indicated by the decline in the plasma concentration of the fusion proteins with time post intravenous injection (Fig. 3a).
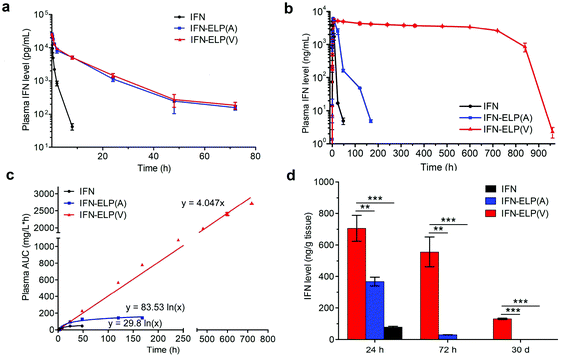 |
| | Fig. 3
In vivo pharmacokinetics and tumor accumulation of IFN, IFN-ELP(V) and IFN-ELP(A). (a) Plasma IFN levels of IFN, IFN-ELP(V) and IFN-ELP(A) versus time after intravenous injections at a dose of 30 μg IFN-equivalent per kg BW (0.1 μM) (n = 3). (b) Plasma IFN levels of IFN, IFN-ELP(V) and IFN-ELP(A) versus time post subcutaneous administration at their MTDs (n = 3). (c) Cumulative AUC of circulating IFN-ELP(V), IFN-ELP(A) and IFN versus time can be fitted with either linear or logarithmic regression (n = 3). (d) IFN levels of IFN, IFN-ELP(V) and IFN-ELP(A) in the tumors 24 h, 72 h and 30 d post administration (n = 3, one-way analysis of variance (ANOVA), **P < 0.01, ***P < 0.001, significant difference for IFN-ELP(V) compared with IFN-ELP(A) and IFN). Data are shown as the mean ± standard error of the mean. | |
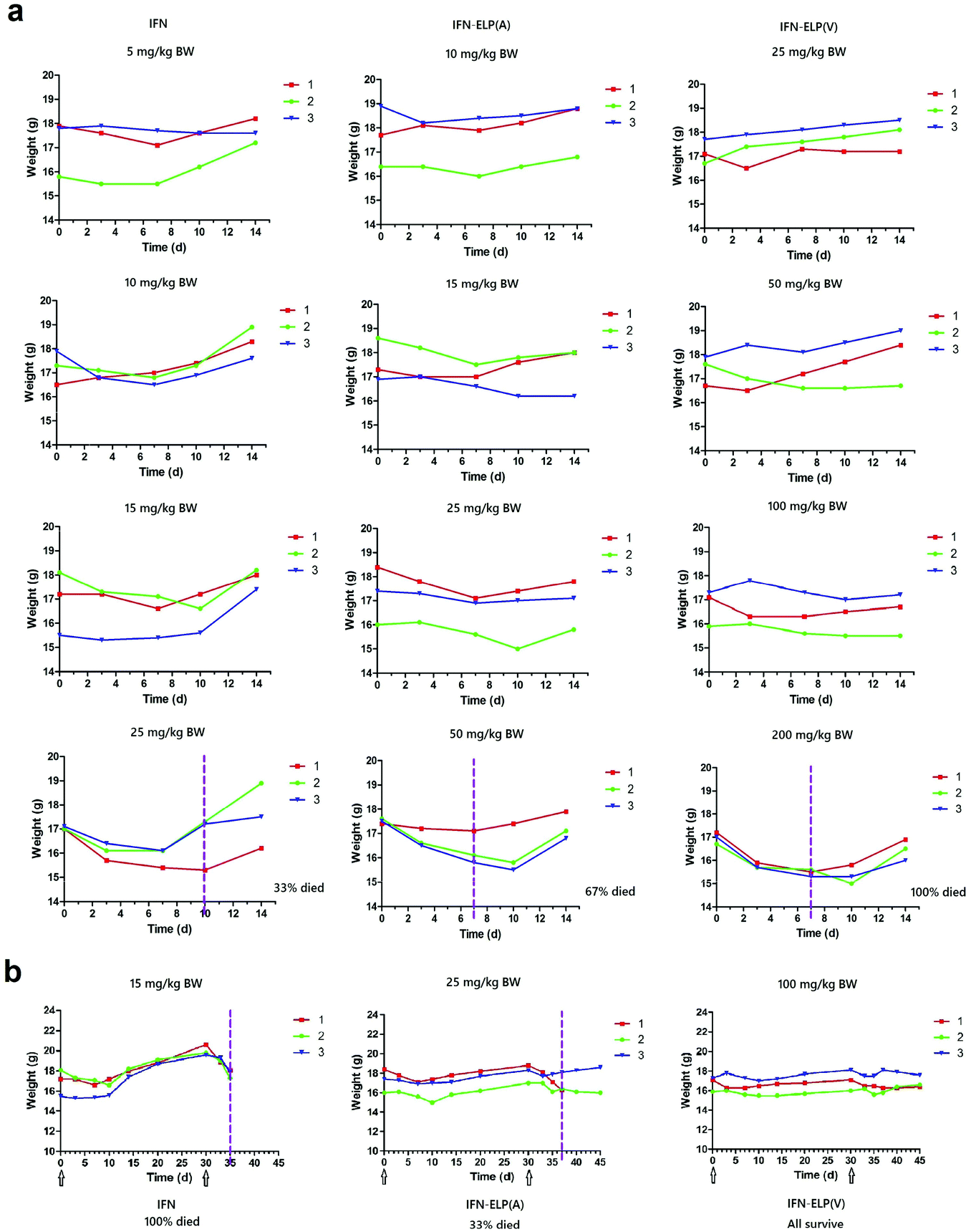
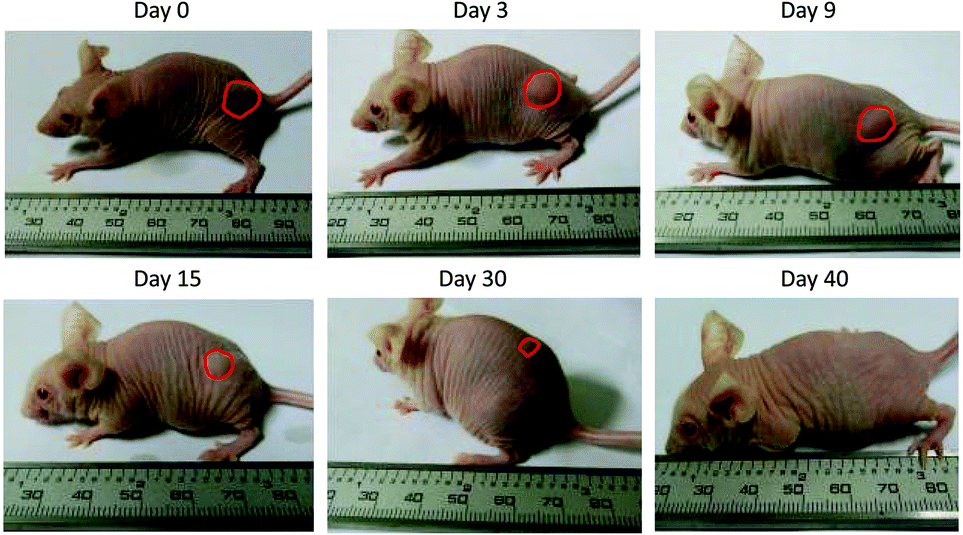
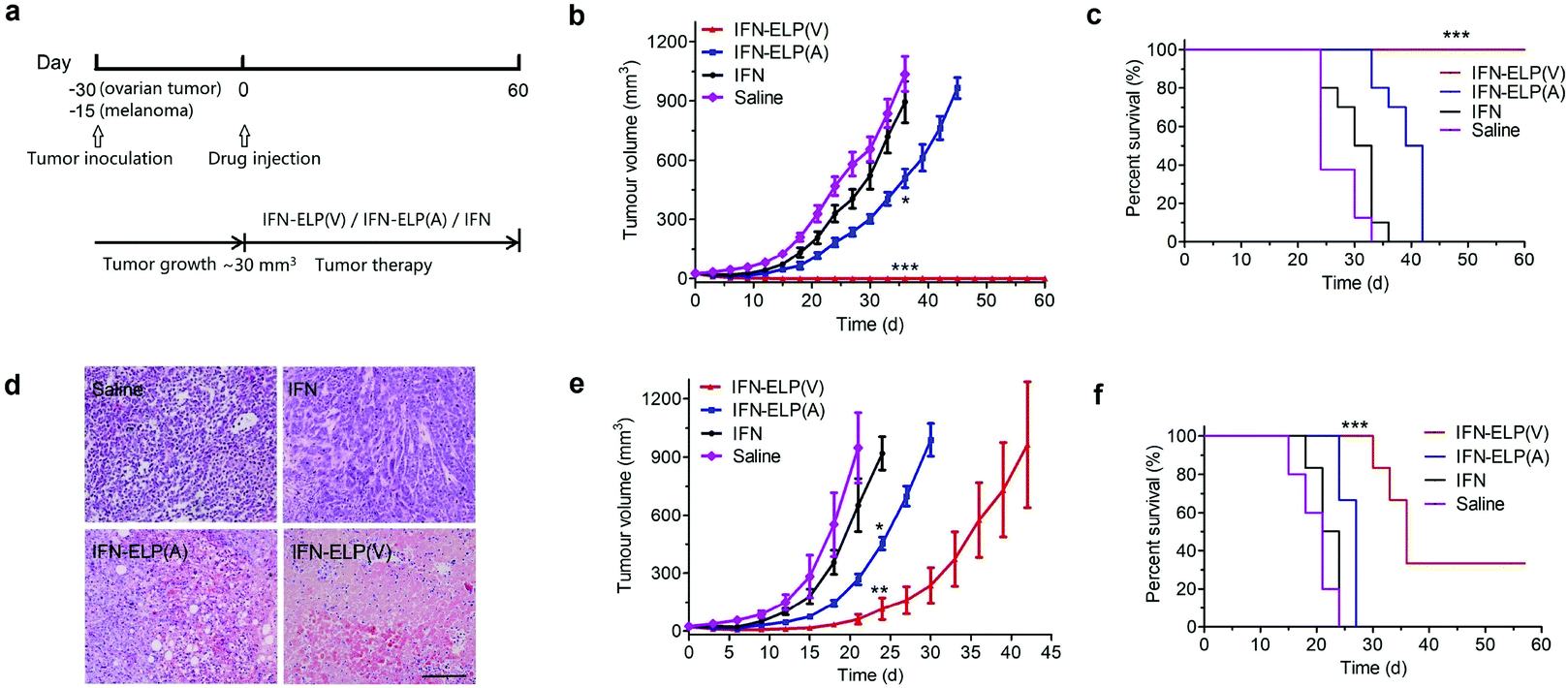
To address this challenge, we subcutaneously injected them at their maximum tolerated doses (MTDs). Interestingly, the MTD of IFN-ELP(V) (100 mg per kg body weight (BW)) is 4.0- and 6.7-fold those of IFN-ELP(A) (25 mg per kg BW) and IFN (15 mg per kg BW), respectively (Fig. 4a). Moreover, a second subcutaneous injection at its MTD was tolerable for IFN-ELP(V) but not for IFN-ELP(A) and IFN (Fig. 4b). These data indicate the remarkable improvement in tolerability by thermoresponsive ELP fusion. According to Fig. 1f, the Tt values of IFN-ELP(V) and IFN-ELP(A) at their MTDs are well below and above body temperature, respectively. As a result, IFN-ELP(V) in situ formed a depot upon a single subcutaneous injection at its MTD, as indicated by the in situ formation of a bump that could last for one month (Fig. 5), while IFN-ELP(A) could not form such a depot at its MTD, as expected. Upon subcutaneous injections, the plasma IFN levels of IFN and IFN-ELP(A) quickly increased to 5457 ± 759 μg L−1 at 2.7 ± 1.2 h and 6131 ± 262 μg L−1 at 4 ± 0 h, respectively, and then dropped rapidly (Fig. 3b), indicating the peak and valley pharmacokinetics. In contrast, the plasma IFN level of IFN-ELP(V) remained nearly persistent until day 30 after it reached 5461 ± 559 μg L−1 at 13.3 ± 9.2 h post the subcutaneous injection, indicating a one-month zero-order sustained release. The striking differences in pharmacokinetics are further supported by the apparent pharmacokinetic parameters of IFN, IFN-ELP(A), and IFN-ELP(V) (Table S2†). Particularly, the apparent half-life (t1/2) of IFN-ELP(V) (497.2 ± 57.0 h) is 47.4- and 261.7-fold those of IFN-ELP(A) (10.5 ± 1.5 h) and IFN (1.9 ± 0.13 h), respectively. This dramatic extension in the half-life indicates that the combination of half-life extension with a thermoresponsive controlled release enabled by thermoresponsive ELP fusion is extremely efficient in improving the apparent half-life of IFN. The apparent AUC of IFN-ELP(V) (3176 ± 247 mg L−1 h−1) is 23.5- and 70.4-fold those of IFN-ELP(A) (135.4 ± 10.2 mg L−1 h−1) and IFN (45.1 ± 7.1 mg L−1 h−1), respectively. This huge increase in the AUC indicates the tremendous improvement in drug exposure by the thermoresponsive ELP fusion. Moreover, the cumulative AUC of IFN-ELP(V) in circulation is linearly correlated with time, thereby confirming the one-month zero-order sustained release of IFN-ELP(V) (Fig. 3c). In contrast, IFN-ELP(A) and IFN followed logarithmic release profiles. Collectively, these data demonstrate that thermoresponsive ELP fusion can eliminate the peak and valley fluctuations in the pharmacokinetics of IFN over a dramatically prolonged period of one month.
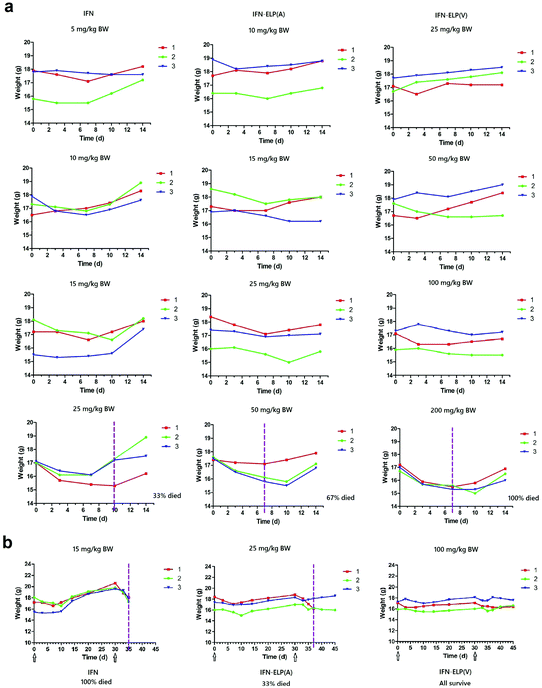 |
| | Fig. 4 Dose escalation in mice (n = 3). (a) Solutions were subcutaneously administered on day 0. (b) A second subcutaneous injection was conducted at 30 d post the first subcutaneous injection. Arrows denote the injection time points. The vertical dashed lines denote the time points at which more than 10% body weight loss was observed. | |
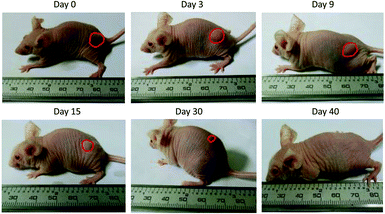 |
| | Fig. 5 Representative images of the IFN-ELP(V) depot formed post a single subcutaneous injection at its MTD. | |
Next, we studied the biodistribution of IFN-ELP(V) in major organs and tissues after a single subcutaneous injection at its MTD. Strikingly, IFN-ELP(V) had high concentrations particularly in tumors and kidneys even 30 d post the injection (Fig. 3d and Fig. S4†), due to its one-month zero-order sustained release kinetics. In contrast, IFN-ELP(A) and IFN had low tumor accumulation and were almost undetectable in all major organs and tissues at 72 h post the injections at their MTDs, due to their short half-lives. These results show that the biodistribution of IFN can greatly be improved by thermoresponsive ELP fusion.
3.4. Antitumor efficacy
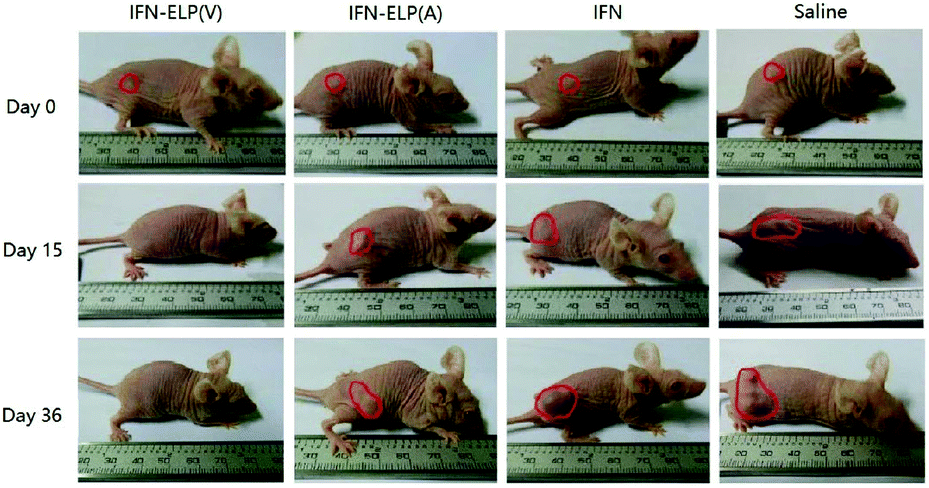
We hypothesized that the dramatic improvements in pharmacokinetics and biodistribution would translate into a great enhancement in pharmacodynamics. The hypothesis was first verified in an ovarian tumor-bearing mouse model with an average tumor size of 30 mm3 (Fig. 6a). After a single subcutaneous injection at its MTD, all the tumors of 10 mice treated with IFN-ELP(V) shrank and disappeared at 15 d post the injection without any tumor recurrence (Fig. 6b and 7). In contrast, the tumor sizes of the mice subcutaneously injected with IFN-ELP(A) and IFN at their MTDs decreased to 13.7 mm3 and 18.1 mm3 at 6 d, respectively, but gradually increased up to 507.6 mm3 and 922.2 mm3 at 36 d, respectively. As a result, the median survival times of the mice treated with IFN-ELP(A) (39 d) and IFN (30 d) are 1.6- and 1.3-fold that of the mice treated with saline (24 d), respectively (Fig. 6c). In contrast, no mouse death was observed for the IFN-ELP(V) treatment. Hematoxylin and eosin (H&E) staining confirmed the antitumor activity of IFN-ELP(V) (Fig. 6d). Obviously, IFN-ELP(V) was more efficient in inducing tumor cell apoptosis than IFN-ELP(A) and IFN, as indicated by the more extensive vacuolization. Based on this exciting result of IFN-ELP(V) in the treatment of ovarian tumor, we further evaluated IFN-ELP(V) in the treatment of malignant melanoma (Fig. 6e). Similar to the treatment of ovarian tumor, IFN-ELP(V) was much superior to IFN-ELP(A) and IFN in suppressing melanoma growth. Notably, 33% of the melanoma-bearing mice were tumor-free without any tumor recurrence post a single subcutaneous injection of IFN-ELP(V) at its MTD. The improved inhibition of melanoma growth was correlated with the prolonged median survival time (Fig. 6f). The median survival times of the mice treated with IFN-ELP(A) (27 d) and IFN (22.5 d) are 1.3- and 1.1-fold that of the mice treated with saline (21 d), respectively. In contrast, 33% of the mice treated with IFN-ELP(V) survived without any tumor burden. Taken together, these in vivo antitumor results show that the pharmacodynamic performance of IFN can be dramatically enhanced by thermoresponsive ELP fusion. The antitumor performance of ELP-fused IFN is mainly attributed to the impact of IFN on host immune systems. IFN is involved in the innate immunoresponse to developing malignancies by actively participating in cancer immunosurveillance.34
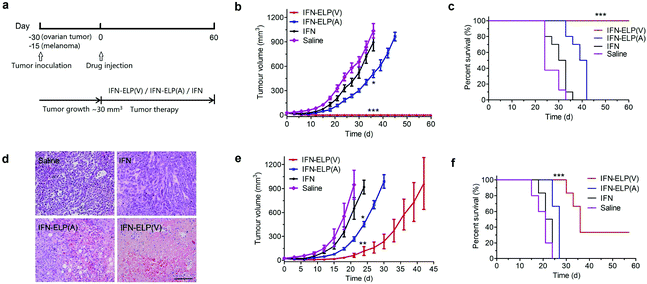 |
| | Fig. 6
In vivo antitumor efficacy of IFN, IFN-ELP(V) and IFN-ELP(A). (a) Schematic illustration of the antitumor experimental procedures. (b) Inhibition of ovarian tumor growth post treatments with IFN, IFN-ELP(V) and IFN-ELP(A) at their MTDs (n = 8–10, *P < 0.01, significant difference for IFN-ELP(A) relative to IFN; ***P < 0.001, significant difference for IFN-ELP(V) relative to IFN). (c) Cumulative survival of the ovarian tumor-bearing mice (n = 8–10, ***P < 0.001, significant difference for IFN-ELP(V) relative to saline, IFN-ELP(A), and IFN). (d) H&E staining of ovarian tumor tissues post administration. Scale bar, 100 μm. (e) Inhibition of melanoma growth post treatments with IFN, IFN-ELP(V) and IFN-ELP(A) at their MTDs (n = 6–8, *P < 0.05, significant difference for IFN-ELP(A) relative to IFN; **P < 0.01, significant difference for IFN-ELP(V) relative to IFN). (f) Cumulative survival of the melanoma-bearing mice (n = 6–8, ***P < 0.001, significant difference for IFN-ELP(V) relative to IFN, IFN-ELP(A) and saline). Data are shown as the mean ± standard error of the mean. | |
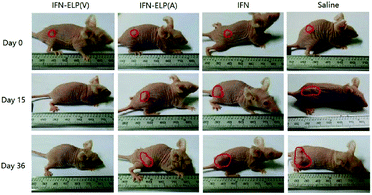 |
| | Fig. 7 Representative images for ovarian tumor growth post treatments with IFN, IFN-ELP(V) and IFN-ELP(A) at their MTDs. | |
3.5. Biological safety
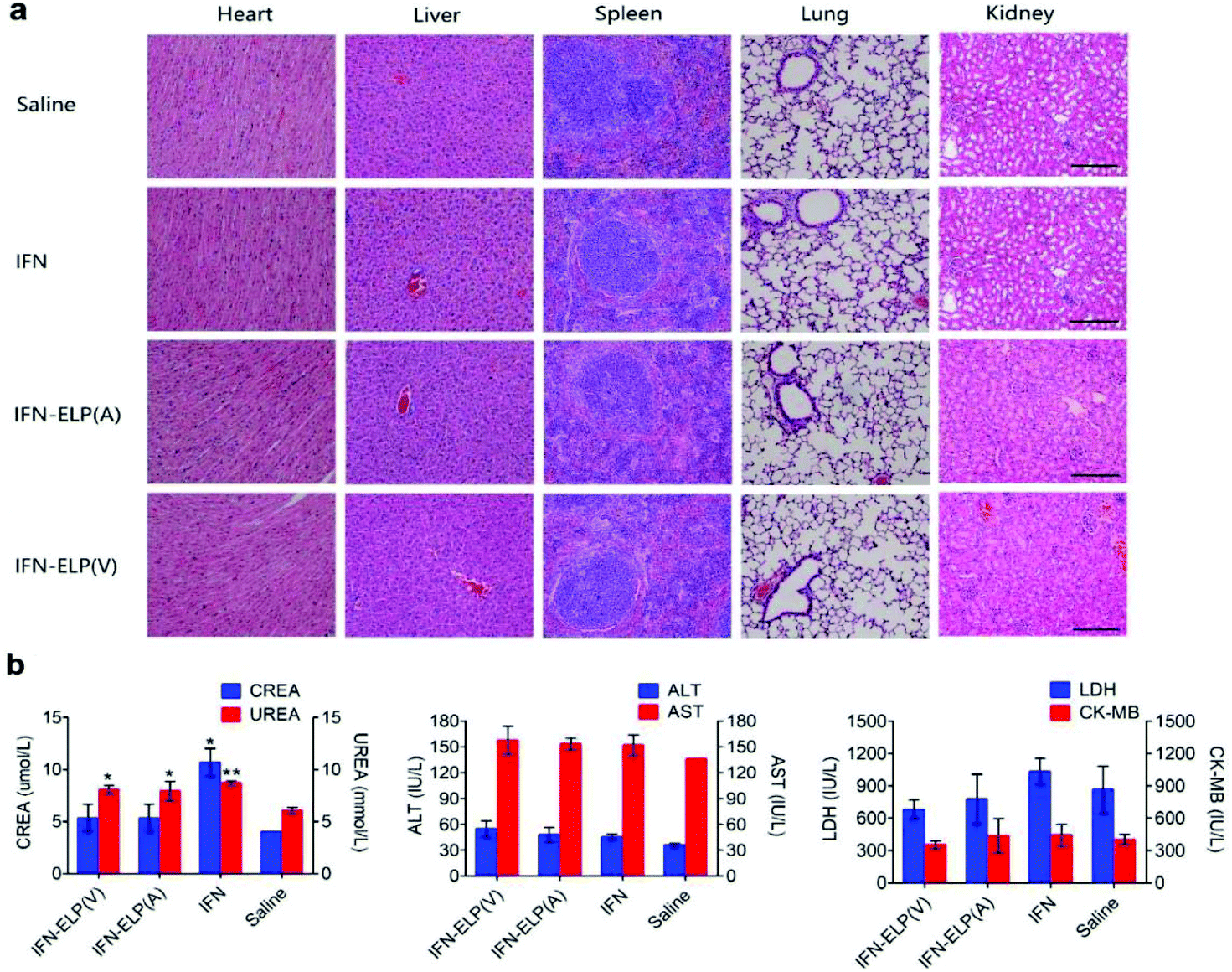
We further examined the biological safety of IFN-ELP(V). No obvious loss of body weight was observed for all the treatments (Fig. S5†). H&E staining indicated that the IFN-ELP(V) and IFN-ELP(A) treatments did not cause obvious histological changes in major organs such as kidney, liver, heart, spleen, and lung (Fig. 8a), whereas the IFN treatment induced kidney damage, as indicated by the extensive granular degeneration of renal tubules and renal interstitial edema. This result was further confirmed by blood biochemistry analysis, in which the levels of kidney function markers such as blood urea nitrogen (UREA) and creatinine (CREA) for the IFN treatment were significantly higher than those for the saline treatment (Fig. 8b). The IFN-ELP(V), IFN-ELP(A) and IFN treatments did not cause significant changes in the levels of all the important hematological markers as compared to the saline treatment (Fig. S6†). Additionally, no hemolytic effects were observed for all the treatments (Fig. S7†). All these biosafety results showed that the systemic toxicity of IFN in mice can be reduced by thermoresponsive ELP fusion.
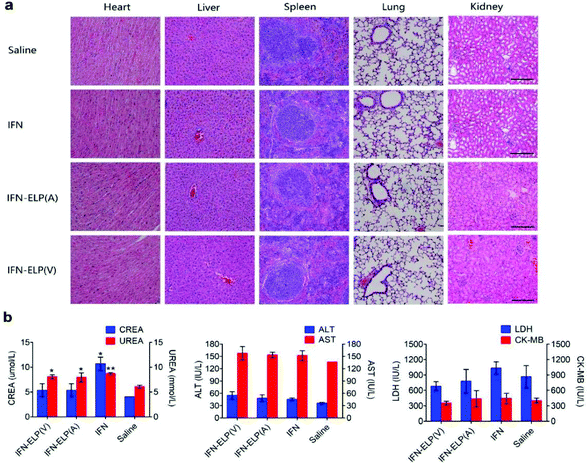 |
| | Fig. 8 Biological safety examination. (a) H&E staining of major tissues of mice post administration. Scale bar, 100 μm. (b) Biochemistry examination for mice post administration. Kidney function markers: CREA, creatinine; UREA, blood urea nitrogen (n = 3, *P < 0.05, **P < 0.01, significant difference for IFN-ELP(V), IFN-ELP(A) and IFN compared with saline). Liver function markers: ALT, alanine aminotransferase; AST, aspartate aminotransferase. Heart function markers: LDH, lactate dehydrogenase; CK-MB, creatine kinase isoenzymes. | |
4. Conclusions
In summary, we have integrated half-life extension with thermoresponsive controlled release to develop a new strategy of thermoresponsive ELP fusion to greatly improve the pharmacological performance of protein therapeutics with short circulation half-lives. A single subcutaneous injection of a thermoresponsive ELP fused IFN offers zero-order sustained release kinetics for a prolonged period of one month in mice, which is, to the best of our knowledge, the longest circulation time for protein delivery systems. Mouse pharmacokinetic data suggest that thermoresponsive ELP fusion would enable once-trimonthly dosing in humans on the basis of allometric scaling by using a power function with the body weight.35–38 Consequently, a single subcutaneous injection of the super-long-acting IFN enables greatly enhanced tumor accumulation and tumor eradication as well as remarkably improved tolerability and biosafety in the mice. Notably, considering that the frequent administration of free IFN (once every two days) for a long time of more than one year is usually required for the treatment of chronic hepatitis B and C, these attributes of the super-long-acting IFN would be particularly beneficial to patients with chronic viral diseases. Moreover, this thermoresponsive polypeptide fusion strategy is applicable to many therapeutic proteins not only to greatly improve the life quality of patients by reducing the injection frequency but also to dramatically enhance the therapeutic efficacy while reducing the side effects.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was financially supported by grants from the National Natural Science Foundation of China (Grant No. 21534006).
References
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- G. Walsh, Nat. Biotechnol., 2010, 28, 917–924 CrossRef CAS PubMed.
- R. J. Wills, Clin. Pharmacokinet., 1990, 19, 390–399 CrossRef CAS PubMed.
- S. D. Putney and P. A. Burke, Nat. Biotechnol., 1998, 16, 153–157 CrossRef CAS PubMed.
- W. Zhao, F. Liu, Y. Chen, J. Bai and W. Gao, Polymer, 2015, 66, A1–A10 CrossRef CAS.
- F. M. Veronese, Biomaterials, 2001, 22, 405–417 CrossRef CAS PubMed.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459–476 CrossRef CAS PubMed.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed.
- X. Liu, J. Sun and W. Gao, Biomaterials, 2018, 178, 413–434 CrossRef CAS PubMed.
- R. E. Kontermann, Curr. Opin. Biotechnol., 2011, 22, 868–876 CrossRef CAS PubMed.
- W. R. Strohl, BioDrugs, 2015, 29, 215–239 CrossRef CAS PubMed.
- Y. S. Huang, Z. Chen, Z. Y. Yang, T. Y. Wang, L. Zhou, J. B. Wu and L. F. Zhou, Eur. J. Pharm. Biopharm., 2007, 67, 301–308 CrossRef CAS PubMed.
- D. R. Nelson, Y. Benhamou, W. L. Chuang, E. J. Lawitz, M. Rodriguez-Torres, R. Flisiak, J. W. F. Rasenack, W. Kryczka, C. M. Lee, V. G. Bain, S. Pianko, K. Patel, P. W. Cronin, E. Pulkstenis, G. M. Subramanian, J. G. Mchutchison and A. S. Team, Gastroenterology, 2010, 139, 1267–1276 CrossRef CAS PubMed.
- D. M. Czajkowsky, J. Hu, Z. Shao and R. J. Pleass, EMBO Mol. Med., 2012, 4, 1015–1028 CrossRef CAS PubMed.
- V. Schellenberger, C. W. Wang, N. C. Geething, B. J. Spink, A. Campbell, W. To, M. D. Scholle, Y. Yin, Y. Yao, O. Bogin, J. L. Cleland, J. Silverman and W. P. C. Stemmer, Nat. Biotechnol., 2009, 27, 1186–1190 CrossRef CAS PubMed.
- M. Schlapschy, U. Binder, C. Borger, I. Theobald, K. Wachinger, S. Kisling, D. Haller and A. Skerra, Protein Eng., Des. Sel., 2013, 26, 489–501 CrossRef CAS PubMed.
- J. Hu, G. Wang, X. Liu and W. Gao, Adv. Mater., 2015, 27, 7320–7324 CrossRef CAS PubMed.
- J. R. McDaniel, J. A. MacKay, F. C. Quiroz and A. Chilkoti, Biomacromolecules, 2010, 11, 944–952 CrossRef CAS PubMed.
- Z. Wang, Q. He, W. Zhao, J. Luo and W. Gao, J. Controlled Release, 2017, 264, 66–75 CrossRef CAS PubMed.
- J. Hu, W. Zhao, Y. Gao, M. Sun, Y. Wei, H. Deng and W. Gao, Biomaterials, 2015, 47, 13–19 CrossRef CAS PubMed.
- D. W. Urry, J. Phys. Chem. B, 1997, 101, 11007–11028 CrossRef CAS.
- D. W. Urry, Trends Biotechnol., 1999, 17, 249–257 CrossRef CAS PubMed.
- S. R. Ong, K. A. Trabbic-Carlson, D. L. Nettles, D. W. Lim, A. Chilkoti and L. A. Setton, Biomaterials, 2006, 27, 1930–1935 CrossRef CAS PubMed.
- D. E. Meyer and A. Chilkoti, Nat. Biotechnol., 1999, 17, 1112–1115 CrossRef CAS PubMed.
- S. R. MacEwan and A. Chilkoti, J. Controlled Release, 2014, 190, 314–330 CrossRef CAS PubMed.
- M. Amiram, K. M. Luginbuhl, X. Li, M. N. Feinglos and A. Chilkoti, J. Controlled Release, 2013, 172, 144–151 CrossRef CAS PubMed.
- K. M. Luginbuhl, J. L. Schaal, B. Umstead, E. M. Mastria, X. Li, S. Banskota, S. Arnold, M. Feinglos, D. D'Alessio and A. Chilkoti, Nat. Biomed. Eng., 2017, 1, 1–14 CrossRef.
- J. H. Chill, R. Nivasch, R. Levy, S. Albeck, G. Schreiber and J. Anglister, Biochemistry, 2002, 41, 3575–3585 CrossRef CAS PubMed.
- S. R. Akabayov, Z. Biron, P. Lamken, J. Piehler and J. Anglister, Biochemistry, 2010, 49, 687–695 CrossRef CAS PubMed.
- C. H. J. Choi, J. E. Zuckerman, P. Webster and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6656–6661 CrossRef CAS PubMed.
- M. Havashida, A. Hoshika, Y. Kanetaka, N. Yanase and J. Mzuquchi, J. Interferon Cytokine Res., 2006, 26, 421–429 CrossRef PubMed.
- S. Foser, A. Schacher, K. A. Weyer, D. Brugger, E. Dietel, S. Marti and T. Schreitmuller, Protein Expression Purif., 2003, 30, 78–87 CrossRef CAS PubMed.
- L. Belloni, L. Allweiss, F. Guerrieri, N. Pediconi, T. Volz, T. Pollicino, J. Petersen, G. Raimondo, M. Dandri and M. Levrero, J. Clin. Invest., 2012, 122, 529–537 CrossRef CAS PubMed.
- L. Zitvogel, L. Galluzzi, O. Kepp, M. J. Smyth and G. Kroemer, Nat. Rev. Immunol., 2015, 15, 405–414 CrossRef CAS PubMed.
- I. Mahmood, Adv. Drug Delivery Rev., 2007, 59, 1177–1192 CrossRef CAS PubMed.
- P. Zou, Y. Yu, N. Zheng, Y. Yang, H. J. Paholak, L. X. Yu and D. Sun, AAPS J., 2012, 14, 262–281 CrossRef PubMed.
- J. Mordenti, S. A. Chen, J. A. Moore, B. L. Ferraiolo and J. D. Green, Pharm. Res., 1991, 8, 1351–1359 CrossRef CAS.
- The persistent circulation time in humans was calculated by using the empirical formula of Thuman = Tmouse(Whuman/Wmouse)0.13 elicited from ref. 35–37.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm01096j |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a
*a