
Impact of bisphenol A influent concentration and reaction time on MnO2 transformation in a stirred flow reactor†
Sarah
Balgooyen
 a,
Gabrielle
Campagnola
b,
Christina K.
Remucal
*ab and
Matthew
Ginder-Vogel
*ab
a,
Gabrielle
Campagnola
b,
Christina K.
Remucal
*ab and
Matthew
Ginder-Vogel
*ab
aEnvironmental Chemistry and Technology Program, University of Wisconsin–Madison, 660 N. Park St., Madison, WI 53706, USA. E-mail: remucal@wisc.edu; Web: http://@remucal Fax: +1 608 262-0454; Tel: +1 608 262-1820
bDepartment of Civil and Environmental Engineering, 660 N. Park St., Madison, WI 53706, USA. E-mail: mgindervogel@wisc.edu; Web: http://@profmattgv Fax: +1 608 262-0454; Tel: +1 608 262-0768
First published on 27th November 2018
Abstract
Bisphenol A (BPA) is an endocrine disrupting compound commonly found in natural waters at concentrations that are considered harmful for aquatic life. Manganese(III/IV) oxides are strong oxidants capable of oxidizing organic and inorganic contaminants, including BPA. Here we use δ-MnO2 in stirred flow reactors to determine if higher influent BPA concentrations, or introduction rates, lead to increased polymer production. A major BPA oxidation product, 4-hydroxycumyl alcohol (HCA), is formed through radical coupling, and was therefore used as a metric for polymer production in this study. The influent BPA concentration in stirred flow reactors did not affect HCA yield, suggesting that polymeric production is not strongly dependent on influent concentrations. However, changes in influent BPA concentration affected BPA oxidation rates and the rate of δ-MnO2 reduction. Lower aqueous Mn(II) production was observed in reactors at higher BPA introduction rates, suggesting that single-electron transfer and polymer production are favored under these conditions. However, an examination of Mn(II) sorption during these reactions indicated that the length of the reaction, rather than BPA introduction rate, caused enhanced aqueous Mn(II) production in reactors with low introduction rates and longer reaction times due to increased opportunity for disproportionation and comproportionation. This study demonstrates the importance of investigating both the organic and inorganic reactants in the aqueous and solid phases in this complex reaction.
Environmental significanceManganese oxides are capable of degrading bisphenol A (BPA) and other phenolic compounds via oxidation in both environmental and engineered systems. Although the oxidation mechanism of BPA is complex, the influent concentration of BPA into a manganese oxide reactor does not alter the oxidation mechanism or products. The concurrent reduction of manganese oxide is strongly driven by the introduction rate of BPA, due to the longer reaction time at low introduction rates. BPA oxidation products include polymers, which can couple with organic matter and other compounds found in the environment forming unknown high molecular weight products. The results of this study show that product distribution observed in a controlled setting will reflect that of the environment, despite the 20–160 μM range of influent BPA concentrations used in this study. |
Introduction
Manganese oxides (MnOx) are one of the strongest naturally-occurring oxidants and can oxidize a wide range of organic contaminants, including phenols.1–4 Previous studies have demonstrated oxidation of phenols by manganese oxides using model organic compounds2,3,5–10 and complex pollutants.11–19 Bisphenol A (BPA) is an industrial plasticizer20 that is commonly found in wastewater,21 landfill leachate,22 and surface water.23,24 In the environment, BPA leads to teratogenic, endocrine, and pleiotropic effects in fish and other aquatic species.25BPA is susceptible to oxidation by manganese oxides.12,18,26,27 Similar to other phenols, BPA undergoes a one-electron transfer with manganese oxides to form a radical species that can form polymeric products through radical coupling or undergo further oxidation through a second one-electron transfer.1–3,10,11,28,29 BPA oxidation is affected by MnO2 concentration, pH, and metal cosolutes, and 11 transformation products have been identified, including 10 phenols and 4 polymers.12 4-Hydroxycumyl alcohol (HCA) is a major product of BPA oxidation and is generated at yields of up to 64% HCA per mole of BPA.30 Note that this calculation is based on direct measurement of HCA and does not consider oxidation of HCA by manganese oxide.30 Since HCA is formed through radical coupling, its production can potentially be used to probe the relative amount of polymeric coupling.
Oxidation rates of organic compounds by manganese oxide are highly dependent on mineral properties. The reaction follows pseudo-first-order kinetics during the initial phase, but the rate of oxidation decreases as the reaction proceeds.11,12,14,31,32 Previous investigations of manganese oxide transformation during organic compound oxidation are limited, but they provide unique insights into changes to the mineral surface. For example, decreased rates of phenol, aniline, and triclosan oxidation by MnOx are associated with decreasing oxidation state and accumulation of reduced manganese species and organic species on the mineral surface.19 Similarly, δ-MnO2 can transform to other phases after accumulating Mn(III) in the presence of high concentrations of fulvic acid.33 Our previous study using Mn(III)-rich MnO2 and BPA shows that changes at the mineral surface are enough to decrease the oxidation rate even without changes in the bulk mineral oxidation state.18
Previous studies typically use batch reactors to characterize the reactivity of manganese oxides.1 These closed systems are experimentally simple and results can be readily compared to previous data. However, stirred flow reactors can provide further benefits, allowing for slow and constant addition of an influent media that can be easily altered. Additionally, batch reactors retain both organic and inorganic reaction products, which can affect the reaction. For example, the addition of Mn(II) can considerably decrease phenol oxidation rate by manganese oxides.12,13,26,27,34 In the environment and in stirred flow reactors, these products are constantly removed. A few studies use stirred flow reactors32,35 or column reactors36,37 to examine manganese oxide reactivity, but none have investigated both the changes in the aqueous and solid phases.
Here calculated HCA yield, aqueous Mn(II) production, and solid phase characterization are used to detect differences in BPA oxidation mechanism by δ-MnO2 in stirred flow reactors as a function of influent BPA concentration. This method is used to test the hypothesis that higher influent BPA concentrations lead to greater polymer production and therefore less overall electron transfer. Radicals formed via single-electron transfer are of concern as they can couple with dissolved organic matter or other compounds in the environment, forming unknown high molecular weight products.38–40 HCA is used as an indicator of conditions that favor single-electron transfer (i.e., polymer production), rather than two sequential electron transfers (i.e., benzoquinone production). Aqueous Mn(II) is produced by reductive dissolution and is used along with solid-phase Mn speciation to quantitatively compare electron transfer across different solution conditions. We use this data, along with measurements from solid phase characterization using X-ray absorption near edge structure (XANES) spectroscopy and X-ray diffraction (XRD), to make inferences about the mechanism of the redox reaction and how the δ-MnO2 structure changes throughout the reaction. By performing these reactions in a stirred flow reactor, this study provides novel insights into the effects of contaminant loading over long time periods, which is more representative of contaminants in a flow-through treatment system.36,41
Materials and methods
Materials
Commercially available chemicals were used as received (ESI Section S1†). HCA was synthesized as described previously.18 Ultrapure water was supplied by a Milli-Q water purification system maintained at 18.2 MΩ cm. BPA and HCA stock solutions were prepared in methanol and stored at 4 °C. Information on preparation and characterization of δ-MnO2 is provided in Section S1.† This synthesis yielded a mineral with an average valence state of 3.94 ± 0.11 v.u. determined by oxalate titration,42 indicating that the mineral is predominantly Mn(IV).HCA characterization
The acid dissociation constant (pKa) of HCA was determined spectrophotometrically at 240 nm by titration of 0.1 HCl into a solution of 100 μM HCA following a previously published method.43Solution conditions
All stirred flow reactions and batch reactors used to determine initial rates of BPA and HCA oxidation were performed at pH 5 in 10 mM sodium acetate to avoid experimental artifacts. Acetate did not affect δ-MnO2 reactivity with BPA over time at pH 5 (Fig. S1†), whereas many common circumneutral pH buffers can form complexes with Mn(II) or reduce manganese oxides.44–47 For example, preliminary experiments with 10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) at pH 7 demonstrated that the buffer decreases MnO2 reactivity (Fig. S1†). Klausen et al. reported that PIPES sorbs to the manganese oxide surface, decreasing the number of reactive sites on the surface.32Batch reactors
Batch reactors were used to determine initial rates of BPA and HCA oxidation by δ-MnO2. Prepared δ-MnO2 slurry (stock concentrations: 30–50 g L−1) was equilibrated in a pH 5 acetate buffered solution for 30 minutes before the addition of BPA or HCA (initial concentration: 40 or 80 μM). BPA and HCA concentrations were determined by high-performance liquid chromatography (HPLC; Agilent 1260) in samples that were quenched in excess ascorbic acid (10.8 mM) in order to completely dissolve the δ-MnO2. Aqueous manganese was quantified by inductively coupled plasma-optical emission spectroscopy (ICP-OES; PerkinElmer Optima 4300 DV) analysis of filtered samples (0.2 μm polytetrafluoroethylene) diluted in 2% nitric acid. Batch reactor experiments were conducted in triplicate.Stirred flow reactors
Each 12.7 mL stirred flow reactor contained 1.58 g L−1 δ-MnO2 slurry and a stir bar (Fig. S2†). A filter (0.1 μm VCWP, Millipore) and filter holder prevented δ-MnO2 from leaving the reactor. The solution was continuously stirred, with BPA in acetate buffer constantly being introduced at a rate of 1 mL min−1 (hydraulic retention time = 12.7 minutes). Each reactor was equilibrated with 10 mM acetate for 30 minutes before introducing BPA. The tubing used for the reactors was Pt-cured Si (2.06 mm I.D. Cole Parmer) and exhibited <7% sorption of BPA and phenol in control experiments (Fig. S3†).Stirred flow reactor effluent was analyzed by HPLC and ICP-OES to quantify BPA, HCA, and aqueous manganese. These samples were not filtered or quenched using excess ascorbic acid since the reaction stops upon exiting the δ-MnO2 reactor. Media with varying BPA concentrations (20–160 μM, corresponding to a BPA introduction rate of 20–160 nmol min−1) were used in stirred flow experiments to determine mechanistic differences due to influent BPA concentrations. The length of the experiment was adjusted so that 20–25 μmol BPA was added to each reactor in total.
Solids were recovered at the end of the reaction. This material was washed in methanol to remove organics, dried at room temperature, and ground before analysis. Average manganese oxidation number (AMON) was determined using XANES spectra collected at beamline 10-BM at the Advanced Photon Source at Argonne National Laboratory (Section S4†). Samples were prepared by diluting 3 mg of manganese oxide into 8 mg of polyvinylpyrrolidone, grinding until homogenous, and pressing into a 7 mm pellet. XANES data was analyzed for AMON using the Combo method.48 XRD patterns were collected (Rigaku Rapid II, Mo Kα source; λ = 0.7093 Å) to determine changes in the order and crystallinity of the mineral.
HCA yield calculation
HCA is a phenolic product of BPA oxidation by manganese oxide that is also susceptible to oxidation by the mineral.18,30 In batch reactors, HCA yield was calculated as described previously:18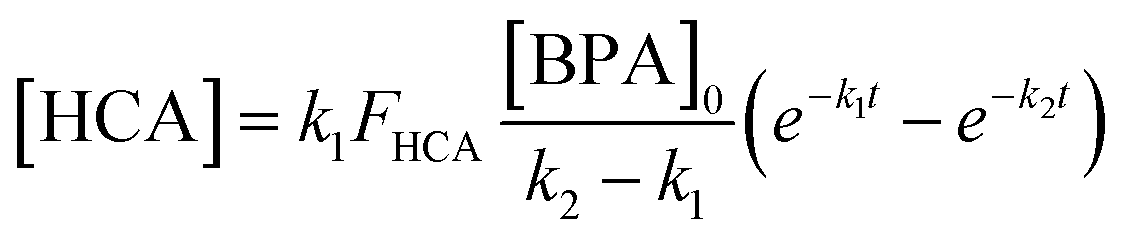 | (1) |
In stirred flow reactors, a steady-state assumption was used to estimate the HCA yield once the reactions reach a plateau in BPA and HCA concentrations using the following equation:
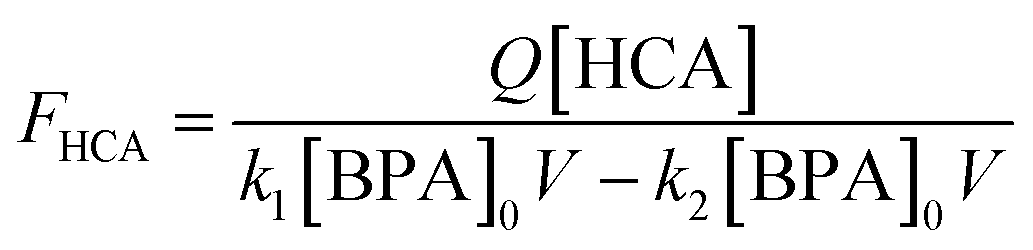 | (2) |
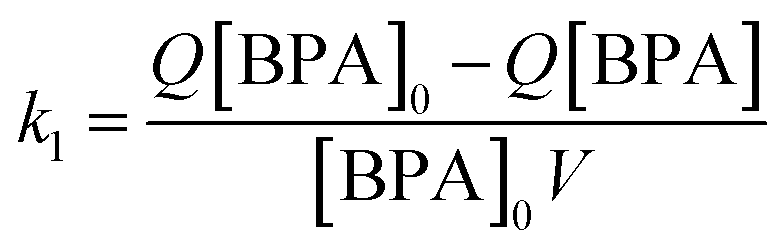 | (3) |
Results and discussion
HCA production and characterization
Upon exposure to δ-MnO2, BPA concentration quickly decreases via oxidation, producing HCA as a major oxidation product (Fig. 1). The production of HCA during BPA oxidation is consistent with its identification and characterization in two previous studies.18,30 BPA oxidation by MnOx produces a multitude of products,12 but at up to 64% yield,30 HCA is the only product detectable by HPLC analysis (Fig. S4†). HCA only accounts for 60% of the carbon atoms present in BPA and it is likely that the C6 moiety formed during HCA production is susceptible to rapid degradation.30 The pKa of HCA is determined to be 10.24 ± 0.05 in this study (Fig. S5†), whereas the pKa values for BPA are 9.6 and 10.2.49 This is an important measurement for HCA since pKa values strongly affect sorption capacity of phenols.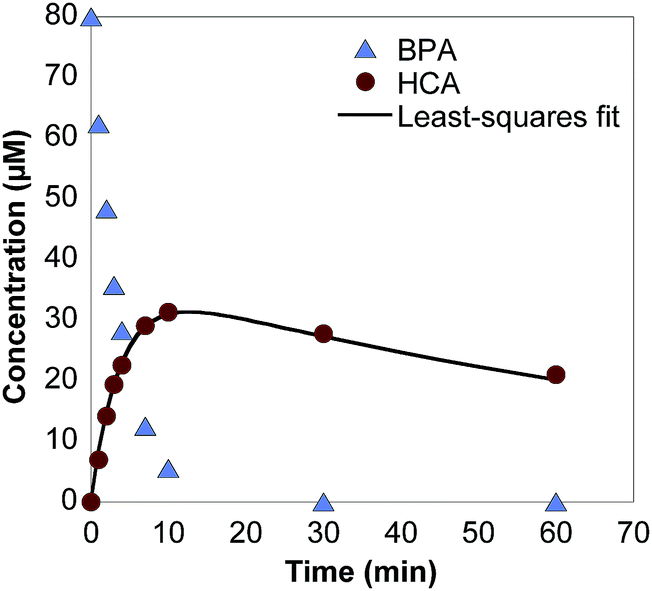 | ||
| Fig. 1 Measured BPA concentrations and measured and theoretical HCA concentrations over time in a batch reactor containing 80 μM BPA and 0.33 g L−1 δ-MnO2 in a pH 5 acetate buffer. | ||
HCA is also susceptible to oxidation by δ-MnO2, albeit at a slower rate than BPA, which is consistent with previous studies.18,30 For example, the pseudo-first-order oxidation rate constants of BPA and HCA at pH 5 are 0.228 min−1 and 0.029 min−1, respectively (Fig. S6; Table S1†). As shown in Fig. 1, HCA concentration increases with time in BPA oxidation reactions, but then reaches a maximum and slowly decreases. Using relative BPA and HCA initial oxidation rate constants determined in separate batch reactors, theoretical HCA yields can be calculated for BPA oxidation reactions in both batch reactors and stirred flow reactors using eqn (1) and (2). For example, the HCA yield in a batch reactor with 0.33 g L−1 δ-MnO2 and 80 μM BPA at pH 5 is 44% (least-squares fit line in Fig. 1).
Characteristics of stirred flow reactors
In these reactions, the BPA solution is introduced into the reactor while the effluent is collected and analyzed. An example data set is presented in Fig. 2. Initially, BPA is completely oxidized by δ-MnO2 and is not detected in the reactor effluent. Similarly, the HCA concentration in the effluent is initially below detection because all HCA produced through BPA oxidation is also completely oxidized by δ-MnO2. As the reaction rate decreases due to changes in the δ-MnO2 during oxidation of BPA and its phenolic transformation products, BPA and HCA concentrations increase in the reactor effluent. After about 40 hours, or 200 μmol BPA introduced, δ-MnO2 is no longer capable of oxidizing BPA during the 12.7 minute hydraulic retention time. As the manganese oxide becomes less reactive, HCA concentrations increase and eventually reach a maximum before returning to zero as BPA ceases reacting with δ-MnO2 and therefore no longer produces HCA. | ||
| Fig. 2 Concentrations of BPA, HCA, and aqueous Mn in the effluent of a stirred flow reactor containing 1.58 g L−1 δ-MnO2, 10 mM acetate buffer (pH 5), and 80 μM BPA over an extended reaction time of 140 hours. | ||
Aqueous Mn(II) is a product of Mn(III/IV) reduction by phenols and is commonly used to quantify reductive dissolution.12–14,19,26,31,32,34 Here, Mn(II) appears in the reactor effluent after the first hour (Fig. 2), showing that reductive dissolution of the mineral is occurring.2,3,50 After six hours, dissolved Mn(II) reaches a maximum and returns to below detection limit as δ-MnO2 stops reacting with BPA and its transformation products. The BPA concentration experiments described below focus on the early stages of this reaction, where 70–100% of BPA is oxidized by δ-MnO2 during the retention time, similar to previous studies investigating As(III)35,51 and aniline32 oxidation by MnO2.
Effect of BPA influent concentration
Four stirred flow reactors with varying concentrations of influent BPA are used to determine the effect of BPA introduction rate on reaction kinetics, aqueous products, and mineral transformation. Influent BPA concentrations range from 20–160 μM, which correspond to BPA introduction rates of 20–160 nmol min−1. All reactors have the same flow rate and retention time but vary in length of reaction time so that each reactor is exposed to the same amount of BPA (20–25 μmol). Due to this difference in time (150–1200 min), data is normalized by plotting as a function of BPA introduced to the reactor (Fig. 3). Data is presented as a function of time in Fig. S7–S8.† | ||
| Fig. 3 (a) BPA and (b) HCA present in the effluent, as well as (c) the ratio of HCA produced to BPA consumed, and (d) aqueous Mn(II) produced in stirred flow reactors containing 1.58 g L−1 δ-MnO2 in 10 mM acetate buffer (pH 5). Reaction times range from 2.5–20 hours. | ||
The BPA oxidation rate decreases with exposure of δ-MnO2 to BPA throughout the shorter time-scale of these experiments, as was observed in the 140 hour experiment (Fig. 2). The BPA concentration in the effluent reaches an apparent plateau in which 5–30% of the BPA is oxidized, depending on the concentration of the influent solution (Fig. 3a). At the lowest BPA introduction rate (20 nmol min−1), BPA appears in the effluent after 11 μmol of BPA are introduced and reaches a plateau almost immediately (i.e., after 13 μmol of BPA are introduced). This trend is also followed for the 40 and 80 nmol min−1 reactors. The plateau that is reached is modeled using a steady-state approximation to calculate BPA oxidation rate constants and HCA yields. At the highest BPA introduction rate (160 nmol min−1), BPA also appears in the effluent after 11 μmol of BPA are introduced but no plateau is observed during the reaction period (25 μmol of BPA introduced).
HCA concentration in the effluent varies with influent BPA concentration, with higher concentrations of BPA leading to higher concentrations of HCA in the effluent and vice versa (Fig. 3b). HCA is found in the effluent earlier in the reaction with lower BPA introduction rates (e.g., after 8 μmol of BPA introduced for 20 nmol min−1 reactor and after 13 μmol of BPA for 160 nmol min−1 reactor). Furthermore, a plateau of HCA concentration is reached sooner in reactors with lower BPA introduction rate (i.e., after 11 μmol of BPA for 20 nmol min−1 reactor and after 25 μmol of BPA for 160 nmol min−1 reactor). However, when plotted as a fraction of BPA consumed in the reactor (Fig. 3c), all reactors produce the same ratio of moles of HCA in effluent per moles of BPA consumed (30.0–33.1%) by the end of the reaction, excluding the 160 nmol min−1 reactor, which does not fully reach a plateau.
The steady-state approximation (Section S5†) and relative initial rates of BPA and HCA oxidation in batch reactors (Table S1; Fig. S6†) are used to calculate BPA oxidation rate constants and HCA yields in stirred flow reactors when the reaction reaches a plateau (e.g., after 12 μmol of BPA introduced for the 20 nmol min−1 reactor). Although the system is not truly at steady-state because δ-MnO2 reactivity changes gradually over extended reaction times (Fig. 2), the steady-state approximation is valid because the BPA and HCA concentrations are not changing from one time point to the next within this shorter timeframe. However, it is not possible to calculate HCA yield with the 160 nmol min−1 introduction rate because this reactor does not reach a distinct plateau by the end of the reaction time. Calculated BPA oxidation rate constants steadily increase with BPA introduction rate in reactors with 20, 40, and 60 nmol min−1 introduction rates (Fig. 4). This observation is in agreement with previous batch reactor studies that show an increase in oxidation rate with increases in either phenol concentration or MnO2 concentration.5,12
 | ||
| Fig. 4 Calculated BPA oxidation rate constants (kBPA) and HCA yields (see eqn (1–3) in stirred flow reactors containing 1.58 g L−1 δ-MnO2 exposed to 20 μmol of BPA in 10 mM acetate buffer (pH 5) at an introduction rate of 20–160 μmol min−1 BPA. Reaction times range from 2.5–20 hours. | ||
HCA yields provide insight into changes in BPA reaction mechanism as a function of BPA concentration. Reactors with 20, 40, and 80 nmol min−1 BPA introduction rates have nearly identical HCA yields between 38–40% at the reaction plateau (Fig. 4). As a major oxidation product formed through radical coupling,30 the fraction of HCA production is theoretically proportional to the fraction of one-electron transfer reactions in this system. Since these polymeric products are more likely to form when there are high concentrations of BPA, we hypothesized that HCA yield would be higher when δ-MnO2 is exposed at a higher BPA introduction rate. However, the data indicates that there is no difference in HCA yield within this BPA concentration range.
Additional experiments were conducted at different BPA influent concentrations to further test whether HCA yields change under different conditions. First, influent BPA concentrations below 20 μM were preliminarily examined but were inconclusive. For example, a trial using 5 nmol min−1 BPA, which corresponds to an initial concentration of 5 μM BPA for 83 hours, shows that BPA is entirely consumed in the reaction (Fig. S9†). Therefore, HCA yields could not be determined at lower BPA influent concentrations due to complete BPA oxidation. Second, a separate experiment using longer reaction times compares HCA yields of 20 nmol min−1 and 160 nmol min−1 introduction rates after they have both reached a plateau (Fig. S10†). The observed yields of 44% and 40% respectively indicate that the higher introduction rate does not yield more HCA than lower introduction rate, further disproving our hypothesis that more polymeric products are produced at higher influent BPA concentrations.
Although the organic data does not indicate a shift in BPA oxidation mechanism, the inorganic data shows a strong trend among the reactors. Aqueous manganese concentrations in stirred flow reactor effluent show that there is a relationship between influent BPA concentration and Mn(II) production (Fig. 3d). At lower BPA introduction rates, Mn(II) is produced earlier in the reaction and in larger quantities than at higher BPA introduction rates. For example, the 20 nmol min−1 introduction rate results in 19.8 μmol total Mn(II) beginning after 11.4 μmol BPA is introduced, while the 160 nmol min−1 introduction rate results in 6.3 μmol total beginning after 18.7 μmol BPA is introduced. Despite the difference in Mn(II) produced, minimal bulk mineralogical changes are observed in the XRD patterns or XANES data. Fig. 5a shows that commonly observed changes, such as reduced tailing of the hkl diffraction band at 37° and the appearance of a dip at ∼47°, only noticeably appear in the 20 nmol min−1 reactor. Analysis of XANES data using the Combo method fits the raw data and provides a calculated AMON for the sample. The AMON of the starting material is 3.85 v.u. (90% Mn(IV), 5% Mn(III), 5% Mn(II)). In samples recovered from stirred flow reactors, the AMON decreased due to reduction, but was the same value of 3.67 ± 0.01 v.u. for all BPA introduction rates (Fig. 5b; Table S2†). Unsurprisingly, no mineral phase changes occurred due to the low pH and relatively low accumulation of reduced Mn. This is consistent with previous work that shows changes in Mn(III)-rich δ-MnO2 reactivity can occur due to mineral transformation at the surface, such as increased interlayer Mn(II/III), and not necessarily changes to the bulk structure.18
 | ||
| Fig. 5 (a) XRD patterns of solids from each reactor in red overlaid by the starting material in black and (b) fitted XANES data of solids recovered from each reactor and the starting material. XANES data was analyzed using the Combo method.48 | ||
The differences in Mn(II) production, despite lack of changes in the bulk average manganese oxidation number, indicate that there are more overall electron transfer reactions occurring at lower BPA introduction rates, resulting in increased reductive dissolution of δ-MnO2. This is further shown by the estimated net electron transfer from organic compounds to δ-MnO2 calculated for each reactor (Table S3†), where electron transfer in 20, 40, 80, and 160 nmol min−1 reactors are estimated to be 79.7, 72.0, 64.9, and 50.7 μmol, respectively. There are several potential explanations for this observation. First, it is possible that two sequential single-electron transfers are favored in reactors with lower BPA introduction rates, forming more Mn(II) through a second single-electron transfer, rather than just one single-electron transfer reaction to produce Mn(III). However, if HCA is used to determine the relative amount of polymeric products formed, the absence of a trend in HCA yield over the 20–80 nmol min−1 reactors indicates that the proportion of single-electron transfer reactions, and therefore polymer production, is consistent over the range of BPA introduction rates. A second explanation is that the differences in reaction lengths allow for more redox reaction to take place, including oxidation of BPA oxidation products (e.g., HCA) and transfer of electrons within the manganese oxide mineral (i.e., disproportionation and comproportionation). This is supported by both data sets, but does not follow the generally accepted concept that more radical coupling and polymeric production will occur at higher concentrations of the target organic compound.52 A third possibility is that HCA does not accurately quantify polymeric production since other polymers can be formed by BPA oxidation.12
One way to narrow down these possibilities is to determine how accurately Mn(II) production quantifies the total number of electrons transferred to δ-MnO2. When Mn(II), Mn(III), and Mn(IV) are present in the same system, the mineral is susceptible to disproportionation and comproportionation (eqn (4)).53–56 A Mn(II) center and Mn(IV) center can exchange electrons, or comproportionate, to form two Mn(III) centers. Conversely, two Mn(III) centers can disproportionate to form Mn(II) and Mn(IV) centers. Due to these reactions, the fraction of reduced Mn(II) species formed in a reaction is not necessarily the same as the reduced species measured in the aqueous phase.
| Mn(II) + Mn(IV) ⇌ 2Mn(III) | (4) |
Desorption experiments
To determine if disproportionation and comproportionation reactions are occurring, Ca2+ is used as a desorption agent to quantify solid-bound Mn(II) concentrations at various points in the reaction. These experiments use stirred flow reactors to introduce buffered BPA, buffer only, and Ca2+ solutions into a δ-MnO2 slurry. Disproportionation and comproportionation reactions can readily occur in mixed-valent manganese oxides,53–57 affecting the distribution of Mn(II), Mn(III), and Mn(IV) over time. These reactions make it difficult to characterize small changes in the mineral structure because they can be attributed to either the reaction of interest (i.e., reaction with BPA) or disproportionation and/or comproportionation. Here, Ca2+ is added to desorb Mn(II) from the δ-MnO2 solid after the oxide reacts with BPA. If aqueous Mn(II) is continually produced after all solid-associated Mn(II) and BPA has been removed, this would indicate that Mn(III) is undergoing disproportionation to form Mn(II) since there is no other reductant present. Preliminary experiments show that BPA and HCA undergo minimal sorption to δ-MnO2 (Section S7; Table S1†), meaning negligible amounts of Mn(II) should be produced by continued organic oxidation reactions in this system.The desorption experiments indicate that Mn(III) disproportionation occurs under our experimental conditions. In Reactor A, exposure of δ-MnO2 to 20 μM BPA (buffered to pH 5 in 10 mM acetate) for 20 hours produces a total of 6.75 μmol aqueous Mn(II). The addition of 25 mM Ca2+ for three hours desorbs an additional 1.54 μmol of Mn(II) that was generated during BPA oxidation (Fig. 6a). We then added 10 mM pH 5 acetate with no BPA for 7 hours, which does not react with δ-MnO2 (Fig. S1†), but allows time for disproportionation of Mn(III) to occur; minimal Mn(II) is produced during this time (i.e., a total of 0.06 μmol). A second introduction of 25 mM Ca2+ for two hours yielded 0.37 μmol additional Mn(II), indicating that Mn(II) production via disproportionation occurs during the time between Ca2+ introductions.
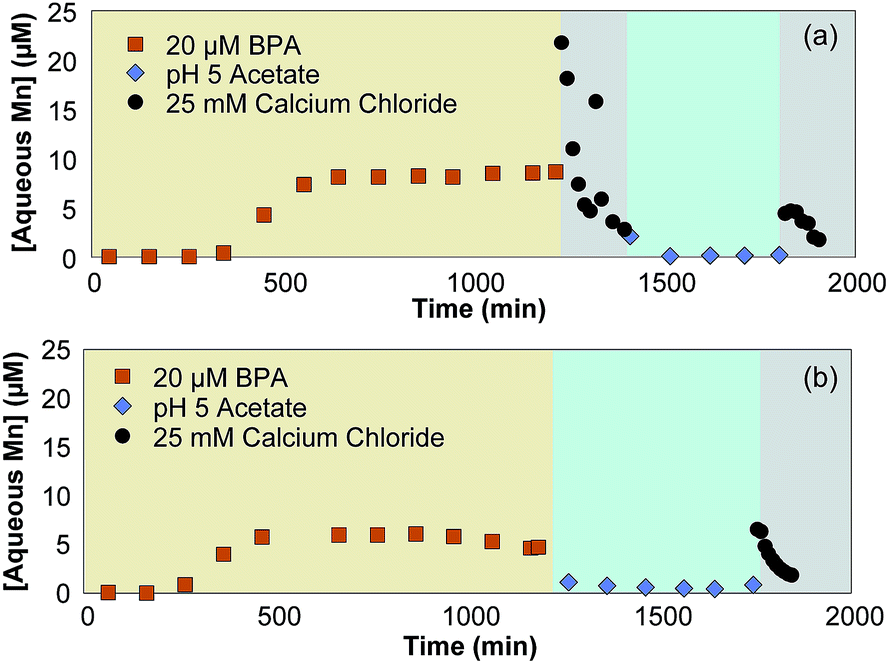 | ||
| Fig. 6 Stirred flow reactors with 1.58 g L−1 δ-MnO2 exposed to (a) 20 μM BPA from 0–1215 min, 25 mM Ca2+ from 1215–1395 min, 10 mM acetate at pH 5 from 1395–1800 min, and 25 mM Ca2+ from 1800–1905 min (Reactor A), and (b) 20 μM BPA from 0–1185 min, 10 mM acetate at pH 5 from 1185–1745 min, and 25 mM Ca2+ from 1745–1845 min (Reactor B). | ||
In Reactor B, δ-MnO2 is exposed to a 20 μM BPA solution (buffered to pH 5 in 10 mM acetate) for 20 hours, 10 mM acetate buffer for 9 hours, and a final 25 mM Ca2+ solution for two hours. Mn(II) is produced during BPA oxidation as described above, but does not desorb during exposure to 10 mM acetate (Fig. 6b). When the Ca2+ solution is introduced, Mn(II) desorbs from the mineral and is found in the effluent. Reactor A produces 1.54 μmol Mn(II), while Reactor B produces a total of 0.3 μmol Mn(II). Since both reactors were exposed to the same amount of reductant (i.e., 24 μmol BPA), this indicates that the Mn(II) produced during BPA oxidation is able to undergo comproportionation with Mn(IV) to form Mn(III) if it is not desorbed from the mineral in a timely fashion.
Three conclusions can be drawn from the desorption experiments. First, Mn(II) does not continue to desorb from the mineral after oxidation with BPA, indicating that desorption of Mn(II) occurs after the mineral reaches saturation either by Mn(II) or organics and is driven by exposure to organics and further production of Mn(II). Second, disproportionation, which is highly dependent on pH, occurs in the system within the timescale of the reaction (i.e., on the order of 7 hours at pH 5). Finally, the amount of Mn(II) desorbed by the first exposure to 25 mM Ca2+ in each reactor (i.e., 1.54 μmol in Reactor A with immediate exposure to Ca2+, and 0.3 μmol in Reactor B with delayed exposure) indicates that comproportionation is occurring in Reactor B. Overall, these desorption experiments show that both disproportionation and comproportionation are possible within the reaction times of our stirred flow experiments. This indicates that the differences in aqueous Mn(II) production (Fig. 3d) are due to the differences in reaction time rather than the BPA introduction rate, as increasing reaction time will lead to a proportional amount of electron transfer between manganese centers.
Conclusions
This study demonstrates the importance of including both organic and inorganic analysis when examining oxidation of organic contaminants by an inorganic substrate. Although there are many studies on the degradation of BPA and other phenols by manganese oxides, most look only at the disappearance of the organic11,12,14,31,32 or the complete reductive dissolution of the mineral.2,11,50,58 Here, we see differences in aqueous Mn(II) production proportional to the introduction rate of BPA into stirred flow reactors. On its own, this information suggests that the introduction rate of BPA determines the redox mechanism, with higher introduction rates leading to more polymeric production via a single-electron transfer, as predicted. However, in tandem with the information from the HCA yield calculations that show no difference in HCA yield with varying introduction rates and the desorption experiments that show that both disproportionation and comproportionation occur in these reactions, we conclude that BPA introduction rate has minimal effect on BPA oxidation mechanism. The difference in aqueous Mn(II) production is likely due to the longer reaction times, which allow for more disproportionation of Mn(III) to occur in reactors with lower BPA introduction rates. This also explains why a plateau is not reached for BPA and HCA concentrations in the shorter 160 nmol min−1 reactor since not as much Mn(II) has formed via disproportionation, which would hinder the reaction. These findings indicate that Mn(III) is prevalent in reacted δ-MnO2, but undergoes disproportionation or further redox reactions over time in extended reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Peter J. Alaimo at Seattle University for synthesis of hydroxycumyl alcohol. Funding for this study was provided by NSF (CBET 1509879) and is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1256259. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation. MRCAT operations are supported by the Department of Energy and the MRCAT member institutions. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.References
- C. K. Remucal and M. Ginder-Vogel, A Critical Review of the Reactivity of Manganese Oxides with Organic Contaminants, Environ. Sci.: Processes Impacts, 2014, 16(6), 1247–1266 RSC.
- A. T. Stone, Reductive Dissolution of Manganese(III/IVIV) Oxides by Substituted Phenols, Environ. Sci. Technol., 1987, 21(10), 979–988 CrossRef CAS PubMed.
- A. T. Stone and J. J. Morgan, Reduction and Dissolution of Manganese(III) and Manganese(IV) Oxides by Organics: 2. Survey of the Reactivity of Organics, Environ. Sci. Technol., 1984, 18(8), 617–624 CrossRef CAS PubMed.
- A. G. Xyla, B. Sulzberger, G. W. Luther III, J. G. Hering, P. Van Cappellen and W. Stumm, Reductive Dissolution of Manganese(III,IV) (Hydr)Oxides by Oxalate: The Effect of pH and Light, Langmuir, 1992, 8(1), 95–103 CrossRef CAS.
- H. J. Ulrich and A. T. Stone, The Oxidation of Chlorophenols Adsorbed to Manganese Oxide Surfaces, Environ. Sci. Technol., 1989, 23(4), 421–428 CrossRef CAS.
- P. S. Nico and R. J. Zasoski, Mn(III) Center Availability as a Rate Controlling Factor in the Oxidation of Phenol and Sulfide on δ-MnO2, Environ. Sci. Technol., 2001, 35(16), 3338–3343 CrossRef CAS PubMed.
- L. Ukrainczyk and M. B. McBride, Oxidation and Dechlorination of Chlorophenols in Dilute Aqueous Suspensions of Manganese Oxides: Reaction Products, Environ. Toxicol. Chem., 1993, 12(11), 2015–2022 CrossRef CAS.
- D. J. Bertino and R. G. Zepp, Effects of Solar Radiation on Manganese Oxide Reactions with Selected Organic Compounds, Environ. Sci. Technol., 1991, 25(7), 1267–1273 CrossRef CAS.
- L. Zhao, Z. Yu, P. Peng, W. Huang and Y. Dong, Oxidative Transformation of Tetrachlorophenols and Trichlorophenols by Manganese Dioxide, Environ. Toxicol. Chem., 2009, 28(6), 1120–1129 CrossRef CAS PubMed.
- L. Ukrainczyk and M. B. McBride, Oxidation of Phenol in Acidic Aqueous Suspensions of Manganese Oxides, Clays Clay Miner., 1992, 40(2), 157–166 CrossRef CAS.
- H. Zhang and C.-H. Huang, Oxidative Transformation of Triclosan and Chlorophene by Manganese Oxides, Environ. Sci. Technol., 2003, 37(11), 2421–2430 CrossRef CAS PubMed.
- K. Lin, W. Liu and J. Gan, Oxidative Removal of Bisphenol A by Manganese Dioxide: Efficacy, Products, and Pathways, Environ. Sci. Technol., 2009, 43(10), 3860–3864 CrossRef CAS PubMed.
- Z. Lu, K. Lin and J. Gan, Oxidation of Bisphenol F (BPF) by Manganese Dioxide, Environ. Pollut., 2011, 159(10), 2546–2551 CrossRef CAS PubMed.
- L. Xu, C. Xu, M. Zhao, Y. Qiu and G. Sheng, Oxidative Removal of Aqueous Steroid Estrogens by Manganese Oxides, Water Res., 2008, 42(20), 5038–5044 CrossRef CAS PubMed.
- H. Xiao, H. Song, H. Xie, W. Huang, J. Tan and J. Wu, Transformation of Acetaminophen Using Manganese Dioxide − Mediated Oxidative Processes: Reaction Rates and Pathways, J. Hazard. Mater., 2013, 250–251, 138–146 CrossRef CAS PubMed.
- K. Lin, W. Liu and J. Gan, Reaction of Tetrabromobisphenol A (TBBPA) with Manganese Dioxide: Kinetics, Products, and Pathways, Environ. Sci. Technol., 2009, 43(12), 4480–4486 CrossRef CAS PubMed.
- Z. Lu and J. Gan, Oxidation of Nonylphenol and Octylphenol by Manganese Dioxide: Kinetics and Pathways, Environ. Pollut., 2013, 180, 214–220 CrossRef CAS PubMed.
- S. Balgooyen, P. J. Alaimo, C. K. Remucal and M. Ginder-Vogel, Structural Transformation of MnO2 during the Oxidation of Bisphenol A, Environ. Sci. Technol., 2017, 51(11), 6053–6062 CrossRef CAS PubMed.
- N. Shaikh, S. Taujale, H. Zhang, K. Artyushkova, A.-M. S. Ali and J. M. Cerrato, Spectroscopic Investigation of Interfacial Interaction of Manganese Oxide with Triclosan, Aniline, and Phenol, Environ. Sci. Technol., 2016, 50(20), 10978–10987 CrossRef CAS PubMed.
- Bisphenol A (BPA) Information & Resources, http://www.bisphenol-a.org/, accessed Sep 6, 2016.
- X. Yu, J. Xue, H. Yao, Q. Wu, A. K. Venkatesan, R. U. Halden and K. Kannan, Occurrence and Estrogenic Potency of Eight Bisphenol Analogs in Sewage Sludge from the U.S. EPA Targeted National Sewage Sludge Survey, J. Hazard. Mater., 2015, 299, 733–739 CrossRef CAS PubMed.
- J. R. Masoner, D. W. Kolpin, E. T. Furlong, I. M. Cozzarelli, J. L. Gray and E. A. Schwab, Contaminants of Emerging Concern in Fresh Leachate from Landfills in the Conterminous United States, Environ. Sci.: Processes Impacts, 2014, 16(10), 2335–2354 RSC.
- A. K. Baldwin, S. R. Corsi, L. A. De Cicco, P. L. Lenaker, M. A. Lutz, D. J. Sullivan and K. D. Richards, Organic Contaminants in Great Lakes Tributaries: Prevalence and Potential Aquatic Toxicity, Sci. Total Environ., 2016, 554–555, 42–52 CrossRef CAS PubMed.
- A. Belfroid, M. van Velzen, B. van der Horst and D. Vethaak, Occurrence of Bisphenol A in Surface Water and Uptake in Fish: Evaluation of Field Measurements, Chemosphere, 2002, 49(1), 97–103 CrossRef CAS PubMed.
- L. Canesi and E. Fabbri, Environmental Effects of BPA: Focus on Aquatic Species, Dose-Response, 2015, 13(3), 1–14 CrossRef CAS PubMed.
- N. Gao, J. Hong, Z. Yu, P. Peng and W. Huang, Transformation of Bisphenol A in the Presence of Manganese Dioxide, Soil Sci., 2011, 176(6), 265–272 CrossRef CAS.
- K. Lin, Y. Peng, X. Huang and J. Ding, Transformation of Bisphenol A by Manganese Oxide-Coated Sand, Environ. Sci. Pollut. Res., 2013, 20(3), 1461–1467 CrossRef CAS PubMed.
- S. Laha and R. G. Luthy, Oxidation of Aniline and Other Primary Aromatic Amines by Manganese Dioxide, Environ. Sci. Technol., 1990, 24(3), 363–373 CrossRef CAS.
- J. Jiang, Y. Gao, S.-Y. Pang, X.-T. Lu, Y. Zhou, J. Ma and Q. Wang, Understanding the Role of Manganese Dioxide in the Oxidation of Phenolic Compounds by Aqueous Permanganate, Environ. Sci. Technol., 2015, 49(1), 520–528 CrossRef CAS PubMed.
- J. Im, C. W. Prevatte, S. R. Campagna and F. E. Löffler, Identification of 4-Hydroxycumyl Alcohol as the Major MnO2-Mediated Bisphenol A Transformation Product and Evaluation of Its Environmental Fate, Environ. Sci. Technol., 2015, 49(10), 6214–6221 CrossRef CAS PubMed.
- K. F. Rubert and J. A. Pedersen, Kinetics of Oxytetracycline Reaction with a Hydrous Manganese Oxide, Environ. Sci. Technol., 2006, 40(23), 7216–7221 CrossRef CAS PubMed.
- J. Klausen, S. B. Haderlein and R. P. Schwarzenbach, Oxidation of Substituted Anilines by Aqueous MnO2: Effect of Co-Solutes on Initial and Quasi-Steady-State Kinetics, Environ. Sci. Technol., 1997, 31(9), 2642–2649 CrossRef CAS.
- Q. Wang, P. Yang and M. Zhu, Structural Transformation of Birnessite by Fulvic Acid under Anoxic Conditions, Environ. Sci. Technol., 2018, 52(4), 1844–1853 CrossRef CAS PubMed.
- T. Zhang, X. Zhang, X. Yan, J. Ng, Y. Wang and D. D. Sun, Removal of Bisphenol A via a Hybrid Process Combining Oxidation on β-MnO2 Nanowires with Microfiltration, Colloids Surf., A, 2011, 392(1), 198–204 CrossRef CAS.
- B. J. Lafferty, M. Ginder-Vogel and D. L. Sparks, Arsenite Oxidation by a Poorly Crystalline Manganese-Oxide 1. Stirred-Flow Experiments, Environ. Sci. Technol., 2010, 44(22), 8460–8466 CrossRef CAS PubMed.
- J. A. Charbonnet, Y. Duan, C. M. van Genuchten and D. L. Sedlak, Chemical Regeneration of Manganese Oxide-Coated Sand for Oxidation of Organic Stormwater Contaminants, Environ. Sci. Technol., 2018, 52(18), 10728–10736 CrossRef CAS PubMed.
- H. Guha, J. E. Saiers, S. Brooks, P. Jardine and K. Jayachandran, Chromium Transport, Oxidation, and Adsorption in Manganese-Coated Sand, J. Contam. Hydrol., 2001, 49(3–4), 311–334 CrossRef CAS PubMed.
- F. Tong, X. Gu, C. Gu, J. Xie, X. Xie, B. Jiang, Y. Wang, T. Ertunc, A. Schäffer and R. Ji, Stimulation of Tetrabromobisphenol A Binding to Soil Humic Substances by Birnessite and the Chemical Structure of the Bound Residues, Environ. Sci. Technol., 2016, 50(12), 6257–6266 CrossRef CAS PubMed.
- K.-H. Kang, J. Dec, H. Park and J.-M. Bollag, Effect of Phenolic Mediators and Humic Acid on Cyprodinil Transformation in Presence of Birnessite, Water Res., 2004, 38(11), 2737–2745 CrossRef CAS PubMed.
- C. Li, B. Zhang, T. Ertunc, A. Schaeffer and R. Ji, Birnessite-Induced Binding of Phenolic Monomers to Soil Humic Substances and Nature of the Bound Residues, Environ. Sci. Technol., 2012, 46(16), 8843–8850 CrossRef CAS PubMed.
- J. E. Grebel, J. A. Charbonnet and D. L. Sedlak, Oxidation of Organic Contaminants by Manganese Oxide Geomedia for Passive Urban Stormwater Treatment Systems, Water Res., 2016, 88, 481–491 CrossRef CAS PubMed.
- J. W. Murray, The Surface Chemistry of Hydrous Manganese Dioxide, J. Colloid Interface Sci., 1974, 46(3), 357–371 CrossRef CAS.
- M. B. McConville, T. D. Hubert and C. K. Remucal, Direct Photolysis Rates and Transformation Pathways of the Lampricides TFM and Niclosamide in Simulated Sunlight, Environ. Sci. Technol., 2016, 50(18), 9998–10006 CrossRef CAS PubMed.
- C. M. H. Ferreira, I. S. S. Pinto, E. V. Soares and H. M. V. M. Soares, (Un)Suitability of the Use of PH Buffers in Biological, Biochemical and Environmental Studies and Their Interaction with Metal Ions – a Review, RSC Adv., 2015, 5(39), 30989–31003 RSC.
- Y. Pan, G. F. Koopmans, L. T. C. Bonten, J. Song, Y. Luo, E. J. M. Temminghoff and R. N. J. Comans, Influence of pH on the Redox Chemistry of Metal (Hydr)Oxides and Organic Matter in Paddy Soils, J. Soils Sediments, 2014, 14(10), 1713–1726 CrossRef CAS.
- S. Ouvrard, M.-O. Simonnot and M. Sardin, Reactive Behavior of Natural Manganese Oxides toward the Adsorption of Phosphate and Arsenate, Ind. Eng. Chem. Res., 2002, 41(11), 2785–2791 CrossRef CAS.
- K. A. Kobe and T. Y. Toribara, Precipitation of Manganous Carbonate, Ind. Eng. Chem., 1939, 31(10), 1272–1274 CrossRef CAS.
- A. Manceau, M. A. Marcus and S. Grangeon, Determination of Mn Valence States in Mixed-Valent Manganates by XANES Spectroscopy, Am. Mineral., 2012, 97(5–6), 816–827 CrossRef CAS.
- Y. Lee, J. Yoon and U. von Gunten, Kinetics of the Oxidation of Phenols and Phenolic Endocrine Disruptors during Water Treatment with Ferrate (Fe(VI)), Environ. Sci. Technol., 2005, 39(22), 8978–8984 CrossRef CAS PubMed.
- A. T. Stone and J. J. Morgan, Reduction and Dissolution of Manganese(III) and Manganese(IV) Oxides by Organics. 1. Reaction with Hydroquinone, Environ. Sci. Technol., 1984, 18(6), 450–456 CrossRef CAS PubMed.
- B. J. Lafferty, M. Ginder-Vogel and D. L. Sparks, Arsenite Oxidation by a Poorly-Crystalline Manganese Oxide. 3. Arsenic and Manganese Desorption, Environ. Sci. Technol., 2011, 45(21), 9218–9223 CrossRef CAS PubMed.
- D. Colombani, Chain-Growth Control in Free Radical Polymerization, Prog. Polym. Sci., 1997, 22, 1649–1720 CrossRef CAS.
- E. J. Elzinga and A. B. Kustka, A Mn-54 Radiotracer Study of Mn Isotope Solid–Liquid Exchange during Reductive Transformation of Vernadite (δ-MnO2) by Aqueous Mn(II), Environ. Sci. Technol., 2015, 49(7), 4310–4316 CrossRef CAS PubMed.
- C. M. Santelli, S. M. Webb, A. C. Dohnalkova and C. M. Hansel, Diversity of Mn Oxides Produced by Mn(II)-Oxidizing Fungi, Geochim. Cosmochim. Acta, 2011, 75(10), 2762–2776 CrossRef CAS.
- J. R. Bargar, B. M. Tebo, U. Bergmann, S. M. Webb, P. Glatzel, V. Q. Chiu and M. Villalobos, Biotic and Abiotic Products of Mn(II) Oxidation by Spores of the Marine Bacillus Sp. Strain SG-1, Am. Mineral., 2005, 90(1), 143–154 CrossRef CAS.
- X. H. Feng, M. Zhu, M. Ginder-Vogel, C. Ni, S. J. Parikh and D. L. Sparks, Formation of Nano-Crystalline Todorokite from Biogenic Mn Oxides, Geochim. Cosmochim. Acta, 2010, 74(11), 3232–3245 CrossRef CAS.
- M. Zhu, M. Ginder-Vogel, S. J. Parikh, X.-H. Feng and D. L. Sparks, Cation Effects on the Layer Structure of Biogenic Mn-Oxides, Environ. Sci. Technol., 2010, 44(12), 4465–4471 CrossRef CAS PubMed.
- H. Zhang, W.-R. Chen and C.-H. Huang, Kinetic Modeling of Oxidation of Antibacterial Agents by Manganese Oxide, Environ. Sci. Technol., 2008, 42(15), 5548–5554 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, Tables S1–S3, and Fig. S1–S10. See DOI: 10.1039/c8em00451j |
| This journal is © The Royal Society of Chemistry 2019 |