
High temperature expulsion of thermolabile groups for pore-space expansion in metal–organic frameworks†
Macguire R.
Bryant
a,
Timothy A.
Ablott
a,
Shane G.
Telfer
 b,
Lujia
Liu
c and
Christopher
Richardson
*a
b,
Lujia
Liu
c and
Christopher
Richardson
*a
aSchool of Chemistry and Biomolecular Science, University of Wollongong, Wollongong, NSW 2522, Australia. E-mail: chris_richardson@uow.edu.au; Fax: +61 2 4221 4287; Tel: +61 2 4221 3254
bMacDiarmid Institute of Advanced Materials and Nanotechnology, Institute of Fundamental Sciences, Massey University, Palmerston North 4442, New Zealand
cDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208-3113, USA
First published on 9th November 2018
Abstract
Direct radiative heating at 200 °C quantitatively converts sulfoxide-tags to desirable vinyl groups on a porous zinc(II) metal–organic framework analogue of IRMOF-9. The transformation results in an expansion of the pore volume of the framework and a higher surface area. The framework topology and crystallinity are preserved during thermolysis, as established by powder X-ray diffraction, thermal analysis and computational studies. Importantly, the volatile by-product is chemically benign and escapes without damaging the framework. In this way, the post-synthetic thermolysis yields a material with identical properties to that prepared by direct synthesis. This work expands the repertoire of post-synthetic thermochemistry for MOFs and demonstrates that high-temperature processes can be compatible with the retention of important framework properties.
Introduction
The concept of tailored molecular building blocks self-assembling in a single step to produce functional porous crystalline materials, such as metal–organic frameworks (MOFs), is immeasurably attractive. A much-vaunted asset in the assembly of MOFs is the chemical mutability of their building blocks. However, direct substitution of building blocks is not always compatible with isoreticular synthesis, that is, when a specific framework topology is targeted. MOF post-synthetic modification (PSM) is a deliberate strategy to modify the framework after its assembly and can overcome problems of chemical incompatibility. Amongst the many variants of MOF PSM, functionalisation of the bridging ligands while maintaining framework topology is enduring.We are involved in a programme of thermally-promoted post-synthetic modification of MOFs.1–4 Provided that the requisite chemical functionality can be incorporated in the framework, thermally-promoted modifications are straightforward to implement as they simply require heating. Most modifications are eliminations and include thermolysis of tert-butoxycarbamates,5–7 hemiaminals,8 carboxylic acids,9 and azides.10–12 Recently, this concept has been extended to deliberate defect introduction by full thermolytic bridging linker removal under oxidising conditions.13,14 From these examples it is evident that post-synthetic elimination chemistry concurrently delivers new chemical functionality and increases pore size by cleaving groups or bridging ligands from the framework.
We considered vinyl tag groups to be an important post-synthetic target given their potential interactions with guest molecules and scope for further derivatisation. Such groups can be incorporated via direct synthesis under mild conditions15,16 but are prone to polymerisation at elevated temperatures in the presence of strong Lewis and Brønsted acids, potentially precluding many MOF synthesis conditions. Thus, a post-synthetic pathway to vinyl groups represents a worthwhile addition to the growing library of thermally-promoted modifications. Sun et al. post-synthetically generated vinyl groups from a hydroxyethyl-functionalised MOF by elimination of water at 250 °C, in what appears to be a Lewis acid induced process by adjacent Zn SBUs; unfortunately, complete loss of framework crystallinity and porosity accompanied the elimination.17 Therefore, we sought to extend our work on thermally-promoted post-synthetic elimination chemistry of alkyl sulfoxides3 to the generation of vinyl tags and report the results here. We show the modification proceeds quantitatively under direct radiative heating in a solventless process with no framework damage, as demonstrated by comparison to material prepared from direct synthesis (Fig. 1).
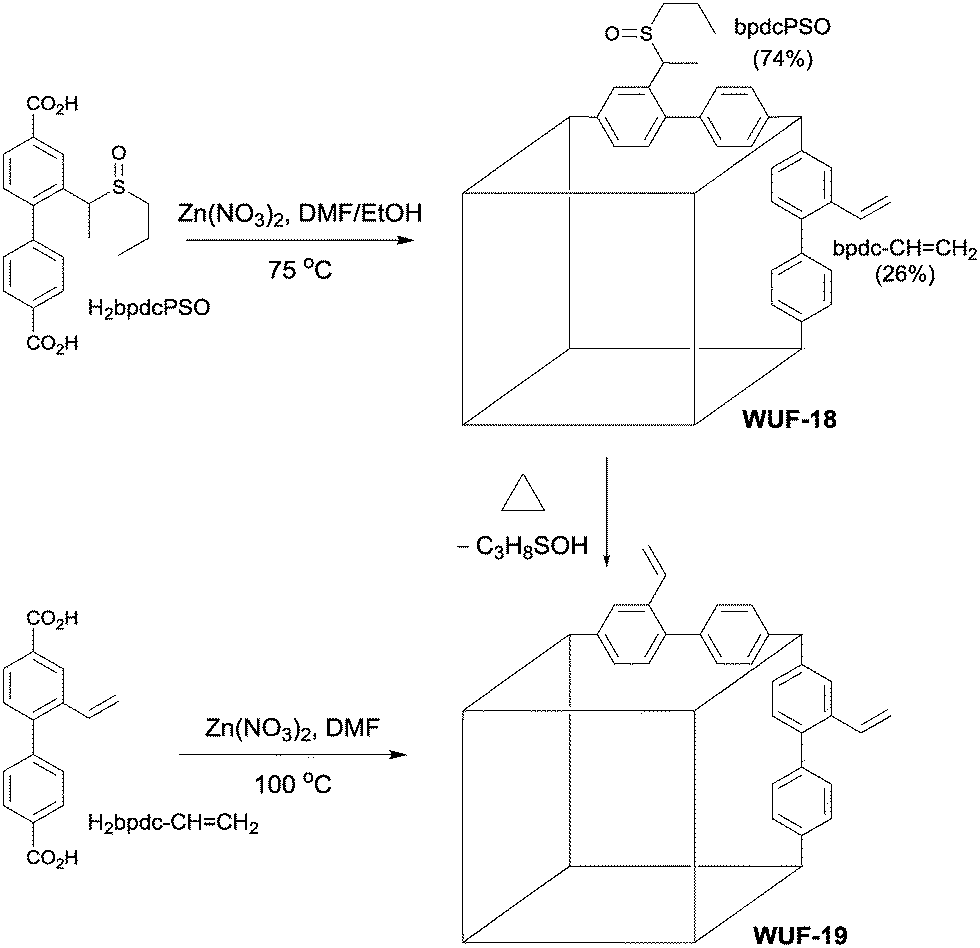 | ||
Fig. 1 The structures of H2bpdcPSO and H2bpdc–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 and the synthetic conditions to form WUF-18 and WUF-19. CH2 and the synthetic conditions to form WUF-18 and WUF-19. | ||
Experimental
All chemicals used were of analytical grade and purchased from either Sigma Aldrich, VWR Australia or Ajax Finechem Pty Ltd. The synthetic procedures for H2bpdcPSO and H2bpdc–CHCH2 are provided in the ESI.†1H NMR and 13C NMR spectra were obtained using a Bruker NMR spectrometer operating at 400 MHz for 1H and 101 MHz for 13C. 1H NMR spectra were referenced to the residual protio peaks at 2.50 ppm (d6-DMSO) or 7.27 ppm (CDCl3). 13C NMR spectra were referenced to the solvent peaks at 39.6 ppm (d6-DMSO) or 77.7 ppm (CDCl3). For 1H NMR analysis, MOF samples (∼5 mg) were digested by adding 35% DCl in D2O (2 μL) and DMSO (500 μL) and waiting until a solution was obtained.
Simultaneous thermogravimetric and differential scanning calorimetry (TG-DSC) data were recorded using a Netzsch STA449F3 at 10 °C min−1 under N2 flow at 20 cm3 min−1 for WUF-18 and WUF-19PSM, and 10 °C min−1 under 5% O2 in N2 at 40 cm3 min−1 for WUF-19.
Powder X-ray diffraction (PXRD) patterns were recorded on a GBC-MMA X-ray diffractometer with samples mounted on 1′′ SiO2 substrates. Experimental settings in the 2θ angle range of 3–30° with 0.02° step size and 1° min−1 scan speed were used for as-synthesised WUF-18 and WUF-19 and 0.04° step size with 3° min−1 scan speed for activated WUF-18 and WUF-19(PSM).
Single crystal diffraction data were collected using a Rigaku Spider diffractometer equipped with a MicroMax MM007 rotating anode generator (Cu Kα radiation, λ = 1.54180 Å), high-flux Osmic multilayer mirror optics, and a curved image-plate detector at 292 K. The data were integrated and scaled and averaged with FS process.18 The crystal structures were solved by direct methods using SHELXS-97 and refined against F2 on all data by full-matrix least-squares with SHELXL-97.19
Gas adsorption studies were carried out using a Quantachrome Autosorb MP instrument and high purity nitrogen (99.999%) gas. Surface areas were determined using Brunauer–Emmett–Teller (BET) calculations. Freeze drying was carried out in a Christ Alpha 1-2 LDplus Freeze Dryer. Elemental microanalysis was performed by the Chemical Analysis Facility, Macquarie University, Australia.
Synthetic procedure for WUF-18
A solution of H2bpdcPSO (10.3 mg, 0.029 mmol) and Zn(NO3)2·6H2O (11.6 mg, 0.040 mmol) dissolved in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMF:EtOH (1.3 cm3) was placed in an oven pre-heated to 75 °C for 48 hours to produce colourless, cubic-shaped crystals. The DMF solution was exchanged five times with fresh DMF at room temperature over three days, then with CH2Cl2 over three days, and then with benzene over three days. The sample was freeze dried for one hour, before being heated at 120 °C for five hours under vacuum to produce an activated sample. Yield 9.1 mg (73%). Found: C, 49.38%; H, 3.96%; N, 0%; S, 5.03%, calc. for [Zn4O(C19H18O5S)2.22(C16H10O4)0.78]·2H2O: C: 49.84%; H: 3.96%; N: 0%; S: 5.39%.
:1 DMF:EtOH (1.3 cm3) was placed in an oven pre-heated to 75 °C for 48 hours to produce colourless, cubic-shaped crystals. The DMF solution was exchanged five times with fresh DMF at room temperature over three days, then with CH2Cl2 over three days, and then with benzene over three days. The sample was freeze dried for one hour, before being heated at 120 °C for five hours under vacuum to produce an activated sample. Yield 9.1 mg (73%). Found: C, 49.38%; H, 3.96%; N, 0%; S, 5.03%, calc. for [Zn4O(C19H18O5S)2.22(C16H10O4)0.78]·2H2O: C: 49.84%; H: 3.96%; N: 0%; S: 5.39%.
Synthetic procedure for WUF-19PSM
Samples were produced by heating WUF-18 at 10 °C min−1 under N2 at 20 cm3 min−1 to 205 °C and holding for 60 minutes in a TG-DSC analyser.Synthetic procedure for WUF-19
A solution of H2bpdc–CHCH2 (29.8 mg, 0.111 mmol) and Zn(NO3)2·6H2O (44.3 mg, 0.148 mmol) dissolved in DMF (2.25 cm3) was placed in an oven pre-heated to 100 °C for 24 hours to produce yellow-coloured crystals. The DMF solution was exchanged three times for fresh DMF at 100 °C, then at room temperature for CH2Cl2 over two days, and then for benzene over two days. The sample was freeze dried for one hour, and then heated at 120 °C for five hours under vacuum to produce an activated sample. Yield 16.8 mg (42%).
Results and discussion
H2bpdcPSO was synthesised in four steps starting from dimethyl 2-formyl-[1,1′-biphenyl]-4,4′-dicarboxylate via a Grignard reaction with methylmagnesium bromide, ZnI2-catalysed thiolation, oxidation with mCPBA and hydrolysis in aqueous base (Scheme 1). The compound was characterised by 1H and 13C NMR spectroscopy.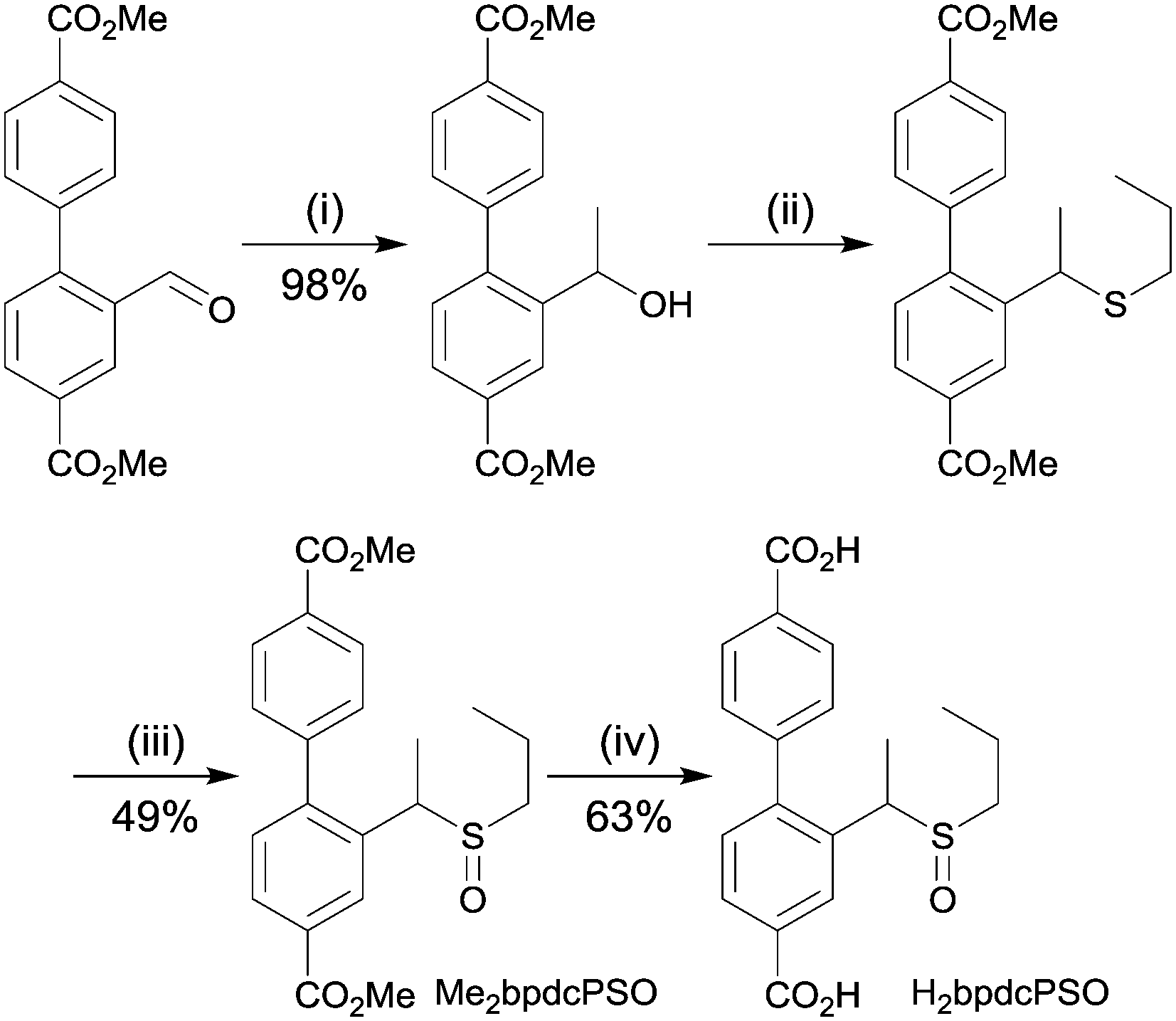 | ||
| Scheme 1 Synthetic pathway to H2bpdcPSO; (i) MeMgBr, THF, 0 °C; (ii) propane thiol, ZnI2, DCE, reflux; (iii) mCPBA, CH2Cl2, 0 °C; (iv) 1 M NaOH, MeOH/THF, rt. | ||
There are two possible syn-eliminations of the sulfoxide tags, as shown in Fig. 2. Path A is favoured, affording a vinyl group and releasing 1-propanesulfenic acid. This conversion is facile in solution and temperature control was important to limit conversion to the vinyl group during ligand synthesis.
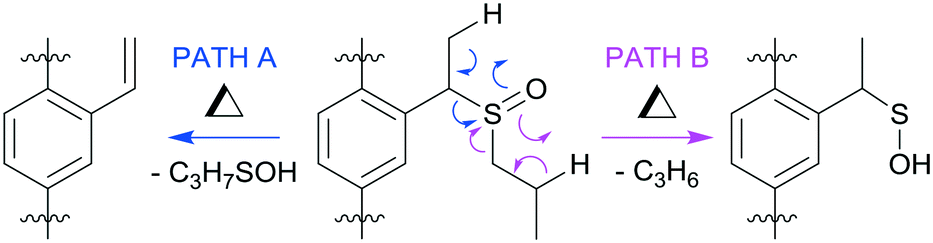 | ||
| Fig. 2 Possible syn-elimination pathways of the sulfoxide tag groups. | ||
For this reason, we applied typical solvothermal conditions for zinc IRMOF formation to Me2bpdcPSO in order to find conditions that minimised conversion of the sulfoxide groups to vinyl groups (Table S1†). The best results were obtained at 75 °C in 3:1 DMF:EtOH where only 10% conversion was found after 24 hours. The reaction of H2bpdcPSO and Zn(NO3)2·6H2O required, however, 48 hours at 75 °C in 3:1 DMF:EtOH solution to produce colourless, blocky crystals of WUF-18. Analysis by 1H NMR spectroscopy of these crystals digested in DCl/d6-DMSO revealed 26% of linkers were bpdc–CHCH2 (Fig. S9†), which is attributable to the longer reaction time. Elemental analysis on activated WUF-18 was consistent with the 1H NMR data in fitting the molecular formula Zn4O(bpdcPSO)2.22(bpdc–CHCH2)0.78·2H2O.
As a basis for comparison in our post-synthetic studies, we prepared Zn4O(bpdc–CHCH2)3 (WUF-19) as yellow crystals via a direct synthesis starting from H2bpdc–CHCH2 (Fig. 1). Analysis by single crystal X-ray diffraction showed WUF-19 crystallises in the space group C2/m as a pair of interpenetrating pcu networks (Fig. 3). We have found this to be a common space group for functionalised IRMOF-9-type compounds.1,4,20–22 A full description of the asymmetric unit of this structure can be found in the ESI.† At their closet point, the frameworks are only 3.5 Å apart but reticulate into a structure with pore sizes of ∼5 and ∼10 Å (Fig. 3). Similar bimodal pore features have been identified for IRMOF-9-type systems.23 The smaller pore is bounded by Zn4O SBUs and entraps a solvate water molecule, while the larger pore system runs parallel to the crystallographic c-axis (Fig. 3b). The vinyl tag groups were not located on Fourier maps, most likely due to positional and rotational disorder, and therefore were implanted in chemically sensible positions to complete the crystallographic model. 1H NMR spectroscopy of digested WUF-19 crystals showed the vinyl groups were incorporated unaltered into the framework.
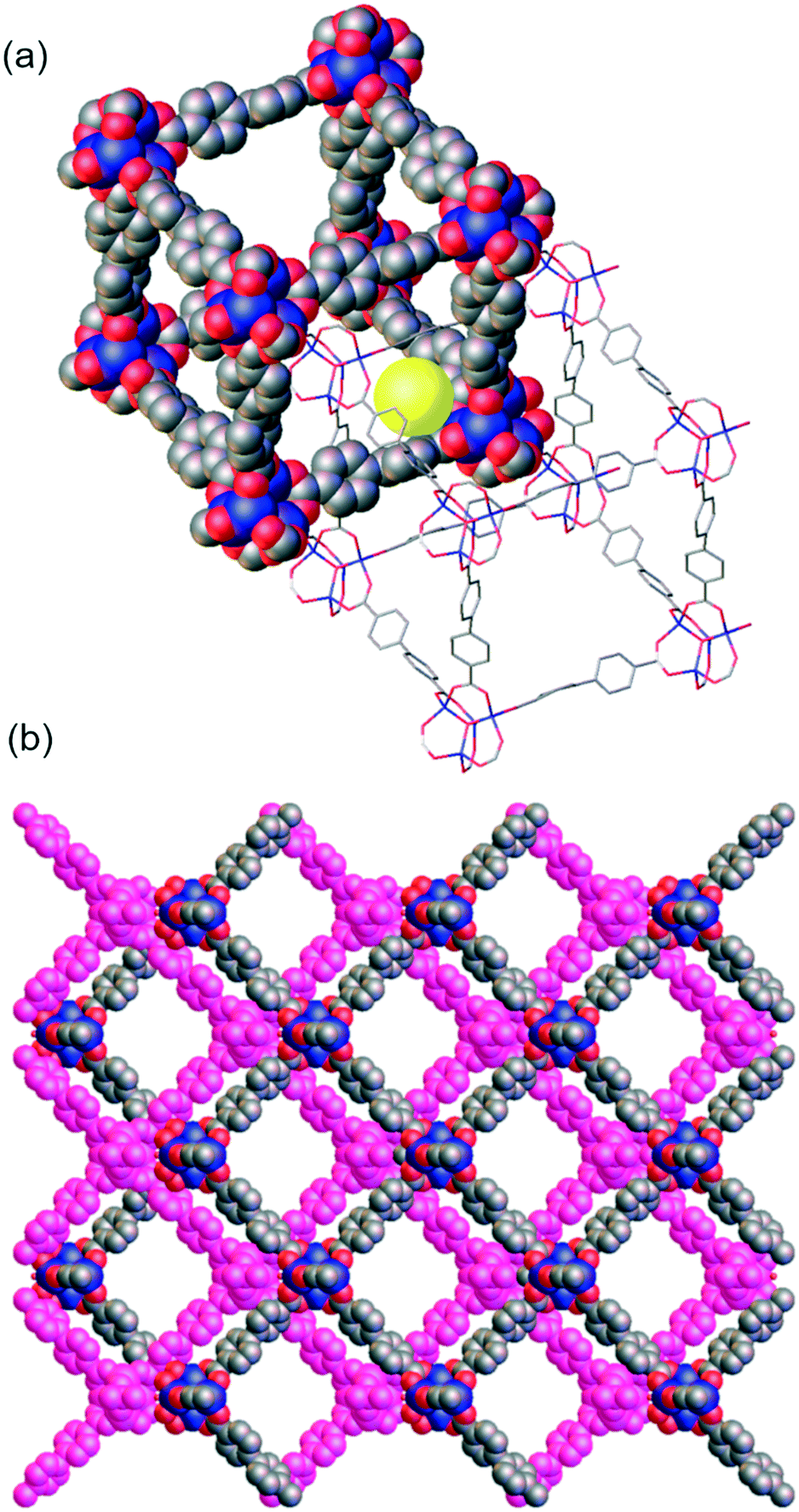 | ||
| Fig. 3 a) A perspective view of the small pore in the interpenetrating networks of WUF-19, b) a view of the larger channels that run parallel to the crystallographic c-axis. Vinyl tag groups and hydrogen atoms are not shown to aid clarity. | ||
The matching PXRD patterns of WUF-18 and WUF-19 with the pattern calculated from the SCXRD analysis (Fig. 4) establishes that bulk samples of these compounds are isoreticular, as expected, and phase pure.
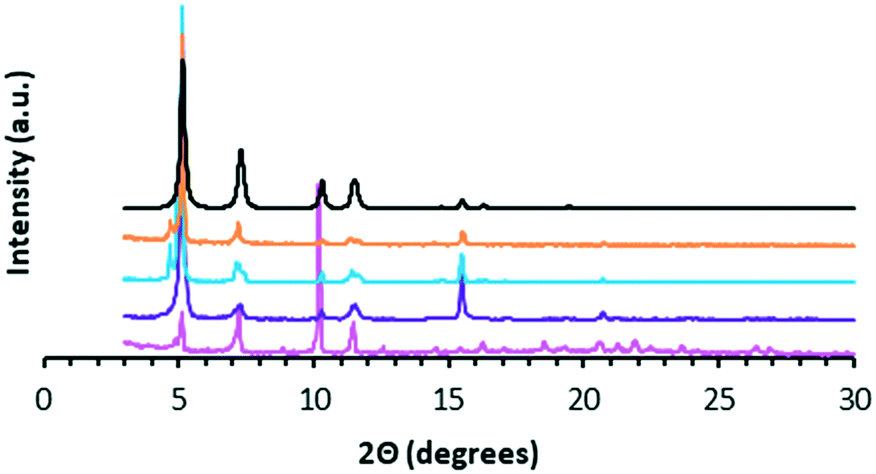 | ||
| Fig. 4 PXRD patterns for ‘as-synthesised’ WUF-18 (magenta), activated WUF-19PSM (purple), ‘as-synthesised’ WUF-19 (blue), activated WUF-19 (orange) and the calculated pattern of WUF-19 (black). | ||
Thermally-promoted post-synthetic chemistry
We used TGA to follow the thermally-promoted post-synthetic chemistry of WUF-18 (Fig. 1). Fig. 5 shows the thermogravimetrogram of activated WUF-18, recorded under a flowing atmosphere of N2. Following a mass loss consistent with desorption of a small amount of surface water (2.0% at 100 °C), there is a loss of 13.6% from 150–350 °C, which is approximately that expected for the elimination of 1-propanesulfenic acid (16.0%). The mass loss observed above 400 °C is due to framework decomposition. In comparison, activated WUF-19 recorded under a flowing atmosphere of 5% O2 in N2 does not show any mass loss until rapid decomposition around 400 °C.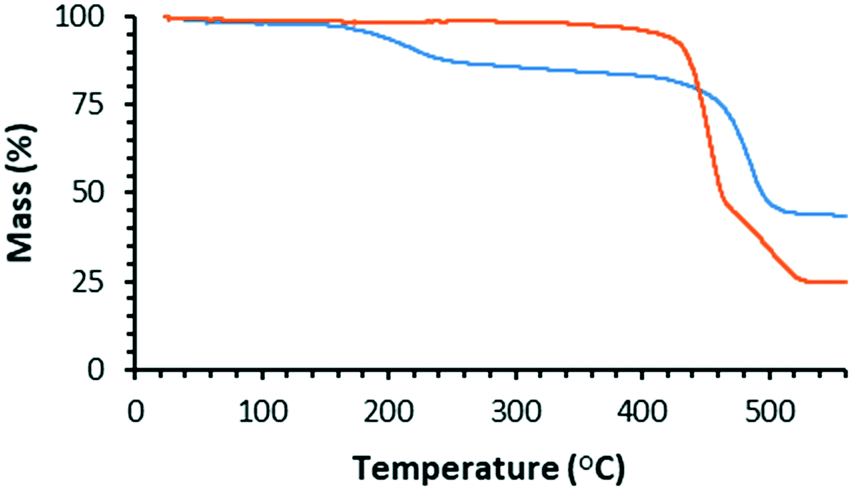 | ||
| Fig. 5 TGA curves for activated WUF-18 (blue) and WUF-19 (orange). | ||
WUF-19PSM was prepared by heating WUF-18 to 205 °C for one hour under N2. Analysis by 1H NMR spectroscopy after digestion in DCl/d6-DMSO confirmed the quantitative nature of the elimination, as the spectrum showed WUF-19PSM was composed solely of bpdc–CHCH2 linkers (Fig. S9†). In addition, WUF-19PSM retains full crystallinity in comparison to the theoretical and experimental PXRD patterns of WUF-18 and WUF-19 (Fig. 4). The excellent maintenance of crystallinity demonstrates that expulsion of 1-propanesulfenic acid does not damage the lattice, despite the high temperature.
Gas adsorption studies
In our experience, gas adsorption isotherms are a more sensitive measure of framework integrity after post-synthetic modifications than PXRD.1–4 With this in mind, we recorded N2 isotherms at 77 K for WUF-18, WUF-19 and WUF-19PSM to ascertain the effects of the modification on surface areas and pore volumes. Fig. 6 shows the adsorption isotherms (see Fig. S14–S16† for full adsorption–desorption isotherms) and the surface areas and pore volumes of the frameworks are summarised in Table 1. Each isotherm displays Type I behaviour typical of microporous MOFs. Significant increases in surface area of over 300 m2 g−1 and in pore volume of ∼0.11 cm3 g−1 are observed moving from WUF18 to the post-modified MOF, WUF-19PSM. This is expected given the elimination of 1-propanesulfenic acid results in smaller sized vinyl groups attached to the framework. Increases in surface area and pore volume are commonly observed when groups were cleaved from MOFs.3,12,24,25 It is also significant that the isotherms, apparent surface areas and pore volumes for WUF-19PSM are virtually identical to WUF-19 prepared by direct synthesis and compare favourably to the geometric surface area calculation of 2070 m2 g−1 calculated using a rolling nitrogen simulation method (see ESI† for details). This demonstrates that this PSM delivers materials directly comparable to direct synthesis and reinforces the gentle nature of this solventless thermal modification.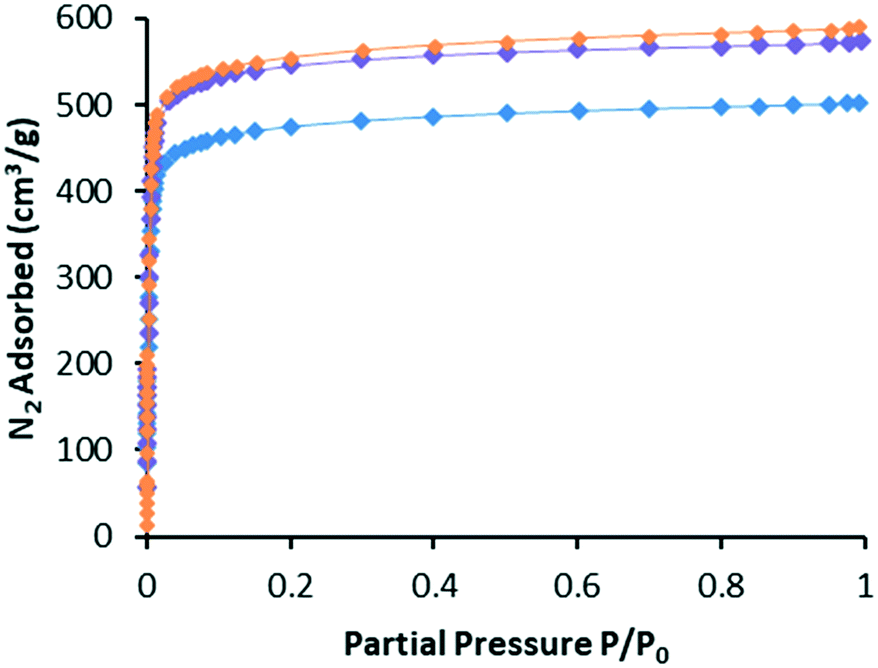 | ||
| Fig. 6 Nitrogen gas adsorption isotherms at 77 K for WUF-18 (blue), WUF-19PSM (purple) and WUF-19 (orange). | ||
Conclusions
In summary, a new, high-temperature, thermally-promoted reaction for MOFs is reported here. This example expands the small but growing library of thermolytic post-synthetic modifications that increase pore volumes and raise surface areas of MOFs. The simple, gentle and reagentless nature of this approach highlights the benefits of post-synthetic thermolysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MRB and TAA acknowledge the Australian Government for Australian Government Research Training Program Awards. CR thanks the University of Wollongong for financial support.Notes and references
- A. D. Burrows, S. O. Hunter, M. F. Mahon and C. Richardson, Chem. Commun., 2013, 49, 990–992 RSC.
- L. Tshering, S. O. Hunter, A. Nikolich, E. Minato, C. M. Fitchett, D. M. D'Alessandro and C. Richardson, CrystEngComm, 2014, 16, 9158–9162 RSC.
- M. R. Bryant and C. Richardson, CrystEngComm, 2015, 17, 8858–8863 RSC.
- T. A. Ablott, M. Turzer, S. G. Telfer and C. Richardson, Cryst. Growth Des., 2016, 16, 7067–7073 CrossRef CAS.
- R. K. Deshpande, J. L. Minnaar and S. G. Telfer, Angew. Chem., Int. Ed., 2010, 49, 4598–4602 CrossRef CAS PubMed.
- A. Sen Gupta, R. K. Deshpande, L. Liu, G. I. N. Waterhouse and S. G. Telfer, CrystEngComm, 2012, 14, 5701 RSC.
- D. J. Lun, G. I. N. Waterhouse and S. G. Telfer, J. Am. Chem. Soc., 2011, 133, 5806–5809 CrossRef CAS PubMed.
- W. Morris, C. J. Doonan and O. M. Yaghi, Inorg. Chem., 2011, 50, 6853–6855 CrossRef CAS PubMed.
- N. Reimer, B. Gil, B. Marszalek and N. Stock, CrystEngComm, 2012, 14, 4119–4125 RSC.
- J. G. Vitillo, T. Lescouet, M. Savonnet, D. Farrusseng and S. Bordiga, Dalton Trans., 2012, 41, 14236–14238 RSC.
- T. Lescouet, J. G. Vitillo, S. Bordiga, J. Canivet and D. Farrusseng, Dalton Trans., 2013, 42, 8249–8258 RSC.
- S. Ganguly, P. Pachfule, S. Bala, A. Goswami, S. Bhattacharya and R. Mondal, Inorg. Chem., 2013, 52, 3588–3590 CrossRef CAS PubMed.
- B. Bueken, N. Van Velthoven, A. Krajnc, S. Smolders, F. Taulelle, C. Mellot-Draznieks, G. Mali, T. D. Bennett and D. De Vos, Chem. Mater., 2017, 29, 10478–10486 CrossRef CAS.
- L. Feng, S. Yuan, L. L. Zhang, K. Tan, J. L. Li, A. Kirchon, L. M. Liu, P. Zhang, Y. Han, Y. J. Chabal and H. C. Zhou, J. Am. Chem. Soc., 2018, 140, 2363–2372 CrossRef CAS PubMed.
- G. Distefano, H. Suzuki, M. Tsujimoto, S. Isoda, S. Bracco, A. Comotti, P. Sozzani, T. Uemura and S. Kitagawa, Nat. Chem., 2013, 5, 335 CrossRef CAS PubMed.
- C. Satheeshkumar, H. J. Yu, H. Park, M. Kim, J. S. Lee and M. Seo, J. Mater. Chem. A, 2018 10.1039/C8TA03803A.
- F. Sun, Z. Yin, Q. Q. Wang, D. Sun, M. H. Zeng and M. Kurmoo, Angew. Chem., Int. Ed., 2013, 52, 4538–4543 CrossRef CAS PubMed.
- Rigaku Corporation: Tokyo, Japan, 1996.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. D. Burrows, C. Frost, M. F. Mahon and C. Richardson, Angew. Chem., Int. Ed., 2008, 47, 8482–8486 CrossRef CAS PubMed.
- A. D. Burrows, C. G. Frost, M. F. Mahon and C. Richardson, Chem. Commun., 2009, 4218–4220 RSC.
- M. R. Bryant, A. D. Burrows, C. J. Kepert, P. D. Southon, O. T. Qazvini, S. G. Telfer and C. Richardson, Cryst. Growth Des., 2017, 17, 2016–2023 CrossRef CAS.
- R. Babarao, C. J. Coghlan, D. Rankine, W. M. Bloch, G. K. Gransbury, H. Sato, S. Kitagawa, C. J. Sumby, M. R. Hill and C. J. Doonan, Chem. Commun., 2014, 50, 3238–3241 RSC.
- T. Gadzikwa, O. K. Farha, C. D. Malliakas, M. G. Kanatzidis, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2009, 131, 13613–13615 CrossRef CAS PubMed.
- K. K. Tanabe, C. A. Allen and S. M. Cohen, Angew. Chem., Int. Ed., 2010, 49, 9730–9733 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, 1H and 13C NMR spectra for H2bpdcPSO and H2bpdc–CHCH2, 1H NMR digestion spectra for WUF-18 and WUF-19PSM, additional TG-DSC data, PXRD data, gas adsorption isotherms and surface area calculations. CCDC 1872644. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce01740a |
| This journal is © The Royal Society of Chemistry 2019 |