
MoIV3-Polyoxomolybdates with frustrated Lewis pairs for high-performance hydrogenation catalysis†‡
Benlong
Luo
ab,
Ruili
Sang
a,
Lifang
Lin
ab and
Li
Xu
 *a
*a
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China. E-mail: xli@fjirsm.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 21st November 2018
Abstract
Bifunctional catalysis of frustrated Lewis pairs (FLPs) is one of the most active frontier areas in catalytic hydrogenation. The first MoIV-γ-Keggin-like cluster H2[(py3MOIV3)2MOVI7O32(OH)4] (γ-1, py = pyridine), which contains unprecedented MoIV–O–MoVI![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O FLPs (POM-FLPs), was prepared in a high yield. The synergistic effect of the POM-FLPs accounts for the high efficiency in catalytic hydrogenation as exemplified by the excellent performance (yield > 99%) of γ-1 as the heterogeneous catalyst in the hydrogenation reduction of nitroarenes to anilines with hydrazine hydrate under mild conditions, revealing the promising potential of such unique MOIV3–POMoVI FLP systems in catalytic hydrogenation.
O FLPs (POM-FLPs), was prepared in a high yield. The synergistic effect of the POM-FLPs accounts for the high efficiency in catalytic hydrogenation as exemplified by the excellent performance (yield > 99%) of γ-1 as the heterogeneous catalyst in the hydrogenation reduction of nitroarenes to anilines with hydrazine hydrate under mild conditions, revealing the promising potential of such unique MOIV3–POMoVI FLP systems in catalytic hydrogenation.
Catalytic hydrogenation is utilized industrially on enormous scales to produce commodity and fine chemicals and pharmaceuticals.1,2 One active frontier area is the bifunctional catalysis of frustrated Lewis pairs (FLPs) built of main-group and/or transition-metal elements.3–6 On the other hand, polyoxometalates (POMs) have been extensively employed in epoxidation, dehydrogenation, photoreductants, water oxidation, electron-transfer and storage mediators in Li-ion batteries among many others,7–10 but their catalytic hydrogenation applications by either high-temperature (>300 °C)11 or electrochemical reduction12 were extremely rare11,12 as a consequence of the absence of Lewis acidic low-valence metal centers to build FLPs.10,13 Recently, we successfully prepared a series of MOIV3–POMoVI hybrids.14 As illustrated in Fig. 1(b), they contain a triangular Mo–Mo-bonded MOIV3 unit wherein each MoIV requires a pair of electrons to achieve its stable 16-electron configuration and hence can serve as a Lewis acid (LA).15–18 The surrounding O–MoVI
O groups have been well established as a Lewis base (LB) induced by the high negative charge and large number of bridging18 and terminal19 oxygen atoms as electron-pair donors. Such unique POM-FLP systems have several advantages: (1) they can simultaneously activate several hydrogen donors and hence provide more protons/hybrids in single catalytic cycles; (2) close proximity between MoIV LAs allows for their cooperative action in binding and activating substrates; (3) the system can be markedly strengthened by the extensive MoIV → MoVI electron transfer that increases MoIV electron-pair accepting and O–MoVIO electron-donating abilities; (4) the peripheral ligands can suffer from functional modification for further applications such as surface decoration20 and engineering.7 In this article, the novel MOIV6-γ-Keggin-like hybrid [(py3MOIV3)2MOVI7O32(OH)4]·2(CH3)2NH2·(CH3)2NH·2H2O (γ-1) was prepared in a repeatable and high yield, and hence employed to examine the catalytic hydrogenation activity of such MOIV3–POMoVI FLP systems for the first time. Expectedly and also excitingly, γ-1 has been found to be an excellent heterogeneous catalyst in the catalytic transfer hydrogenation of nitroarenes to anilines, which are vital building blocks and central intermediates for the manufacture of dyes, pigments, agrochemicals, and pharmaceuticals,21 under mild conditions as will be described in this work.
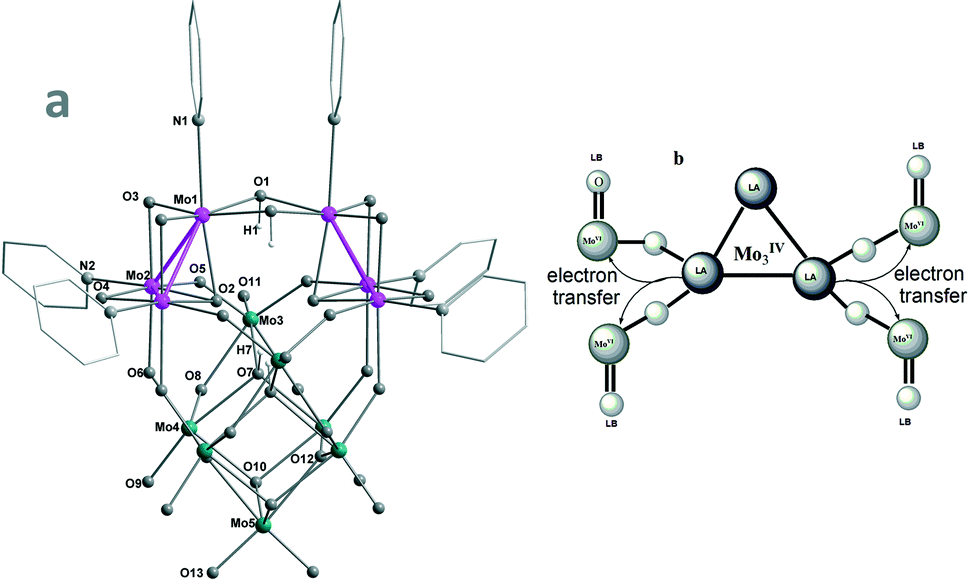 | ||
| Fig. 1 (a, left) Structure of the γ-Keggin-like dianion (γ-1a); (b, right) MoIV–O–MoVIO FLP systems in MOIV3–POMoVI hybrids. | ||
The structure of the hybrid [(py3MOIV3)2MOVI7O32(OH)4]2− (γ-1a) (Fig. 1(a), S1 and S2†) is of crystallographic C2v symmetry with two mirror planes defined by Mo1–Mo1′–Mo5 and Mo3–Mo3′–Mo5, respectively. It can be described as resulting from the 60° rotation of one py3MOIV3O4 of the asymmetric isomer cluster [(py3MOIV3)2MOVI7O32(OH)4]2− (β-2), which was prepared from the uncontrollable oxidation of the [Mo3O4(H2O)9]4+ precursor in an unrepeatable and low yield.14a It features the two (μ2-OH)2-bridged [py3MoIV3O4] incomplete cuboidal units stabilized by the strong triangular Mo–Mo bonds (Mo1–Mo2, 2.5680(1); Mo2–Mo2′, 2.5215(2) Å) and confirmed by XPS (Fig. S3†) as well as the low bond valence sum (BVS) values (Table S3,† Mo1, 4.33; Mo2, 4.35). Each MoIV has an octahedral coordination geometry (without counting Mo–Mo bonds) with an apical pyridine trans to the capping oxygen (O2) to provide the required electron pair. Each pyridine is loosely coordinated to MoIV (2.1408(1)–2.3664(1) Å) and hence easily replaced to activate MoIV LAs. The two [MOIV3O4] LAs are supported by the MOVI7O26 unit featuring the multiple MoVIO and MoVIO2 LBs. The electrophilic addition of the strongly Lewis-basic MoVIO2 (Mo5) occurs at the four μ2-O trans to [MoIV(μ2-OH)]2. The low BVS values of the edge-bridged O1 (1.34) and capping O7 (1.33) reveal that they appear as OH groups. The resulting γ-Keggin-like dianion (γ-1a) is charge-balanced by two protonated Me2NH from the decomposition of DMF. Of note is that reported γ-Keggin anions, one of the five Baker–Figgis–Keggin isomers,22 are all stabilized by interstitial MO4 groups.23 γ-1 represents the first example of empty γ-Keggin-like clusters. The facile formation of γ-1 in a high yield can be partly rationalized on the fact that the electrostatic repulsion in MoVI-γ-Keggin24 is highly alleviated in MoIV-γ-1.
The DFT calculations of γ-1a, β-2a and α-[H4MOVI12O40]4− clearly reveal the MoIV–MoIV-bonded HOMO of the former two (Fig. S4c and S5†), which is higher in energy than that of the latter by about 0.97 eV. Also notable is the decreased HOMO–LUMO gap by 0.3 eV (Fig. S4b†). The markedly increased HOMO energy levels of the 12e-reduced γ-1a and β-2a make them more catalytically active as a consequence of the easier electron transfer from frontier orbitals. The DFT calculations (Table S4†) reveal remarkable MoIV → MoVI electron transfer in γ-1, especially from the tetravalent Mo1(+0.164)/Mo2(+0.125) to hexavalent Mo4(−0.166)/Mo5(−0.246), which appreciably enhances their Lewis acidity (MoIV) and basicity (MoVIO) and hence contributes their synergistic action to be described below.
Unlike previously reported β-2, the insoluble γ-1 was obtained in a repeatable and high yield and selected as the heterogeneous catalyst with the advantages of easy separation and recovery and hence recycling. The catalytic hydrogenation activity to be examined is the reduction of nitroarenes to anilines. Hydrazine hydrate (NH2NH2·H2O), a stable hydrogen transfer reagent, is employed since in principle after hydrogen transfer, the only by-products are water and molecular nitrogen.25 Compared to its unstable anhydrous form, hydrazine hydrate is relatively safe and easy to handle. As shown in Fig. 2 and Table S1,† it is a highly selective and very effective catalyst in the hydrogenation reduction of nitrobenzene to aniline under mild conditions. γ-1 exhibits time-independent perfect selectivity in the catalytic transfer hydrogenation reduction reaction. More than 80% nitroarenes were converted to their corresponding anilines in about thirty minutes. Within two hours, the complete conversion of nitroarenes into the desired anilines in ethanol at 80 °C was achieved. γ-1 can be reused by simple centrifugation with more than 99% conversion and 98% yield in the hydrogenation reduction of nitrobenzene to aniline after the first use as the heterogeneous catalyst shown in Fig. S11 and Table S1.† The examination of molybdenum in reaction solution after centrifugation shows that the concentration of Mo is about 14 mg L−1 corresponding to 0.7% weight of γ-1, clearly indicative of the heterogeneous catalysis. To figure out if the catalytic reactions started from the substitution of the coordinated pyridine to activate MoIV LAs, the reaction solution after the first centrifugation is detected to show the presence of about one-third the amount of pyridine of γ-1 and only traces of pyridine are found after the second centrifugation. This observation reveals that almost all surface pyridines of γ-1 were substituted after the first cycle, indicative of the fact that the catalytic reactions begin with the substitution of the surface pyridine to activate MoIV LAs. This observation was also supported by the IR spectra of 2, 3 and 4 (Fig. S12†) in the second, third and fourth cycle, respectively, wherein the pyridine IR peaks are not well recognized as in the case of the IR spectra of fresh and 1 (1607 cm−1, 1449 cm−1, and 644 cm−1) which may account for the weakened structural integrity and the lowered conversion in the second, third and fourth cycle, as shown in Fig. S11.† Remarkably, as displayed in Fig. 2(left), the larger amount of hydrazine and prolonged catalytic reaction time can afford yields of more than 98% in the second, third, fourth and fifth cycle.
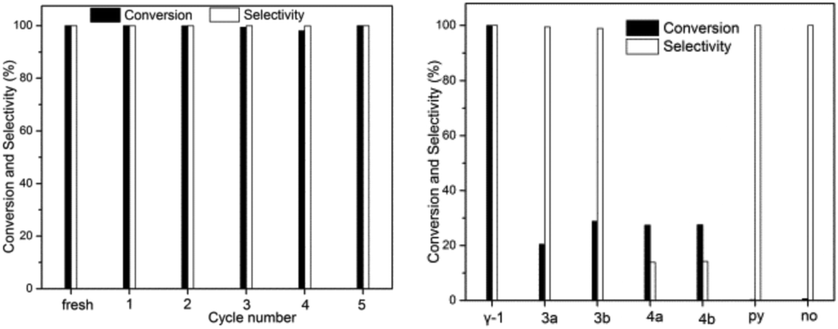 | ||
| Fig. 2 Reusable profiles (left) and catalyst-dependent performance (right) of γ-1 for the reduction of nitrobenzene to aniline (reaction conditions see the ESI† for more details). | ||
The scope of this catalytic system has also been examined by testing a variety of nitroarenes with different substituents. As shown in Table S1,† the nitroarenes with both electron-donating (H, CH3) and electron-withdrawing (Br, CO2CH3) groups are smoothly reduced to the desired anilines with a quantitative yield. Meanwhile, bromide-substituted aniline is obtained in excellent yield without the formation of any dehalogenation products. γ-1 is also able to reduce the nitro group with excellent selectivity in a substrate bearing a reducible ester moiety to afford a more demanding functionalized aniline.
Fig. S13† shows the solvent-dependent catalytic performance of γ-1 under the same reaction conditions. These Lewis basic solvents are of decreasing MoIV-coordinating capacity: pyridine > CH3CN > H2O > C2H5OH ~ isopropanol > THF > ethyl acetate, of which water, ethanol and isopropanol are the protic ones and only H2O can't dissolve nitroarene. The appropriate and reversible solvent→MoIV dative coordination by replacing the surface pyridine13–15 plays an essential role in both activating and in situ stabilizing MoIV LAs. As revealed in Fig. S13,† both water incapable of dissolving nitroarenes and THF/ethyl acetate with very poor coordination ability exhibit the lowest activity. The selectivity and, to a much less extent, conversion can be significantly improved by using pyridine and acetonitrile of enhanced datively coordinating capacity. The moderate yields for both pyridine and acetonitrile should be assigned to the fact that they are not protic solvents, which is supported by the observation that the protic isopropanol with the required coordination capacity markedly enhances the yield (90%). However, its performance is still not perfect because of its steric bulkiness. This has been confirmed by the perfect performance (yield, 99.8%) of the smaller ethanol molecules (Fig. S13†). These observations reveal the essential roles of protic, dissolving and datively coordinating capacities in a solvent, which acts cooperatively to deliver both hydrogen transfer reagent (hydrazine) and nitroarenes to the active MoIV LAs by forming hydrogen bonds and coordination bonds (PhNO2⋯HOEt→MoIV3 and H2N⋯HOEt→MoIV3) for subsequent hydrazine heterolysis and H+/H− transfer. Another important role of the protic solvent is to facilitate the dissociation of the coordinated pyridine driven by the C5H5N⋯HOEt hydrogen bond for subsequent solvent substitution.
A comparison of the catalytic performance of γ-1 and the triangular MoIV oxoclusters [MOIV3O2(O2CCH3)6L3]ZnCl4 (L = H2O, 3a; py, 3b), [Pro4N]2[MOIV3O4(C2O4)3L3] (L = H2O, 4a; py, 4b; Pro = CH3CH2CH2) offers further insights into the hydrogenation mechanism. As shown in Fig. 2(right), the right two MoIV-free blank systems show negligible reactivity. Surprisingly, the discrete [MOIV3O4]-type clusters of 4 have poor selectivity (10%), somewhat similar to the cases of THF and ethyl acetate of poor coordination capacity. This probably results from the presence of strong electron-pair donors C2O42− in 4 that deactivate MoIV LAs. The replacement of C2O42− in 4 with acetate groups in 3 remarkably enhances the selectivity (>90%). The low conversions in both 3a and 3b are assignable to the absence of both enhanced MoIV LAs and more efficient edge-bridging and terminal oxygen LBs that are believed to account for the perfect performance of γ-1 (yield, 99.8%). As revealed by the DFT calculations (Table S4†), the MoIV Lewis acidity is significantly boosted by MoIV → MoVI electron transfer, which, together with the numerous bridging/terminal oxygen atoms with the correspondingly enhanced Lewis basicity, leads to the excellent efficiency of γ-1 in the hydrogenation reduction of nitroarenes to anilines.
The comparative investigation into the catalytic performance described above offers us invaluable information to understand the hydrogenation reduction mechanism. It is commonly recognized that bifunctional hydrogenation usually starts with the LP-promoted heterolysis of hydrogen donors, which produces a hydride and a proton stabilized by Lewis acidic and basic sites, respectively. The related examples include [CpMo(CO)(κ3-P2N2)]+ with the non-frustrated N→Mo Lewis pair, which can efficiently catalyze H2 heterolysis to form MoH− and NH+.5b The density functional theory calculations suggest the stepwise hydrogen transfer via the prior cleavage of the N–H bond rather than the N–N bond,26a while [MOIV3S4] clusters have a strong tendency to accept a hydride for transfer hydrogenation.26b,c The heterolytic H → N → H cleavage mechanism of hydrazine by γ-1 bearing the multiple py→MoIV Lewis pairs with removable pyridines is thus proposed. The MoIV3 Lewis acidic active sites, which are further promoted by the MoIV → MoVI electron transfer, actuate the N → H heterolysis to form MoIVH−. Meanwhile, the MoVIO and even stronger OMoVIO Lewis basic sites have a pronounced capacity to drive the heterolytic H → N cleavage and stabilize the protons, which is in turn promoted by the MoIV → MoVI electron transfer. With the above considerations in mind, we propose the following catalytic cycle exemplified by the [pyMoIV(μ2-OH)]2 unit of γ-1 involved in the hydrogenation reduction of nitrobenzene to aniline, as depicted in Scheme 1. Initially, the two pyridine ligands are replaced by hydrazine (N2H4) to form [MoIV(μ2-OH)]2(μ-N2H4) (Scheme 1b). The subsequent dual N → H heterolysis of the bridging N2H4 through transition state c promoted by the synergistic action of the two MoIV LAs leads to the N → H electron transfer to form the MoIVH− hydride intermediates. Meanwhile, the surrounding O–MoVIO and hydrazine LBs facilitate the H → N heterolytic breakage to form MoVIOH+ and/or H+2N2H4 that are boosted and stabilized by O–MoVIO (Scheme 1d, Fig. S6 and S7†). This step is accelerated by the formation of a N2H2N2 six-membered ring and N2 release. The last multi-step (d → b, Scheme S1†) closes the cycle via the hydride and proton transfer to produce aniline which starts the next one. The very low azobenzene → aniline conversion (11.6%) by γ-1 and the undetected former reveals the direct route (Scheme S1†). The other two couples of the MoIV2 LAs (Mo2–Mo2′ and its equivalent) are proposed to suffer from another two similar catalytic cycles, as shown in Scheme 1, to provide a total of four hydrides and four protons. As a consequence, every catalytic cycle produces six hydrides and six protons to reduce two PhNO2 to PhNH2 concomitant with the formation of four H2O, as proposed in Scheme 1d. The countercations [H2N(CH3)2]+ may also be involved in the proton transfer process.
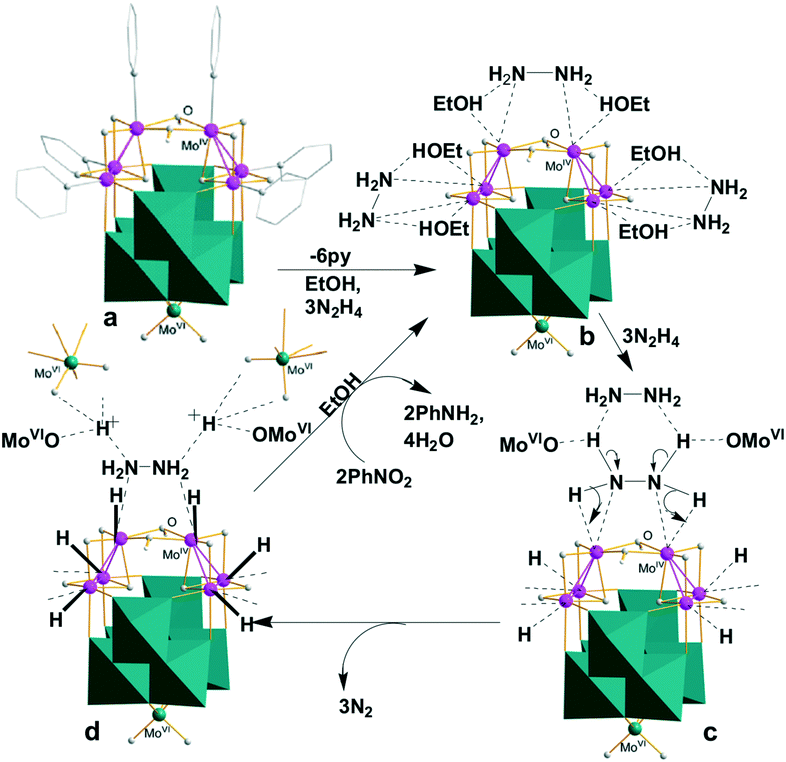 | ||
| Scheme 1 Proposed catalytic cycle for γ-1 involved in the reduction of nitrobenzene. (a) Structure of γ-1a (b) hydrazine coordination to Mo(IV) LAs (c) Mo(IV)/Mo(VI) promoted H → N → H heterolysis of hydrazine (d) HMo(IV) hydrides and protonated Mo(VI)OH intermediates. | ||
Conclusions
The MoIV6-γ-Keggin-like hybrid (γ-1) with the unprecedented MoIV3–O–MoVIO FLPs was prepared in a high yield. The high efficiency of such unique FLP systems in catalytic hydrogenation has been established by the excellent performance of γ-1 as the heterogeneous catalyst in the hydrogenation reduction of nitroarenes to anilines under mild conditions, revealing the promising potential of the unique POM-FLP systems in catalytic hydrogenation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21873102 and 21073192) and the Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDB 20000000.Notes and references
- H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH, Weinheim, 5th edn, 2010 Search PubMed (a) Modern Reduction Methods, ed. P. G. Andersson and I. J. Munslow, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2008 Search PubMed; (b) Catalytic Hydrogenation, ed. L. Cerveny, Elsevier, Amsterdam, 1986 Search PubMed.
- (a) A. E. Allen and D. W. C. MacMillan, Chem. Sci., 2012, 3, 633–658 RSC; (b) M. K. Karunananda and N. P. Mankad, ACS Catal., 2017, 7, 6110–6119 CrossRef CAS; (c) D. H. Paull, C. J. Abraham, M. T. Scerba, E. Alden-Danforth and T. Lectka, Acc. Chem. Res., 2008, 41, 655–663 CrossRef CAS PubMed; (d) W. P. Deng, P. Wang, B. J. Wang, Y. L. Wang, L. F. Yan, Y. Y. Li, Q. H. Zhang, Z. X. Cao and Y. Wang, Green Chem., 2018, 20, 735–744 RSC.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- (a) A. Stute, G. Kehr, C. G. Daniliuc, R. Fröhlich and G. Erker, Dalton Trans., 2013, 42, 4487–4499 RSC; (b) L. J. Hounjet, C. Bannwarth, C. N. Garon, C. B. Caputo, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2013, 52, 7492–7495 CrossRef CAS PubMed.
- (a) S. R. Flynn and D. W. Wass, ACS Catal., 2013, 3, 2574–2581 CrossRef CAS; (b) S. G. Zhang, A. M. Appel and R. M. Bullock, J. Am. Chem. Soc., 2017, 139, 7376–7387 CrossRef CAS PubMed.
- D. W. Stephan, Science, 2016, 354, 7229 CrossRef PubMed.
- (a) M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48 CrossRef; (b) C. L. Hill, Chem. Rev., 1998, 98, 1–2 CrossRef CAS PubMed.
- (a) D. L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed; (b) H. N. Miras, J. Yan, D. L. Long and L. Cronin, Chem. Soc. Rev., 2012, 41, 7403–7430 RSC.
- (a) S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed; (b) Y. F. Song and R. Tsunashima, Chem. Soc. Rev., 2012, 41, 7384–7402 RSC; (c) R. F. Renneke, M. Pasquali and C. L. Hill, J. Am. Chem. Soc., 1990, 112, 6585–6594 CrossRef CAS; (d) E. Haviv, L. J. W. Shimon and R. Neumann, Chem. – Eur. J., 2017, 23, 92–95 CrossRef CAS PubMed; (e) C. Zhang, X. Lin, Z. Zhang, L. S. Long and W. Lin, Chem. Commun., 2014, 50, 11591–11594 RSC.
- N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 0112 CrossRef CAS.
- (a) H. Benaissa, P. N. Davey, E. F. Kozhevnikova and I. V. Kozhevnikov, Appl. Catal., A, 2008, 351, 88–92 CrossRef CAS; (b) H. Benaissa, P. N. Davey, Y. Z. Khimyak and I. V. Kozhevnikov, J. Catal., 2008, 253, 244–252 CrossRef CAS; (c) F. Lin and Y.-H. Chin, J. Catal., 2016, 341, 136–148 CrossRef CAS.
- L. MacDonald, B. Rausch, M. D. Symes and L. Cronin, Chem. Commun., 2018, 54, 1093–1096 RSC.
- (a) K. Piepgrass and M. T. Pope, J. Am. Chem. Soc., 1987, 109, 1586–1587 CrossRef CAS; (b) M. H. Dickman, T. Ozeki, H. T. Evans-Jr., C. Rong, G. B. Jameson and M. T. Pope, J. Chem. Soc., Dalton Trans., 2000, 149–154 RSC.
- (a) W. P. Chen, R. L. Sang, Y. Wang and L. Xu, Chem. Commun., 2013, 49, 5883–5885 RSC; (b) X. Xu, B. L. Luo, L. L. Wang and L. Xu, Dalton Trans., 2018, 47, 3218–3222 RSC; (c) J. Zhao and L. Xu, Eur. J. Inorg. Chem., 2011, 26, 4096–4102 CrossRef.
- (a) D. T. Richens, The Chemistry of Aqua Ions, John Wiley & Sons, Chichester, 1997 Search PubMed; (b) D. T. Richens, Chem. Rev., 2005, 105, 1961–2002 CrossRef CAS PubMed.
- (a) A. Bino, F. A. Cotton and Z. Dori, J. Am. Chem. Soc., 1978, 100, 5252–5253 CrossRef CAS; (b) R. K. Murmam and M. E. Shelton, J. Am. Chem. Soc., 1980, 102, 3984–3985 CrossRef; (c) F. A. Cotton, D. O. Marler and W. Schwotzer, Inorg. Chem., 1984, 23, 3671–3673 CrossRef CAS; (d) M. Brorson, A. Hazell, C. J. H. Jaconson, I. Schmidt and J. Villadsen, Inorg. Chem., 2000, 39, 1346–1350 CrossRef CAS PubMed; (e) K. Nakata, A. Nagasawa, N. Soyama, Y. Sasaki and T. Ito, Inorg. Chem., 1991, 30, 1575–1579 CrossRef CAS; (f) B.-L. Ooi and A. G. Sykes, Inorg. Chem., 1988, 27, 310–315 CrossRef CAS; (g) P. Kathirgamanathan, A. B. Soares, D. T. Richens and A. G. Sykes, Inorg. Chem., 1985, 24, 2950–2954 CrossRef CAS.
- S. F. Gheller, T. W. Hambey, R. T. Brownlee, M. J. Oconnor, M. R. Show and A. G. Wedd, J. Am. Chem. Soc., 1983, 105, 1527–1532 CrossRef CAS.
- (a) L. Xu, D. Yan, H. Liu, J. S. Huang and Q. E. Zhang, J. Chem. Soc., Chem. Commun., 1993, 1507–1510 CAS; (b) L. Xu, H. Liu, J. S. Huang and Q. E. Zhang, Chem. – Eur. J., 1997, 3, 226–231 CrossRef CAS PubMed; (c) L. Xu, Z. F. Li and J. S. Huang, Inorg. Chem., 1996, 35, 5097–5100 CrossRef CAS PubMed; (d) L. Xu, H. Liu, D. C. Yan, J. S. Huang and Q. E. Zhang, J. Chem. Soc., Dalton Trans., 1994, 2099–2107 RSC.
- (a) K. Sugahara, T. Kimura, K. Kamata, K. Yamaguchi and N. Mizuno, Chem. Commun., 2012, 48, 8422–8424 RSC; (b) K. Sugahara, N. Satake, K. Kamata, T. Nakajima and N. Mizuno, Angew. Chem., Int. Ed., 2014, 53, 13248–13252 CrossRef CAS PubMed.
- X. F. Yi, N. V. Izarova, M. Stuckart, D. Guerin, L. Thomas, S. Lenfant, D. Vuillaume, J. V. Leusen, T. Duchoň, S. Nemsăk, S. D. M. Bourone, S. Schmitz and P. Kögerler, J. Am. Chem. Soc., 2017, 139, 14501–14510 CrossRef CAS PubMed.
- (a) R. S. Downing, P. J. Kunkeler and H. V. Bekkum, Catal. Today, 1997, 37, 121–136 CrossRef CAS; (b) N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, New York, 2001 CrossRef; (c) A. Stephen, Amines: Synthesis, Properties and Applications, Cambridge University Press, Cambridge, UK, 2006, pp. 205–215 Search PubMed.
- L. C. W. Baker and J. S. Figgis, J. Am. Chem. Soc., 1970, 92, 3794–3797 CrossRef CAS.
- (a) B. Botar, Y. V. Geletii, P. Kögerler, D. G. Musaev, K. Morokuma, I. A. Weinstock and C. L. Hill, J. Am. Chem. Soc., 2006, 128, 11268–11277 CrossRef CAS PubMed; (b) Y. Kikukawa, K. Suzuki, K. Yamaguchi and N. Mizuno, Inorg. Chem., 2013, 52, 8644–8652 CrossRef CAS PubMed; (c) A. Tézé, E. Cadot, V. Béreau and G. Hervé, Inorg. Chem., 2001, 40, 2000–2004 CrossRef.
- M. T. Pope, Inorg. Chem., 1976, 15, 2008–2010 CrossRef CAS.
- A. Furst, R. C. Berlo and S. Hooton, Chem. Rev., 1965, 65, 51–68 CrossRef CAS.
- (a) C. F. Zhang, J. M. Lu, M. R. Li, Y. H. Wang, Z. Zhang, H. J. Chen and F. Wang, Green Chem., 2016, 18, 2435–2442 RSC; (b) A. G. Algarra, M. G. Basallote, M. J. Fernández-Trujillo, M. Feliz, E. Guillamón, R. Llusar, I. Sorribes and C. Vicent, Inorg. Chem., 2010, 49, 5935–5942 CrossRef CAS PubMed; (c) I. Sorribes, G. Wienhóer, C. Vicent, K. Junge, R. Llusar and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 7794–7798 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, chemicals, instrumentation, molecular and crystal structure figures, XPS, DFT calculations, catalysis (PDF) and crystal data (CIF). CCDC 1575811. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cy01771a |
| ‡ Dedicated to Prof. Jin-Shun Huang on the occasion of his 80th birthday. |
| This journal is © The Royal Society of Chemistry 2019 |