
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Maintaining homogeneity during a sol–gel transition by an autocatalytic enzyme reaction†
Santanu
Panja
 and
Dave J.
Adams
*
and
Dave J.
Adams
*
School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
First published on 23rd November 2018
Abstract
Kinetic control over supramolecular gelation by increasing the pH can be achieved using an enzymatic reaction. This method allows us to produce homogeneous hydrogels with superior and improved mechanical properties as compared to gels obtained from simple addition of base.
Hydrogels derived from the self-assembly of small organic molecules have potential in many areas including structuring, sensors, optoelectronics, catalysis, and cell culture.1–7 These gels are formed by non-covalent interactions such as hydrogen bonding, π–π stacking, and van der Waals interactions between the molecules. As the interactions are individually weak, tuning of the gel properties is possible by changing the molecular environment, which can be used to produce a wide variety of materials.4–8 However, the final gel properties often depend on the method by which the self-aggregation is triggered i.e. upon the kinetics of gelation. In many cases, gels which form slowly are more homogeneous, and often exhibit superior and improved mechanical properties compared to the gels obtained under kinetic trapping.6–10
Gels can be formed by applying a trigger to a solution or suspension of the gelator molecules which result in a significant decrease in their solubility. The most common triggers that are used for hydrogelation include temperature, pH, light, the use of a co-solvent, and an increase in ionic strength.1,3–5,10–16 The choice of gelator determines the trigger that is appropriate. The desired application will also determine the appropriateness of the trigger. pH-Triggered gels are very common.5 In many cases, the pH changes are driven by the addition of a mineral acid or a strong base, resulting in rapid gelation. This can result in gels with irreproducible properties. Since the change in pH results in a decrease in the gelator's solubility, addition of a strong acid or base means that mixing competes with gelation, meaning that preparing homogeneous gels can be difficult in many cases. For example, in the case of acid-triggered Fmoc-dipeptide gels, this is a real issue.15 Related work showed that the gel properties depended heavily on the mixing rates.17 To improve on this, methods that allow a more homogeneous pH decrease were developed, which lead to significantly more homogeneous gels.15,18 Similarly, Thornton et al. showed that a slow enzymatic trigger at a fixed pH resulted in improved properties over a fast pH change for a Fmoc-amino acid gelator.19
In comparison, for gels where an increase in pH triggers gelation, there are more limited options. Beyond the simple addition of aliquots of strong base, Stupp's group have used the diffusion of gaseous ammonia into a vial.20 Whilst this is suitable for some systems, there are limitations in terms of the volume of gel that will be suitable, and there are kinetic issues here since gelation will begin from the gas/liquid interface. Similarly, Nakanishi and co-workers introduced a hot aqueous-urea solution to generate NH3in situ to prepare silica aerogels.21 An alternative approach is to generate ammonia locally by the enzymatic hydrolysis of urea by urease. The urease-catalysed hydrolysis of urea produces NH3, which in turn results in an increase in the pH of the medium.22 The rate of the enzymatic reaction depends upon the initial pH of the solution and the nature of the acid. This method has been used to prepare temporally-controlled polymer gels by Jee et al.22 but not exploited in low molecular weight gels. Here, we utilise this method to form hydrogels by increasing the pH. We show that the kinetics of gelation affects the microstructure of the gels and the mechanical properties.
For the gelators, we investigated a small library of Fmoc-derivatives. Among the many classes of gelators, Fmoc-derivatives have gained attention because of their propensity towards hydrogel formation.9,15,23–28 The Fmoc group is of course widely used as a protecting group during peptide synthesis. It is extremely stable under acidic conditions,29 but unstable at higher pH (typically > pH 10.5).24 Unsurprisingly, when Fmoc-derivatives are used as gelators, these tend to form gels at low pH, with the time at higher pH minimised.
Presumably because of the lack of stability at high pH, Fmoc-based gelators that form gels at high pH are rare.28 Rajbhandary et al. have recently reported hydrogelation of Fmoc-derivatised cationic gelators but gelation was triggered by NaCl.27 Mandal and co-workers reported gelation of some acid-functionalized Fmoc-derivatives in DMSO/H2O at basic pH,28 although the stability in the assembled state at high pH was only explored by FTIR and it is known that DMSO can facilitate Fmoc cleavage even under base-free conditions.29 Whilst there are potential issues in using Fmoc-derivatives for gels at higher pH, we will be targeting a pH lower than 10.5. Aggregation is also known to improve the stability of Fmoc-amino acids to deprotection.30
Initially for screening, the gelation ability of a small library (Fig. 1) was examined by addition of an aliquot of a solution of sodium hydroxide (1 molar equivalent) to aqueous solutions of the compounds (Table S1, ESI†). This resulted in sudden switching of the local pH from acidic (pH 3–5) to basic (pH 9–10.3) that led to the formation of kinetically trapped self-assembled systems. Only Fmoc-2 and Fmoc-3 formed gels (Fig. 1). The resulting gels were turbid and inhomogeneous, presumably due to the kinetics of gelation being faster than the kinetics of mixing as described above. Under identical conditions, the other compounds remained either insoluble in water at low pH (Fmoc-HZ) or resulted in precipitation upon addition of base (Fmoc-4, Fmoc-5 and Fmoc-6).
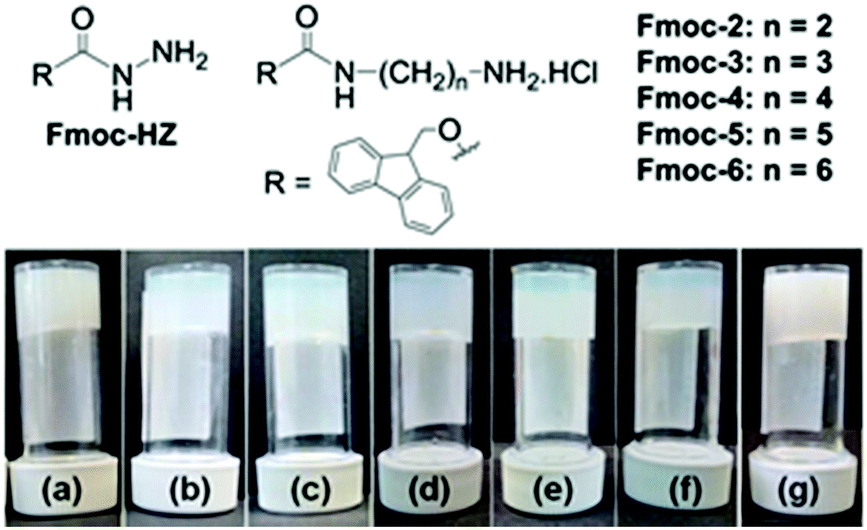 | ||
| Fig. 1 (top) Library of Fmoc-derivatives used here. (bottom) Photographs of hydrogels of Fmoc-2 formed by (a) NaOH and (b)–(f) urea–urease reaction. (b) No acid, [urease] = 0.1 mg mL−1; (c) pH 3.9 (HCl, [urease] = 0.1 mg mL−1); (d) pH 3.9 (AcOH, [urease] = 0.1 mg mL−1); (e) pH 3.9, (HCl, [urease] = 0.03 mg mL−1); (f) pH 3.9 (AcOH, [urease] = 0.03 mg mL−1). In all cases, final concentration of Fmoc-2 is 2 mg mL−1, initial urea concentration is 0.01 M. (g) Photograph of hydrogel of Fmoc-3 (5 mg mL−1) formed by addition of NaOH. | ||
In developing a method for triggering gelation at high pH, the apparent pKa of the gelator needs to be considered. The value of the changed pH in the medium should exceed the apparent pKa of the gelator to allow the formation of the corresponding conjugate base that undergoes self-aggregation. Hence, the apparent pKa of Fmoc-2 and Fmoc-3 were determined and were found to fall in the range of 8.4–8.7 (Fig. S1, ESI†). We then used the urease-catalysed hydrolysis reaction of urea to produce NH3 which results in an increase in the pH of the medium above pH 9 (Fig. 2a).22 The rate of the reaction depends upon the initial pH of the solution and the acid used to lower the pH.22 Initially, we used dilute HCl (0.1 M) to adjust the pH of the urease solution. In the absence of gelator, for solutions initially at pH 5.0 and pH 6.0, the increase in pH upon addition of urea was similar to that of the solution of urease only and reached a plateau at pH 9.2–9.3 within a few minutes (Fig. 2a). However, when the initial pH of the solution was adjusted to pH 3.9 using dilute HCl (0.1 M) or AcOH (0.1 M), the rate of increase in pH was significantly lower (Fig. 2a) and produced a sigmoidal curve for the pH–time profile in both cases. There are slight differences in the profile of pH change depending on the acid used as expected from the effect of in situ generated buffer.22
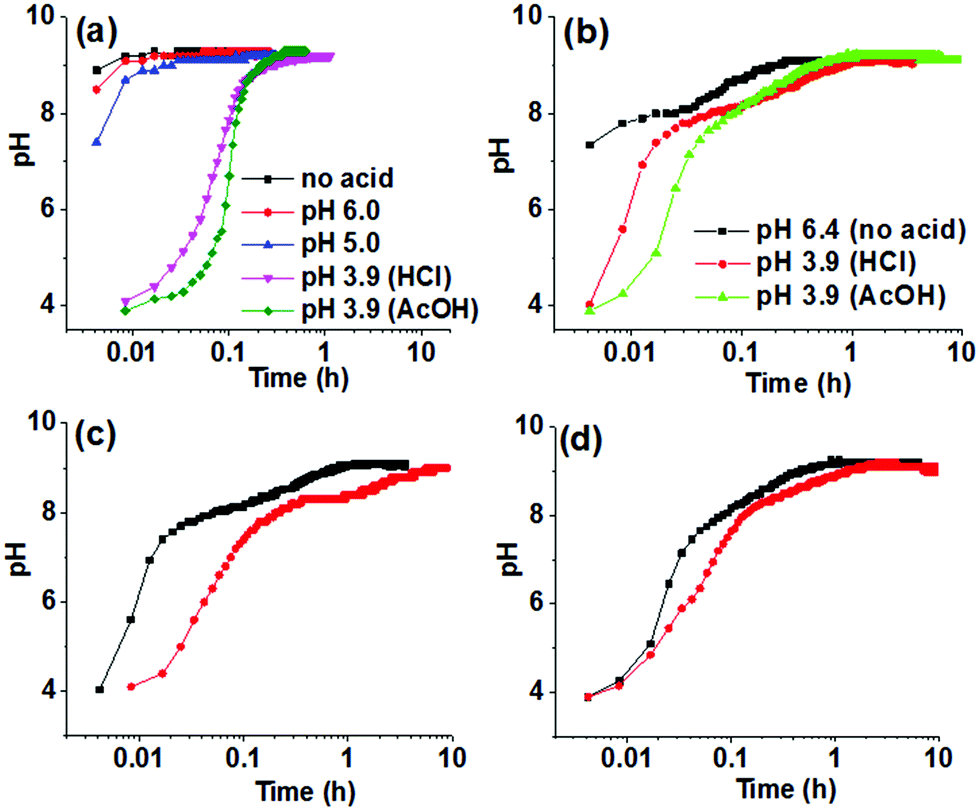 | ||
| Fig. 2 Change in pH with time from the urease–urea reaction in (a) the absence and (b–d) the presence of Fmoc-2 under different conditions at 25 °C. In (b), we compare solutions where the pH has either not been adjusted, or adjusted with either HCl or AcOH. For (c) and (d), the initial pH of the solutions was adjusted to pH 3.9 by using HCl and AcOH, respectively. For (a) and (b) concentration of urease is 0.1 mg mL−1. For (c) and (d) concentration of urease is 0.1 mg mL−1 (black data) and 0.03 mg mL−1 (red data). In all cases, concentration of Fmoc-2 is 2 mg mL−1, initial concentration of urea is 0.01 M. | ||
When adding Fmoc-2 or Fmoc-3, the pH–time profiles were no longer sigmoidal curves. In the presence of Fmoc-2, irrespective of the nature of the acid used to adjust the initial pH of the solutions, a slow but steady increase in pH with time was observed with the propagation of the enzyme-catalyzed reaction (Fig. 2b). When AcOH was used to adjust the pH, the rate of pH change was slower than when HCl was used within the pH range 3.9–8.3 but it was slightly higher from pH 8.3 to pH 9.1. This is probably due to the higher basicity of the conjugate base AcO− compared to Cl−, which was generated within the reaction medium because of the neutralization of the AcOH. The AcO− ions could play the role of a supportive base in solution4 and along with NH3 result in the acceleration of the pH of the medium slightly faster than the HCl case. In all cases, we obtained gels that were more transluscent in comparison to the NaOH-triggered gel (Fig. 1). Interestingly, Fmoc-3 which formed gels when NaOH was used to change the pH did not form gels when using the enzymatic reaction. We ascribe this to the fact that the final pH of the medium (pH 8.6) did not exceed the apparent pKa of Fmoc-3 (8.6–8.7) even after 24 hours (Fig. S2, ESI†).
To further control the rate of gelation, we decreased the concentration of urease keeping other parameters unchanged. A significant decrease in the rate of pH change was recorded with time for both the solutions containing either HCl or AcOH initially (Fig. 2c and d). Here also the pH–time profiles show the existence of two different rate zones below and above the apparent pKa of Fmoc-2, and the rate of increase of pH with time was slow within the pH range 8.3–9.0. This was more pronounced for the solution where HCl was used to adjust the initial pH as compared to AcOH. This indicates the persistence of a new acid–base equilibrium at pH 8.4, where the gelator becomes the free amine and allows gelation (Scheme S1, ESI†). These gels showed no significant change in their visual appearance as compared to the gels obtained at high urease concentrations (Fig. 1). In the absence of any acid, turbidity appeared within 1–2 min, and gelation took place for around 15 minutes (as confirmed by the inversion of vial method). When we decreased the initial pH using HCl or AcOH, gelation occurred for 75 and 50 minutes respectively. A decrease in the concentration of urease further reduced the gelation rate and extended the overall time until gelation more than 4 times. Table S2 (ESI†) summarises the pH–time data for Fmoc-2 under different conditions.
To probe the development of the gels, we carried out rheological time sweep experiments and the data were correlated with pH–time profiles. First, the NaOH-triggered gels were analysed. For Fmoc-2 and Fmoc-3, gelation begins immediately after the addition of NaOH (pH > 9.3) and the gel stiffness (as measured by the storage modulus, G′) gradually increases with time (Fig. S3, ESI†). However, analysis of tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ(G′′/G′) (G′′ is the loss modulus) suggests that no plateau was reached even after 16 hours. In comparison, the hydrogelation of Fmoc-2 by the urea-urease reaction showed a different behaviour (Fig. S4, ESI†). In all cases, gelation begins (shown by G′ > G′′) at ∼pH 8.4, i.e. when the pH had reached the apparent pKa (8.4–8.5) of Fmoc-2. Noticeably, the time required for the initialization of gelation (G′ > G′′) depends upon the initial reaction conditions. As observed, the appearance of G′ > G′′ was delayed proportionally with the decrease in the rate of pH change. With time, both the storage modulus (G′) and the loss modulus (G′′) increased and the tanδ values reached a plateau after a time span that is comparable with the pH–time profiles (Table S2, ESI†).
δ(G′′/G′) (G′′ is the loss modulus) suggests that no plateau was reached even after 16 hours. In comparison, the hydrogelation of Fmoc-2 by the urea-urease reaction showed a different behaviour (Fig. S4, ESI†). In all cases, gelation begins (shown by G′ > G′′) at ∼pH 8.4, i.e. when the pH had reached the apparent pKa (8.4–8.5) of Fmoc-2. Noticeably, the time required for the initialization of gelation (G′ > G′′) depends upon the initial reaction conditions. As observed, the appearance of G′ > G′′ was delayed proportionally with the decrease in the rate of pH change. With time, both the storage modulus (G′) and the loss modulus (G′′) increased and the tanδ values reached a plateau after a time span that is comparable with the pH–time profiles (Table S2, ESI†).
The final mechanical properties of the gels were affected by the kinetics of hydrogel formation. Gels formed using the different triggers exhibited significant differences in G′ and G′′ values (Fig. 3 and Fig. S5, ESI†). For all gels, the storage modulus (G′) was significantly higher than the corresponding loss modulus (G′′) as expected for a gel. At the minimum gelation concentration (mgc), the NaOH-triggered gel of Fmoc-3 exhibited a higher G′ than the gel formed from Fmoc-2. In the strain sweeps for all gels, both G′ and G′′ were essentially constant at low strain, but deviated from linearity after a certain applied stress. Deformation starts (critical strain) at a slightly lower strain for the NaOH-triggered gels (∼0.5% strain) compared to the enzyme-triggered hydrogels (0.7–0.9% strain). The enzyme-triggered gels could withstand a higher strain with higher crossover points (yield point), where G′′ > G′ (>390% strain), than the NaOH-triggered hydrogels (<100% strain) (Table S3, ESI†).
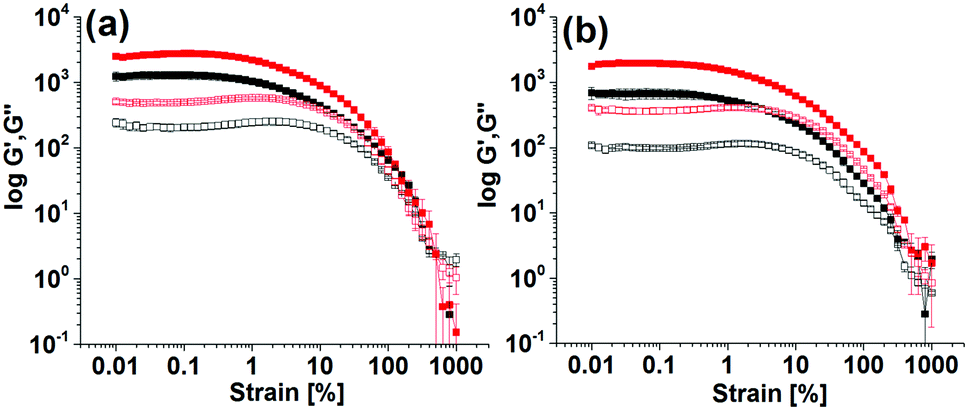 | ||
| Fig. 3 Strain sweep experiments of the enzyme catalyzed hydrogels of Fmoc-2 under different conditions. Initial pH of the solutions adjusted to 3.9 by using (a) HCl and (b) AcOH. In all cases, the closed symbols represent G′, the open symbols G′′. Concentration of urease is 0.1 mg mL−1 (black data) and 0.03 mg mL−1 (red data). Concentration of Fmoc-2 is 2 mg mL−1, initial concentration of urea is 0.01 M. | ||
The enzyme-triggered gels of Fmoc-2 exhibited different rheological properties depending on the exact formation conditions (Fig. 3 and Fig. S5, ESI†). At high enzyme concentrations while using HCl to lower the pH produced gels with higher stiffness (high G′), use of AcOH resulted in substantial increases in gel strength with higher crossover points, as compared to the gel formed without using any acids (Table S3, ESI†). As the urease concentration was decreased, gelation was slower and there was a significant increase (>2 times) in G′ and G′′ (Fig. 3). These gels also showed an increased resistance to strain. The gels were essentially frequency independent (Fig. S6, ESI†). Hence, in comparison to the NaOH-triggered gel, hydrogelation by the urea–urease reaction with low enzyme concentration (irrespective of the acid used) resulted in gels with superior mechanical properties in terms of both gel stiffness and gel strength (Table S3, ESI†). We also prepared gels of Fmoc-2 by adding NaOH and NH4OH slowly over 1 hour (Fig. S7, ESI†). These gels were found to be less stiff and less strong compared to those obtained by the single addition of NaOH. Comparison of the rheological data (Table S3, ESI†) emphasises therefore that a slow and homogeneous pH change is necessary to prepare gels with improved mechanical properties. The data also show that the viscoelastic properties of the gels can be tuned by using different triggers for gelation.
The microstructure of the respective gels was characterised by confocal microscopy imaging. In all cases, fibres are formed (Fig. 4 and Fig. S8, ESI†). The fibres in the network are generally arranged in spherulitic domains. Interestingly, when the gelation rate is high, the gels contained more spherulitic domains, which are less interlinked (Fig. 4a–d). Gels which were formed more slowly exhibit a higher density of long fibres (Fig. 4e and f). This difference in the network structure correlates with the lower stiffness (G′) of the gel as mentioned above.
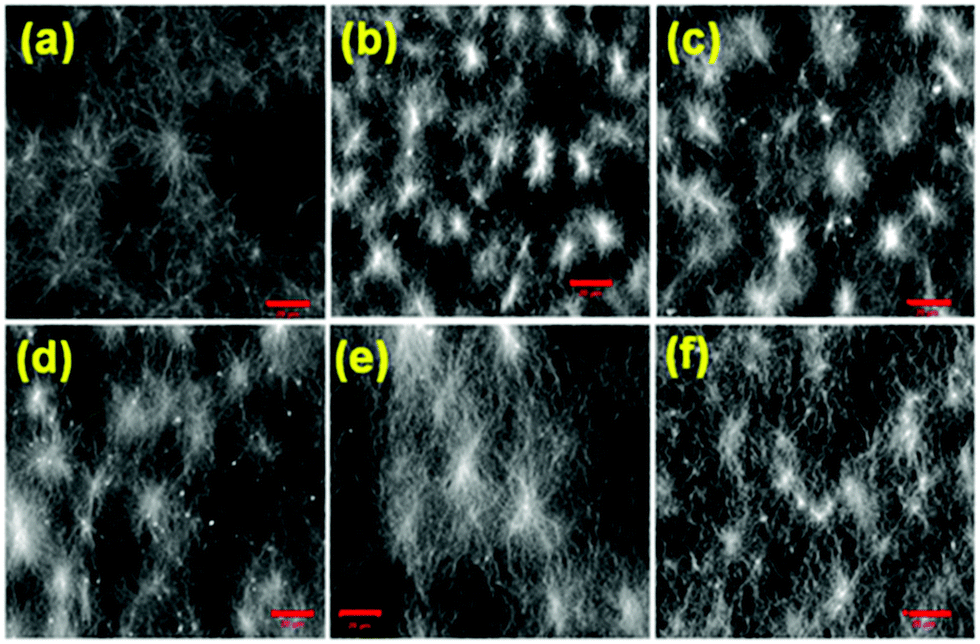 | ||
| Fig. 4 Confocal microscopy images of the hydrogels of Fmoc-2 (conc. = 2 mg mL−1) from (a) NaOH and (b–f) urea–urease reaction with initial conditions of (b) no acid; (c) pH 3.9 (HCl), [urease] = 0.1 mg mL−1; (d) pH 3.9 (AcOH), [urease] = 0.1 mg mL−1; (e) pH 3.9 (HCl), [urease] = 0.03 mg mL−1; (f) pH 3.9 (AcOH), [urease] = 0.03 mg mL−1. Scale bar: 20 μm. | ||
The rate of assembly is likely to affect the quality of the molecular packing.31 The absorption and emission spectra of Fmoc-2 and Fmoc-3 were compared in their respective solution and gel states. In the case of Fmoc-2, on moving from solution to gel states, the absorption bands at 264 nm and 299 nm were slightly red shifted (1–4 nm) (Fig. S9, ESI†). By fluorescence, gelation resulted in around a 10 nm red shift in the monomer emission at 327 nm along with appearance of the excimer bands in the 400–500 nm region (Fig. S9, ESI†). The excimer peaks can be ascribed to overlapping of the fluorenyl groups.32,33 Interestingly, the relative intensity of the excimer peaks increases as the rate of gelation decreases (Fig. S10, ESI†), suggesting that the structures are packed more effectively when gelation is slow.31 Red-shifted absorption and emission spectra were also observed for the NaOH induced gel of Fmoc-3 (Fig. S11, ESI†). Excimer emission was recorded in the same region but with a higher intensity compared to Fmoc-2. This is may be due to the more flexible hydrophobic chain in Fmoc-3 which controls the orientation of the fluorenyl group and allows effective aromatic overlap between the molecules.24
Finally, as noted, the chemical stability of the gelators is a potential concern since the Fmoc might be deprotected at elevated pH. To probe this, we prepared gels, allowed these to stand for 2–3 hours, and then reacidified the samples. The water was removed by freeze-drying and the 1H NMR spectra recorded in d6-DMSO (Fig. S12 and S13, ESI†). These showed that Fmoc-2 and Fmoc-3 were stable under gelation conditions, with no evidence of deprotection as would be evidenced by a peak at 6.27 ppm corresponding to the olefinic protons of dibenzofulvene.29 The FT-IR data for the gels directly freeze-dried at high pH back up this stability, showing the presence of the intact carbamate (Fig. S14, ESI†). However, the NMR spectra recorded at high pH resulted in artefactual data as DMSO facilitates deprotection of the carbamate (Fig. S12 and S13, ESI†).29
In conclusion, we have successfully used the autocatalytic reaction between urease and urea to drive the self-assembly of Fmoc-based cationic amphiphiles at high pH. Kinetic control over gelation is achieved by modulation of the reaction conditions allowing us to prepare homogeneous gels with superior mechanical properties. The chemical stability of the Fmoc gelator at basic pH in the assembled state is not affected. This method complements the methods for slow pH decrease, and we envisage that this will be of great use for the field.
SP thanks the Royal Society and SERB of India for a Newton International Fellowship. DJA thanks the EPSRC for a Fellowship (EP/L021978/1). We thank E. R. Draper and A. M. Fuentes-Caparrós for help with rheometers and confocal microscope and Annette Taylor (Sheffield) for helpful discussions.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133 CrossRef CAS.
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404 RSC.
- C. D. Jones and J. W. Steed, Chem. Soc. Rev., 2016, 45, 6546 RSC.
- S. Panja, S. Ghosh and K. Ghosh, New J. Chem., 2018, 42, 6488 RSC.
- X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165 CrossRef CAS PubMed.
- J. Mayr, C. Saldías and D. D. Díaz, Chem. Soc. Rev., 2018, 47, 1484 RSC.
- E. R. Draper and D. J. Adams, Chem, 2017, 3, 390 CAS.
- M. D. Segarra-Maset, V. J. Nebot, J. F. Miravet and B. Escuder, Chem. Soc. Rev., 2013, 42, 7086 RSC.
- E. R. Draper, K. L. Morris, M. A. Little, J. Raeburn, C. Colquhoun, E. R. Cross, T. O. McDonald, L. C. Serpell and D. J. Adams, CrystEngComm, 2015, 17, 8047 RSC.
- X. Zhang, Y. Wang, Y. Hua, J. Duan, M. Chen, L. Wang and Z. Yang, Chem. Commun., 2018, 54, 755 RSC.
- J. Boekhoven, J. M. Poolman, C. Maity, F. Li, L. van der Mee, C. B. Minkenberg, E. Mendes, J. H. van Esch and R. Eelkema, Nat. Chem., 2013, 5, 433 CrossRef CAS PubMed.
- J. S. Foster, J. M. Żurek, N. M. S. Almeida, W. E. Hendriksen, V. A. A. le Sage, V. Lakshminarayanan, A. L. Thompson, R. Banerjee, R. Eelkema, H. Mulvana, M. J. Paterson, J. H. van Esch and G. O. Lloyd, J. Am. Chem. Soc., 2015, 137, 14236 CrossRef CAS.
- J. Raeburn, T. O. McDonald and D. J. Adams, Chem. Commun., 2012, 48, 9355 RSC.
- Y. Gao, Z. Yang, Y. Kuang, M.-L. Ma, J. Li, F. Zhao and B. Xu, Pept. Sci., 2010, 94, 19 CrossRef CAS.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856 RSC.
- M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615 CrossRef CAS.
- W. Helen, P. de Leonardis, R. V. Ulijn, J. Gough and N. Tirelli, Soft Matter, 2011, 7, 1732 RSC.
- E. R. Draper, L. L. E. Mears, A. M. Castilla, S. M. King, T. O. McDonald, R. Akhtar and D. J. Adams, RSC Adv., 2015, 5, 95369 RSC.
- K. Thornton, A. M. Smith, C. L. R. Merry and R. V. Ulijn, Biochem. Soc. Trans., 2009, 37, 660 CrossRef CAS PubMed.
- A. Mata, L. Hsu, R. Capito, C. Aparicio, K. Henrikson and S. I. Stupp, Soft Matter, 2009, 5, 1228 RSC.
- G. Hayase, S. Nagayama, K. Nonomura, K. Kanamori, A. Maeno, H. Kaji and K. Nakanishi, J. Australas. Ceram. Soc., 2017, 5, 104 CrossRef.
- E. Jee, T. Bánsági, A. F. Taylor and J. A. Pojman, Angew. Chem., Int. Ed., 2016, 55, 2127 CrossRef CAS.
- S. Debnath, A. Shome, D. Das and P. K. Das, J. Phys. Chem. B, 2010, 114, 4407 CrossRef CAS.
- S. Fleming, S. Debnath, P. W. J. M. Frederix, T. Tuttle and R. V. Ulijn, Chem. Commun., 2013, 49, 10587 RSC.
- R. Orbach, I. Mironi-Harpaz, L. Adler-Abramovich, E. Mossou, E. P. Mitchell, V. T. Forsyth, E. Gazit and D. Seliktar, Langmuir, 2012, 28, 2015 CrossRef CAS.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37 CrossRef CAS.
- A. Rajbhandary, D. M. Raymond and B. L. Nilsson, Langmuir, 2017, 33, 5803 CrossRef CAS.
- S. M. M. Reddy, G. Shanmugam, N. Duraipandy, M. S. Kiran and A. B. Mandal, Soft Matter, 2015, 11, 8126 RSC.
- S. Höck, R. Marti, R. Riedl and M. Simeunovic, Chimia, 2010, 64, 200 CrossRef.
- R. Vijay and P. L. Polavarapu, J. Phys. Chem. A, 2012, 116, 10759 CrossRef CAS.
- A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik, J. Boekhoven, J. H. van Esch, S. Santabarbara, N. T. Hunt and R. V. Ulijn, Nat. Chem., 2010, 2, 1089 CrossRef CAS.
- Y. Zhang, H. Gu, Z. Yang and B. Xu, J. Am. Chem. Soc., 2003, 125, 13680 CrossRef CAS PubMed.
- Z. Yang, H. Gu, Y. Zhang, L. Wang and B. Xu, Chem. Commun., 2004, 208 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8cc08501c |
| This journal is © The Royal Society of Chemistry 2019 |