
Versatile iron–catechol-based nanoscale coordination polymers with antiretroviral ligand functionalization and their use as efficient carriers in HIV/AIDS therapy†
Rubén
Solórzano
ab,
Olivia
Tort
 c,
Javier
García-Pardo
a,
Tuixent
Escribà
c,
Julia
Lorenzo
d,
Mireia
Arnedo
c,
Daniel
Ruiz-Molina
a,
Ramon
Alibés
b,
Félix
Busqué
*b and
Fernando
Novio
*a
c,
Javier
García-Pardo
a,
Tuixent
Escribà
c,
Julia
Lorenzo
d,
Mireia
Arnedo
c,
Daniel
Ruiz-Molina
a,
Ramon
Alibés
b,
Félix
Busqué
*b and
Fernando
Novio
*a
aCatalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and BIST, Campus UAB, Bellaterra, 08193 Barcelona, Spain. E-mail: fernando.novio@icn2.cat
bDepartament de Química, Universitat Autònoma de Barcelona (UAB), Campus UAB, Cerdanyola del Vallès 08193, Barcelona, Spain
cLaboratory of Retrovirology and Viral Immunopathogenesis, Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
dInstitut de Biotecnologia i de Biomedicina and Departament de Bioquímica i Biologia Molecular. Universitat Autónoma de Barcelona, 08193 Bellaterra, Barcelona, Spain
First published on 15th November 2018
Abstract
A novel chemical approach integrating the benefits of nanoparticles with versatility of coordination chemistry is reported herein to increase the effectiveness of well-known HIV antiretroviral drugs. The novelty of our approach is illustrated using a catechol ligand tethered to the known antiretroviral azidothymidine (AZT) as a constitutive building block of the nanoparticles. The resulting nanoscale coordination polymers (NCPs) ensure good encapsulation yields and equivalent antiretroviral activity while significantly diminishing its cytotoxicity. Moreover, this novel family of nanoparticles also offers (i) long-lasting drug release that is dissimilar inside and outside the cells depending on pH, (ii) triggered release in the presence of esterases, activating the antiviral activity in an on–off manner due to a proper chemical design of the ligand and (iii) improved colloidal stabilities and cellular uptakes (up to 50-fold increase). The presence of iron nodes also adds multifunctionality as possible contrast agents. The present study demonstrates the suitability of NCPs bearing pharmacologically active ligands as an alternative to conventional antiretroviral treatments.
Introduction
The lifelong treatment of human immunodeficiency virus (HIV) infection still faces several drawbacks: (1) daily dosing can be cumbersome for the patient; (2) the existence of cellular reservoirs of latent HIV viruses that escape treatment;1–4 (3) limited capacity of antiretroviral (ARV) drugs to access tissue reservoirs and sanctuaries, i.e., central nervous system, lymph nodes or lungs;5 (4) low solubility and poor bioavailability of the antiviral drugs; and (5) drug resistance development after continued treatment and the corresponding side effects.6,7 With no definitive cure, patients are forced to suffer these lifelong side effects; consequently, the target for HIV drug administration is concentrated on decreasing long-term toxicity and the development of new treatment strategies that do not rely on daily medication by employing new nanoscale platforms for drug delivery.8–13 Nanocarriers can also facilitate drug transport and permeation in all infected CD4+ cell reservoirs throughout the body, present even in patients who have had undetectable HIV RNA plasma levels for years,14 thus facilitating its elimination.So far, different combinations of nanostructured ARV drugs have been reported using protein-based lactoferrin,15 polymeric nanoparticles16–18 or lipids.19,20 In most of these examples, modest to low drug loading contents, usually oscillating around 5 wt%, are achieved. More effective encapsulation processes have been shown on nanoscale metal–organic frameworks (nano-MOFs)21–24 with antiretroviral drug loadings up to 42 wt%23,25 or in drug solid nanoparticles (up 70 wt%) obtained using emulsion-templated freeze-drying.26,27 However, in spite of these pioneering results, minimization of side effects, improvement in biodistribution or the development of novel formulation allowing for metabolization into a pharmacologically active drug, mainly intracellularly, still represent a real challenge. Therefore, the development of novel nanoformulations has become the foremost objective in HIV therapy.
Nanoscale coordination polymers (NCPs) embody a novel family of hybrid nanoparticles combining metal ions and organic ligands that can use drugs as constitutive building blocks (chemical entrapment)28–31 through physical encapsulation.32–34 Chemical entrapment allows better fine-tuning of the release kinetics (up to many hours) as well as better formulation with increased encapsulation yields.35 The use of active metal drugs, such as Pt(IV), as polymeric nodes of coordination polymers is the most representative and successful example of chemical entrapment.29,36–38 Chemical entrapment through tethering of active drugs as chelating ligands has been less explored in spite of the fact that its excellent efficiency has already been reported.39 Moreover, the chemical flexibility of organic synthesis may allow for the design of drugs (ligands) cleaved under physiological conditions.40
As a model compound for these studies, we have selected the broadly studied ARV drug azidothymidine (AZT) that selectively inhibits the HIV-1 reverse transcriptase (Fig. 1a). AZT has been frequently used in the past as an effective antiretroviral drug that rapidly metabolizes in the liver to the inactive glucoronide form, resulting in poor bioavailability with high residual toxic effects when administered orally.41 Therefore, this antiretroviral drug represents an adequate example to validate our approach. Specifically, we proposed functionalization of AZT at the 5′-OH position with a chelating catechol group (catAZT, Fig. 1a). Functionalization at this position has already been shown to be successful for the synthesis of AZT prodrugs with chemical and/or enzymatic hydrolysis.42,43 Afterwards, this ligand (catechol-functionalized azidothymidine) in combination with an iron salt and a bidentate ligand (1,4-bis(imidazol-1-ylmethyl)benzene; bix) is used to form the polymeric material that precipitates as nanoparticles (Fig. 1a). The resulting material contains catAZT ligands coordinated to the metal centres and bix ligands that act as linker ligands connecting the metal ions. Finally, the release of the drug is expected to occur following a two-step mechanism: (I) release of catAZT from the nanoparticles in a pH-controlled process and (II) release of free AZT drug from the catechol unit in the presence of corresponding enzymes (Fig. 1b). For comparison purposes and to test the possible effects of the material without an antiretroviral drug, the use of analogous but inactive thymidine (THY) is also proposed (Fig. 1a).
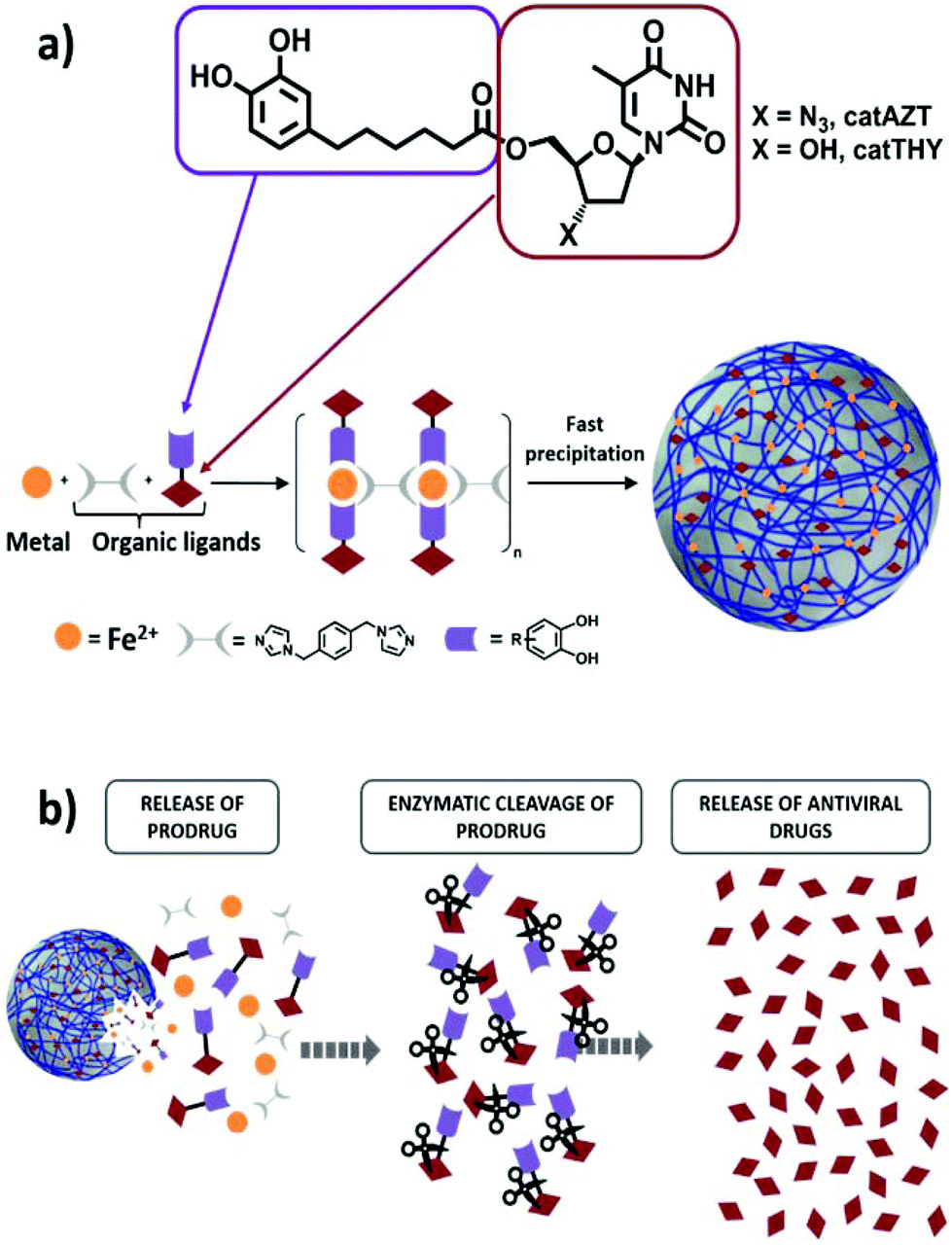 | ||
| Fig. 1 (a) Schematic representation of a catechol ligand linked to the antiviral drug AZT by an enzymatically cleavable bond via the carboxylic group. Posterior incorporation of catAZT or catTHY into a mixture containing iron and a bridging bis-imidazol ligand results in nanoparticle formation induced by rapid precipitation of the formed coordination polymer. (b) Schematic representation of the antiviral drug release process from the nanoparticle constructs to the AZT drug. It involves two main steps: (I) release of the catAZT ligand most likely due to particle degradation and (II) enzymatic cleavage of the free catechol derivative containing AZT. | ||
Results and discussion
Synthesis and characterization of catAZT and catTHY
The synthesis of catAZT was achieved through a 5-step synthetic sequence with 32% overall yield. Commercially available 3,4-dibenzyloxybenzaldehyde (please see Fig. 2a and ESI S1† for details) reacted with 4-carboxybutyltriphenylphosphonium bromide and sodium hydride in dry toluene to afford the olefin 2 with 88% yield as a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of Z and E isomers. The use of other bases and solvents such as potassium tert-butoxide in dry THF proved to be less efficient for this process. Simultaneous hydrogenation of the alkene moiety and removal of the benzyl protecting group at high pressure of H2 under Pd/C catalyst in EtOAc led to catechol 3 with almost quantitative yield. Protection of the hydroxyl groups of 3 as their tert-butyldiphenylsilyl (TBDPS) derivatives was accomplished using TBDPSCl and 1,8-diazabicyclo[5.4.0] undec-7-ene (DBU) in dry acetonitrile, affording the common intermediate 4 with 64% yield. Next, the nucleoside analogue AZT was tethered to this compound using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5b] pyridinium 3-oxid hexafluorophosphate (HATU) as a coupling reagent and N,N-diisopropylethylamine (DIPEA) as base in THF, leading to derivative 5 with 86% yield. Final removal of the protecting silyl groups of 5 was achieved using triethylamine trihydrofluoride, affording the target compound catAZT in 68% yield. Synthesis of catTHY, changing azidothymidine by thymidine, was also achieved from the common intermediate 4. However, initial attempts to carry out the coupling reaction of 4 with thymidine using conventional coupling reagents (HATU, EDCl/HOBt or CDI) under standard conditions were unsuccessful. After some experimentation, it was found that the reaction of thymidine with 4 under Mitsunobu conditions (Ph3P and DBAB) delivered the 5′-substituted compound 6 with 56% yield. After coupling, treatment with triethylamine trihydrofluoride removed the silyl protecting groups to afford catTHY in 68% yield (please see Fig. 2a and ESI S1† for details).
:1 mixture of Z and E isomers. The use of other bases and solvents such as potassium tert-butoxide in dry THF proved to be less efficient for this process. Simultaneous hydrogenation of the alkene moiety and removal of the benzyl protecting group at high pressure of H2 under Pd/C catalyst in EtOAc led to catechol 3 with almost quantitative yield. Protection of the hydroxyl groups of 3 as their tert-butyldiphenylsilyl (TBDPS) derivatives was accomplished using TBDPSCl and 1,8-diazabicyclo[5.4.0] undec-7-ene (DBU) in dry acetonitrile, affording the common intermediate 4 with 64% yield. Next, the nucleoside analogue AZT was tethered to this compound using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5b] pyridinium 3-oxid hexafluorophosphate (HATU) as a coupling reagent and N,N-diisopropylethylamine (DIPEA) as base in THF, leading to derivative 5 with 86% yield. Final removal of the protecting silyl groups of 5 was achieved using triethylamine trihydrofluoride, affording the target compound catAZT in 68% yield. Synthesis of catTHY, changing azidothymidine by thymidine, was also achieved from the common intermediate 4. However, initial attempts to carry out the coupling reaction of 4 with thymidine using conventional coupling reagents (HATU, EDCl/HOBt or CDI) under standard conditions were unsuccessful. After some experimentation, it was found that the reaction of thymidine with 4 under Mitsunobu conditions (Ph3P and DBAB) delivered the 5′-substituted compound 6 with 56% yield. After coupling, treatment with triethylamine trihydrofluoride removed the silyl protecting groups to afford catTHY in 68% yield (please see Fig. 2a and ESI S1† for details).
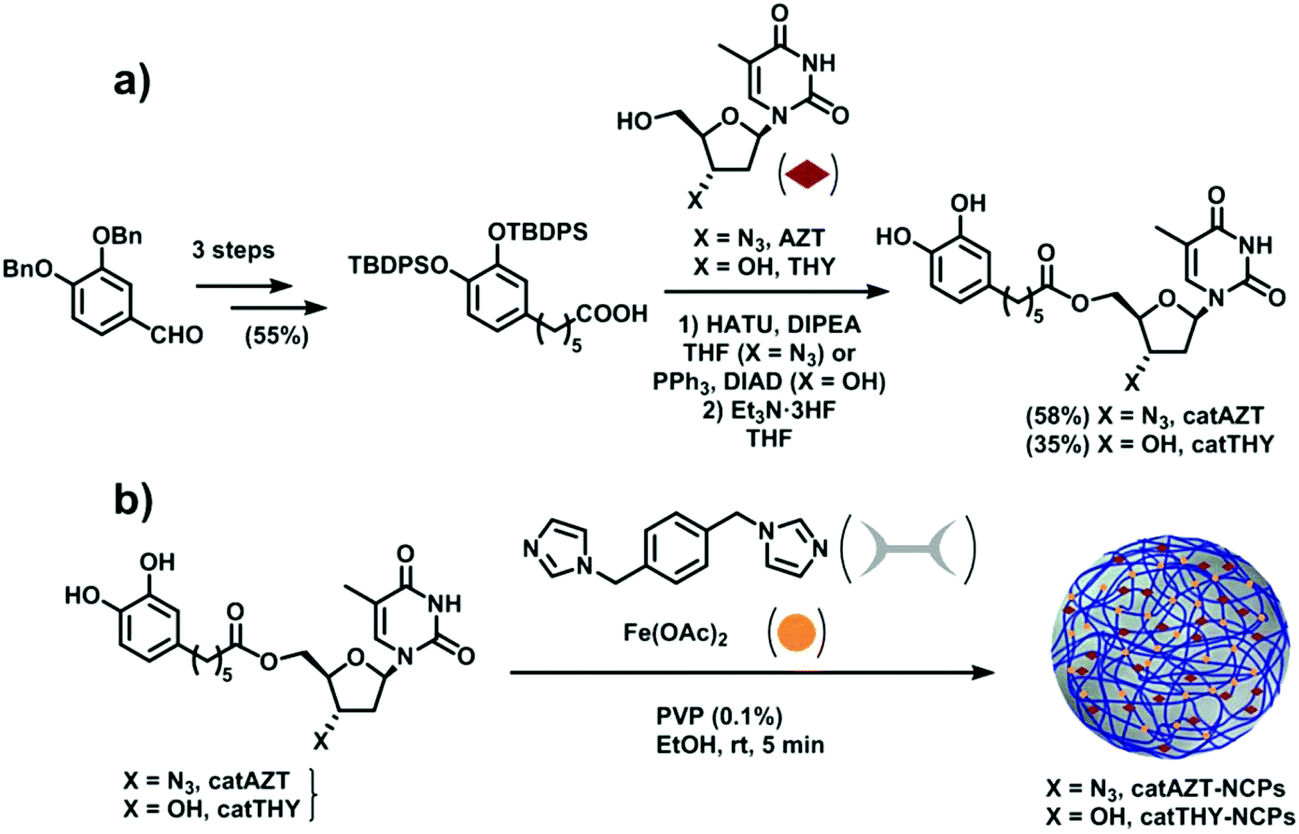 | ||
| Fig. 2 (a) Schematic of catAZT and catTHY syntheses. (b) Preparation of catAZT and catTHY-containing nanoparticles. | ||
Next, experiments were conducted to determine the kinetics of enzymatic hydrolysis of catAZT in PBS/BSA (0.5 mM) at 37 °C. As shown in Fig. 3, in the absence of pig liver esterase (PLE) and independent of the pH used, there was almost no hydrolysis of the ligand (∼35% in 72 h, pH 7.4). On the contrary, addition of PLE under the same experimental conditions resulted in fast release of the antiviral drug (ca. 90% in 1 h). The release profiles for both pH 5.1 and 7.4 offered similar results, indicating weak dependence of the enzymatic activity on pH.
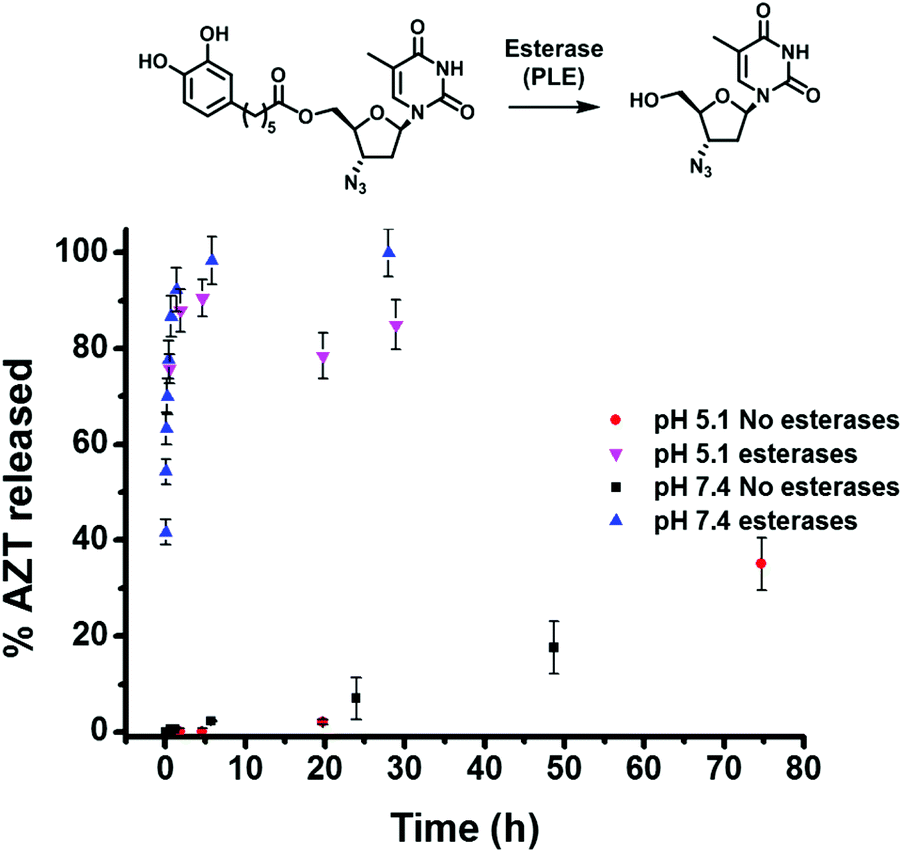 | ||
| Fig. 3 Hydrolysis kinetics of catAZT in the absence (no esterases) or presence (esterases) of pig liver esterases (PLE) at the indicated pH values. In all cases, experiments were performed in PBS/BSA 0.5 mM buffer at 37 °C. Top: Schematic representation of the enzymatic cleavage of catAZT by pig liver esterases (PLE). | ||
Synthesis and characterization of nanoparticles
Ligand catAZT was used to prepare the corresponding antiretroviral nanoparticles (ARV-NCP) catAZT-NCP following a well established methodology32 involving a direct reaction of the catechol ligand and 1,4-bis(imidazol-1-ylmethyl)benzene (bix) with iron(II) acetate using polyvinylpyrrolidone (PVP) as a stabilizing agent (Fig. 2b). After vigorous stirring at room temperature, a dark-purple precipitate was collected by centrifugation, washed several times with ethanol, and dried under vacuum. The synthetic procedure is detailed in the Experimental section, and complete nanoparticle characterization is summarised in ESI S2.† SEM images revealed the formation of spherical catAZT-NCP nanoparticles with an average diameter of 147 ± 33 nm (Fig. 4a), whereas dynamic light scattering (DLS) studies showed an average hydrodynamic radius of 202 ± 10 nm (PDI = 0.07) in ethanol (ESI S2 and Fig. S1†). | ||
| Fig. 4 (a) Representative SEM image of catAZT-NCPs. Inset: Histogram of particle sizes extracted from SEM micrographs (200 particles, mean size 147 ± 33 nm). (b) FTIR spectra of (i) Bix, (ii) catAZT and (iii) catAZT-NCPs. (c) Effect of catAZT-NCPs and free AZT on the cell viability of human primary CD4+ T lymphocytes. CD4+ T cells were incubated for 24 h in the presence of indicated concentrations of catAZT-NCPs or AZT. Cell viability is expressed as percentage compared to an untreated control. Values are presented as mean ± standard error of the mean (SEM) (n = 3). (d) 1H MRI T1 and T2 phantom maps of catAZT-NCPs in a PBS/agarose 1% solution at pH 7.4 at different concentrations (0, 1, 5, 10, and 25 mM, referred to the Fe concentration). | ||
The X-ray powder diffraction (XRD) pattern was characteristic of an amorphous material (ESI S2 and Fig. S2†). The Fourier-transform infrared (FTIR) spectra confirmed coordination of the bix and prodrug ligands to the metal (Fig. 4b). Characteristic bands for the asymmetric azido stretching were observed for both catAZT (2108 cm−1) and catAZT-NCPs (2106 cm−1). Characteristic bands for Bix (1508, 1228 and 1103 cm−1) were also observed in the FTIR spectrum of catAZT-NCPs (1519, 1263 and 1097 cm−1, respectively). Finally, elemental analysis, NMR and ICP data confirmed the presence of different elements with a general chemical formula of [Fe(catAZT)1.5(bix)0.6(AcO)(H2O)2.4] (ESI S2, Table S1 and Fig. S3†). The stoichiometric deviation from theoretical results, which is quite common for NCPs, arises from the out-of-equilibrium conditions used for the synthesis of nanoparticles. In any case, the reaction is very reproducible, as shown by elemental analysis and complete chemical characterization of at least three different and independent synthetic batches. HPLC quantification of the AZT prodrug showed a loading content of 25% by weight (44% of catAZT), which is in agreement with the expected value from the previous chemical formula (for detailed characterization, please see ESI S2 and Fig. S4–S5†). ICP measurements for the iron metal ion (5.46%) were also in agreement with that deduced from the formula of elemental analysis (5.50%). Although the original source of iron is an Fe2+ salt, the complex was stabilized as high-spin Fe3+, as shown by Mössbauer spectroscopy (ESI S2 and Fig. S6†). This electronic modification results from a redox interplay between the metal ion and electroactive catechol ligands in air, as previously reported.44
Complementarily, model catTHY-NCP nanoparticles were also synthesized and tested for comparison purposes. catTHY-NCP nanoparticles showed comparable physicochemical features to catAZT-NCPs; for example, an average diameter of 87 ± 26 nm was measured by SEM micrographs and we obtained an average hydrodynamic radius of 170 ± 2 nm (PDI = 0.14) in ethanol by DLS (for complete characterization, please see ESI S3†).
When the CD4+ T cells were treated with free AZT, a clear cytotoxic effect was observed at concentration of 10 μM (or higher), with an IC50 value of 64 μM. To our surprise, when the same cells were treated with catAZT-NCPs, irrelevant cytotoxic effects were detected even at the highest concentration tested (500 μM). At this concentration, the nanoparticles maintained a cell viability of 70%, whereas for free AZT, it was <10% (IC50 > 500 μM). These results confirmed that nanostructuring reduced the toxicity of AZT remarkably on primary CD4+ T lymphocytes.
Drug release profile
The release profile of AZT from catAZT-NCP nanoparticles involves two main steps: (1) release of the catAZT ligand from the nanoparticles and (2) its transformation into the active AZT prodrug upon enzymatic cleavage by esterases (Fig. 5a). First experiments were conducted to determine the kinetics of catAZT release (step 1) using HPLC for quantification after incubation of the nanoparticles at 37 °C in PBS/BSA 0.5 mM buffer solution at two different pHs 5.1 and 7.4. First experiments in the absence of esterases at pH 7.4 showed no detectable presence of catAZT outside the dialysis bag, indicating high stability of the coordination polymer at this pH (for more details, please see Experimental section). Nevertheless, significant amounts were found at the lowest pH of 5.1, even at early stages of the release, due to the lower stability of coordinative bond between iron and catechol at low pH values. This pH-triggered release of the catAZT ligand could favour the release inside the cells (i.e., acidic pH present in lysosomes), decreasing toxicity-associated side effects. Independent of pH and in the absence of esterases, the presence of free AZT was low (≈15% at 72 h, Fig. 5b). Overall, the release rates are sufficient for NCPs to circulate throughout the body at physiological pH, enhancing the biodistribution.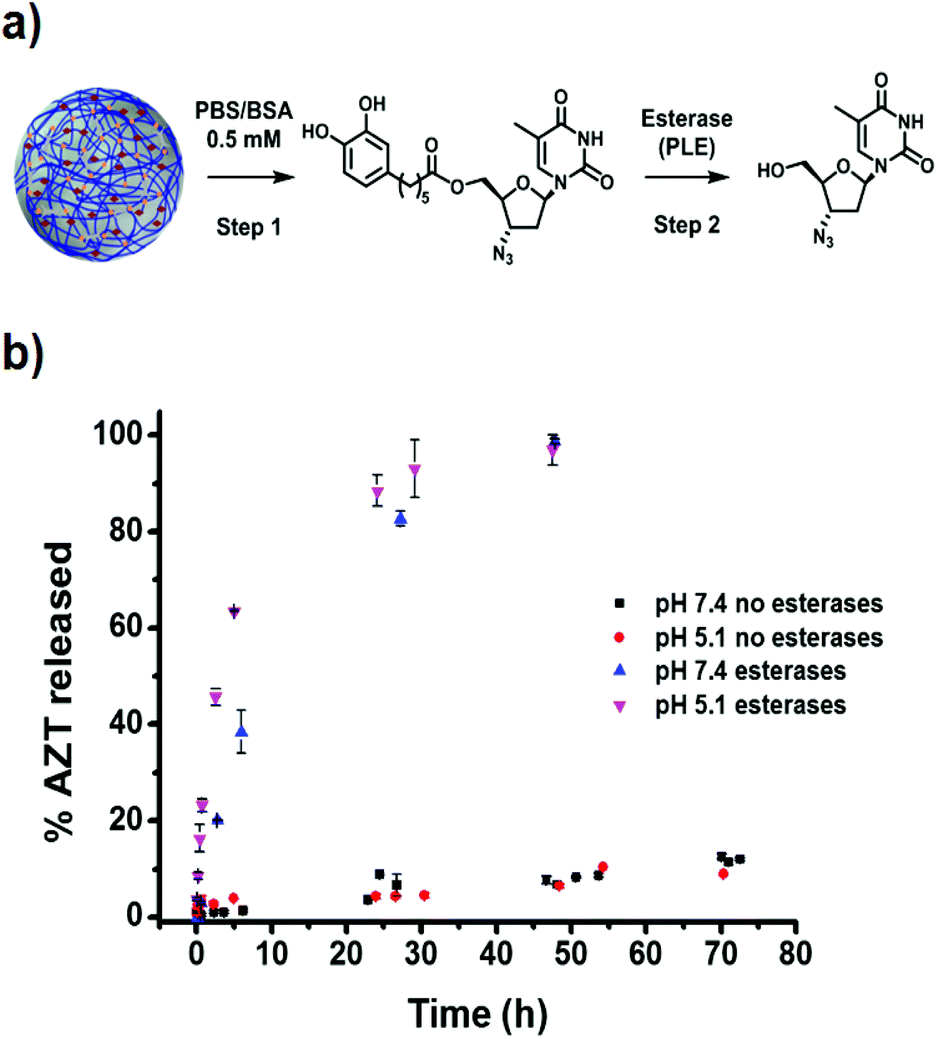 | ||
| Fig. 5 (a) Schematic representation of different release steps for the catAZT prodrug (step 1) and subsequent release of AZT (step 2). (b) AZT release kinetics from catAZT-NCPs in the absence (no esterases) or in the presence (esterases) of pig liver esterase (PLE) at pH 5.1 or 7.4. In all cases, the experiments were performed in PBS/BSA buffer at 37 °C (please see Experimental section for details). | ||
Completely different results were obtained in the presence of the model esterase PLE (180 U L−1). In this case, AZT release appeared to be faster, reaching almost 100% after 50 hours, with estimated half-lives of ∼4.5 h (pH 5.1) and ∼10 h (pH 7.4). These results confirmed that the enzymatic hydrolysis was faster, as previously described for the free catAZT ligand. Moreover, it was confirmed that nanostructuring protects the catAZT ligand from rapid hydrolysis. Meanwhile, for free catAZT, complete AZT release was achieved at 10 h in the presence of esterases (Fig. 3); 100% hydrolysis of catAZT loaded into NCPs required much longer time (ca. 50 h). This experimental observation corroborates the protective ability of NCPs; the release of catAZT ligand from the nanoparticles was proposed as the limiting step for the prodrug hydrolysis.
HIV-antiviral activity of catAZT-NCPs
The antiviral activity of all synthesized nanoparticles and control molecules was tested on a model MT-2 human lymphocytic cell line (purchased from NIH AIDS Reagent Program) by means of an MTT assay in a biohazard P3 laboratory specially prepared for it (for more details, please see Experimental section). MT-2 cells is an established cell line of CD4+ T cells, which is easier to manipulate and obtain than primary human cells; thus, it is suitable for first line studies of anti-HIV effect and therefore broadly used to test the efficacy of experimental antiviral agents due to its high reproducibility.52–54 HIV-1 exerts a profound cytopathic (CPE) effect against CD4+ T lymphocytes. Once infected, the lymphocytes accumulate viral DNA and actively produce HIV proteins, which results in the concomitant lysis of such infected cells by apoptosis. MT-2 cells are profoundly sensitive to the CPE effect of HIV-1.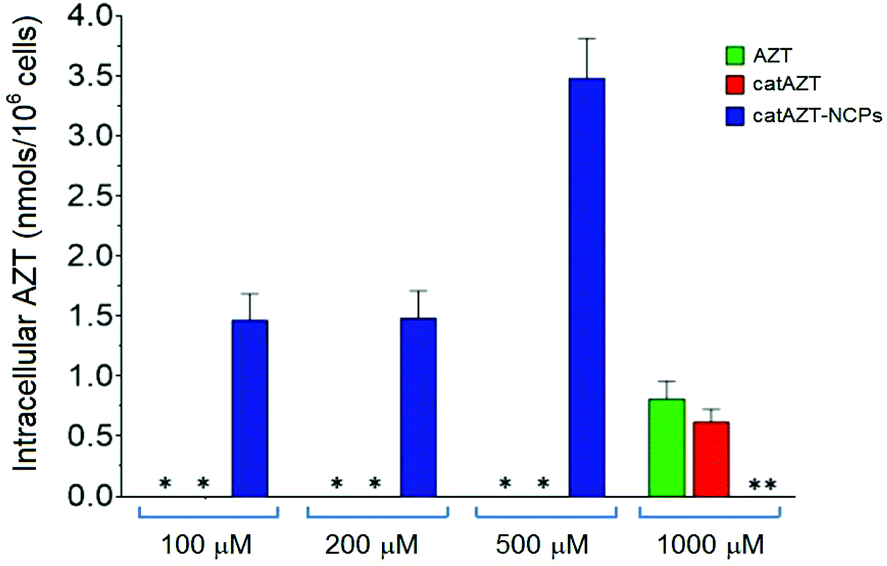 | ||
| Fig. 6 Intracellular levels of AZT in MT-2 cells after 4 h of incubation in the presence of different concentrations (100 μM, 200 μM, 500 μM, referred to AZT equivalent concentrations) of AZT, catAZT or catAZT-NCPs. The asterisk (*) indicates those conditions with levels of AZT in the samples under limit of detection of the method (<0.070 nmols per 106 cells). For AZT and catAZT, an additional concentration of 1000 μM was assayed. Please note that this concentration was not assayed for catAZT-NCPs (**) because nanoparticles were not stable at concentrations higher than 10 mg ml−1. | ||
Considering the detection limit of the HPLC method used for quantification as a reference (<0.070 nmols per 106 cells), we can easily estimate that treatment with the nanostructured material resulted in >50-fold increase at 500 μM in comparison with free AZT. This could potentially be due to the mechanism of uptake. Nanoparticles are generally taken up by endocytosis mechanisms57–60 as opposed to small molecules, which are taken up by other pathways. In the case of AZT, it has been reported to enter the cells through membranes via passive diffusion and/or by uptake transporters.61,62 Similar results were observed in a recent report, in which a nanoformulation of the antiviral drug dolutegravir resulted in higher uptake by human monocyte-derived macrophages in comparison with the free molecule.63 It is worth mentioning that cellular dolutegravir levels reached maximum at 4 h after treatment with 100 μM of the nanoformulated drug.
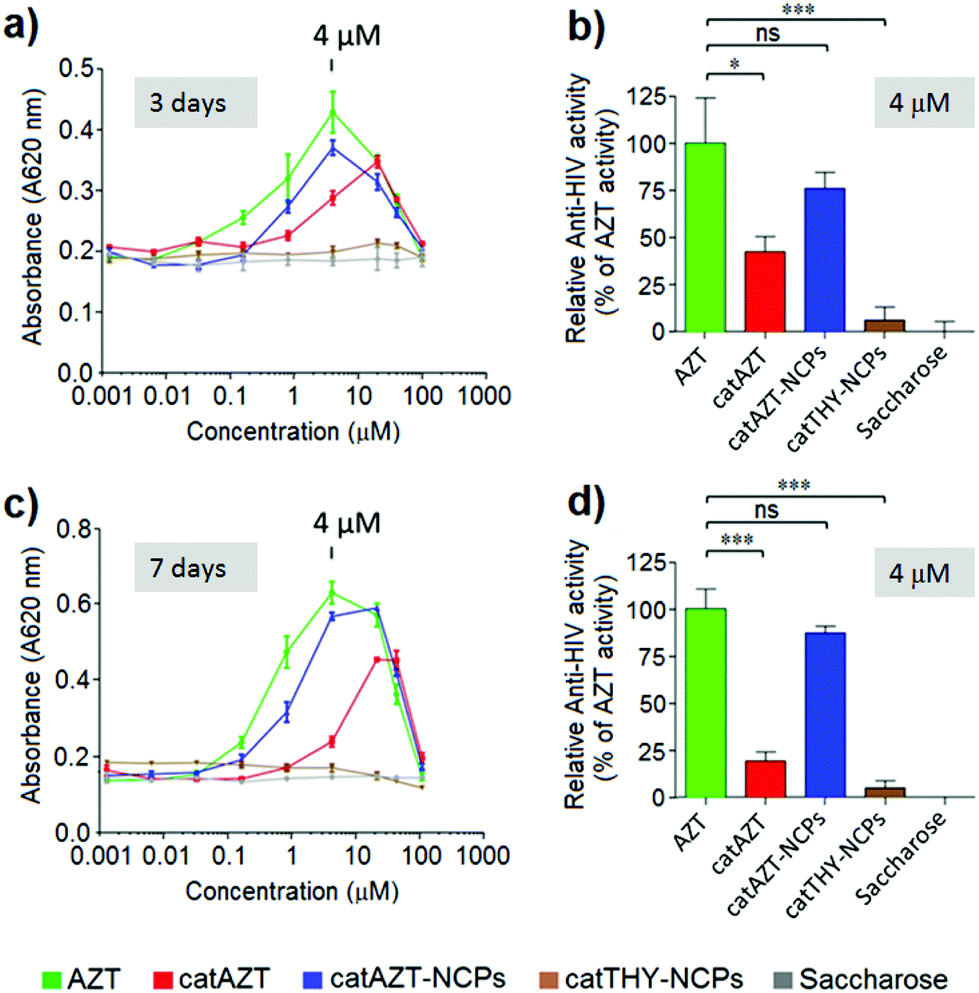 | ||
| Fig. 7 HIV-antiviral activity. Cell viability of different concentrations of AZT, catAZT, catAZT-NCPs, and catTHY-NCPs on HIV-infected MT-2 cell culture after 3 day (a, b) and 7 day incubation (c, d). The cytopathic effect of HIV (decrease in cell viability) is observed after incubating HIV-infected MT-2 cells with the compounds with no antiviral effect (catTHY-NCPs and saccharose). The antiviral effect of the compounds was indirectly measured as an increase in cell viability (increase in absorbance at 620 nm), as shown when incubating HIV-infected MT-2 cells in the presence of AZT, catAZT-NCPs or catAZT (a, c). (b, d) Relative Anti-HIV activity expressed as percentage compared to the absence of activity (saccharose); maximal anti-viral activity in the assay was determined for the most effective concentration of AZT (4 μM) after (b) 3 day or (d) 7 day incubation. An unpaired t-test was performed to compare anti-HIV efficacies of the compounds at 4 μM compared to the reference compound AZT. Please note that the anti-viral efficacy of catAZT-NCPs was similar to that of AZT, whereas the effects of the prodrug catAZT and the control catTHY-NCPs were significantly lower. ns, non-significant; * p < 0.05; ** p < 0.01; *** p < 0.0001. | ||
Conclusions
We have designed and synthesized novel iron–catechol-based nanoscale coordination polymers that incorporate a prodrug molecule tethered to a catechol ligand. To illustrate the potential of this nanoplatform, we have used a catechol ligand (catAZT) linked through an enzymatically cleavable ester bond to the well-known antiretroviral AZT. The presence of a chelating catechol group allowed reproducible incorporation of catAZT with high payloads within the coordination polymer nanoparticles of 147 ± 5 nm average size. Following this approach, we have successfully reproduced the effective antiretroviral activity of free AZT prodrug, whereas nanostructuring allows the following significant advantages: (I) stabilization of the drug in physiological media as a colloidal suspension; (II) control over the release properties of the drug by pH and the presence of enzymes, (III) the nanoparticles retain inherent multifunctionality due to the presence of iron ions with MRI responses, (IV) significant reduction in AZT toxicity and (V) enhancement of the cellular uptake (up to 50-fold increase). catAZT -NCPs exhibited good anti-HIV activity in cell assays to equivalent levels than free AZT but over longer periods of time.According to previous results, the tethering of active drugs as coordinating ligands represents a novel but promising family of carriers to optimize pharmacological characteristics of known antiretrovirals with controlled release while substantially minimizing side effects derived from systemic toxicity effects. Studies are ongoing nowadays to extend this novel approach to other diseases facing similar challenges. It is expected that these new class of nanocarriers will have the capacity to address challenges associated with delivering drug combinations, increasing bioavailability in tissue sanctuaries and latently infected cells, and improving cellular uptake, contributing to the development of the next generation of pharmacological strategies for HIV treatment.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by project MAT2015-70615-R, MAT2012-35324, CTQ2013-44161-R, CTQ2016-75363-R and BIO2016-78057-R from the Spanish Government and FEDER funds. Authors also thank Instituto de Salud Carlos III (ISCIII), Madrid, Spain - Red de Investigación en SIDA (RIS), ISCIII-RETIC (RD16/0025/0002). R. S. thanks the Ministerio de Educación, Cultura y Deporte for the predoctoral grant FPU14/03170. The ICN2 is funded by the CERCA programme/Generalitat de Catalunya and supported by the Severo Ochoa programme of the Spanish Ministry of Economy, Industry and Competitiveness (MINECO, grant no. SEV-2013-0295). MR studies were carried out at the joint NMR facility of UAB and CIBER-BBN, Unit 25 of NANBIOSIS, with a 7T horizontal.Notes and references
- D. A. Kulpa and N. Chomont, J. Virus Erad., 2015, 1, 59–66 Search PubMed.
- T.-W. Chun, S. Moir and A. S. Fauci, Nat. Immunol., 2015, 16, 584–589 CrossRef CAS PubMed.
- C. Schwartz, S. Bouchat, C. Marban, V. Gautier, C. Van Lint, O. Rohr and V. Le Douce, Biochem. Pharmacol., 2017, 146, 10–22 CrossRef CAS PubMed.
- A. Sosnik, Nanomedicine, 2010, 5, 833–837 CrossRef PubMed.
- R. Rose, D. J. Nolan, E. Maidji, C. A. Stoddart, E. J. Singer, S. L. Lamers and M. S. McGrath, AIDS Res. Hum. Retroviruses, 2018, 34, 3–8 CrossRef PubMed.
- B. K. Titanji, D. Pillay and C. Jolly, J. Gen. Virol., 2017, 98, 821–834 CrossRef CAS PubMed.
- A. F. Capetti, M. Micale, L. Carenzi, F. Niero, S. Landonio, S. Vimercati, G. Dedivitiis and G. Rizzardini, Medicine, 2017, 96, e5728 CrossRef CAS PubMed.
- R. R. Adhikary, P. More and R. Banerjee, Nanoscale, 2015, 7, 7520–7534 RSC.
- J. Lisziewicz and E. R. Toke, Nanomedicine, 2013, 9, 28–38 CrossRef CAS PubMed.
- S. D. Mahajan, R. Aalinkeel, W.-C. Law, J. L. Reynolds, B. B. Nair, D. E. Sykes, K.-T. Yong, I. Roy, P. N. Prasad and S. A. Schwartz, Int. J. Nanomed., 2012, 7, 5301–5314 CrossRef CAS PubMed.
- J. das Neves, M. M. Amiji, M. F. Bahia and B. Sarmento, Adv. Drug Delivery Rev., 2010, 62, 458–477 CrossRef CAS PubMed.
- J. das Neves, R. Nunes, F. Rodrigues and B. Sarmento, Adv. Drug Delivery Rev., 2016, 103, 57–75 CrossRef CAS PubMed.
- J. das Neves, B. Sarmento and A. Sosnik, Adv. Drug Delivery Rev., 2016, 103, 1–4 CrossRef PubMed.
- J. D. Siliciano, J. Kajdas, D. Finzi, T. C. Quinn, K. Chadwick, J. B. Margolick, C. Kovacs, S. J. Gange and R. F. Siliciano, Nat. Med., 2003, 9, 727–728 CrossRef CAS PubMed.
- P. Kumar, Y. S. Lakshmi and A. K. Kondapi, Pharm. Res., 2017, 34, 257–268 CrossRef CAS PubMed.
- W. Li, F. Yu, Q. Wang, Q. Qi, S. Su, L. Xie, L. Lu and S. Jiang, AIDS, 2016, 30, 827–838 CrossRef CAS PubMed.
- T. Chaowanachan, E. Krogstad, C. Ball and K. A. Woodrow, PLoS One, 2013, 8, e61416 CrossRef CAS PubMed.
- Y. Jiang, S. Cao, D. K. Bright, A. M. Bever, A. K. Blakney, I. T. Suydam and K. A. Woodrow, Mol. Pharm., 2015, 12, 4363–4374 CrossRef CAS PubMed.
- A. Dalpiaz, L. Ferraro, D. Perrone, E. Leo, V. Iannuccelli, B. Pavan, G. Paganetto, S. Beggiato and S. Scalia, Mol. Pharm., 2014, 11, 1550–1561 CrossRef CAS PubMed.
- J. P. Freeling, J. Koehn, C. Shu, J. Sun and R. J. Y. Ho, AIDS, 2014, 28, 2625–2627 CrossRef CAS PubMed.
- J. Della Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- V. Agostoni, T. Chalati, P. Horcajada, H. Willaime, R. Anand, N. Semiramoth, T. Baati, S. Hall, G. Maurin, H. Chacun, K. Bouchemal, C. Martineau, F. Taulelle, P. Couvreur, C. Rogez-Kreuz, P. Clayette, S. Monti, C. Serre and R. Gref, Adv. Healthcare Mater., 2013, 2, 1630–1637 CrossRef CAS PubMed.
- M. T. Marcos-Almaraz, R. Gref, V. Agostoni, C. Kreuz, P. Clayette, C. Serre, P. Couvreur and P. Horcajada, J. Mater. Chem. B, 2017, 5, 8563–8569 RSC.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
- M. Giardiello, N. J. Liptrott, T. O. McDonald, D. Moss, M. Siccardi, P. Martin, D. Smith, R. Gurjar, S. P. Rannard and A. Owen, Nat. Commun., 2016, 7, 13184 CrossRef CAS PubMed.
- T. O. McDonald, M. Giardiello, P. Martin, M. Siccardi, N. J. Liptrott, D. Smith, P. Roberts, P. Curley, A. Schipani, S. H. Khoo, J. Long, A. J. Foster, S. P. Rannard and A. Owen, Adv. Healthcare Mater., 2014, 3, 400–411 CrossRef CAS PubMed.
- W. J. Rieter, K. M. Pott, K. M. L. Taylor and W. B. Lin, J. Am. Chem. Soc., 2008, 130, 11584–11585 CrossRef CAS PubMed.
- L. Xing, Y. Y. Cao and S. A. Che, Chem. Commun., 2012, 48, 5995–5997 RSC.
- R. C. Huxford, K. E. deKrafft, W. S. Boyle, D. M. Liu and W. B. Lin, Chem. Sci., 2012, 3, 198–204 RSC.
- N. N. Adarsh, C. Frias, T. M. P. Lohidakshan, J. Lorenzo, F. Novio, J. Garcia-Pardo and D. Ruiz-Molina, Chem. Eng. J., 2018, 340, 94–102 CrossRef CAS.
- F. Novio, J. Lorenzo, F. Nador, K. Wnuk and D. Ruiz-Molina, Chem. – Eur. J., 2014, 20, 15443–15450 CrossRef CAS PubMed.
- L. Xing, H. Q. Zheng, Y. Y. Cao and S. A. Che, Adv. Mater., 2012, 24, 6433–6437 CrossRef CAS PubMed.
- I. Imaz, M. Rubio-Martinez, L. Garcia-Fernandez, F. Garcia, D. Ruiz-Molina, J. Hernando, V. Puntes and D. Maspoch, Chem. Commun., 2010, 46, 4737–4739 RSC.
- C. B. He, D. M. Liu and W. B. Lin, Chem. Rev., 2015, 115, 11079–11108 CrossRef CAS PubMed.
- R. C. Huxford-Phillips, S. R. Russell, D. M. Liu and W. B. Lin, RSC Adv., 2013, 3, 14438–14443 RSC.
- D. Liu, C. Poon, K. Lu, C. He and W. Lin, Nat. Commun., 2014, 5, 4182 CrossRef CAS PubMed.
- C. Poon, C. He, D. Liu, K. Lu and W. Lin, J. Controlled Release, 2015, 201, 90–99 CrossRef CAS PubMed.
- L. Amorin-Ferre, F. Busque, J. L. Bourdelande, D. Ruiz-Molina, J. Hernando and F. Novio, Chem. – Eur. J., 2013, 19, 17508–17516 CrossRef CAS PubMed.
- F. Nador, F. Novio and D. Ruiz-Molina, Chem. Commun., 2014, 50, 14570–14572 RSC.
- M. R. Blum, S. H. Liao, S. S. Good and P. de Miranda, Am. J. Med., 1988, 85, 189–194 CrossRef CAS PubMed.
- A. Khandazhinskaya, E. Matyugina and E. Shirokova, Expert Opin. Drug Metab. Toxicol., 2010, 6, 701–714 CrossRef CAS PubMed.
- S. R. Ribone, E. M. Schenfeld, M. Madrid, A. B. Pierini and M. A. Quevedo, New J. Chem., 2016, 40, 2383–2392 RSC.
- D. H. Jo, Y.-M. Chiou and L. Jr Que, Inorg. Chem., 2001, 40, 3181–3190 CrossRef CAS PubMed.
- T. L. Moore, L. Rodriguez-Lorenzo, V. Hirsch, S. Balog, D. Urban, C. Jud, B. Rothen-Rutishauser, M. Lattuada and A. Petri-Fink, Chem. Soc. Rev., 2015, 44, 6287–6305 RSC.
- P. Aggarwal, J. B. Hall, C. B. McLeland, M. A. Dobrovolskaia and S. E. McNeil, Adv. Drug Delivery Rev., 2009, 61, 428–437 CrossRef CAS PubMed.
- M. G. Anhorn, H. C. Mahler and K. Langer, Int. J. Pharm., 2008, 363, 162–169 CrossRef CAS PubMed.
- D. T. Chiu and P. H. Duesberg, Genetica, 1995, 95, 103–109 CrossRef CAS PubMed.
- M. Borges, S. Yu, A. Laromaine, A. Roig, S. Suarez-Garcıa, J. Lorenzo, D. Ruiz-Molina and F. Novio, RSC Adv., 2015, 5, 86779–86783 RSC.
- S. Suárez-García, N. Arias-Ramos, C. Frias, A. P. Candiota, C. Arús, J. Lorenzo, D. Ruiz-Molina and F. Novio, ACS Appl. Mater. Interfaces, 2018, 10, 38819–38832 CrossRef PubMed.
- Z. He, P. Zhang, Y. Xiao, J. Li, F. Yang, Y. Liu, J.-R. Zhang and J.-J. Zhu, Nano Res., 2018, 11, 929–939 CrossRef CAS.
- M. Tsiang, G. S. Jones, J. Goldsmith, A. Mulato, D. Hansen, E. Kan, L. Tsai, R. A. Bam, G. Stepan, K. M. Stray, A. Niedziela-Majka, S. R. Yant, H. Yu, G. Kukolj, T. Cihlar, S. E. Lazerwith, K. L. White and H. Jin, Antimicrob. Agents Chemother., 2016, 60, 7086–7097 CAS.
- L. M. Bedoya, M. Beltran, P. Obregon-Calderon, J. Garcia-Perez, H. E. de la Torre, N. Gonzalez, M. Perez-Olmeda, D. Aunon, L. Capa, E. Gomez-Acebo and J. Alcami, AIDS, 2016, 30, 2767–2776 CrossRef CAS PubMed.
- R. M. Oguariri, L. Dai, J. W. Adelsberger, A. Rupert, R. Stevens, J. Yang, D. Huang, R. A. Lempicki, M. Zhou, M. W. Baseler, H. C. Lane and T. Imamichi, J. Biol. Chem., 2013, 288, 17812–17822 CrossRef CAS PubMed.
- R. M. Mainardes, M. P. D. Gremiao, I. L. Brunetti, L. M. Da Fonseca and N. M. Khalil, J. Pharm. Sci., 2009, 98, 257–267 CrossRef CAS PubMed.
- S. Mandal, Y. Zhou, A. Shibata and C. J. Destache, AIP Adv., 2015, 5, 084803 CrossRef PubMed.
- A. S. Nowacek, R. L. Miller, J. McMillan, G. Kanmogne, M. Kanmogne, R. L. Mosley, Z. Ma, S. Graham, M. Chaubal, J. Werling, B. Rabinow, H. Dou and H. E. Gendelman, Nanomedicine, 2009, 4, 903–917 CrossRef CAS PubMed.
- A. Beduneau, Z. Ma, C. B. Grotepas, A. Kabanov, B. E. Rabinow, N. Gong, R. L. Mosley, H. Dou, M. D. Boska and H. E. Gendelman, PLoS One, 2009, 4, e4343 CrossRef PubMed.
- A. Vonarbourg, C. Passirani, P. Saulnier, P. Simard, J. C. Leroux and J. P. Benoit, J. Biomed. Mater. Res., Part A, 2006, 78, 620–628 CrossRef CAS PubMed.
- Y. Tabata and Y. Ikada, Biomaterials, 1988, 9, 356–362 CrossRef CAS PubMed.
- T. P. Zimmerman, W. B. Mahony and K. L. Prus, J. Biol. Chem., 1987, 262, 5748–5754 CAS.
- E. Errasti-Murugarren and M. Pastor-Anglada, Pharmacogenomics, 2010, 11, 809–841 CrossRef CAS PubMed.
- B. Sillman, A. N. Bade, P. K. Dash, B. Bhargavan, T. Kocher, S. Mathews, H. Su, G. D. Kanmogne, L. Y. Poluektova, S. Gorantla, J. McMillan, N. Gautam, Y. Alnouti, B. Edagwa and H. E. Gendelman, Nat. Commun., 2018, 9, 443 CrossRef PubMed.
- T. Zhou, et al. , Biomaterials, 2018, 151, 53–65 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8bm01221k |
| This journal is © The Royal Society of Chemistry 2019 |