
Transition-metal-free α-arylation of nitroketones with diaryliodonium salts for the synthesis of tertiary α-aryl, α-nitro ketones†
Yang
An
a,
Xiao-Ming
Zhang
 a,
Ze-Yu
Li
a,
Wen-Hui
Xiong
a,
Run-Dong
Yu
a and
Fu-Min
Zhang
*ab
a,
Ze-Yu
Li
a,
Wen-Hui
Xiong
a,
Run-Dong
Yu
a and
Fu-Min
Zhang
*ab
aState Key Laboratory of Applied Organic Chemistry & College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, P. R. China. E-mail: zhangfm@lzu.edu.cn
bKey Laboratory of Drug-Targeting of Education Ministry and Department of Medicinal Chemistry, West China School of Pharmacy, Sichuan University, Chengdu, 610041, P. R. China
First published on 28th November 2018
Abstract
Transition-metal-free α-arylation of α-nitroketones with diaryliodonium salts has been realized for the first time. As an application of this methodology, a concise synthesis of the clinical drug tiletamine was also achieved via a three-step procedure from 2-nitrocyclohexanone without the isolation of intermediates.
α-Arylation of nitroalkanes has been an important but challenging topic in modern organic synthesis,1 and the exploration of effective arylation reagents has been a central focus in this research field, with three types of arylation reagents having been developed so far: (1) nitroaromatic or polyfluoroaromatic compounds bearing strong electron-withdrawing groups,2 (2) organometallic arylation reagents,3 and (3) aryl halides which have recently been independently developed by Muratake and Buchwald and then extensively explored by Kozlowski and others.4 Despite the development of the arylation reagents mentioned above, some inherent drawbacks of these methodologies, such as a narrow substrate scope,2 pre-preparation of the salts of nitroalkanes,3 use of highly toxic reagents with simultaneous generation of environmentally unfriendly waste,3 and relatively expensive catalysts,4 are evident and need to be addressed.
As an ideal arylation reagent, diaryliodonium salt has attracted attention from organic synthetic chemists due to its unique chemical properties as an environmentally-benign, easy-to-prepare, and air-stable reagent that shows good compatibility in both transition-metal-catalyzed and transition-metal-free reactions.5 Retrospectively, the arylation of linear nitroalkanes (6 examples) and dinitroalkanes (2 examples) with diaryliodonium salts was only sporadically reported by Kornhlum and Taylor, and Park and Clapp6a,b in the 1960s, respectively. The corresponding reaction mechanism involving a single electron transfer was later proposed by Singh in 1982.6c Very recently, this attractive arylation approach has been further explored by Olofsson and coworkers, who not only expanded the substrate scope of the reaction to include various cyclic nitroalkanes and ethyl 2-nitropropanoates, but also investigated the possible reaction mechanism.6d However, the arylation of highly active cyclic α-carbonyl nitroalkanes for the construction of synthetically valuable tertiary α-aryl, α-nitro cyclic ketones has not been explored to date.
Tertiary α-aryl, α-nitro cyclic ketones are present in some bioactive compounds, such as herbicides (Fig. 1).7 More importantly, these continuous functionalities could be further derived to produce other valuable products, such as 1,2-amino alcohol, α-nitro alcohol and other important synthetic intermediates which have been widely applied in both catalytic asymmetric reactions and the syntheses of natural products, clinical drugs, and functional molecules. Particularly, tertiary α-aryl, α-amino ketone motifs, which exist in numerous natural alkaloids and clinical medicines (Fig. 1),7 could be smoothly synthesized by the selective reduction of the nitro group. Although this unique scaffold is of valuable importance in current organic synthesis, to the best of our knowledge, only three synthetic approaches have been reported up to now: (1) nitration of highly active 2-aryl-1,3-cycloketones using fuming HNO3,8a (2) arylation of 2-nitrocyclohexanones with triacetoxyl-(3-tert-butylphenyl)-Lead derived from tributylphenylstannane,8b and (3) general nitration of 2-aryl cyclic ketones using ceric ammonium nitrate (CAN) recently developed by our group.9b The former two approaches showed some shortcomings, such as harsh reaction conditions, a narrow substrate scope and the use of highly toxic organic phenylstannane and inorganic Hg(OAc)2 and Pb(OAc)4 reagents (only one example in the second procedure), while the third approach required the assistance of a copper salt (Scheme 1). Therefore, the exploration of novel synthetic approaches toward this unique moiety, especially under the transition-metal-free conditions, is still in high demand. Inspired by the recent elegant work of Olofsson,6d and in connection with our research interest in the efficient construction of aza-quaternary carbon centers of α-substituted cyclic ketones as well as the synthetic application of diaryliodonium salts,9 herein we reported an efficient and general approach toward tertiary α-aryl, α-nitro ketones and its application in the concise synthesis of tiletamine.
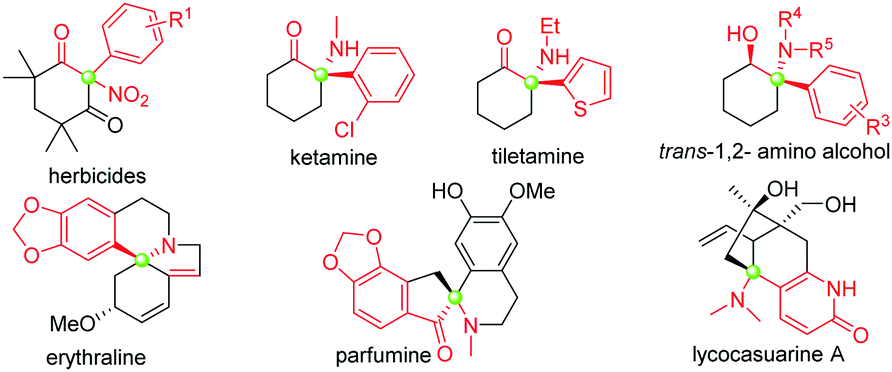 | ||
| Fig. 1 Representative functional molecules related to tertiary α-nitro, α-aryl cyclic ketone moieties. | ||
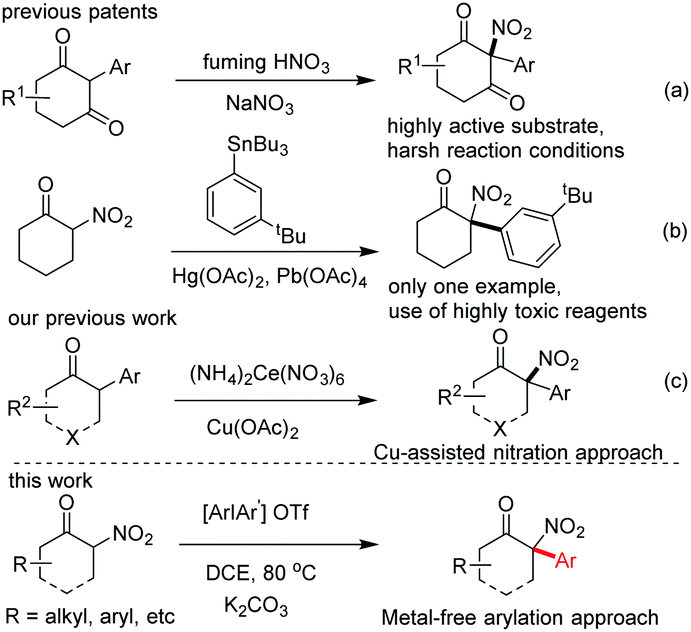 | ||
| Scheme 1 The overview of approaches toward the construction of tertiary α-aryl, α-nitro scaffolds. | ||
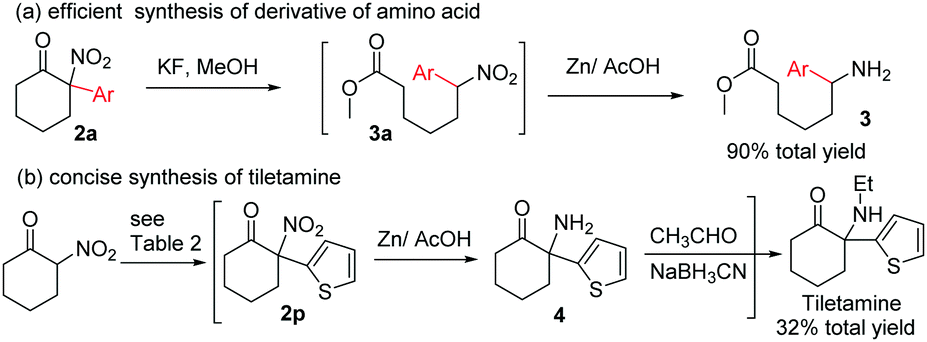 | ||
| Scheme 2 The synthetic application of the resulting products. | ||
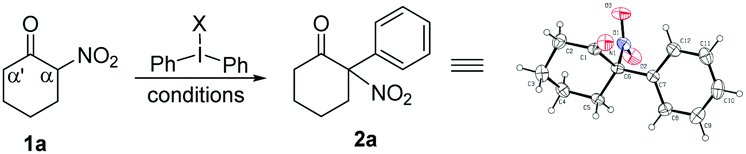
Initially, we selected 2-nitrocyclohexanone as a model substrate and diphenyliodonium triflate as the corresponding arylation partner to investigate the optimal conditions for the arylation reaction. Despite a seemingly easy transformation, some potential problems could probably exist.10 (1) Cleavage of the carbon–carbon bond between the carbonyl and the nitro group under basic reaction conditions has been previously documented;10a (2) the steric effect needs to be considered, because the introduction of a bulky aryl group at the sterically more hindered α position (rather than the α′ position) of the carbonyl group would be less favourable.10b,c (3) O-Arylation would possibly compete with the desired C-arylation.10d After some initial attempts, the desired product 2a (confirmed by X-ray analysis)11 was isolated in 25% yield when the reaction was conducted in toluene using Cs2CO3 as the base at 60 °C (entry 1), while raising the temperature of the reaction would result in a slight increase of the product yield (entry 2).12 Next, various other solvents were screened, and it was found that the reaction in 1,2-dichloroethane (DCE) could produce product 2a in a better yield (entries 3–6).12 Further investigation of different reaction temperatures in DCE revealed an optimal result at 80 °C (entries 7–9). Then, the examination of bases showed that K2CO3 was the best choice,12 which afforded the desired product 2a in 69% yield (entry 10), but a further increase of the amount of K2CO3 could not enhance the yield of 2a. To our delight, the concentration of two reactants also had an influence on the reaction results, and an increased 73% yield could be achieved when the model reaction was performed in 3 mL DCE (entry 11). Subsequently, the equivalent of diphenyliodonium triflate was tested.12 When 1.2 equiv. diphenyliodonium triflate was applied, the expected product 2a was isolated with the best 80% yield (entry 12). Finally, replacement of diphenyliodonium triflate with other diphenyliodonium salts bearing different counter anions was investigated, albeit with no better results obtained.12 Therefore, the reaction parameters listed in entry 12 in Table 1 were selected as the optimal conditions for the next investigation.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Reagent | Base | Solvent | Temp. | Yieldb (%) |
| a Unless noted specially, the reaction was performed using 2-nitrocyclohexanone (0.20 mmol) and diaryliodonium salts (0.20 mmol) in 2.0 mL solvent at the indicated temperature. b Isolated yield of product 2a. c The reaction was performed in 3.0 mL DCE. d 1.2 eq. diphenyliodonium salt was applied. | |||||
| 1 | Ph2IOTf | Cs2CO3 | Toluene | 60 | 25 |
| 2 | Ph2IOTf | Cs2CO3 | Toluene | 110 | 30 |
| 3 | Ph2IOTf | Cs2CO3 | DME | 60 | 21 |
| 4 | Ph2IOTf | Cs2CO3 | DCM | 60 | 22 |
| 5 | Ph2IOTf | Cs2CO3 | CH3CN | 60 | 31 |
| 6 | Ph2IOTf | Cs2CO3 | DCE | 60 | 43 |
| 7 | Ph2IOTf | Cs2CO3 | DCE | 70 | 45 |
| 8 | Ph2IOTf | Cs2CO3 | DCE | 80 | 55 |
| 9 | Ph2IOTf | Cs2CO3 | DCE | 90 | 52 |
| 10 | Ph2IOTf | K2CO3 | DCE | 80 | 69 |
| 11c | Ph2IOTf | K2CO3 | DCE | 80 | 73 |
| 12c,d | Ph2IOTf | K2CO3 | DCE | 80 | 80 |
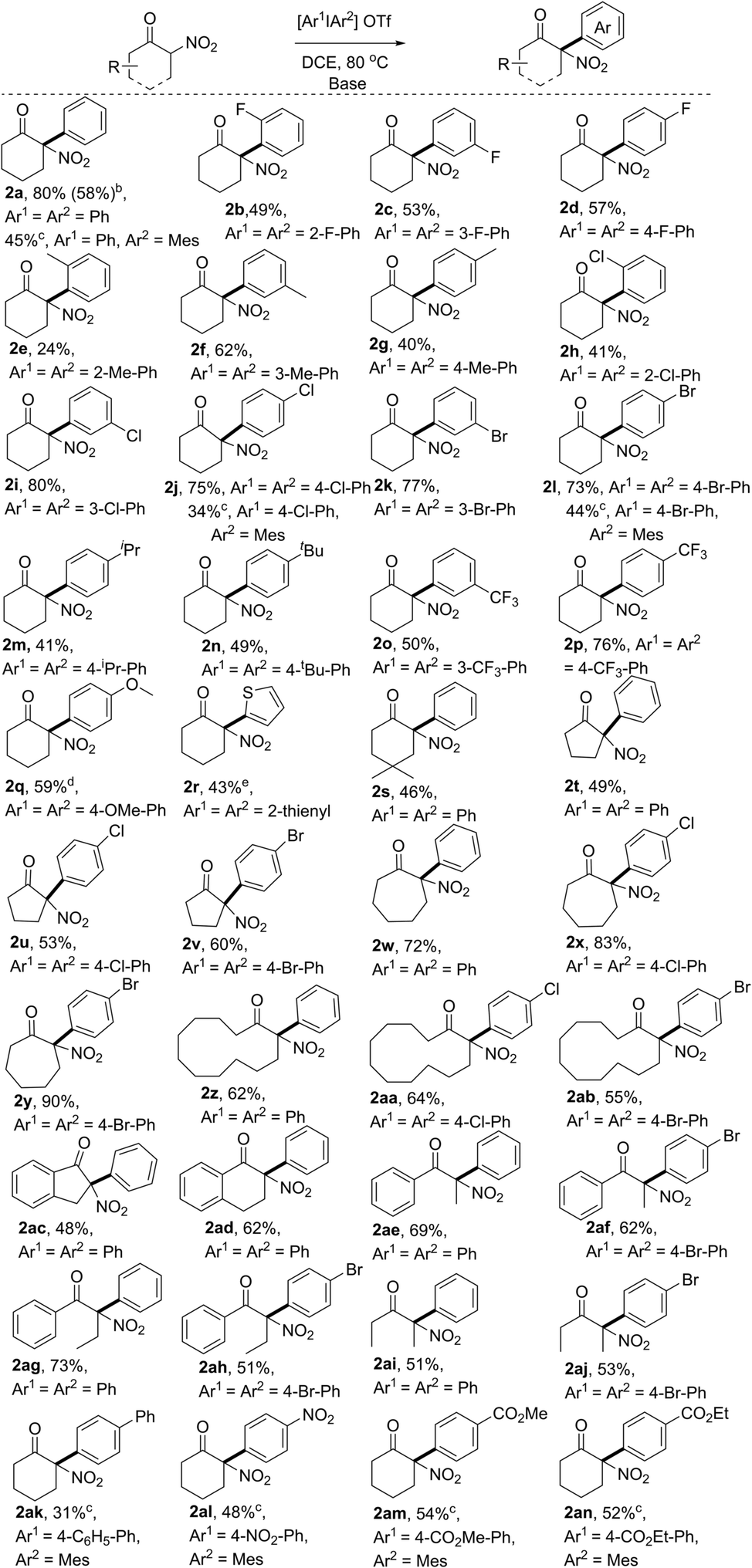
With the optimal reaction conditions in hand, we first explored the scope of substrates using various symmetric diaryliodonium salts. The reactions of these diaryliodonium salts containing o-F, m-F, p-F, o-Me, m-Me, p-Me, o-Cl, m-Cl, or p-Cl substituents performed well, affording the corresponding 2-nitro-, 2-aryl-cyclohexanone derivatives 2b–2j in moderate to good yields. Notably, the steric effects of diaryliodonium salts were evident, because products 2e and 2h were isolated in 24% and 41% yield, respectively, a decreased yield compared to those of their meta- or para-substituted analogues 2f/2g and 2i/2j, while the difference in isolated yields of products 2b–2d with a fluorine atom on arene was negligible. The steric influence was also confirmed by the reaction with Mes2IOTf, which produced the O-arylation enol ethers without isolation of the C-arylation product.12 So other diaryliodonium salts bearing meta- and para-substituents were next tested, and the results showed that the arylation products 2k–2q could be isolated in 41–77% yields. Either electron-withdrawing (p-Br, p-CF3, m-Br, and m-CF3,) or electron-donating (p-iPr, p-tBu, and p-OMe) groups on arene would have negligible influence on the reaction outcomes. It should be noted that dithienyliodonium salt was also proved to be a suitable substrate, affording the desired product 2r in moderate yield, a core carbon skeleton of drug tiletamine.13 Moreover, substituted 2-nitrocyclohexanone was also an ideal substrate, which produced the corresponding 2s in good yield. Then, nitroketones with various ring sizes were also investigated, with 2-nitrocyclopentanone, 2-nitrocycloheptanone, and 2-nitrocyclododecanone providing 2t–2ab in good yields. Notably, benzocyclic nitroketones reacted smoothly, furnishing the desired products 2ac and 2ad in 48% and 62% yield, respectively. Finally, linear nitroketones, either with aryl or alkyl substituents, were also compatible with this transformation, resulting in the formation of arylation products 2ae–2aj in satisfactory yields.
The unsymmetric diaryliodonium salts bearing a “dummy” ligand have a unique property in the control of aryl group transfer in many arylation reactions involving diaryliodonium salts.14 However, when phenyl(mesityl)iodonium triflate was subjected to the optimal conditions, only poor results were initially obtained. After a slight modification of the reaction conditions,12 product 2a could also be isolated in 45% yield (Table 2). Subsequently, other aryl(mesityl)iodonium triflates with different substitution patterns (like Cl, Br, Ph, NO2, and esters) were tested, and could be efficiently converted to the corresponding products 2a, 2j, 2l, and 2ak–2an with acceptable yields. Notably, the application of unsymmetric diaryliodonium salts provided a more concise approach to access α-aryl nitroketones using the strategy of the “dummy” ligand. It is noted that the resulting arylation products bearing halo atoms (F, Cl, and Br), OMe, NO2, and ester groups could be easily transformed to more complex functional molecules through coupling reactions, reduction, substitution, addition, and so on. Hence, the current approach not only provided a new approach to access the synthetically valuable tertiary α-aryl, α-nitro moieties, but also provided potential possibilities to produce more complex molecules bearing this moiety.
| a Unless noted specially, reactions were performed on a 0.20 mmol scale, and the isolated yields were listed. b Reaction was performed on a 7.0 mmol scale. c Reaction on the 0.20 mmol scale using unsymmetric diaryliodonium salts. d The Ar2IBF4 was used to replace Ar2IOTf in CH2Cl2. e Reaction was performed in CH2Cl2. |
|---|
|
We next turned our attention to the synthetic application of the present transformation. A scalable synthesis of product 2a was first conducted. When 2-nitrocyclohexanone (1.00 g, 7 mmol) was subjected to the optimal reaction conditions, product 2a was isolated in 58% yield (0.89 g), albeit with a longer reaction time (3.5 h) (Table 2).12 This result indicated that the current transformation could be applied to the preparation of tertiary α-aryl, α-nitro cyclic scaffolds on a gram-scale. Subsequently, derivation of product 2a was performed. Amino acid derivative 3 could be obtained in 90% total yield through a ring-opening/reduction procedure without the isolation of intermediate 3a, indicating that the resulting products could be used to produce aryl substituted linear α,ω-amino acid derivatives,15 which were not easily accessed using classical synthetic methods (Scheme 2).16
To further demonstrate the synthetic value of the resulting tertiary α-aryl, α-nitro ketones, tiletamine, a clinically used dissociative anesthetic agent for the treatment of various animal diseases, was selected as a target molecule. Arylation of 2-nitrocyclohexanone with dithienyliodonium triflate afforded product 2r, which after reduction of the NO2 group followed by reductive amination of the resulting primary amine produced tiletamine. This three-step manipulation obviates the need for intermediate isolation and thus is efficient in the synthesis of tiletamine11,13 from 2-nitrocyclohexanone, with a 32% total yield (Scheme 2).
In conclusion, we have developed an efficient transition-metal-free approach for the synthesis of α-aryl, α-nitro ketones through arylation of α-nitroketones. The current transformation features mild reaction conditions, the use of environmentally-friendly diaryliodonium salts, and a broad substrate scope, and could produce synthetically valuable products. Moreover, a concise three-step synthetic procedure for a clinical drug tiletamine was achieved starting from 2-nitrocyclohexanone.
We are grateful to the NSFC (No. 21772076 and 21502080) and PCSIRT of MOE (No. IRT-15R28) for supporting this work.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Selected books and review: (a) The Nitro Group in Organic Synthesis, ed. N. Ono, Wiley-VCH, New York, 2001 Search PubMed; (b) R. A. Aitken and K. M. Aitken, Nitroalkanes, in Science of Synthesis, ed. K. Banert, Thieme, Stuttgart, 2010, vol. 41, pp. 9–258 Search PubMed; (c) S. Z. Zard, Helv. Chim. Acta, 2012, 95, 1730 CrossRef CAS.
- (a) N. Kornblum, L. Cheng, R. C. Kerber, M. M. Kestner, B. N. Newton, H. W. Pinnick, R. G. Smith and P. A. Wade, J. Org. Chem., 1976, 41, 1560 CrossRef CAS; (b) R. K. Norris and D. Randles, Aust. J. Chem., 1979, 32, 2413 CrossRef CAS; (c) B. J. Barnes, P. J. Newcombe, R. K. Norris and K. Wilson, J. Chem. Soc., Chem. Commun., 1985, 1408 RSC; (d) W. Danikiewicz and M. Makosza, Tetrahedron Lett., 1985, 26, 3599 CrossRef CAS; (e) N. Ono, T. X. Jun and A. Kaji, Synthesis, 1987, 821 CrossRef CAS; (f) T. Kawakami and H. Suzuki, Tetrahedron Lett., 1999, 40, 1157 CrossRef CAS; (g) N. Colgin, N. J. Tatum, E. Pohl, S. L. Cobb and G. Sandford, J. Fluorine Chem., 2012, 133, 33 CrossRef CAS; (h) R. Vaidyanathaswamy, K. Radha, M. Dharani, T. S. Raguraman and R. Anand, J. Fluorine Chem., 2012, 144, 33 CrossRef CAS; (i) J. I. Day and J. D. Weaver, J. Org. Chem., 2017, 82, 6801 CrossRef CAS.
- (a) D. H. R. Barton, J. C. Blazejewski, B. Charpiot, D. J. Lester, W. B. Motherwell and M. T. B. Papoula, J. Chem. Soc., Chem. Commun., 1980, 827 RSC; (b) R. S. Fornicola, E. Oblinger and J. Montgomery, J. Org. Chem., 1998, 63, 3528 CrossRef CAS; (c) T. Arnauld and D. H. R. Barton, J. Org. Chem., 1999, 64, 6915 CrossRef CAS; (d) R. P. Kozyrod and J. T. Pinhey, Tetrahedron Lett., 1981, 22, 783 CrossRef CAS; (e) R. P. Kozyrod and J. T. Pinhey, Tetrahedron Lett., 1982, 23, 5365 CrossRef CAS; (f) R. P. Kozyrod and J. T. Pinhey, Aust. J. Chem., 1985, 38, 713 CrossRef CAS; (g) R. P. Kozyrod and J. T. Pinhey, Aust. J. Chem., 1985, 38, 1155 CrossRef CAS; (h) H. Kurosawa, M. Sato and H. Okada, Tetrahedron Lett., 1982, 23, 2965 CrossRef CAS; (i) R. K. Norris and D. Randles, Aust. J. Chem., 1979, 32, 2413 CrossRef CAS.
- (a) H. Muratake and H. Nakai, Tetrahedron Lett., 1999, 40, 2355 CrossRef CAS; (b) J. M. Fox, X. Huang, A. Chieffi and S. L. Buchwald, J. Am. Chem. Soc., 2000, 122, 1360 CrossRef CAS; (c) E. M. Vogl and S. L. Buchwald, J. Org. Chem., 2002, 67, 106 CrossRef CAS; (d) H. Muratake, M. Natsume and H. Nakai, Tetrahedron, 2004, 60, 11783 CrossRef CAS; (e) R. R. Walvoord, S. Berritt and M. C. Kozlowski, Org. Lett., 2012, 14, 4086 CrossRef CAS; (f) R. R. Walvoord and M. C. Kozlowski, J. Org. Chem., 2013, 78, 8859 CrossRef CAS; (g) K. F. VanGelder and M. C. Kozlowski, Org. Lett., 2015, 17, 5748 CrossRef CAS; (h) J. Xu, X. Li, J. Wu and W.-M. Dai, Tetrahedron, 2014, 70, 6384 CrossRef CAS; (i) A. A. Mikhaylov, A. D. Dilman, R. A. Novikov, Y. A. Khoroshutina, M. I. Struchkova, D. E. Arkhipov, Y. V. Nelyubina, A. A. Tabolin and S. L. Ioffe, Tetrahedron Lett., 2016, 57, 11 CrossRef CAS; (j) J. Brals, J. D. Smith, F. Ibrahim, F. Gallou and S. Handa, ACS Catal., 2017, 7, 7245 CrossRef CAS; (k) V. V. Bakharev, A. A. Gidaspov and E. V. Kachanovskaya, Russ. J. Org. Chem., 2007, 43, 454 CrossRef CAS; (l) A. V. Gulevskaya, V. V. Goryunenko and A. F. Pozharskii, Chem. Heterocycl. Compd., 2000, 36, 975 CrossRef CAS.
- For recently selected reviews, see: (a) M. Fañanás-Mastral, Synthesis, 2017, 1905 CrossRef; (b) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328 CrossRef CAS; (c) M. Brown, U. Farid and T. Wirth, Synlett, 2013, 424 CAS; (d) Z. Xiao and C. Xia, Chin. J. Org. Chem., 2013, 33, 2119 CrossRef CAS; (e) M. S. Yusubov, A. V. Maskaev and V. V. Zhdankin, ARKIVOC, 2011, i, 370 Search PubMed; (f) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052 CrossRef CAS; (g) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS.
- (a) N. Kornblum and H. J. Taylor, J. Org. Chem., 1963, 28, 1424 CrossRef CAS; (b) K. P. Park and L. B. Clapp, J. Org. Chem., 1964, 29, 2108 CrossRef CAS; (c) P. R. Singh and R. K. Khanna, Tetrahedron Lett., 1982, 23, 5355 CrossRef CAS; (d) C. Dey, E. Lindstedt and B. Olofsson, Org. Lett., 2015, 17, 4554 CrossRef CAS.
- (a) N. K. Garg, D. D. Caspi and B. M. Stoltz, J. Am. Chem. Soc., 2005, 127, 5970 CrossRef CAS; (b) D. B. Freeman, A. A. Holubec, M. W. Weiss, J. A. Dixon, A. Kakefuda, M. Ohtsuka, M. Inoue, R. G. Vaswani, H. Ohki, B. D. Doan, S. E. Reisman, B. M. Stoltz, J. J. Day, R. N. Tao, N. A. Dieterich and J. L. Wood, Tetrahedron, 2010, 66, 6647 CrossRef CAS . For recent reviews, see: ; (c) S. A. Chambers, J. M. DeSousa, E. D. Huseman and S. D. Townsend, ACS Chem. Neurosci., 2018, 9, 2307 CrossRef CAS; (d) A. Hager, N. Vrielink, D. Hager, J. Lefranc and D. Trauner, Nat. Prod. Rep., 2016, 33, 491 RSC . For recently selected examples, see: ; (e) E. E. Blackham and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2017, 56, 6613 CrossRef CAS PubMed; (f) H. Umihara, T. Yoshino, J. Shimokawa, M. Kitamura and T. Fukuyama, Angew. Chem., Int. Ed., 2016, 55, 6915 CrossRef CAS; (g) M. Paladino, J. Zaifman and M. A. Ciufolini, Org. Lett., 2015, 17, 3422 CrossRef CAS; (h) J. Zhang and A. Zhang, Chem. – Eur. J., 2009, 15, 11119 CrossRef CAS PubMed; (i) P. Zanos, R. Moaddel, P. J. Morris, P. Georgiou, J. Fischell, G. I. Elmer, M. Alkondon, P. Yuan, H. J. Pribut, N. S. Singh, K. S. S. Dossou, Y. Fang, X.-P. Huang, C. L. Mayo, I. W. Wainer, E. X. Albuquerque, S. M. Thompson, C. J. Thomas, C. A. Zarater Jr and T. D. Gould, Nature, 2016, 533, 481 CrossRef CAS.
- (a) J. Scutt, C. J. Mathews and M. Muehlebach, Novel Herbicides, WO 20090150093 A1, 2009 Search PubMed; (b) V. John, M. Maillard, J. Tucker, J. Aquino, B. Jagodzinska, L. Brogley, J. Tung, S. Bowers, D. Dressen, G. Probst and N. Shah, Substituted Hydroxyethylamine Aspartyl Protease Inhibitors, WO 2005087751 A2, 2005 Search PubMed.
- (a) S.-Z. Tang, H.-L. Bian, Z.-S. Zhan, M.-E. Chen, J.-W. Lv, S. Xie and F.-M. Zhang, Chem. Commun., 2018, 54, 12377 RSC; (b) Z.-Q. Zhang, T. Chen and F.-M. Zhang, Org. Lett., 2017, 19, 1124 CrossRef CAS; (c) T. Chen, R. Peng, W. Hu and F.-M. Zhang, Org. Biomol. Chem., 2016, 14, 9859 RSC; (d) J.-L. Pan, T. Chen, Z.-Q. Zhang, Y.-F. Li, X.-M. Zhang and F.-M. Zhang, Chem. Commun., 2016, 52, 2382 RSC.
- (a) X. Jiang, B. Gan, J. Liu and Y. Xie, Synlett, 2016, 2737 CAS; (b) G. Giorgi, S. Maiti, P. López-Alvarado and J. C. Menéndez, Org. Biomol. Chem., 2011, 9, 2722 RSC; (c) G. Giorgi, F. J. Arroyo, P. López-Alvarado and J. C. Menéndez, Tetrahedron, 2011, 67, 5582 CrossRef CAS; (d) Z.-F. Xu, C.-X. Cai, M. Jiang and J.-T. Liu, Org. Lett., 2014, 16, 3436 CrossRef CAS PubMed.
- Compounds 2a (CCDC 1859421), 2al (CCDC 1859431), and tiletamine (CCDC 1859419) contains the supplementary crystallographic data for this paper†.
- For details, please see the ESI†.
- (a) S. Sato, N. Takeda, M. Ueda and O. Miyata, Synthesis, 2016, 882 CAS; (b) Y. A. Lapin and I. H. Sanchez, US Pat., 005969159A, 1999 Search PubMed; (c) Y. A. Lapin and I. H. Sanchez, US Pat., 006147226A, 2000 Search PubMed; (d) R. F. Parcell and A. Arbor, US Pat., 3522273, 1970 Search PubMed.
- For recently selected examples, see: (a) Y. Masumoto, K. Miyamoto, T. Iuchi, M. Ochiai, K. Hirano, T. Saito, C. Wang and M. Uchiyama, J. Org. Chem., 2018, 83, 289 CrossRef CAS; (b) D. R. Stuart, Chem. – Eur. J., 2017, 23, 15852 CrossRef CAS; (c) J. Malmgren, S. Santoro, N. Jalalian, F. Himo and B. Olofsson, Chem. – Eur. J., 2013, 19, 10334 CrossRef CAS.
- (a) R. Ballini and M. Petrini, Synth. Commun., 1986, 16, 1781 CrossRef CAS; (b) R. Ballini, M. Petrini and G. Rosini, Tetrahedron, 1990, 46, 7531 CrossRef CAS; (c) R. Ballini, Synlett, 1999, 1009 CrossRef CAS; (d) R. Ballini, F. Papa and C. Abate, Eur. J. Org. Chem., 1999, 87 CrossRef CAS; (e) A. A. Tabolin, A. Y. Sukhorukov, S. L. Ioffe and A. D. Dilman, Synthesis, 2017, 3255 CAS.
- (a) T. R. Demmin and M. M. Rogić, J. Org. Chem., 1980, 45, 2737 CrossRef CAS; (b) A. Laurent, P. Jacquault, J.-L. D. Martino and J. Hamelin, J. Chem. Soc., Chem. Commun., 1995, 1101 RSC; (c) J. H. Sattler, M. Fuchs, F. G. Mutti, B. Grischek, P. Engel, J. Pfeffer, J. M. Woodley and W. Kroutil, Angew. Chem., Int. Ed., 2014, 53, 14153 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1859419, 1859421 and 1859431. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc08920e |
| This journal is © The Royal Society of Chemistry 2019 |