
Differences in photochemistry between seawater and freshwater for two natural organic matter samples†
Laura T.
Stirchak
a,
Kyle J.
Moor
b,
Kristopher
McNeill
 b and
D. James
Donaldson
*ac
b and
D. James
Donaldson
*ac
aDepartment of Chemistry, University of Toronto, Canada. E-mail: jdonalds@chem.utoronto.ca
bInstitute for Biogeochemistry and Pollutant Dynamics, ETH Zurich, Switzerland
cDepartment of Physical and Environmental Sciences, University of Toronto, Canada
First published on 17th December 2018
Abstract
We report changes in the excitation and resolved fluorescence spectra, inferred triplet formation and singlet oxygen formation abilities of two different Natural Organic Matter samples (NOM) in seawater vs. freshwater or NaCl solution. In artificial seawater solution (but not in NaCl solution), the natural water-derived NOM samples Suwannee River Natural Organic Matter (SRNOM) and Nordic Reservoir Natural Organic Matter (NRNOM) display large enhancements in fluorescence intensity. Nearly identical spectra are seen when seawater is replaced by solutions of Mg2+ at its seawater concentration, consistent with magnesium binding to ligand sites of the natural organic matter giving rise to different photophysics. Fluorescence anisotropy measurements show a decrease in anisotropy of SRNOM and NRNOM in seawater, also consistent with Mg2+ binding. Different effects of Mg2+ are seen when the different NOM samples are illuminated: NRNOM exhibits increased formation of its triplet state and also quenching of its triplet by oxygen, compared to its photochemistry in the absence of Mg2+, while SRNOM exhibits a reduction in triplet formation in the presence of Mg2+. These observations imply that the photochemistry of NOM in seawater may be very different from what is expected based on freshwater or NaCl solution measurements.
Environmental significance statementNatural organic matter (NOM) in surface waters is an important photosensitizing agent, forming reactive triplet states and reactive oxidizing species (·OH, HO2, 1O2). Seawater components, such as halides and metals, have the potential to quench the photoexcited NOM and form complexes, possibly altering the formation of reactive species. In this paper we examine the changes in formation of the triplet state and its quenching by dissolved O2 (forming in part 1O2) in seawater vs. freshwater for two commercially available NOM samples. Results suggest that changes to the formation of the triplet state, and its quenching by dissolved O2, are dependent upon the presence of Mg2+ and the identity of NOM. Thus, the photochemistry of NOM appears to be different in seawater versus freshwater. |
Introduction
Natural organic matter (NOM) is ubiquitous in aquatic environments, but it originates from a variety of different sources, which can affect its chemical characteristics.1–8 The presence of chromophoric moieties, such as aromatic and carbonyl groups, gives rise to NOM participating in photochemical reactions either as a photosensitizer, or as a reactant in its excited triplet state.9–15 Its indirect photolysis leads to the formation of photochemically produced reactive intermediates (PPRI), such as H2O2,16–18 hydroxyl radical (·OH),19–24 singlet oxygen (1O2),17,25–28 and the excited triplet state, 3NOM*.27,29–31Many studies have investigated the production pathways of PPRIs using different NOM samples in freshwater. The production of PPRIs varies depending upon the identity of the NOM sample. Recently, a few studies have looked at the effect of ionic strength and halides on the production and steady state concentrations of different reactive intermediates, as an approach to understanding how the photochemical pathways might differ in seawater versus freshwater.32,33 These papers were complementary to a paper by al Housari et al., who noted that the concentrations of ·OH, 1O2 and 3NOM* were all higher in estuarine than freshwater samples. The difference between this study and the previously mentioned studies is that the organic matter samples used in these experiments were sampled in the Camargue region of southern France.34 Since halides generally quench the fluorescence of organic species in solution, the rate of intersystem crossing may therefore be increased, leading to higher reported concentrations of 3NOM* and 1O2 when halides are present.33 However, it was determined that with increasing ionic strength, a higher [3NOM*] was formed, and this was due to slower loss mechanisms of the excited triplet.32
Although these studies provide valuable insights into PPRI production, their focus on halides neglects the possible influences of other seawater components. For example, many metals bind to humic and fulvic acids.35–40 Models have been developed to predict the binding strength of various NOM samples to different metal ions.41–43 Many studies have also looked at the competition between metals for different binding sites,39,44–46 noting that varying metals, such as Fe3+, Ca2+, Cu2+ and Mg2+, appear to bind at different sites within the organic matrix.44 Studies have also looked at the effect of metal–NOM complexes on the fluorescence of NOM in solution. Here, the specific effects seem to depend upon the identity of the metal.47–52 It has also been found that metals can facilitate the formation of NOM aggregates, potentially altering the formation kinetics of PPRI species.53–57
The present study aims to expand on this earlier work by comparing the fluorescence, fluorescence anisotropy and PPRI production from two organic matter samples in freshwater vs. seawater, with the aim of exploring whether sea-surface NOM photochemistry differs from that observed from freshwater surfaces. Specifically, we measured excitation and fluorescence spectra,1O2 production and 3NOM* production from two natural water-derived organic matter samples, Suwannee River and Nordic Reservoir natural organic matter, in freshwater, NaCl solution and artificial seawater. The same experiments were run with the controversial model humic substance Aldrich humic acid (AHA)58 to compare with the other two NOM samples. Although it is not considered to be a good proxy for natural organic matter, AHA has been used as such by the atmospheric chemistry community.11,59–61 We present the AHA experimental details and results can be found in the ESI† for completeness and as further proof that AHA is not a good model humic substance.
Experimental
Materials
Nordic Reservoir Natural Organic Matter 1R108N (NRNOM) and Suwannee River Natural Organic Matter 2R101N (SRNOM), were purchased from the International Humic Substances Society and were used without any workup. Sodium chloride (ASP), sodium bicarbonate (EMD), sodium sulfate (Caledon), FeCl3 anhydrous (ACP), and magnesium chloride anhydrous (Sigma) were used as delivered. Instant Ocean Sea Salt (IO) was purchased from the company Instant Ocean. 2,4,6-Trimethylphenol (TMP), used as a probe for the excited triplet state, was purchased from Acros Organics.Optical measurements
Stock NOM solutions were made at concentrations of 16 mg L−1 NRNOM and 16 mg L−1 SRNOM using Milli-Q deionized water. Solutions with NaCl ranged from 0.05 M to 2 M. Solutions with IO ranged from 2.9 to 116 g L−1. Sodium bicarbonate (3.9, 19.6 and 38.7 mM) and sodium sulfate (0.34, 1.7 and 3.4 mM) were only made with SRNOM. Magnesium chloride solutions were made at 7.6, 38 and 76 mM. FeCl3 solutions were made at concentrations of 0.4 and 1 μM. All solutions were stored in volumetric flasks the dark and used within 24 hours. Some solutions were bubbled with nitrogen for 20 minutes and then tightly sealed until their use, to eliminate dissolved oxygen.Absorbance spectra were measured between 200 and 700 nm using Milli-Q deionized water as a reference. Each solution was run in triplicate and the results averaged. Steady-state excitation and resolved fluorescence spectra were acquired using a commercial fluorimeter. Excitation spectra were measured between 250 and 470 nm, using an emission wavelength of 480 nm, and resolved fluorescence spectra were excited at 405 nm and measured between 410–600 nm. The slit widths for both types of spectrum gave spectral resolution of 3.0 nm for excitation and 5.0 for emission. All fluorescence experiments were run in triplicate and the three spectra averaged. A background spectrum (without NOM) was measured for each type of solution in Milli-Q deionized water. Fluorescence anisotropy experiments used an excitation wavelength of 405 nm and monitored fluorescence between 410 and 600 nm. The instrument automatically used vertical–horizontal and vertical–vertical polarizer arrangements to collect the spectra, and the software calculated the anisotropy at each wavelength. Each solution was run in triplicate and the three spectra averaged.
Triplet NOM production
The production of triplet state NOM during illumination was monitored by following the loss of TMP due to its rapid reaction with triplet-state organics.9,14,62 Because the chromatographic column used could not tolerate high ionic strengths, these experiments were done using the same concentration of Mg2+ as is present in “0.5 M” Instant Ocean (see below). A mass of 0.85 mg of 2,4,6-trimethylphenol (TMP) was added to each of the NOM solutions to create stock solutions of 25 μM TMP. All solutions were stirred and gently heated for 45 minutes to ensure the TMP dissolved. Masses of 0.1812 and 0.3623 g of MgCl2 were added to 100 mL volumetric flasks along with the stock solutions of each NOM sample to make 19 mM and 38 mM solutions of MgCl2, respectively. The latter concentration of magnesium chloride corresponds to its concentration in the 26 g L−1 IO solutions. All solutions were made the night before being tested and were stored at room temperature in the dark.Three solutions were used for the TMP kinetics experiments: 0, 19 and 38 mM MgCl2, each containing 25 μM TMP and a NOM sample (16 mg L−1 SRNOM or 16 mg L−1 NRNOM). Each solution was poured into 10 mL quartz cuvettes and illuminated in a Solar Simulator (Atlas Suntest CPS) (see ESI† for information on the solar simulator). Experiments designed to remove ambient oxygen were run following a nitrogen purge of the solutions and with nitrogen flowing through the solar simulator during illumination. Samples were removed at 5, 10, 20, 30 and 45 minutes, with four replicates per solution. The rest of the three solutions were used as the controls, or 0 minutes of illumination samples. After illumination, the illuminated solutions were introduced to a PerkinElmer Series 200 HPLC equipped with a diode array detector (Shimadzu UV-Vis detector SPD-10A), to measure the remaining TMP concentrations. An isocratic solvent gradient of 70% acetonitrile, HPLC grade, and 30% 0.1% phosphoric acid was used. The HPLC method was borrowed from previous studies.32 A second set of experiments was performed to test the effect of oxygen on the triplet production. For these experiments, the solutions were made in the same way as described above, except there was no nitrogen purge of the solutions. Samples were also only removed at the 0 and 45 illumination time points and were analyzed using the HPLC method described above.
Time-resolved 1O2 phosphorescence measurements
In order to generate sufficient signal, higher concentrations of NOM were needed for these studies. At least for SRNOM, the results showed no dependence on NOM concentration, between 48 and 134 mg L−1. Trial experiments using concentrations lower than 48 mg L−1 showed too much emission from the NOM itself to resolve the oxygen phosphorescence with adequate S/N. Solutions were therefore made using stock solutions of 48 mg L−1 NRNOM and 48 mg L−1 SRNOM. Magnesium concentrations used were 22.8, 114.3 and 228 mM, while NaCl was used at a concentration of 0.68 M. IO was used at concentrations of 0.3, 1.5 and 3 M IO, which were calculated assuming a molar mass of IO the same as that of NaCl. All solutions were made at least the night before the experiments were run.The experimental setup for the 1O2 phosphorescence experiments was based on a previously published experimental design.63 Excitation pulses at 380 and 401 nm were generated by converting the primary 795 nm output of a laser (Solstice, Spectra-Physics, pulse width < 100 fs, 1 kHz repetition rate) using a TOPAS optical parametric amplifier. NOM samples were excited with a beam set to a power of 40 mW. Background spectra were collected by bubbling solutions with argon for approximately 15 minutes before the experiment began during the experiment. Only one background was taken per set of NOM samples. All other solutions were purged with neat oxygen for approximately 3 minutes before the experiment and then continuously purged during the experiment. The dissolved oxygen concentration was measured using a PreSens fiber optic micro-optode. All solutions were stirred during the entirety of the experiment.
Phosphorescence from 1O2 formed via energy transfer from 3NOM* was monitored at 90° to the excitation laser beam. Emitted photons passed through a 1270 nm bandpass filter and were focused onto a 1 mm optical fiber attached to a near-IR PMT (Hamamatsu, model H10330-45). Photon counts were integrated over a period of 720 seconds. All data was exported to Origin for fitting and analysis.
Results
The results may be summarized as follows:(A). NRNOM and SRNOM samples show a factor-of-two increase in fluorescence intensity in artificial seawater compared to freshwater. There is no difference in fluorescence intensity observed between NaCl solution and freshwater for any of the samples studied here. Corresponding fluorescence anisotropy measurements indicate a small decrease in anisotropy for the SRNOM and NRNOM samples in artificial seawater compared to freshwater, but no such change in NaCl solution. We attribute these seawater-induced changes in fluorescence features to the presence of Mg2+ at its normal seawater concentration in the artificial seawater solutions.
(B). The presence of Mg2+ at its normal seawater concentration changes the formation rate of the triplet NOM (as inferred from triplet reactivity studies) in different ways: SRNOM solutions with added Mg2+ show a decrease in the triplet formation rate compared to those without additives, while NRNOM displays a modest increase.
(C). These different influences of Mg2+ on NOM triplet formation are reflected in the quenching of the triplet by dissolved O2, which at least in part produces 1O2 (1Δ), as inferred from 1O2 formation kinetics. With small additions of Mg2+ to the NOM freshwater solutions, NRNOM shows a modest increase in the total quenching rate constant by oxygen, whereas SRNOM displays a decrease. This same effect is apparent as well in artificial seawater solutions of the NOM samples.
Below, we present these results in detail and discuss their potential significance to seawater photochemistry.
Fluorescence
This work was motivated by the surprising observation that solutions of simple halide salts, such as NaCl or NaBr, do not quench the fluorescence of the NOM samples used here. More surprisingly, when Instant Ocean (IO) was used at the same concentration as NaCl, the fluorescence of both NOM samples was greatly enhanced. Fig. 1 displays this result for excitation of SRNOM, and Fig. 2 shows resolved fluorescence spectra for both NOM samples in NaCl and IO solutions. Fig. S1 and S2 in the ESI† display excitation and resolved emission spectra, respectively, as a function of added salt concentration. Absorption spectra of the NOM samples (displayed in Fig. S3†) show little to no intensity increase upon addition of IO, regardless of concentration, suggesting that the higher fluorescence intensity is not due to greater absorbance of these samples in the presence of IO. Solutions purged with N2 for 20 minutes prior to measurement showed identical intensities to those measured under ambient air, indicating that oxygen is not quenching the fluorescence.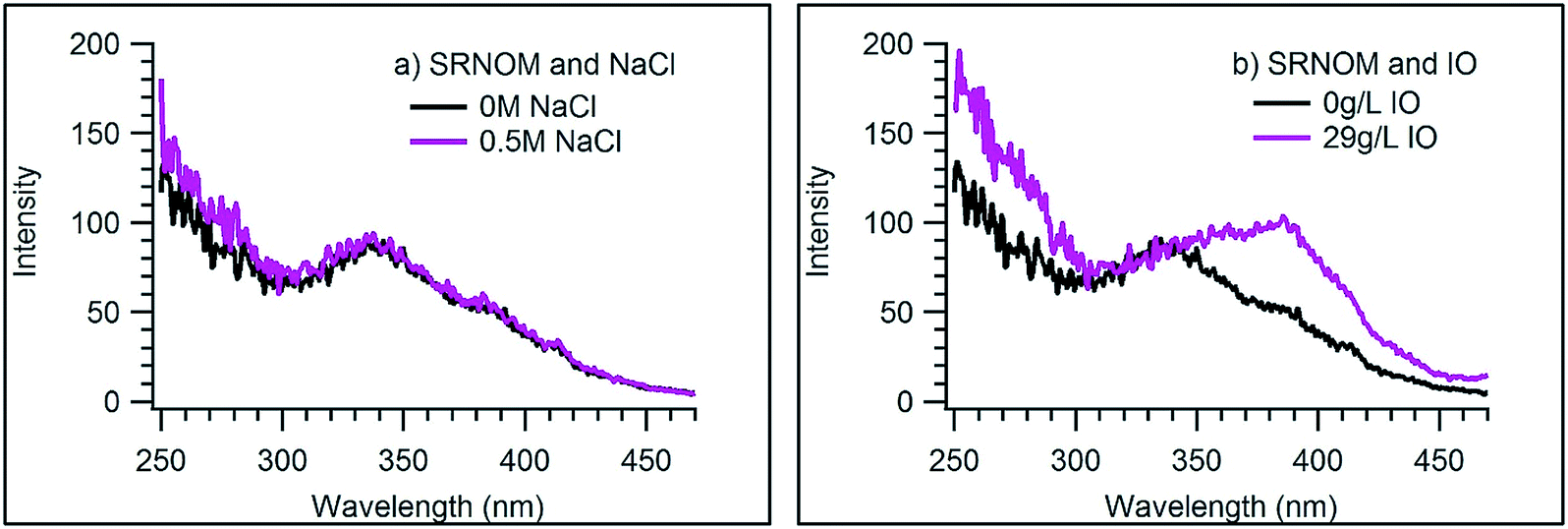 | ||
| Fig. 1 Excitation spectra of 16 mg L−1 SRNOM with 0.5 M NaCl (a) and 29 g L−1 IO (b). For both plots, the black trace is the NOM sample in water and the magenta trace represents the 0.5 M NaCl and 29 g L−1 IO solutions. | ||
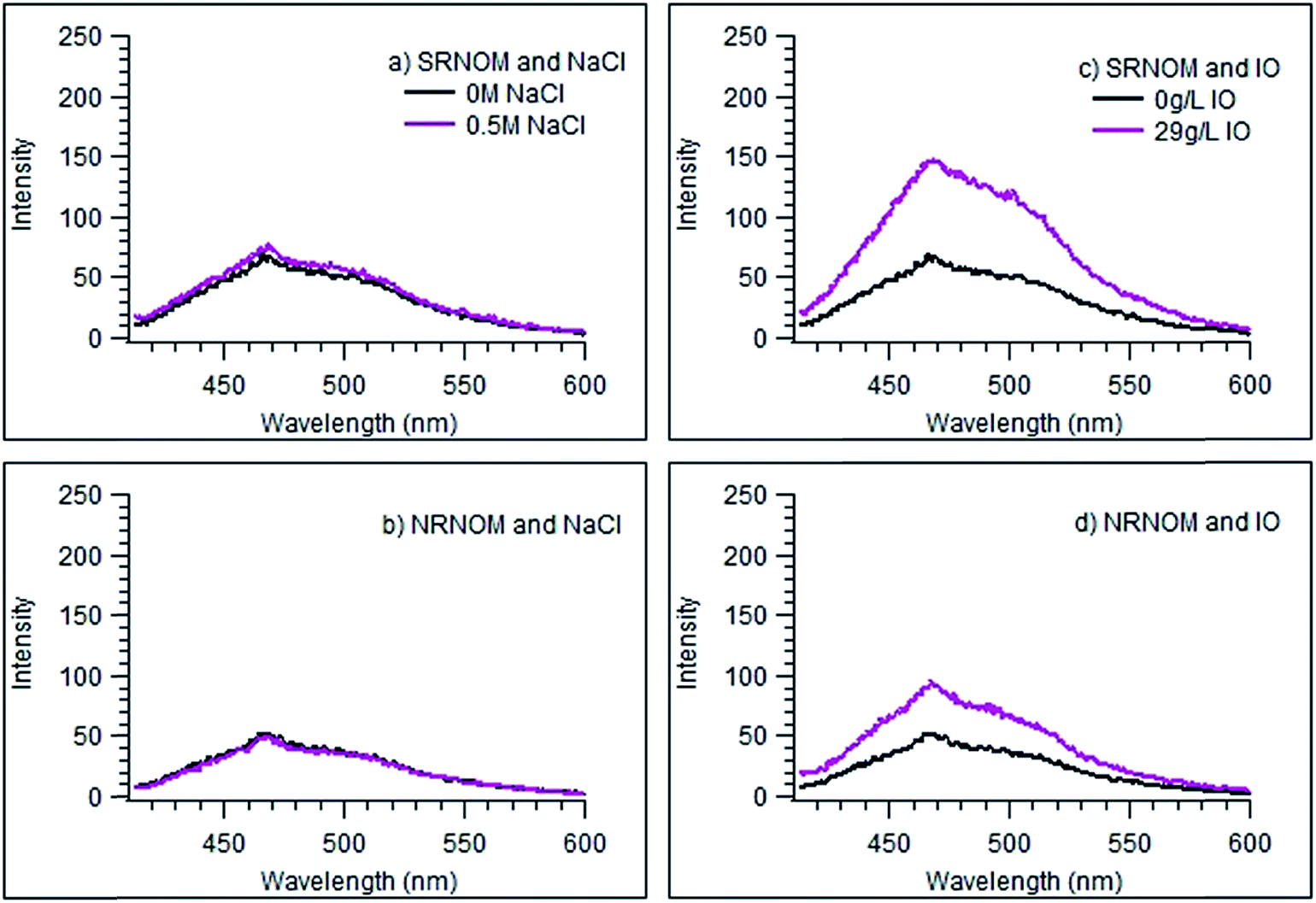 | ||
| Fig. 2 Emission plots of SRNOM and NRNOM with additions of NaCl and Instant Ocean (IO). Plots a and b are the emission spectra for SRNOM and NRNOM respectively with an addition of 0.5 M NaCl. Plots c and d are the emission spectra for SRNOM and NRNOM respectively with an addition of 29 g L−1 of IO. For all plots, the black trace is the NOM sample in water. The magenta trace is the 0.5 M and 29 g L−1 solution. | ||
The markedly different behaviours seen in NaCl and IO solution point to a role for other seawater components in the fluorescence intensity enhancement. The effect of pH on the fluorescence of the NOM samples was tested by measuring spectra as a function of solution pH, over a range of roughly 4 to 9.5, using NaOH to basify the (otherwise freshwater) solutions. No effect on the emission intensities was observed, as shown in Fig. S4 in the ESI.† Likewise, the addition of HCO3−, at its concentration in IO,64 to the freshwater solution causes no change to the fluorescence emission intensity or shape for SRNOM (Fig. S5a†). We conclude that a change in the pH is not responsible for the large change in fluorescence intensity seen with IO. Likewise, neither sulfate nor Fe3+ show any fluorescence-changing ability in the NOM samples, at least up to their concentrations in seawater. The results of these studies are given in the ESI (Fig. S5b and S6† respectively).
By contrast, the addition of Mg2+, which has a significant concentration in seawater (ca. 50 mM), to the NOM solutions resulted in changes to the spectra similar to those seen in IO solution. Fig. 3 shows the emission spectrum of SRNOM in a solution of 38.1 mM Mg2+, corresponding to the amount of magnesium ion present in 29 g L−1 IO solution, whose fluorescence spectrum is displayed in Fig. 2d. A full set of spectra is shown in Fig. S7.† Similar to what was seen with IO, there was no significant change in absorbance of the NOM solutions in the presence of magnesium ion (shown in Fig. S3†).
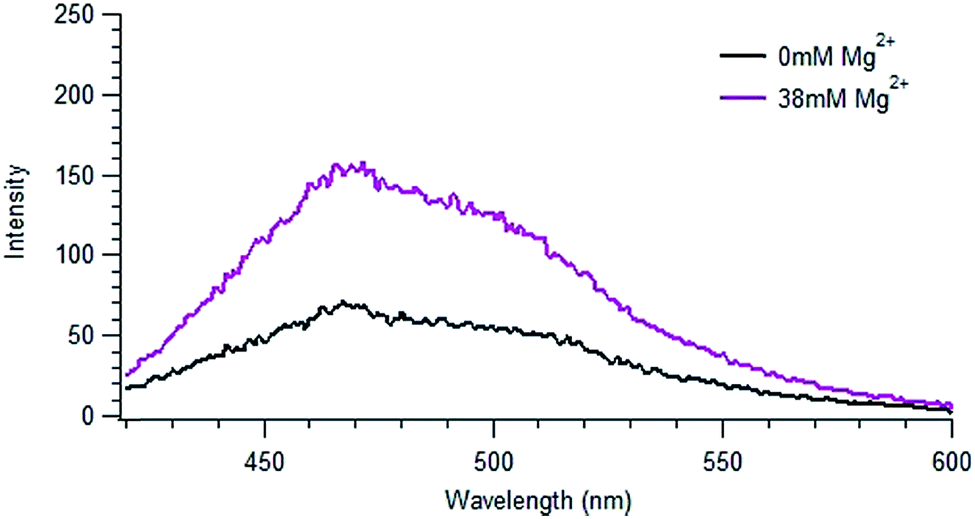 | ||
| Fig. 3 Emission spectrum of SRNOM in a solution of 38.1 mM MgCl2 using an excitation wavelength of 405 nm. The concentration of Mg2+ present in solution corresponds to the magnesium present in the IO solution shown in Fig. 1. | ||
The spectra obtained in IO and Mg2+ solutions look very similar, suggesting that Mg2+ may be the species responsible for the fluorescence enhancement. Just as observed in IO solutions, the total fluorescence intensities of both SRNOM and NRNOM roughly doubled with the addition of Mg2+, both under N2 and air. We conclude that magnesium ion is largely responsible for the effect of IO seen in the emission spectra for both samples. No effect of oxygen at the concentration present in ambient air was observed.
Fluorescence anisotropy
This technique uses polarized light to excite a fluorophore, with the resulting fluorescence intensities measured parallel (I∥) and perpendicular (I⊥) to the plane of excitation radiation. The anisotropy (r), is defined as: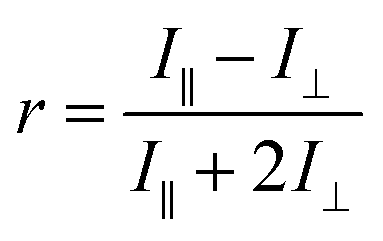
The fluorescence anisotropy of a molecule depends upon the molecular rate of rotation during the excited lifetime of the molecule, as well as the electronic transition moment for the emission and excitation processes. When a molecule binds to a metal, there may be a change in the conformation of the complex; this in turn may give rise to a change in the rotational diffusion rate and hence the measured anisotropy. For example, if the molecule binds to the metal in such a way that it coils around the metal ion, decreasing its effective size, the rate of rotation may increase, and the anisotropy value will decrease.
The fluorescence anisotropies of the NOM samples were measured as a function of wavelength in the presence and absence of added NaCl or IO as described above. Fig. 4 illustrates the difference between these “anisotropy spectra” collected in the presence of 100 mM of added salt and those measured in the absence of added salts. The only clear difference from zero in any of these spectra is observed for SRNOM and (to a lesser extent) NRNOM in the presence of IO – exactly the same combination that gives rise to enhanced emission intensities. For these two samples, the anisotropies appear to decrease somewhat in the presence of IO, suggesting a more tightly-wrapped structure. This is entirely consistent with binding of Mg2+ being responsible for both the enhanced emission and the reduced anisotropy observed in the SRNOM and NRNOM samples.
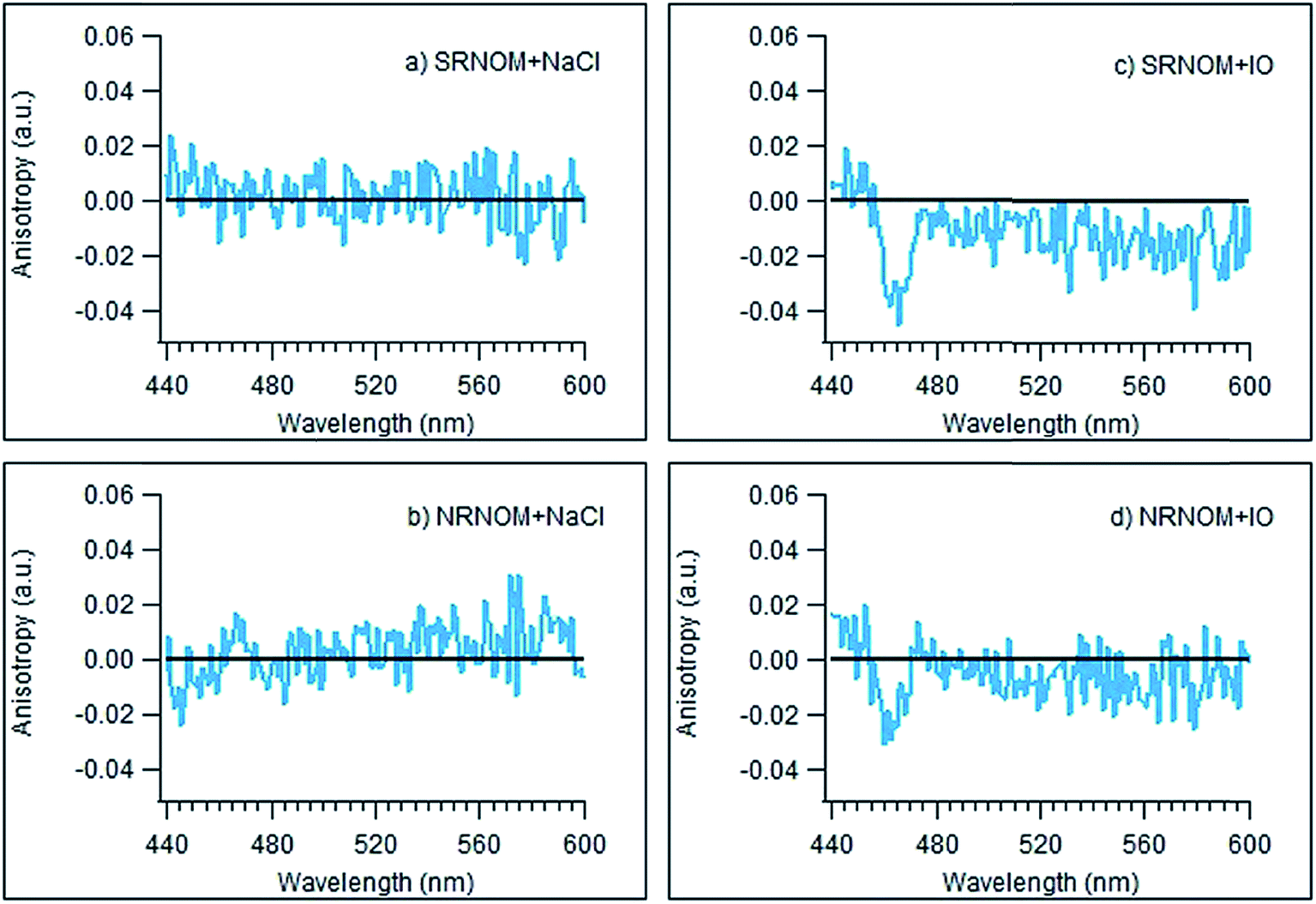 | ||
| Fig. 4 Difference spectra of the fluorescence anisotropy of the two NOM samples measured in the presence of 100 mM NaCl minus that measured without added NaCl (a and b) and those measured in the presence and absence of added IO (c and d) minus those without added IO. | ||
Formation of 3NOM*
Having concluded that Mg2+ is largely responsible for the changes in NOM fluorescence observed in artificial seawater, we tested its effect on the formation of triplet NOM, using the reaction with 2,4,6-trimethylphenol (TMP) as a probe for triplet formation. TMP reacts very rapidly with triplet state organics and is routinely used to probe for triplet NOM.9,14,65 Although reaction with TMP may not capture the total picture of triplet state production,29,66 our interest here is in exploring differences in reactivity between freshwater and seawater, rather than to determine an absolute triplet state concentration. As we shall discuss in the following section, the formation of singlet oxygen (assumed via reaction with the triplet) displays the same freshwater–seawater differences we report for the TMP result. Therefore, we are confident that the TMP results presented below provide insight into how triplet formation may depend on the NOM environment.The loss of TMP from an initial concentration of 25 μM in illuminated NOM solutions was measured in a solar simulator. Under our conditions, the loss of TMP is due to reaction with 3NOM* as well as with any 1O2 which may be formed via energy transfer to oxygen from 3NOM*. Its loss rate is given by:
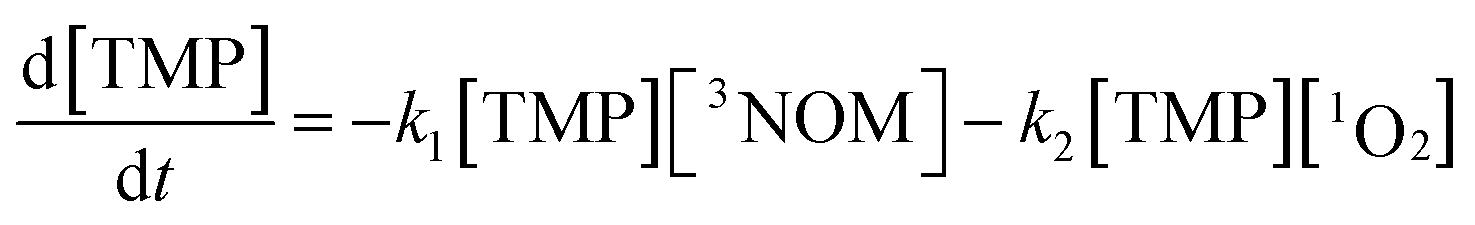
By conducting experiments under nitrogen only, the second term vanishes and the loss rate of TMP may be expressed by pseudo-first order kinetics, with an effective rate constant given by keff = k1[3NOM*]ss. The values of keff determined from the slopes of the corresponding kinetic plots (displayed in Fig. S8†) for different concentrations of Mg2+ are given in Table 1. No loss of TMP is observed in the dark, demonstrating that no excited triplet state is formed in the absence of light, and that no dark reactions occur between the NOM, TMP and magnesium.
| k eff (min−1) | |||
|---|---|---|---|
| 0 mM Mg2+ | 19 mM Mg2+ | 38 mM Mg2+ | |
| SRNOM | 0.057 ± 0.001 | 0.037 ± 0.001 | 0.034 ± 0.001 |
| NRNOM | 0.016 ± 0.002 | 0.021 ± 0.003 | 0.023 ± 0.001 |
Assuming that k1 is roughly the same for the two NOM samples,67 their relative efficiencies of triplet formation is reflected by the different keff values. Under N2, and in the absence of Mg2+, SRNOM forms more triplet than NRNOM by a factor of three. For both NOM samples, the presence of Mg2+ changes the formation rate of the triplet state. SRNOM shows a large monotonic decrease in keff, as a function of increasing magnesium concentration, while for NRNOM there is an increase in the formation rate of the triplet as [Mg2+] increases. It is clear that the addition of magnesium ion can either lower or raise the steady state concentration of 3NOM*, at least under an N2 atmosphere.
The effect of oxygen was explored at the higher [Mg2+] by measuring the TMP loss after 45 minutes of illumination in experiments conducted under ambient air. Table 2 reports the fraction of TMP reacted for each of the different conditions, with uncertainties calculated from the standard deviation of the measured TMP concentrations for nine measurements. Of note, under ambient air we measure a ∼40% loss in the amount of 3SRNOM* in Mg2+ solution compared to freshwater, and a ∼25% gain in the amount of 3NRNOM* triplet formed under the same conditions.
| N2 | Air | |||
|---|---|---|---|---|
| No Mg2+ | With Mg2+ | No Mg2+ | With Mg2+ | |
| SRNOM | 0.73 ± 0.01 | 0.68 ± 0.01 | 0.85 ± 0.13 | 0.50 ± 0.14 |
| NRNOM | 0.60 ± 0.02 | 0.71 ± 0.01 | 0.34 ± 0.03 | 0.42 ± 0.04 |
The different behaviour of NRNOM from that displayed by SRNOM extends to the influence of oxygen in the absence of magnesium ion. Whereas the presence of dissolved O2 increases the amount of triplet formed in SRNOM, it suppresses the amount of triplet formed in NRNOM. In the presence of Mg2+, both natural NOM samples show a significant decrease in the amount of triplet formation in solutions containing dissolved oxygen vs. those saturated with N2. These observations suggest different roles for Mg2+ in the photochemical pathways accessible to different NOM samples. However, overall it seems that in the presence of Mg2+, the effect of oxygen is to reduce the fraction of TMP reacted over that seen under N2 in all NOM samples. This observation is consistent with dissolved oxygen competing with TMP for reaction with 3NOM*. Next, we report on explicit measurements of the complementary 1O2 formation kinetics.
Singlet oxygen formation kinetics
The effect of Instant Ocean and NaCl on the formation and loss of singlet oxygen by the three NOM samples was investigated by measuring the intensity of 1O2 phosphorescence as a function of time following pulsed laser excitation of the NOM. As mentioned above, in order to ensure that sufficient 1O2 was formed so that its phosphorescence was greater than NOM fluorescence, the NOM concentrations were tripled relative to those used in the fluorescence and TMP experiments described above, and the laser excitation wavelength was set to 380 nm. The NaCl and IO concentrations were scaled up to maintain the same NOM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :salt ratio as in the fluorescence and triplet experiments.
:salt ratio as in the fluorescence and triplet experiments.
All solutions were saturated with O2 by bubbling neat oxygen for 3 minutes before each experiment began. After three minutes, the concentration of oxygen in solution was measured to be approximately 1300 μmol L−1 O2 using a PreSens fiber optic micro-optode for all solutions, indicating that there is little to no ionic strength dependence on the O2 solubility under these conditions.
The 1O2 phosphorescence signal was measured as a function of time for the solutions bubbled with O2 for 3 minutes, and for solutions bubbled with argon for 15 minutes. Time-resolved intensity traces from the solutions bubbled with argon were used as background and subtracted from the O2-containing solution traces to remove noise. The argon traces were originally done for all the solutions, but ionic strength had no effect on the background signal, so the same background collection was applied to all phosphorescence traces.
Fig. 5 shows a sample background-corrected plot of the 1O2 phosphorescence intensity versus time after the excitation of 44 mg L−1 SRNOM with no added salt. Such curves were fitted to a simple kinetic model to determine the formation and loss rate constants of 1O2 for both NOM samples with the two salts. A full kinetic scheme and analysis is presented in a paper by Erickson, Moor et al.67 A simplified kinetic scheme arises by assuming that there is no singlet oxygen present at time zero and the quenching of 3NOM* to NOM‡ (via ISC) is slow compared to its reaction with O2, allowing singlet oxygen concentrations to be modelled by considering only a few processes:
| 3NOM* + 3O2 → NOM‡ + 1O2 |
| 1O2 → 3O2 |
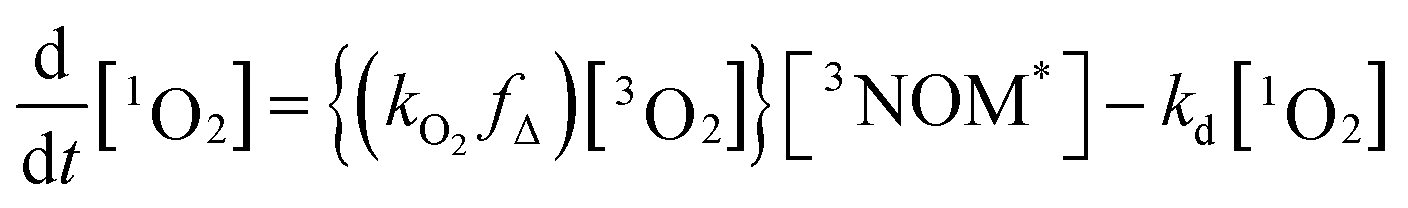
where kO2 is the rate coefficient for quenching of 3NOM* by ground state oxygen, fΔ represents the fraction of quenching events that produces 1O2, and kd is the rate constant for loss of singlet oxygen (primarily via collisions with solvent – vide infra). The lifetime of 3NOM* in de-oxygenated aqueous solution is reported to be between 20 and 80 μs;27 using the measured oxygen concentrations and assuming a kO2 of about 109 M−1 s−1 (ref. 67) yields a triplet lifetime of approximately 285 ns. We conclude that the reaction between 3NOM* and O2 is much faster than the other loss processes of NOM in these experiments, meaning we can set
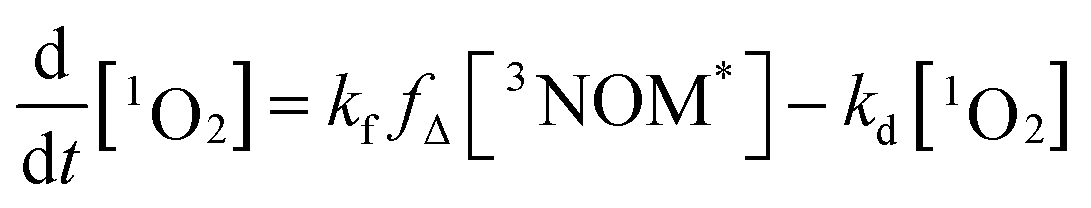
| [3NOM*] = [3NOM*]t=0e−kft |
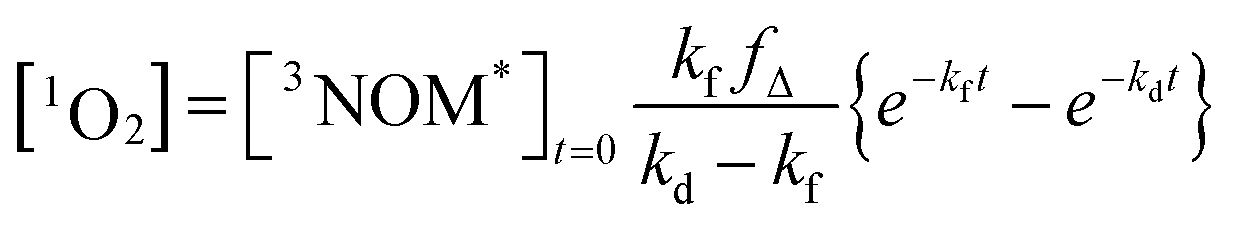 | (1) |
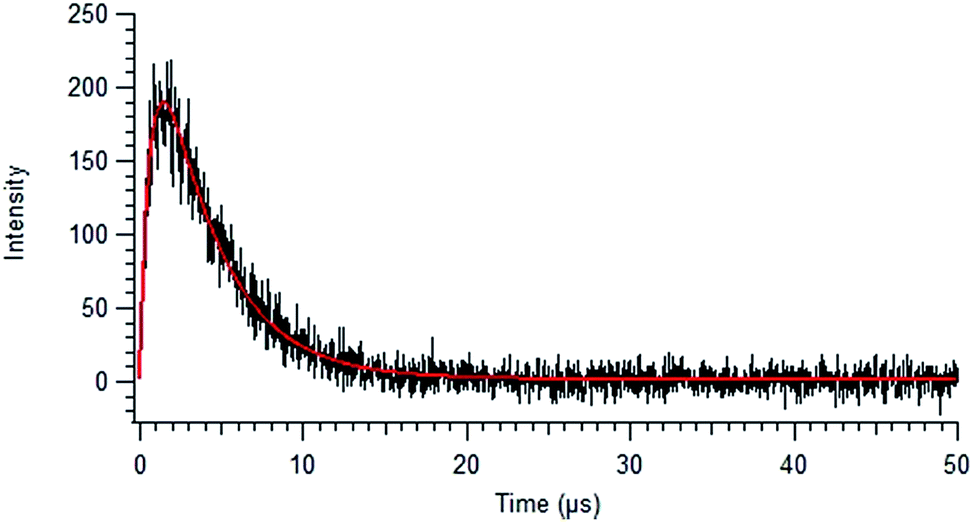 | ||
| Fig. 5 Background-corrected singlet oxygen phosphorescence intensity versus time curve, collected for 44 mg L−1 SRNOM using an excitation wavelength of 380 nm. The solid red line shows the fit to eqn (2). | ||
In Fig. 5 is displayed a fit of the experimental data fit to the expression:
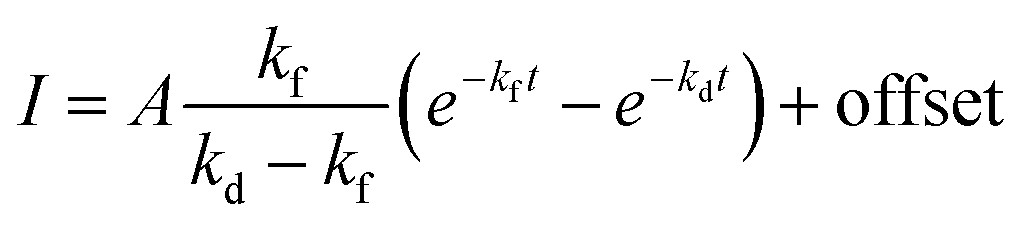 | (2) |
Fig. 6 displays the dependence of kf on [Mg2+] for both NOM samples. At low magnesium ion concentration, kf decreases sharply with added Mg2+ for SRNOM, and stays fairly constant, perhaps increasing, for NRNOM. This trend is seen as well in IO solutions, displayed in Fig. S9.† A gradual overall decrease in kf with increasing salt content is observed, but the behaviour at low [IO] mimics that at low [Mg2+], as displayed in Fig. 6.
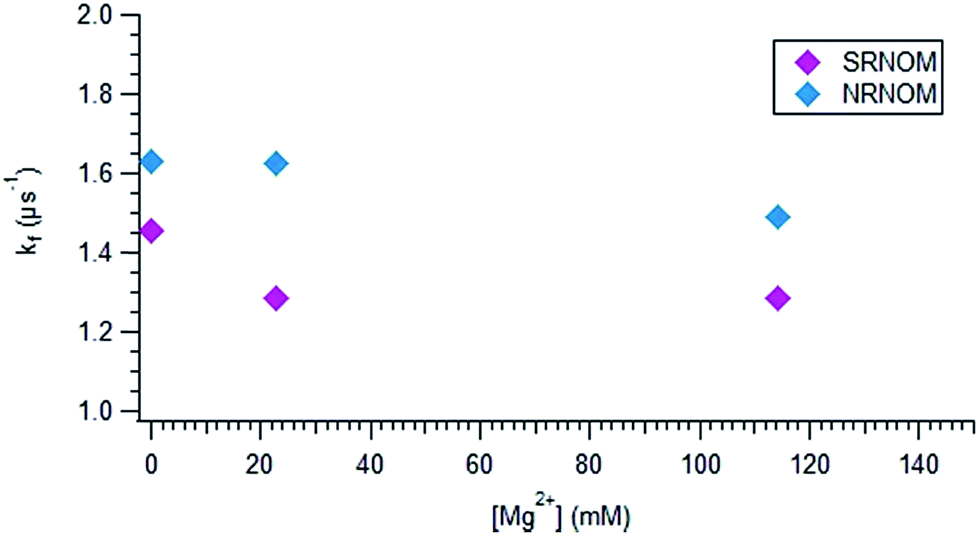 | ||
| Fig. 6 Plot of kfversus the concentration of magnesium for SRNOM (purple diamonds) and NRNOM (blue diamonds). | ||
Discussion
This work set out to determine whether photochemistry involving NOM is, or could be, different in seawater than in freshwater. Our results indicate that this is a very real possibility, with both the formation of the reactive NOM triplet state and singlet oxygen production showing different behaviour in the two types of aqueous environment.The observation, illustrated in Fig. 1 and 2, that simple halide salts do not quench fluorescence from any of the NOM samples is certainly interesting. Halides are well known as quenchers of fluorescence from many organics, so much so that the extent of this quenching is the basis for halide detection in solution.68 Indeed, for some NOM (i.e. plant-extracted chlorophyll), there is a strong reactive quenching observed in the presence of halide ions.69 Likewise, dissolved oxygen has no effect on the fluorescence spectra for any of the NOM samples, in any of the aqueous solutions studied here. We conclude that neither dissolved oxygen nor halide ions have strong interactions with the excited singlet state(s) of these NOM samples.
However, as shown in Fig. 2, 3 and S7,† the presence of magnesium ions at sea water concentrations, either alone or in a sea salt matrix, enhances the fluorescence of SRNOM and NRNOM. In agreement with Yan et al.,70 we see no significant change in the absorbance intensities or spectra of the NOM samples in solutions containing Mg2+. These observations may be explained by Mg2+ binding to carboxylic acid sites in the NOM samples, with the resulting complexes showing shifts in the excitation maxima to longer wavelengths.44,51,70,71 It may be that because both NRNOM and SRNOM are reported to contain carboxylic functional groups,72,73 which is why we observe a fluorescence intensity enhancement in the excitation and emission spectra of SRNOM and NRNOM in seawater and in Mg2+-containing solution.
Our hypothesis about the importance of the carboxylic acid functional group to the fluorescence enhancement is supported by changes seen to the fluorescence anisotropy curves of NRNOM and SRNOM in the presence of IO, displayed in Fig. 4. For these NOM samples, there is a reduction in the anisotropy values in the presence of IO, consistent with Mg2+ binding to carboxylate functionalities forming a more compact, spherical structure.
At sea water concentrations, Mg2+ also changes the formation rate of 3NOM*, as evidenced by its effect on keff, the pseudo-first order rate constant for TMP loss displayed in Fig. S8.† The results shown in Tables 1 and 2 indicate keff decreases for SRNOM samples in the presence of Mg2+, and increases for NRNOM, both under nitrogen or ambient air. If we assume that the reaction rate constant of 3NOM* is the same for both NOM samples,67 this finding implies that there is more 3NRNOM* and less 3SRNOM* formed in seawater than in freshwater, as a consequence of binding by Mg2+. Previous studies have reported an increase in [3NOM*]ss with increasing ionic strength, typically using sea water halides,32,33 an effect attributed to a decrease in the reaction rate with TMP with increasing ionic strength. However, in the present work, the ionic strength of the Mg2+ solutions was always very low, so it seems unlikely to have been a factor in the observed changes in keff. We hypothesize that the changes we observe in the formation of triplet NOM arise from changes in intersystem crossing, induced by binding of Mg2+.
Oxygen does not quench the fluorescence of any of the NOM samples in fresh water or in sea water. However, as indicated in Table 2, it does affect the triplet formation for all three NOM samples in each medium. In freshwater under ambient air, triplet formation is somewhat enhanced for SRNOM compared to that seen under N2 but inhibited for NRNOM. By contrast, in Mg2+ solution, both NOM samples show lower triplet formation under ambient air relative to solutions that were bubbled with N2. It appears that oxygen might play a complex role in both inducing intersystem crossing and in quenching the resulting triplet state. Nevertheless, given the object of this study is to compare NOM reactivity between freshwater and seawater environments on Earth, we concentrate on comparing the results under ambient air, with and without Mg2+present. Fig. 6 and S9† show that with small amounts of Mg2+ present, the changes in the triplet quenching rate constant by oxygen – yielding in part O2 (1Δ) − between freshwater and seawater qualitatively track the changes seen in the triplet production. For NRNOM, there is a small enhancement in this rate constant with increasing [Mg2+], and for SRNOM there is a decrease. This close tracking is certainly expected, given the work presented in Erickson, Moor et al.67 We do not measure fΔ, and so cannot directly relate the triplet quenching by oxygen to 1O2 production. Nevertheless, it seems reasonable to assume that the changes in quenching rate constant relate to changes in singlet oxygen production. In a natural, sunlit environment, the steady-state concentration of singlet oxygen is given by:
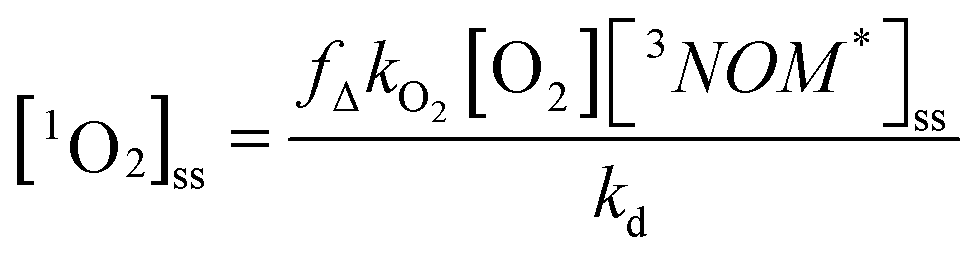
Thus it appears that in seawater, the photoproduction of reactive species from NOM can be quite different from that observed in freshwater samples. This difference is likely related to the binding of trace cationic species, such as Mg2+, to specific sites in the natural organic material. Comparing the general functional group composition of the three samples studied here may yield some insight into why and how these differences may come about. Jiang et al. report an extensive comparison of the different functional groups present in a wide range of NOM samples.74 SRNOM contains a significantly smaller fraction of monocyclic aromatics and phenols than NRNOM, and about half the amount of N-containing fragments. The two samples contain roughly equal amounts of benzene carboxylic acids and fatty acid methyl esters. These findings point to a similar Mg2+ binding potential for the two NOM samples (via the acidic moieties), as we infer from our fluorescence results, but possibly very different triplet behavior. It may be that binding to Mg2+ may effectively quench the triplet in SRNOM compared to NRNOM, due to its lesser fraction of aromatic and phenolic fragments.
Conclusions
This work provides evidence that NOM photochemistry in seawater may be quite different from that observed in freshwater. Here, we find that the halide salts present in seawater play little role in such differences; rather, it is trace species – in particular Mg2+ – that are responsible for changes in fluorescence, triplet state yields and singlet oxygen formation. The changes in formation of 1O2 qualitatively track those of 3NOM* production. NOM samples with higher aromatic, phenolic and nitrogen-containing content may show increased 3NOM* and 1O2 production in seawater compared to freshwater.One caveat of this study is that the two NOM samples used in this study are terrestrial in composition, and are therefore not completely representative of marine NOM. Past studies, some using high resolution mass spectrometry,75–78 have shown that marine NOM, while containing some similar chemical formulas as terrestrial NOM, tends to have a lower abundance of oxygen containing formulas.75–79 This suggests that marine NOM has fewer carboxylic groups and would therefore have fewer ligands to bind to Mg2+. Thus, changes to the PPRI formation of marine NOM may be less pronounced than is suggested here. In fact, the conclusions from this paper are likely important for estuaries and coastal waters rather than open ocean.
Conflicts of interest
There are no conflicts of interest to declareAcknowledgements
This work was funded by NSERC, to whom we are grateful. LS thanks the Centre for Global Change Science at the University of Toronto for a Graduate Student Research Award. K. J. M. gratefully acknowledges support from the ETH Zurich Postdoctoral Fellowship Program.References
- J. A. Rice and P. MacCarthy, Statistical evaluation of the elemental composition of humic substances, Org. Geochem., 1991, 17, 635–648 CrossRef CAS.
- J. A. Leenheer and J.-P. Croué, Characterizing aquatic dissolved organic matter, Environ. Sci. Technol., 2003, 37, 18A–26A CrossRef CAS PubMed.
- J. A. Leenheer, T. I. Noyes, C. E. Rostad and M. L. Davisson, Characterization and origin of polar dissolved organic matter from the Great Salt Lake, Biogeochemistry, 2004, 69, 125–141 CrossRef CAS.
- R. Gonçalves-Araujo, C. A. Stedmon, B. Heim, I. Dubinenkov, A. Kraberg, D. Moiseev and A. Bracher, From fresh to marine waters: characterization and fate of dissolved organic matter in the Lena River Delta region, Siberia, Front. Mar. Sci., 2015, 2, 1–13 Search PubMed.
- S. Inamdar, S. Singh, S. Dutta, D. Levia, M. Mitchell, D. Scott, H. Bais and P. McHale, Fluorescence characteristics and sources of dissolved organic matter for stream water during storm events in a forested mid-Atlantic watershed, J. Geophys. Res., 2011, 116, G03043 CrossRef.
- M. R. Provenzano, V. D'Orazio, M. Jerzykiewicz and N. Senesi, Fluorescence behaviour of Zn and Ni complexes of humic acids from different sources, Chemosphere, 2004, 55, 885–892 CrossRef CAS PubMed.
- D. J. Burdige, S. W. Kline and W. Chen, Fluorescent dissolved organic matter in marine sediment pore waters, Mar. Chem., 2004, 89, 289–311 CrossRef CAS.
- L. G. Larsen, G. R. Aiken, J. W. Harvey, G. B. Noe and J. P. Crimaldi, Using fluorescence spectroscopy to trace seasonal DOM dynamics, disturbance effects, and hydrologic transport in the Florida Everglades, J. Geophys. Res.: Biogeosci., 2010, 115, 1–14 Search PubMed.
- S. Canonica, U. Jans, K. Stemmler and J. Hoigné, Transformation kinetics of phenols in water: photosensitization by dissolved natural organic material and aromatic ketones, Environ. Sci. Technol., 1995, 29, 1822–1831 CrossRef CAS PubMed.
- Y. P. Chin, P. L. Miller, L. Zeng, K. Cawley and L. K. Weavers, Photosensitized degradation of bisphenol A by dissolved organic matter, Environ. Sci. Technol., 2004, 38, 5888–5894 CrossRef CAS PubMed.
- K. Stemmler, M. Ammann, C. Donders, J. Kleffmann and C. George, Photosensitized reduction of nitrogen dioxide on humic acid as a source of nitrous acid, Nature, 2006, 440, 195–198 CrossRef CAS PubMed.
- J. Wenk, M. Aeschbacher, M. Sander, U. Von Gunten and S. Canonica, Photosensitizing and inhibitory effects of ozonated dissolved organic matter on triplet-induced contaminant transformation, Environ. Sci. Technol., 2015, 49, 8541–8549 CrossRef CAS PubMed.
- P. Calza, D. Vione and C. Minero, The role of humic and fulvic acids in the phototransformation of phenolic compounds in seawater, Sci. Total Environ., 2014, 493, 411–418 CrossRef CAS PubMed.
- S. Canonica and M. Freiburghaus, Electron-rich phenols for probing the photochemical reactivity of freshwaters, Environ. Sci. Technol., 2001, 35, 690–695 CrossRef CAS PubMed.
- J. Wenk, S. N. Eustis, K. McNeill and S. Canonica, Quenching of excited triplet states by dissolved natural organic matter, Environ. Sci. Technol., 2013, 47, 12802–12810 CrossRef CAS PubMed.
- Y. Zhang, R. Del Vecchio and N. V. Blough, Investigating the mechanism of hydrogen peroxide photoproduction by humic substances, Environ. Sci. Technol., 2012, 46, 11836–11843 CrossRef CAS PubMed.
- R. M. Dalrymple, A. K. Carfagno and C. M. Sharpless, Correlations between dissolved organic matter optical properties and quantum yields of singlet oxygen and hydrogen peroxide, Environ. Sci. Technol., 2010, 44, 5824–5829 CrossRef CAS PubMed.
- Y. Lester, C. M. Sharpless, H. Mamane and K. G. Linden, Production of photo-oxidants by dissolved organic matter during UV water treatment, Environ. Sci. Technol., 2013, 47, 11726–11733 CrossRef CAS PubMed.
- B. Sur, M. Rolle, C. Minero, V. Maurino, D. Vione, M. Brigante and G. Mailhot, Formation of hydroxyl radicals by irradiated 1-nitronaphthalene (1NN): oxidation of hydroxyl ions and water by the 1NN triplet state, Photochem. Photobiol. Sci., 2011, 10, 1817–1824 RSC.
- L. Sun, H. Chen, H. A. Abdulla and K. Mopper, Estimating hydroxyl radical photochemical formation rates in natural waters during long-term laboratory irradiation experiments, Environ. Sci.: Processes Impacts, 2014, 16, 757–763 RSC.
- D. Vione, G. Falletti, V. Maurino, C. Minero, E. Pelizzetti, M. Malandrino, R. Ajassa, R.-I. Olariu and C. Arsene, Sources and sinks of hydroxyl radicals upon irradiation of natural water samples, Environ. Sci. Technol., 2006, 40, 3775–3781 CrossRef CAS PubMed.
- G. McKay and F. L. Rosario-Ortiz, Temperature dependence of the photochemical formation of hydroxyl radical from dissolved organic matter, Environ. Sci. Technol., 2015, 49, 4147–4154 CrossRef CAS PubMed.
- S. E. Page, J. R. Logan, R. M. Cory and K. McNeill, Evidence for dissolved organic matter as the primary source and sink of photochemically produced hydroxyl radical in arctic surface waters, Environ. Sci.: Processes Impacts, 2014, 16, 807–822 RSC.
- G. McKay, K. D. Couch, S. P. Mezyk and F. L. Rosario-Ortiz, Investigation of the coupled effects of molecular weight and charge-transfer interactions on the optical and photochemical properties of dissolved organic matter, Environ. Sci. Technol., 2016, 50, 8093–8102 CrossRef CAS PubMed.
- W. R. Haag and J. Hoigné, Singlet oxygen in surface waters. 3. Photochemical formation and steady-state concentrations in various types of waters, Environ. Sci. Technol., 1986, 20, 341–348 CrossRef CAS PubMed.
- W. R. Haag, J. Hoigné, E. Gassman and A. M. Braun, Singlet oxygen in surface waters- part 1: furfuryl alcohol as a trapping agent, Chemosphere, 1984, 13, 631–640 CrossRef CAS.
- C. M. Sharpless, Lifetimes of triplet dissolved natural organic matter (DOM) and the effect of NaBH4 reduction on singlet oxygen quantum yields: Implications for DOM photophysics, Environ. Sci. Technol., 2012, 46, 4466–4473 CrossRef CAS PubMed.
- A. Fede and A. M. Grannas, Photochemical production of singlet oxygen from dissolved organic matter in ice, Environ. Sci. Technol., 2015, 49, 12808–12815 CrossRef CAS PubMed.
- K. McNeill and S. Canonica, Triplet state dissolved organic matter in aquatic photochemistry: reaction mechanisms, substrate scope, and photophysical properties, Environ. Sci.: Processes Impacts, 2016, 18, 1381–1399 RSC.
- L. C. Bodhipaksha, C. M. Sharpless, Y. P. Chin, M. Sander, W. K. Langston and A. A. MacKay, Triplet photochemistry of effluent and natural organic matter in whole water and isolates from effluent-receiving rivers, Environ. Sci. Technol., 2015, 49, 3453–3463 CrossRef CAS PubMed.
- E. De Laurentiis, M. Minella, V. Maurino, C. Minero, M. Brigante, G. Mailhot and D. Vione, Photochemical production of organic matter triplet states in water samples from mountain lakes, located below or above the tree line, Chemosphere, 2012, 88, 1208–1213 CrossRef CAS PubMed.
- K. M. Parker, J. J. Pignatello and W. A. Mitch, Influence of ionic strength on triplet-state natural organic matter loss by energy transfer and electron transfer pathways, Environ. Sci. Technol., 2013, 47, 10987–10994 CrossRef CAS PubMed.
- C. M. Glover and F. L. Rosario-Ortiz, Impact of halides on the photoproduction of reactive intermediates from organic matter, Environ. Sci. Technol., 2013, 47, 13949–13956 CrossRef CAS PubMed.
- F. al Housari, D. Vione, S. Chiron and S. Barbati, Reactive photoinduced species in estuarine waters. Characterization of hydroxyl radical, singlet oxygen and dissolved organic matter triplet state in natural oxidation processes, Photochem. Photobiol. Sci., 2010, 9, 78–86 RSC.
- J. G. Hering and F. M. Morel, Humic acid complexation of calcium and copper, Environ. Sci. Technol., 1988, 22, 1234–1237 CrossRef CAS PubMed.
- E. Tipping, C. Rey-Castro, S. E. Bryan and J. Hamilton-Taylor, Al(III) and Fe(III) binding by humic substances in freshwaters, and implications for trace metal speciation, Geochim. Cosmochim. Acta, 2002, 66, 3211–3224 CrossRef CAS.
- M. Chen, W. X. Wang and L. Guo, Phase partitioning and solubility of iron in natural seawater controlled by dissolved organic matter, Global Biogeochem. Cycles, 2004, 18, 1–12 CAS.
- A. K. Pandey, S. D. Pandey and V. Misra, Stability constants of metal-humic acid complexes and its role in environmental detoxification, Ecotoxicol. Environ. Saf., 2000, 47, 195–200 CrossRef CAS PubMed.
- H. Van Dijk, Cation binding of humic acids, Geoderma, 1971, 5, 53–67 CrossRef CAS.
- M. Yan and G. V. Korshin, Comparative examination of effects of binding of different metals on chromophores of dissolved organic matter, Environ. Sci. Technol., 2014, 48, 3177–3185 CrossRef CAS PubMed.
- D. G. Kinniburgh, C. J. Milne, M. F. Benedetti, J. P. Pinheiro, J. Filius, L. K. Koopal and W. H. Van Riemsdijk, Metal ion binding by humic acid: application of the NICA-Donnan model, Environ. Sci. Technol., 1996, 30, 1687–1698 CrossRef CAS.
- E. Tipping and M. A. Hurley, A unifying model of cation binding by humic substances, Geochim. Cosmochim. Acta, 1992, 56, 3627–3641 CrossRef CAS.
- E. Tipping, Humic ion-binding model VI: an improved description of the interactions of protons and metal ions with humic substances, Aquat. Geochem., 1998, 4, 3–48 CrossRef CAS.
- Y. Lu and H. E. Allen, Characterization of copper complexation with natural dissolved organic matter (DOM) - link to acidic moieties of DOM and competition by Ca and Mg, Water Res., 2002, 36, 5083–5101 CrossRef CAS PubMed.
- K. G. J. Nierop, B. Jansen and J. M. Verstraten, Dissolved organic matter, aluminium and iron interactions: precipitation induced by metal/carbon ratio, pH and competition, Sci. Total Environ., 2002, 300, 201–211 CrossRef CAS PubMed.
- J. J. Alberts and Z. Filip, Metal binding in estuarine humic and fulvic acids: FTIR analysis of humic acid-metal complexes, Environ. Technol., 1998, 19, 923–931 CrossRef CAS.
- D. K. Ryan and J. H. Weber, Copper(II) complexing capacities of natural waters by fluorescence quenching, Environ. Sci. Technol., 1982, 16, 866–872 CrossRef CAS PubMed.
- T. Ohno, A. Amirbahman and R. Bro, Parallel factor analysis of excitation-emission matrix fluorescence spectra of water soluble soil organic matter as basis for the determination of conditional metal binding parameters, Environ. Sci. Technol., 2008, 42, 186–192 CrossRef CAS PubMed.
- M. J. Pullin, C. Anthony and P. A. Maurice, Effects of iron on the molecular weight distribution, light absorption, and fluorescence properties of natural organic matter, Environ. Eng. Sci., 2007, 24, 987–997 CrossRef CAS.
- J. D. Wiley, The effect of magnesium on natural fluorescence during estuarine mixing, and implications for tracer applications, Mar. Chem., 1984, 15, 19–46 CrossRef.
- V. I. Esteves, E. B. Santos and A. C. Duarte, Study of the effect of pH, salinity and DOC on fluorescence of synthetic mixtures of freshwater and marine salts, J. Environ. Monit., 1999, 1, 251–254 RSC.
- A. Seritti, E. Morelli, L. Nannicini, A. Giambelluca and G. Scarano, Fluorescence emission characteristics of naturally occurring organic matter in relation to metal complexation studies, Sci. Total Environ., 1994, 148, 73–81 CrossRef CAS.
- T. Andjelković, J. M. Perović, M. M. Purenović and D. Andjelković, Destabilization and aggregation of aqueous humic acids solution by metal ions, Facta Univ., Ser.: Phys., Chem. Technol., 2004, 3, 79–85 CrossRef.
- N. Kloster, M. Brigante, G. Zanini and M. Avena, Aggregation kinetics of humic acids in the presence of calcium ions, Colloids Surf., A, 2013, 427, 76–82 CrossRef CAS.
- R. R. Engebretson and R. Von Wandruszka, Kinetic aspects of cation-enhanced aggregation in aqueous humic acids, Environ. Sci. Technol., 1998, 32, 488–493 CrossRef CAS.
- J. Peuravuori, R. Koivikko and K. Pihlaja, Characterization, differentiation and classification of aquatic humic matter separated with different sorbents: synchronous scanning fluorescence spectroscopy, Water Res., 2002, 36, 4552–4562 CrossRef CAS.
- R. Von Wandruszka, C. Ragle and R. Engebretson, The role of selected cations in the formation of pseudomicelles in aqueous humic acid, Talanta, 1997, 44, 805–809 CrossRef CAS PubMed.
- R. L. Malcolm and P. MacCarthy, Limitations in the use of commercial humic acids in water and soil research, Environ. Sci. Technol., 1986, 20, 904–911 CrossRef CAS PubMed.
- K. Stemmler, M. Ndour, Y. Elshorbany, J. Kleffmann, B. D'Anna, C. George, B. Bonn and M. Ammann, Light induced conversion of nitrogen dioxide into nitrous acid on submicron humic acid aerosol, Atmos. Chem. Phys., 2007, 7, 4237–4248 CrossRef CAS.
- C. L. Badger, I. George, P. T. Griffiths, C. F. Braban, R. A. Cox and J. P. D. Abbatt, Phase transitions and hygroscopic growth of aerosol particles containing humic acid and mixtures of humic acid and ammonium sulphate, Atmos. Chem. Phys., 2006, 6, 755–768 CrossRef CAS.
- Z. Krivacsy, G. Kiss, B. Varga, I. Galambos, Z. Sarvari, A. Gelencser, A. Molnar, S. Fuzzi, M. C. Facchini, S. Zappoli, A. Andracchio, T. Alsberg, H. C. Hansson and L. Persson, Study of humic-like substances in fog and interstitial aerosol by size-exclusion chromatography and capillary electrophoresis, Atmos. Environ., 2000, 34, 4273–4281 CrossRef CAS.
- S. Halladja, A. Ter Halle, J. P. Aguer, A. Boulkamh and C. Richard, Inhibition of humic substances mediated photooxygenation of furfuryl alcohol by 2,4,6-trimethylphenol. Evidence for reactivity of the phenol with humic triplet excited states, Environ. Sci. Technol., 2007, 41, 6066–6073 CrossRef CAS PubMed.
- E. Appiani, R. Ossola, D. E. Latch, P. R. Erickson and K. McNeill, Aqueous singlet oxygen reaction kinetics of furfuryl alcohol: effect of temperature, pH, and salt content, Environ. Sci.: Processes Impacts, 2017, 19, 507–516 RSC.
- A. Laskin, D. J. Gaspar, W. Wang, S. W. Hunt, J. P. Cowin, S. D. Colson and B. J. Finlayson-pitts, Reactions at interfaces as a source of sulfate formation in sea-salt particles, Science, 2003, 301, 340–344 CrossRef CAS PubMed.
- K. S. Golanoski, S. Fang, R. Del Vecchio and N. V. Blough, Investigating the mechanism of phenol photooxidation by humic substances, Environ. Sci. Technol., 2012, 46, 3912–3920 CrossRef CAS PubMed.
- F. L. Rosario-Ortiz and S. Canonica, Probe Compounds to Assess the Photochemical Activity of Dissolved Organic Matter, Environ. Sci. Technol., 2016, 50, 12532–12547 CrossRef CAS PubMed.
- P. R. Erickson, K. J. Moor, J. J. Werner, D. E. Latch, W. A. Arnold and K. McNeill, Singlet oxygen phosphorescence as a probe for triplet-state dissolved organic matter reactivity, Environ. Sci. Technol., 2018, 52, 9170–9178 CrossRef CAS PubMed.
- C. D. Geddes, Optical halide sensing using fluorescence quenching: theory, simulations and applications - a review, Meas. Sci. Technol., 2001, 12, R53–R88 CrossRef CAS.
- D. I. Reeser, C. George and D. J. Donaldson, Photooxidation of Halides by Chlorophyll at the Air - Salt Water Interface, J. Phys. Chem. A, 2009, 113, 0–4 CrossRef PubMed.
- M. Yan, Y. Lu, Y. Gao, M. F. Benedetti and G. V. Korshin, In situ investigation of interactions between magnesium ion and natural organic matter, Environ. Sci. Technol., 2015, 49, 8323–8329 CrossRef CAS PubMed.
- E. Iskrenova-Tchoukova, A. G. Kalinichev and R. J. Kirkpatrick, Metal cation complexation with natural organic matter in aqueous solutions: molecular dynamics simulations and potentials of mean force, Langmuir, 2010, 26, 15909–15919 CrossRef CAS PubMed.
- N. W. Green, D. McInnis, N. Hertkorn, P. A. Maurice and E. M. Perdue, Suwannee River natural organic matter: isolation of the 2R101N reference sample by reverse osmosis, Environ. Eng. Sci., 2015, 32, 38–44 CrossRef CAS.
- J. A. Leenheer, R. L. Wershaw and M. M. Reddy, Strong-acid, carboxyl-group structures in fulvic acid from the Suwannee River, Georgia. 1. Minor structures, Environ. Sci. Technol., 1995, 29, 393–398 CrossRef CAS PubMed.
- T. Jiang, J. Kaal, J. Liang, Y. Zhang, S. Wei, D. Wang and N. W. Green, Composition of dissolved organic matter (DOM) from periodically submerged soils in the Three Gorges Reservoir areas as determined by elemental and optical analysis, infrared spectroscopy, pyrolysis-GC–MS and thermally assisted hydrolysis and methylation, Sci. Total Environ., 2017, 603–604, 461–471 CrossRef CAS PubMed.
- R. L. Sleighter and P. G. Hatcher, Molecular characterization of dissolved organic matter (DOM) along a river to ocean transect of the lower Chesapeake Bay by ultrahigh resolution electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry, Mar. Chem., 2008, 110, 140–152 CrossRef CAS.
- B. P. Koch, M. Witt, R. Engbrodt, T. Dittmar and G. Kattner, Molecular formulae of marine and terrigenous dissolved organic matter detected by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry, Geochim. Cosmochim. Acta, 2005, 69, 3299–3308 CrossRef CAS.
- E. C. Minor, J. P. Simjouw, J. J. Boon, A. E. Kerkhoff and J. Van der Horst, Estuarine/marine UDOM as characterized by size-exclusion chromatography and organic mass spectrometry, Mar. Chem., 2002, 78, 75–102 CrossRef CAS.
- D. J. Repeta, T. M. Quan, L. I. Aluwihare and A. Accardi, Chemical characterization of high molecular weight dissolved organic matter in fresh and marine waters, Geochim. Cosmochim. Acta, 2002, 66, 955–962 CrossRef CAS.
- M. Zark and T. Dittmar, Universal molecular structures in natural dissolved organic matter, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8em00431e |
| This journal is © The Royal Society of Chemistry 2019 |