
Phosphine-free cobalt catalyst precursors for the selective hydrogenation of olefins†
Pim
Puylaert
 a,
Andrea
Dell'Acqua
a,
Fatima
El Ouahabi
a,
Anke
Spannenberg
a,
Thierry
Roisnel
b,
Laurent
Lefort
c,
Sandra
Hinze
a,
Sergey
Tin
a and
Johannes Gerardus
de Vries
*a
a,
Andrea
Dell'Acqua
a,
Fatima
El Ouahabi
a,
Anke
Spannenberg
a,
Thierry
Roisnel
b,
Laurent
Lefort
c,
Sandra
Hinze
a,
Sergey
Tin
a and
Johannes Gerardus
de Vries
*a
aLeibniz Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Straße 29a, 18055 Rostock, Germany. E-mail: johannes.devries@catalysis.de
bUniversité de Rennes 1, UMR “Sciences Chimiques de Rennes”, UR1-CNRS 6226, Campus de Beaulieu, CS 74205, 35042 Rennes Cedex, France
cInnoSyn B.V., Urmonderbaan 22, 6167 RD, Geleen, The Netherlands
First published on 23rd November 2018
Abstract
Cobalt(II) complexes bearing phosphine-free tridentate NNS ligands were prepared. Depending on the ligand, dimeric or monomeric complexes were isolated. Monomeric Co(NNMeS)Cl2 selectively catalysed the hydrogenation of olefins in the presence of reducible moieties such as ketones. Further investigation showed that this complex functions as a nanoparticle precursor under the reaction conditions.
The field of homogeneous hydrogenation has, over the last decade, undergone a major shift away from noble metals such as Rh, Ir, and Ru towards base metals like Fe, Co, and Mn.1 Strongly electron-donating, stabilising pincer ligands have played a key role in the development of such catalysts, to the point where pincer complexes of most base metals have now been applied for hydrogenation reactions.2,3
The impact on the environment and the costs of phosphine- or carbene-based ligands, however, should not be underestimated, and can even outweigh the advantages of using base metals. Phosphine-based ligands also suffer from shorter shelf-lives and can be hard to prepare on a large scale, due to their sensitivity. In this light, we recently reported the application of flexible alkylthio-N-(pyridine-2-yl)methyl-1-ethanamine (NNS) tridentate ligands and their ruthenium complexes in the selective hydrogenations of α,β-unsaturated aldehydes, ketones, and even esters to the corresponding allylic alcohols.4 Notable examples of similar ligands used in hydrogenations were published earlier by Gusev, Dub and Gordon (Scheme 1).5,6 There is a clear incentive for combining such cheap, readily accessible ligands with base metals in order to reap the benefits provided by both. Midya, Balaraman and co-workers recently reported the acceptorless dehydrogenative coupling of aminoalcohols and alcohols to obtain heterocycles, which was catalysed by a dimeric cobalt(II) SNS complex (Scheme 2).7 At the time, we were evaluating the use of our NNS ligands with cobalt for catalytic hydrogenations, the results of which we report here.
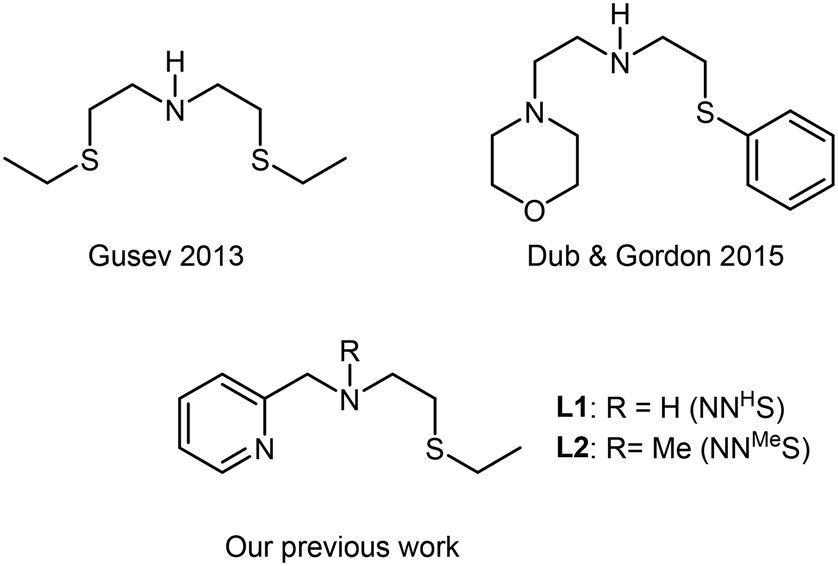 | ||
| Scheme 1 Previously reported N,S-tridentate ligands used for hydrogenations. | ||
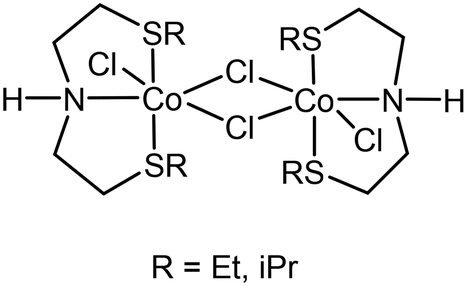 | ||
| Scheme 2 Co-SNS dimers reported by Balaraman.7 | ||
The addition of 1 equivalent of ligand to a solution of CoCl2 in ethanol afforded a clean reaction to the Co(II)NNS complexes 1 and 2 depicted in Scheme 3. Interestingly, the secondary amine ligand L1 yielded the dimeric, chloride-bridged complex 1, with the tridentate ligand coordinated in a fac manner, while monomeric complex 2 was obtained with the N-methylated ligand L2. Crystals suitable for X-ray diffraction analysis were obtained for both complexes, and their solid-state molecular structures are shown in Fig. 1. Interestingly, it appears that N-methylation of the ligand provided sufficient steric hindrance to prevent complex 2 from dimerising.
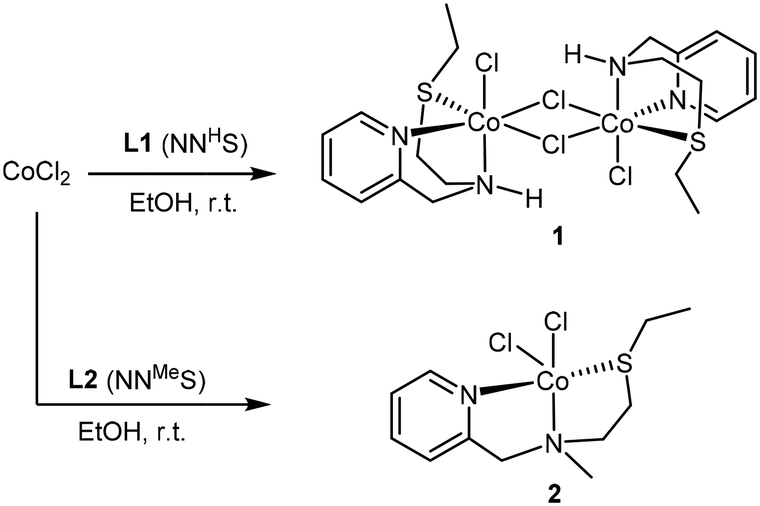 | ||
| Scheme 3 Synthesis of Co(NNS) complexes 1 and 2. | ||
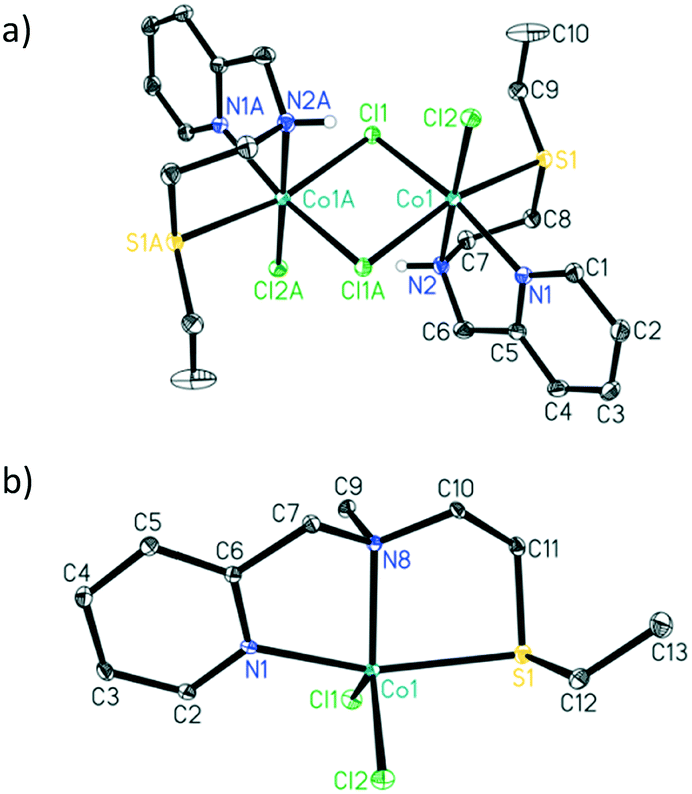 | ||
| Fig. 1 ORTEP drawings (30% probability ellipsoids, hydrogen atoms, except the N–H hydrogen for 1, were omitted for clarity). Selected bond lengths (Å) and angles (°) for a) complex 1: Co1–S1 2.5162(4), Co1–N1 2.1159(11), Co1–N2 2.1704(11), Co1–Cl1 2.4186(3), Co1–Cl1A 2.4991(3), Co1–Cl2 2.3633(3); N1–Co–N2 78.41(4), N1–Co1–S1 83.76(3), N2–Co1–S1 83.72(3), Cl1–Co1–Cl1A 91.547(12), Cl2–Co1–Cl1 95.210(13), C6–N2–C7 115.33(11) and b) complex 2: Co1–S1 2.5612(8), Co1–N1 2.102(2), Co1–N8 2.122(2), Co1–Cl1 2.2785(8), Co1–Cl2 2.2885(8); N1–Co1–N8 78.14(8), N1–Co1–S1 161.69(6), N8–Co1–S1 84.06(6), Cl1–Co1–Cl2 118.99(3), C7–N8–C10 111.2(2).† | ||
We investigated the obtained complexes in the catalytic hydrogenation of olefins. Several activation pathways were envisioned. Firstly, a strong base is often added to activate hydrogen via a bifunctional mechanism.6b,8 This pathway should enable the hydrogenation of polarised, unsaturated moieties such as carbonyl groups.8 Another strategy to activate Co(II) complexes is their reduction, be it in situ or ex situ, to the corresponding Co(I) complexes, which has recently been reported to lead to increased activities.9 The reaction conditions reported by Balaraman et al. for their dehydrogenative coupling (reflux in m-xylene) made us suspect that the dimeric complex may well be very stable in solution, and therefore would require elevated temperatures to dissociate into an active, monomeric form.7 In order to test if the dimer could be dissociated at lower temperatures, we included the addition of monodentate ligand into the screening of activating additives. In an initial high-throughput screening (HTS), the hydrogenation of 1-octene, acetophenone and 1-octen-3-ol were investigated (see ESI†). After these initial experiments, 1-octen-3-ol (3a) was selected as a model substrate since its isomerisation to the corresponding ketone (4b) was observed during the HTS.10 Consequently, we used this substrate to explore the activity of the Co catalysts in both hydrogenation and isomerisation. The results of the study of the activation of the Co precatalyst by different additives are summarised in Table 1. Interestingly, the best results were obtained using 2 together with a reductant (NaBH4, entry 18). Subsequently, several reaction solvents were investigated, showing that isopropanol gave the highest yield and selectivity (Table S3†).
|
|
||||
|---|---|---|---|---|
| #a | Cat. | Base | Additive | Yield 4a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4bb (%) :4bb (%) |
| a Reaction conditions: 0.33 mmol 1-octen-3-ol, 1.0 mL THF, 1 mol% 1 or 2 mol% 2, 5 mol% of additive and base, 50 bar H2, 100 °C, 16 h reaction time. b Determined by GC using dodecane as internal standard. c 1 mol% of 2. | ||||
| 1 | 1 | — | — | 0:0 |
| 2 | 1 | KOtBu | — | 0:1 |
| 3 | 1 | NaOH | PPh3 | 0:0 |
| 4 | 1 | NaOH | NaBH4 | 12:0 |
| 5 | 1 | NaOH | Zn | 3:0 |
| 6 | 2 | — | — | 0:0 |
| 7 | 2 | KOtBu | — | 0:1 |
| 8 | 2 | KOtBu | PPh3 | 41:0 |
| 9 | 2 | KOtBu | NaBH4 | 20:0 |
| 10 | 2 | NaOEt | PPh3 | 94:3 |
| 11 | 2 | NaOEt | NaBH4 | 94:1 |
| 12 | 2 | NaOH | PPh3 | 90:5 |
| 13 | 2 | NaOH | NaBH4 | 95:1 |
| 14 | 2 | NaOEt | Zn | 12:0 |
| 15c | 2 | NaOH | PPh3 | 41:4 |
| 16c | 2 | NaOH | NaBH4 | 72:2 |
| 17 | 2 | NaOH | — | 0:0 |
| 18 | 2 | — | NaBH4 | 95:0 |
For all reactions where full conversion with 2 was obtained, a black residue was observed after the experiment. This suggested the formation of cobalt nanoparticles, either after consumption of the substrate or during the reaction. In the latter case, the hydrogenation was possibly catalysed by cobalt nanoparticles formed in situ.11 To investigate this possibility, we observed the reaction progress by monitoring the hydrogen uptake, and performed two types of poisoning experiments.
The hydrogen uptake curve at 1 mol% catalyst loading (Fig. 2a) shows an induction period of around 30 minutes, during which no hydrogen was consumed. After this time, the reaction proceeded to full conversion and >99% yield through a roughly sigmoidal curve, which is often indicative for nanoparticle-catalysed reactions.12 Mercury poisoning experiments provided no conclusive evidence (details can be found in the ESI†). The hydrogenation reaction still proceeded to a certain extent. However, it has been reported that Co nanoparticles do not readily form an amalgam with mercury.12,13 Consequently, we performed a second poisoning experiment with PMe3 instead, the results of which are shown in Fig. 2b.14 After the induction period, a sub-stoichiometric amount of PMe3 (0.15 eq. w.r.t. 2) was injected into the autoclave using excess of H2 pressure, leading to an immediate stop of the hydrogen consumption. This is a strong indication that the reaction is catalysed by cobalt nanoparticles, which are poisoned when their surface gets saturated with PMe3.
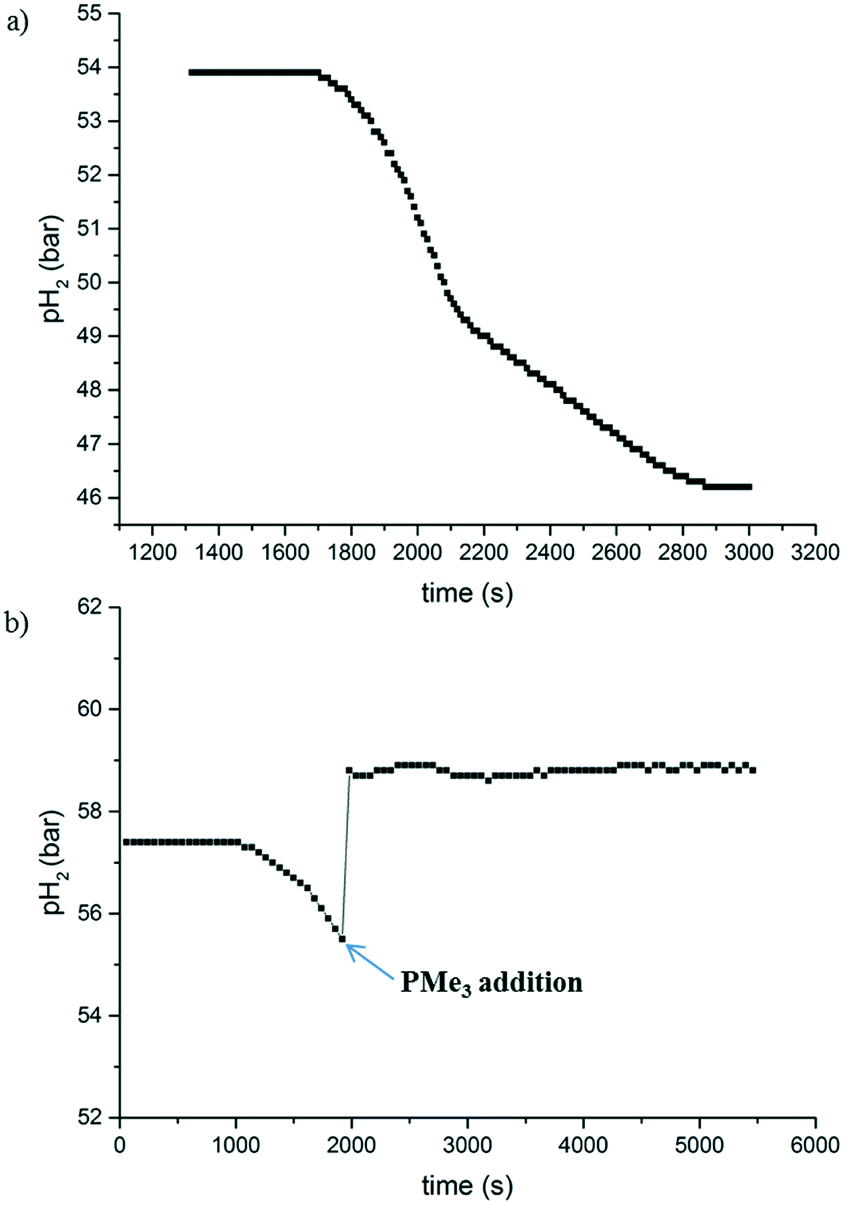 | ||
| Fig. 2 a) Hydrogen uptake curve at 1 mol% catalyst loading under normal reaction conditions (corrected for heating to 100 °C). b) Hydrogen uptake under the same conditions, where a PMe3 solution was injected after the induction period, which initially increased the total pressure, but then remained stable. At this point a conversion of 65% had been achieved (determined by GC, dodecane as internal standard). | ||
A variety of olefins were reduced with full conversions, and the products isolated in good to excellent yields, as illustrated in Fig. 3.
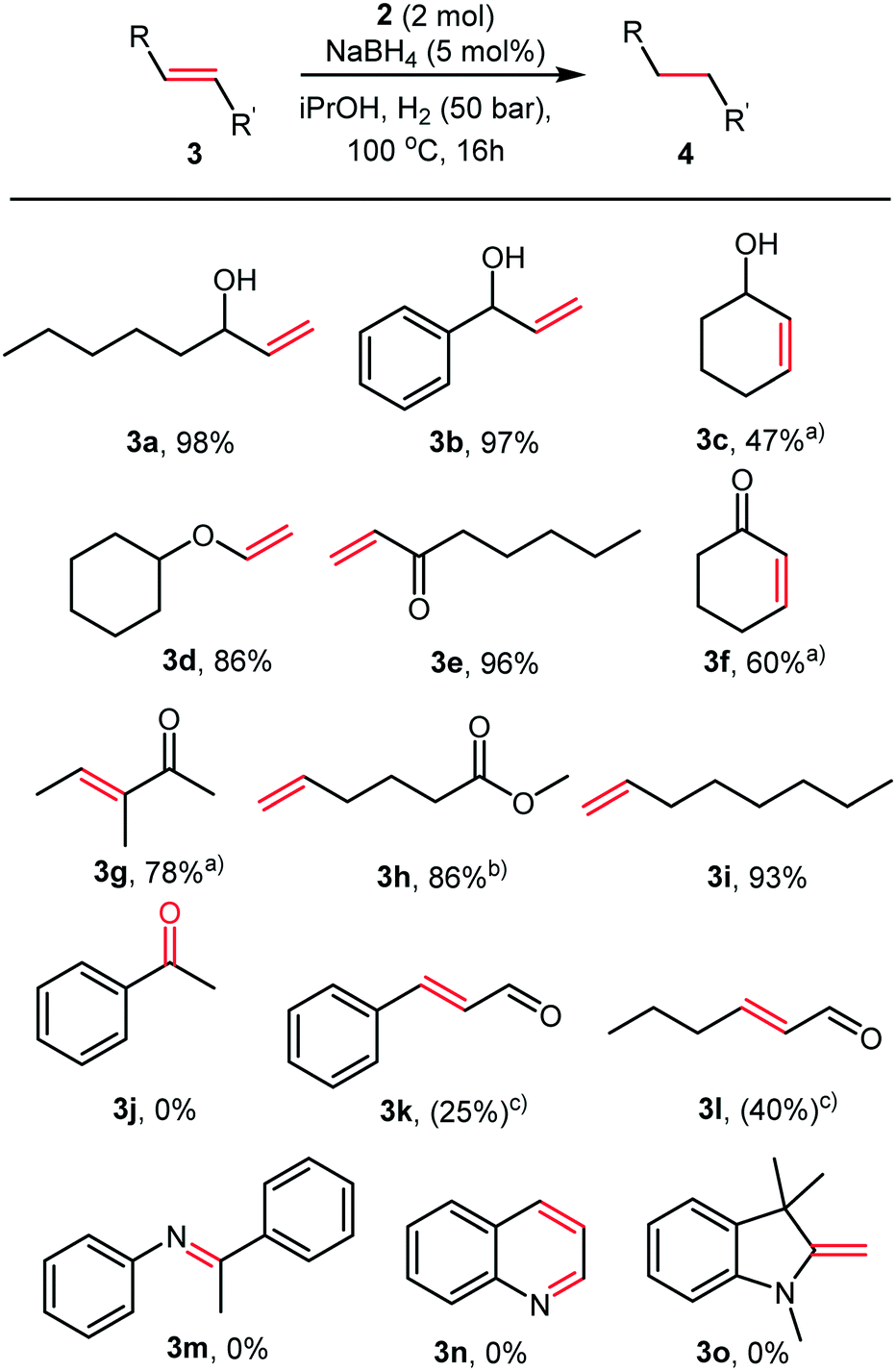 | ||
| Fig. 3 Substrate scope and limitations of the hydrogenation of olefins catalysed by 2. Reaction conditions: substrate (5 mmol), iPrOH (15 mL), 2 (33.9 mg, 2 mol%), NaBH4 (9.5 mg, 5 mol%), 50 bar H2, 100 °C, 16 h reaction time. a) Full conversions, but isolated yields are low due to volatility of the products being comparable to that of iPrOH. b) The isolated product is the isopropyl ester. c) The reactions never reached full conversions, and as such the products were not isolated. | ||
During our initial HTS, we observed that our catalysts were inactive in the hydrogenation of acetophenone. Therefore, we decided to explore the selectivity of the Co nanoparticles for olefins in the presence of carbonyl functionalities (Fig. 3). Ketones (3j) and esters (3h) were not reduced even under these harsh conditions. Excellent chemoselectivities were observed for α,β-unsaturated ketones (3f–3g), with no reduction of the carbonyl even after 16 hours. Aldehydes appear to inhibit the reaction, only 25 and 40% conversion was observed in the hydrogenation of cinnamaldehyde (3k) and hexen-2-al (3l). Even here, the sole product observed was the saturated aldehyde. No conversion was observed in the attempted hydrogenation of an imine (3m), quinoline (3n) or enamine (3o). These reactivities contrast with the activity of cobalt pincer complexes containing strongly donating phosphine ligands such as those recently described by Junge et al., which hydrogenated esters readily.9a The cobalt nanoparticles recently reported by the Jacobi von Wangelin group reduced carbonyl groups efficiently, and under far milder reaction conditions.11a,b In our case, it is likely that L2 stabilises the nanoparticles and partially deactivates them, leading to chemoselectivity towards olefinic double bonds.
Conclusions
Two new cobalt(II) complexes bearing non-phosphorus NNS ligands were synthesised, characterised and their reactivities in catalytic hydrogenation were investigated. The dimeric complex 1 was nearly inactive for hydrogenation, but complex 2 proved useful as catalyst precursor for the selective hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in the presence of other reducible moieties.
C bonds in the presence of other reducible moieties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Dr. Ralf Jackstell and Mr. Rui Sang for assistance with the PMe3 poisoning experiment, and to Mrs. Susann Buchholz, Mrs. Susanne Schareina, Mr. Andreas Koch, and Dr. Christine Fischer (all at the Likat) for their indispensable analytical support. ADA acknowledges the European Union Erasmus+ Traineeship Programme for the exchange scholarship.Notes and references
- The Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Elsevier, Wiley-VCH, Weinheim, 2007, vol. 1–3 Search PubMed.
- (a) P. A. Dub and T. Ikariya, ACS Catal., 2012, 2, 1718–1741 CrossRef CAS; (b) S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289–302 CrossRef CAS; (c) S. Werkmeister, J. Neumann, K. Junge and M. Beller, Chem. – Eur. J., 2015, 21, 12226–12250 CrossRef CAS PubMed; (d) G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC; (e) F. Kallmeier and R. Kempe, Angew. Chem., Int. Ed., 2018, 57, 46–60 CrossRef CAS PubMed; (f) W. Liu, B. Sahoo, K. Junge and M. Beller, Acc. Chem. Res., 2018, 51, 1858–1869 CrossRef CAS PubMed.
- (a) Q. Knijnenburg, A. D. Horton, H. van der Heijden, T. M. Kooistra, D. G. H. Hetterscheid, J. M. M. Smits, B. de Bruin, P. H. M. Budzelaar and A. W. Gal, J. Mol. Catal. A: Chem., 2005, 232, 151–159 CrossRef CAS; (b) G. Zhang, B. L. Scott and S. K. Hanson, Angew. Chem., Int. Ed., 2012, 51, 12102–12106 CrossRef CAS PubMed; (c) G. Zhang and S. K. Hanson, Chem. Commun., 2013, 49, 10151–10153 RSC; (d) M. R. Friedfeld, G. W. Margulieux, B. A. Schaefer and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 13178–13181 CrossRef CAS PubMed; (e) D. Gärtner, A. Welther, B. Rezaei Rad, R. Wolf and A. Jacobi von Wangelin, Angew. Chem., Int. Ed., 2014, 53, 3722–3726 CrossRef PubMed; (f) T. J. Korstanje, J. I. van der Vlugt, C. J. Elsevier and B. de Bruin, Science, 2015, 350, 298–302 CrossRef CAS PubMed; (g) S. Roesler, J. Obenauf and R. Kempe, J. Am. Chem. Soc., 2015, 137, 7998–8001 CrossRef CAS PubMed; (h) D. Srimani, A. Mukherjee, A. F. G. Goldberg, G. Leitus, Y. Diskin-Posner, Y. Ben David and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12357–12360 CrossRef CAS PubMed; (i) Z. Shao, S. Fu, M. Wei, S. Zhou and Q. Liu, Angew. Chem., Int. Ed., 2016, 55, 14653–14657 CrossRef CAS PubMed; (j) K. Tokmic, C. R. Markus, L. Zhu and A. R. Fout, J. Am. Chem. Soc., 2016, 138, 11907–11913 CrossRef CAS PubMed; (k) P. Büschelberger, D. Gärtner, E. Reyes-Rodrigues, F. Kreyenschmidt, K. Koszinowski, A. Jacobi von Wangelin and R. Wolf, Chem. – Eur. J., 2016, 23, 3139–3151 CrossRef PubMed; (l) J. Yuwen, S. Chakraborty, W. W. Brennessel and W. D. Jones, ACS Catal., 2017, 7, 3735–3740 CrossRef; (m) M. Mastalir, G. Tomsu, E. Pittenauer, G. Allmaier and K. Kirchner, Org. Lett., 2016, 18, 3462–3465 CrossRef CAS PubMed.
- (a) P. Puylaert, R. van Heck, Y. Fan, A. Spannenberg, W. Bauman, M. Beller, J. Medlock, W. Bonrath, L. Lefort, S. Hinze and J. G. de Vries, Chem. – Eur. J., 2017, 23, 8473–8481 CrossRef CAS PubMed; (b) B. S. Stadler, P. Puylaert, J. Diekamp, R. van Heck, Y. Fan, A. Spannenberg, S. Hinze and J. G. de Vries, Adv. Synth. Catal., 2018, 360, 1151–1158 CrossRef CAS.
- D. Spasyuk, S. Smith and D. G. Gusev, Angew. Chem., Int. Ed., 2013, 52, 2538–2542 CrossRef CAS PubMed.
- (a) P. A. Dub, B. L. Scott and J. C. Gordon, Organometallics, 2015, 34, 4464–4479 CrossRef CAS; (b) P. A. Dub, B. L. Scott and J. C. Gordon, Dalton Trans., 2016, 45, 1560–1571 RSC.
- S. P. Midya, V. G. Landge, M. K. Sahoo, J. Rana and E. Balaraman, Chem. Commun., 2018, 54, 90–93 RSC.
- (a) T. Zell and R. Langer, ChemCatChem, 2018, 10, 1930–1940 CrossRef CAS; (b) P. A. Dub, N. J. Henson, R. L. Martin and J. C. Gordon, J. Am. Chem. Soc., 2014, 136, 3505–3521 CrossRef CAS PubMed; (c) P. A. Dub and J. C. Gordon, ACS Catal., 2017, 7, 6635–6655 CrossRef CAS.
- (a) K. Junge, B. Wendt, A. Cingolani, A. Spannenberg, Z. Wei, H. Jiao and M. Beller, Chem. – Eur. J., 2017, 24, 1046–1052 CrossRef PubMed; (b) M. R. Friedfeld, H. Zhong, R. T. Ruck, M. Shevlin and P. J. Chirik, Science, 2018, 360, 888–893 CrossRef CAS PubMed.
- (a) R. W. Goetz and M. Orchin, J. Am. Chem. Soc., 1963, 85, 1549–1550 CrossRef CAS; (b) T. Xia, Z. Wei, B. Spiegelberg, H. Jiao, S. Hinze and J. G. de Vries, Chem. – Eur. J., 2018, 24, 4043–4049 CrossRef CAS PubMed.
- (a) S. Sandl, F. Schwarzhuber, S. Pöllath, J. Zweck and A. Jacobi von Wangelin, Chem. – Eur. J., 2018, 24, 3403–3407 CrossRef CAS PubMed; (b) P. Büschelberger, E. Reyes-Rodrigues, C. Schöttle, J. Treptow, C. Feldmann, A. Jacobi von Wangelin and R. Wolf, Catal. Sci. Technol., 2018, 8, 2648–2653 RSC; (c) V. Rysak, A. Descamps-Mandine, P. Simon, F. Blanchard, L. Burylo, M. Trentesaux, M. Vandewalle, V. Collière, F. Agbossou-Niedercorn and C. Michon, Catal. Sci. Technol., 2018, 8, 3504–3512 RSC; (d) During the course of writing this publication, Beller reported on the use of a biowaste-derived heterogeneous cobalt catalyst which also exhibits selectivity for olefins: F. K. Scharnagl, M. F. Hertrich, F. Ferretti, C. Kreyenschulte, H. Lund, R. Jackstell and M. Beller, Sci. Adv., 2018, 4, eaau1248 CrossRef PubMed.
- (a) J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS; (b) R. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS PubMed.
- C. Guminki, J. Phase Equilib., 1993, 14, 643–644 CrossRef.
- For example: J. F. Sonnenberg, N. Coombs, P. A. Dube and R. H. Morris, J. Am. Chem. Soc., 2012, 134, 5893–5899 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Typical procedure for olefin hydrogenation: oven-dried 4 mL glass vials were filled in the glovebox with 2 (2.2 mg, 0.006 mmol) and NaBH4 (0.6 mg, 0.016 mmol). Solvent (1 mL), dodecane (150 μL), and 1-octen-3-ol were added (50 μL, 0.33 mmol), and the PTFE septum pierced with a needle. The vial was transferred to an autoclave, which was flushed with Ar and then pressurised with H2 to 50 bar, and heated to 100 °C overnight. Full experimental details and characterisation for isolated products can be found in the ESI. CCDC 1864013 and 1864014. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cy02218f |
| This journal is © The Royal Society of Chemistry 2019 |