
Selenoneine and ergothioneine in human blood cells determined simultaneously by HPLC/ICP-QQQ-MS†
Nina
Kroepfl
 a,
Kevin A.
Francesconi
a,
Tanja
Schwerdtle
b and
Doris
Kuehnelt
*a
a,
Kevin A.
Francesconi
a,
Tanja
Schwerdtle
b and
Doris
Kuehnelt
*a
aInstitute of Chemistry, Analytical Chemistry, NAWI Graz, University of Graz, Universitaetsplatz 1, A-8010 Graz, Austria. E-mail: doris.kuehnelt@uni-graz.at; Tel: +43 316 380 5316
bInstitute of Nutritional Science, University of Potsdam, Arthur-Scheunert-Allee 114-116, 14558 Nuthetal, Germany
First published on 28th November 2018
Abstract
The possible relevance to human health of selenoneine and its sulfur-analogue ergothioneine has generated interest in their quantitative determination in biological samples. To gain more insight into the similarities and differences of these two species, a method for their simultaneous quantitative determination in human blood cells using reversed-phase high performance liquid chromatography (RP-HPLC) coupled to inductively coupled plasma triple quadrupole mass spectrometry (ICP-QQQ-MS) is presented. Spectral interferences hampering the determination of sulfur and selenium by ICPMS are overcome by introducing oxygen to the reaction cell. To access selenoneine and ergothioneine in the complex blood matrix, lysis of the cells with cold water followed by cut-off filtration (3000 Da) is performed. Recoveries based on blood cells spiked with selenoneine and ergothioneine were between 80% and 85%. The standard deviation of the method was around 0.10 mg S per L for ergothioneine (corresponding to relative standard deviations (RSD) between 10–1% for ergothioneine concentrations of 1–10 mg S per L) and 0.25 μg Se per L for selenoneine (RSDs of 25–2% for concentrations of 1–10 μg Se per L). The method was applied to blood cell samples from three volunteers which showed selenoneine and ergothioneine concentrations in the range of 3.25 to 7.35 μg Se per L and 0.86 to 6.44 mg S per L, respectively. The method is expected to be of wide use in future studies investigating the dietary uptake of selenoneine and ergothioneine and their relevance in human health.
Introduction
The essentiality of the micronutrient selenium and the macronutrient sulfur was established in the middle of the 20th century.1,2 In spite of the disparity in the required quantities of these two essential elements, strong similarities in respect to their chemical properties and naturally occurring compounds can be observed.3 For instance, the sulfur amino acids cysteine and methionine, as well as their selenium-analogues selenocysteine and selenomethionine are found in proteins.4,5 The more recent discovery of selenoneine (2-selenyl-Nα,Nα,Nα-trimethyl-L-histidine), the selenium-analogue of the sulfur-containing antioxidant ergothioneine (for structures see Fig. 1), further demonstrated the similarities between the two elements' compounds and their properties.6–8 Despite differences in the auto-oxidative behaviour of selenoneine and ergothioneine,6,9 both compounds were discussed to show antioxidant activity and might, therefore, be of relevance to human health.10,11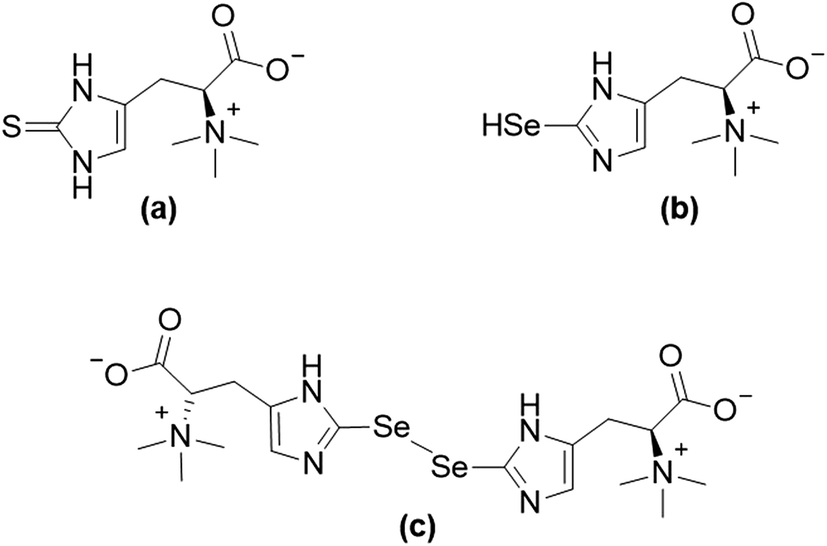 | ||
| Fig. 1 Structures of ergothioneine (a), and selenoneine in its reduced form (b) and its oxidized form (c). | ||
Since selenoneine and ergothioneine are not synthesized by mammals, their primary source for humans is diet. Ergothioneine can be found in mushrooms, beans, wheat, oats and organ meats.12 Its distribution in food is uneven12 and individual differences in the ergothioneine concentrations in body fluids might be due to variations in the diet, long washout periods and genetic differences.11 Foods rich in selenoneine on the other hand are mainly marine fish species like tuna, swordfish and sardine.13
To gain more insight into the similarities of ergothioneine and selenoneine regarding their uptake from food, reliable analytical methods for their simultaneous determination are needed. As selenoneine and ergothioneine are both found in human blood,11,14–17 predominantly in blood cells, a readily accessible sample for monitoring their dietary uptake, their simultaneous quantitative determination in this matrix is of interest. While high performance liquid chromatography (HPLC) coupled to inductively coupled plasma mass spectrometry (ICPMS) is the most common technique for selenium speciation analysis in biological samples,18 application of HPLC/ICPMS for sulfur speciation analysis has long been hampered by spectral interferences.19 These difficulties have been overcome by introducing high-resolution mass spectrometry, by applying reaction/collision cell techniques, or recently by the use of ICP-triple quadrupole-MS (ICP-QQQ-MS).19,20
We present a method using ICP-QQQ-MS in oxygen reaction mode for the quantitative determination of sulfur and selenium in the form of SO+ and SeO+, thereby overcoming interferences typically hindering the analysis. The use of reversed-phase HPLC allows the separation of several sulfur and selenium species from the target analytes ergothioneine and selenoneine, thereby introducing a promising tool for the simultaneous determination of these two potentially health relevant species, in human blood cells.
Experimental
Chemicals, reagents and standards
Analytical grade chemicals and ultrapure water (18.2 MΩ cm, Academic water purification system, Millipore GmbH, Vienna, Austria) were used for the preparation of solutions throughout. Ammonium formate (≥95%), formic acid (≥98%, p.a.), nitric acid (65%, p.a.), hydrochloric acid (fuming, 37%, p.a.), tris-(hydroxymethyl)-aminomethane (TRIS) (UltraQualität, PUFFERAN, ≥99.9%) and tris-(2-carboxyethyl)phosphine hydrochloride (TCEP, >98%, for biochemistry) were purchased from Carl Roth GmbH + Co. KG (Karlsruhe, Germany). Iodoacetamide (IAM, BioUltra) was obtained from Sigma Aldrich (Steinheim, Germany). Methanol (HPLC gradient grade) and acetonitrile (HPLC grade) were purchased from VWR International (Fontenay-sous-Bois, France) and Chem Lab NV (Zedelgem, Belgium), respectively. Further purification of nitric acid was achieved by sub-boiling distillation. The buffer for cell lysis (50 mM TRIS–HCl, pH 7.5) was prepared by weighing in the appropriate amount of TRIS, dissolving it in water and adjusting the pH with HCl. TCEP and IAM solutions used for the direct derivatization procedure of blood cell lysates were prepared fresh before use by dissolving the appropriate amount of reagent in water. To prepare the mobile phases for chromatographic separations of sulfur and selenium species, the appropriate amount of ammonium formate was dissolved in water, the required amount of methanol was added and the pH was adjusted with formic acid. For preparation of the mobile phase containing TCEP, the appropriate amount of reducing agent was dissolved in the mobile phase.Single element standard solutions of sulfur (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mg S per L ± 0.2% in H2O), selenium (1000 mg Se per L ± 0.2% in 2% HNO3), germanium (1000 mg Ge per L in 2% HNO3 and 0.5% HF) and indium (1000 mg In per L ± 0.2% in 2% HNO3) for total element determination by ICP-QQQ-MS were purchased from Carl Roth GmbH + Co. KG. A standard solution of (NH4)2SO4 in water (10000 mg S per L, Carl Roth GmbH + Co. KG) was additionally used for quality control in sulfur speciation analysis. Sulfur and selenium standard solutions used for HPLC/ICP-QQQ-MS analysis were obtained as previously reported.21–23 Standards were prepared fresh daily from aqueous stock standard solutions, kept at 4 °C, by dilution with water.
000 mg S per L ± 0.2% in H2O), selenium (1000 mg Se per L ± 0.2% in 2% HNO3), germanium (1000 mg Ge per L in 2% HNO3 and 0.5% HF) and indium (1000 mg In per L ± 0.2% in 2% HNO3) for total element determination by ICP-QQQ-MS were purchased from Carl Roth GmbH + Co. KG. A standard solution of (NH4)2SO4 in water (10000 mg S per L, Carl Roth GmbH + Co. KG) was additionally used for quality control in sulfur speciation analysis. Sulfur and selenium standard solutions used for HPLC/ICP-QQQ-MS analysis were obtained as previously reported.21–23 Standards were prepared fresh daily from aqueous stock standard solutions, kept at 4 °C, by dilution with water.
Selenoneine, isolated from Schizosaccharomyces pombe was prepared in-house.24 Additionally, a water extract of canned tuna (skipjack tuna in brine, Katsuwonus pelamis, caught in the West-Pacific Ocean, Indonesia), purchased from a local distributor in Graz was prepared. Briefly, canned tuna in brine was homogenized with a food processor and 1 g was extracted with 10 mL of water by sonication in an ultrasonic bath (4 × 5 min) followed by centrifugation (10 min, 3700 × g) and syringe filtration (0.2 μm nylon (PA) membrane; ProFill, Markus Bruckner Analysentechnik, Linz, Austria), to remove the solid residues from the extract. Se-methylselenoneine was extracted and purified from fresh tuna muscle as previously reported.25
Instrumentation
For the preparation of a water extract of canned tuna a Transsonic 700 H ultrasonic bath (Elma, Singen, Germany; ultrasonic power RMS 160 W, ultrasonic maximum peak power 640 W) was used. Separation of blood plasma from blood cells was achieved through centrifugation utilizing a Hermle Z 200 A centrifuge (Hermle Labortechnik, Wehingen, Germany) and a D3024R High Speed Refrigerated Micro-Centrifuge (SCILOGEX, LLC., Rocky Hill, Connecticut, USA) was used for centrifugation during all further sample preparation steps, except for derivatization, where a Hettich Mikroliter 2043 centrifuge (Hettich, Tuttlingen, Germany) was used. Microwave-assisted acid digestion was performed in an UltraClave IV High Performance Microwave Reactor (MLS GmbH, Leutkirch, Germany). Solvents used to precipitate proteins from samples were evaporated to dryness in a Christ RVC 2-33 CD plus centrifugal lyophilizer equipped with a Christ CT 02-50 SR cooling trap (Martin Christ Gefriertrocknungsanlagen GmbH, Osterode am Harz, Germany) and a MD 4C diaphragm vacuum pump (Vacuubrand GmbH + Co. KG, Wertheim, Germany). Quantitative determination of sulfur and selenium (total element and speciation analysis) was performed on an 8800-ICP-QQQ-MS system (Agilent Technologies, Waldbronn, Germany) equipped with a MicroMist nebulizer. Sample solutions were introduced to the ICP-QQQ-MS for total element analysis with a SPS 4 autosampler (Agilent Technologies). Sulfur and selenium speciation analysis was performed after reversed-phase high-performance liquid chromatography using an Agilent HPLC system (Agilent Technologies) consisting of Agilent 1100 and 1200 modules (degasser G 1379A, quaternary pump G 1311A, autosampler G 1367C with thermostat G 1330B, thermostated column compartment G 1316A). The Atlantis dC18 4.6 × 150 mm column (Waters Corporation, Milford, USA), protected by its respective guard-column, was directly coupled to the nebulizer of the ICP-QQQ-MS system. Verification of the presence of target species in lysates of blood cells was performed by high resolution molecular mass spectrometry using a Q Exactive Orbitrap Mass Spectrometer (Thermo Scientific, Waltham, USA) equipped with a heated electrospray ionization (HESI) source (Thermo Scientific) after reversed-phase HPLC on a Dionex Ultimate 3000 HPLC system (Thermo Scientific) consisting of a Rapid Separation (RS) pump, RS autosampler and RS column compartment.Samples
Whole blood from 3 volunteers (volunteer 1–25 year-old female, volunteer 2–31 year-old male and volunteer 3–50 year-old male) was collected in K3E EDTA Vacuette Tubes (9 mL, Greiner Bio-One, Frickenhausen, Germany). To separate blood plasma from red blood cells, whole blood was centrifuged for 10 minutes at 470 × g. Plasma was removed, the buffy coat was discarded and aliquots (500 μL) of the red blood cells (estimated number of approximately 2.5×109 cells per aliquot)26 were stored in disposable, conical micro-centrifuge tubes (1.5 mL, for high G-force; VWR International, Radnor, Pennsylvania, USA) at −80 °C until analysis, which was performed in duplicates unless stated otherwise. Informed consent was given by the three volunteers and all procedures were in accordance with the Declaration of Helsinki.Determination of total selenium and sulfur
For determination of total selenium and sulfur, 0.4 mL of blood cells were acid-digested as previously reported.22 Prior to analysis an internal standard solution containing Ge and In in 2% HNO3 was added to all samples (final concentration 25 μg per L).ICP-QQQ-MS was operated in oxygen reaction mode with a flow rate of ca. 0.3 mL O2 per min as a cell gas. For signal enhancement, 1% CO2 in Ar was used as optional gas at a flow rate of 12% of the carrier gas flow.27 Monitoring of sulfur and selenium occurred in the mass shift mode after the reaction with oxygen as detailed previously,21 using integration times of 0.05 s and 0.3 s for sulfur and selenium, respectively (typical ICP-QQQ-MS settings are given in ESI, Table S1†). Quantification was performed by external calibration using m/z 32 → 48 and m/z 78 → 94 for determination of sulfur and selenium, respectively. Quality control of total selenium determination was performed using plasma samples from a round-robin exercise (low selenium plasma: reference value, 62.1 μg Se per L; range of tolerance, 50.1–74.1 μg Se per L, found values, 62.8/63.5 μg Se per L; high selenium plasma: reference value, 237.8 μg Se per L; range of tolerance, 206.3–269.3 μg Se per L, found values, 247.5/248.1 μg Se per L).
Determination of selenoneine and ergothioneine by HPLC/ICP-QQQ-MS and high resolution molecular mass spectrometry.
To access small selenium and sulfur species in human blood cells, several lysis procedures were tested. For lysis using liquid nitrogen, blood cell aliquots (500 μL) were thawed and subjected to 10 freeze-thaw cycles in liquid nitrogen. Alternatively, cell lysis was induced by taking 200 μL aliquots of blood cells and thoroughly mixing with varying amounts of cold water (1:1 up to 1:6.5 v/v) or 50 mM TRIS–HCl buffer pH 7.5 (1:1 v/v). In an additional experiment, lysis of the cells with cold water (1:1 v/v), was performed in combination with sonication in an ultrasonic bath for 10 minutes. Each of the tested cell lysis procedures was followed by centrifugation of the lysates for 10 minutes (21000 × g, 4 °C) to remove the cell debris; lysates were then subjected to derivatization and/or protein removal.
Blood cell lysates were derivatized following the procedure described by Klein et al.14 with minor modification. Briefly, an aliquot (500 μL) of the lysate of blood cells obtained by freeze-thaw cycles in liquid nitrogen was treated with 17 μL of a 0.45 M aqueous TCEP solution (reducing agent) and 46 μL of a 0.5 M aqueous IAM solution (alkylating agent), and the mixture was incubated for 2 hours at room temperature. The samples were centrifuged (5 minutes, 7400 × g) and then subjected to protein removal.
The first of two approaches tested to remove proteins from the blood cell lysates was adapted from a method reported for the investigation of small selenium species in serum.22 Briefly, blood cell lysate (500 μL) was mixed with an equal volume of acetonitrile, and the precipitated proteins were separated by centrifugation (10 minutes, 21000 × g, 4 °C). A 500 μL portion of the resulting supernatant was removed and evaporated to dryness in a centrifugal lyophilizer overnight; the dry residue was re-dissolved in 100 μL mobile phase before HPLC/ICP-QQQ-MS analysis. The second approach to protein removal was adapted from the procedure reported by Flouda et al.28 and consisted of cut-off filtration (molecular weight cut-off: 3000 Da). For this purpose, 300 μL of the blood cell lysate were transferred to a Vivaspin 500 cut-off filter (MWCO 3000 Da, Sartorius, Stonehouse, Gloucestershire, UK) and centrifuged for 60 minutes at 4 °C and 15000 × g. The filtrate was directly subjected to HPLC/ICP-QQQ-MS analysis.
Determination of selenoneine and ergothioneine in lysates of blood cells was performed by reversed-phase HPLC/ICP-QQQ-MS; two mobile phases, without or with TCEP, were employed (for chromatographic conditions see ESI, Table S1†). Ergothioneine and selenoneine were quantified against ergothioneine and selenomethionine, since selenoneine is not commercially available, assuming similar instrument response for both selenium species. Instrument settings for the ICP-QQQ-MS were the same as detailed above for total sulfur and selenium measurements.
For verifying the presence of the target species in the lysates of blood cells, high resolution molecular mass spectrometry with electrospray ionization in positive ion mode was performed after reversed-phase chromatography (ESI, Table S1,† chromatographic condition 1 – mobile phase without TCEP). The source settings were as follows: gas temperature 500 °C, gas flow rates 65 (sheath) and 20 (aux) instrument units, spray voltage 3500 V and capillary temperature 300 °C. A resolution (full width half maximum) of 70000, an automatic gain control (AGC) target of 3 × 106, a maximum injection time (IT) of 100 ms and a full scan range of m/z 170–400 and m/z 370–600 were used.
Stability, spiking recovery and precision
The stability of selenoneine and ergothioneine during sample preparation was assessed by measuring (HPLC) lysed blood cells kept at room temperature for one hour before cut-off filtration and analysis. Results were compared to selenoneine and ergothioneine concentrations in lysed blood cells, directly subjected to cut-off filtration after cell lysis. To gain information on the stability of the target compounds during HPLC measurements, lysates of blood cells were re-measured after being kept at 4 °C in the autosampler of the HPLC system for one and two days.The applicability of the method for the quantitative determination of selenoneine and ergothioneine in human blood cells was evaluated by spiking blood cells with aqueous solutions of commercially available ergothioneine and selenoneine isolated from S. pombe. Subsequently, lysis using an equal volume of both the sample and cold water followed by cut-off filtration as detailed above was performed on the spiked blood cells. Amounts of ergothioneine added to native samples were 1.6 mg S per L for blood cells from volunteers 1 and 2, and 6.6 mg S per L for volunteer 3; these concentrations of spiked ergothioneine were based on ergothioneine concentrations found in lysates of the native blood cells of the three volunteers. Selenoneine was spiked in a concentration of 45 μg Se per L to all three samples of native blood cells.
Duplicate samples from all volunteers were prepared fresh on three consecutive days and measured directly after sample preparation using either one of the two chromatographic conditions described above. The standard deviation of the method was estimated for a series of 9 duplicate analyses (3 duplicates for each of the 3 volunteers) for each of the two chromatographic conditions. The limit of detection (LoD) for selenoneine was estimated by visual investigation of the two lowest selenomethionine calibration standard solutions (concentrations 0.1 and 0.5 μg Se per L).
Results and discussion
Determination of selenoneine and ergothioneine in human blood
ICP-QQQ-MS in oxygen reaction mode is a powerful tool for removing interferences occurring in single quadrupole ICPMS measurements of various analyte elements like sulfur or selenium.19,29 Hence, it was used to overcome spectral interferences on 32S and 78Se mainly deriving from oxygen (16O16O+, 14N18O+) and argon (40Ar38Ar+, 38Ar40Ca+) by employing a mass shift of +16 (32S+ → 32S16O+ and 78Se+ → 78Se16O+), and thereby allowing simultaneous determination of sulfur and selenium species at physiological concentrations on m/z 32 → 48 and 78 → 94, respectively.20,29 Reversed-phase chromatography prior to ICP-QQQ-MS detection allowed the separation of ergothioneine and selenoneine from several sulfur and selenium species in cell cultures and mushroom extracts.21 The chromatographic conditions (Gemini C6-Phenyl 4.6 × 150 mm column with a mobile phase at alkaline pH) used in that study, however, were not compatible with the complex blood matrix; although the retention and separation of ergothioneine and selenoneine were initially acceptable, the column resolution rapidly declined after several injections of the blood cell lysates. Therefore, we sought an alternative approach ensuring stable separation of the target species even after the injection of many samples with a heavy matrix load such as human blood cells.When we changed to an acidic mobile phase, ergothioneine was baseline separated from all tested sulfur species, and selenoneine was similarly separated from the tested selenium species, except for methylselenocysteine (Fig. 2), a species previously tentatively identified in human serum.22 When lysates of blood cells from three volunteers were analyzed with the acidic chromatographic setup, several peaks, one of which was matching the retention time of ergothioneine, were detected on m/z 32 → 48 and a peak matching the retention time of selenoneine was detected on m/z 78 → 94 (Fig. 3). The presence of methylselenocysteine in the blood cell lysates was excluded by spiking (Fig. 4) and the identity of the selenium peak as selenoneine was confirmed by high resolution molecular mass spectrometry (ESI, Fig. S1†). The same analysis also confirmed the presence of ergothioneine in the blood cell lysates (ESI, Fig. S2†). It is worth noting that the HPLC/ICP-QQQ-MS method was capable of distinguishing between selenoneine and methylselenocysteine even when baseline separation was not achieved.
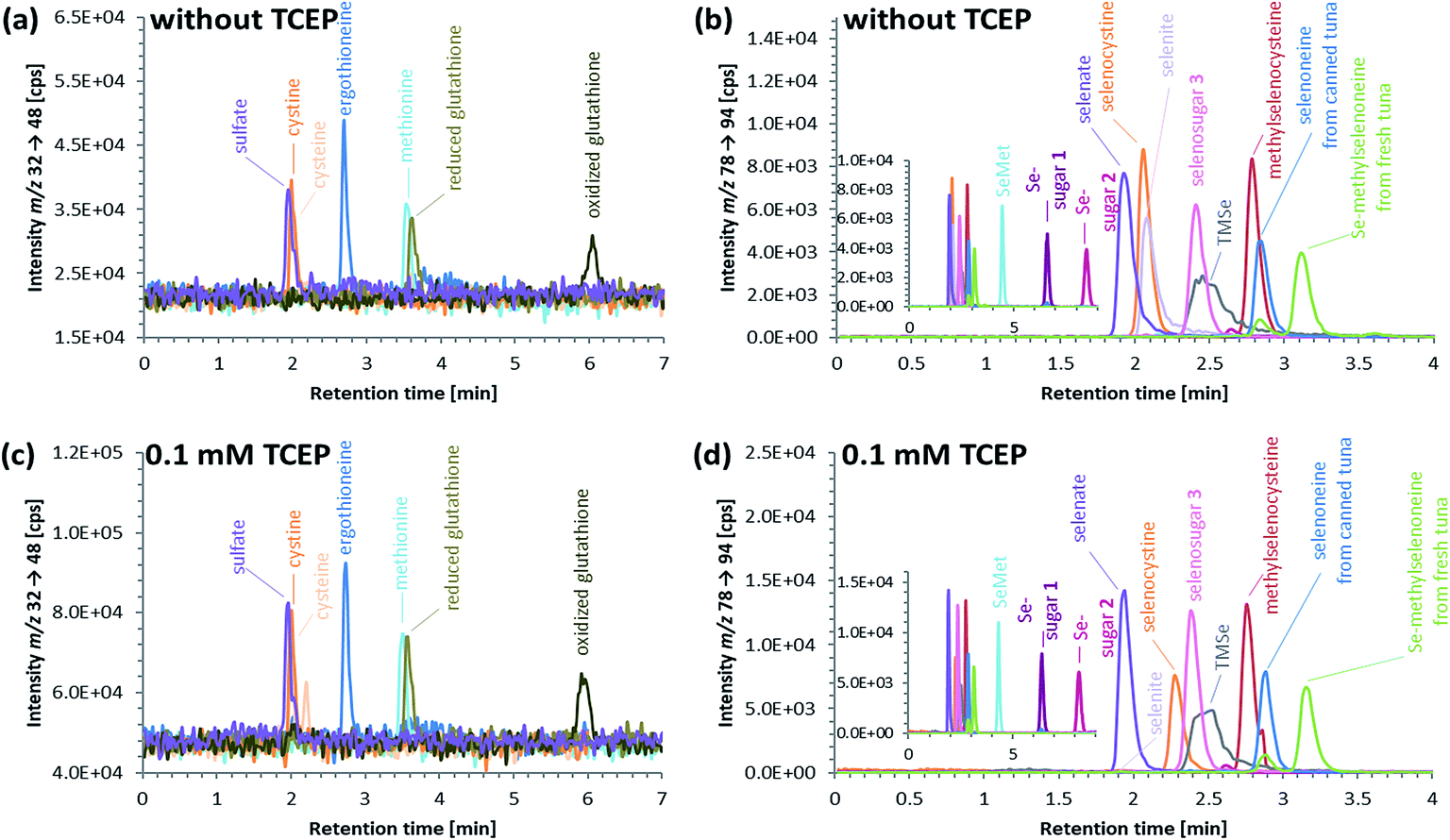 | ||
| Fig. 2 HPLC/ICP-QQQ-MS elution profile of various sulfur ((a) and (c)) and selenium ((b) and (d)) species (integration times: 0.05 s for sulfur and 0.3 s for selenium); sulfur (50 μg S per L) or selenium (50 μg Se per L) standard solutions, selenoneine in a water extract of canned tuna (30 μg Se per L) and Se-methylselenoneine isolated from a water extract of fresh tuna muscle (30 μg Se per L); analogous sulfur and selenium species are shown in the same color; column: Atlantis dC18 4.6 × 150 mm; mobile phase: (a) & (b) 20 mM ammonium formate, 3% MeOH, pH 3.0 or (c) & (d) 20 mM ammonium formate, 3% MeOH, 0.1 mM TCEP, pH 3.0; flow rate: 1.0 mL per minute; injection volume: 10 μL. | ||
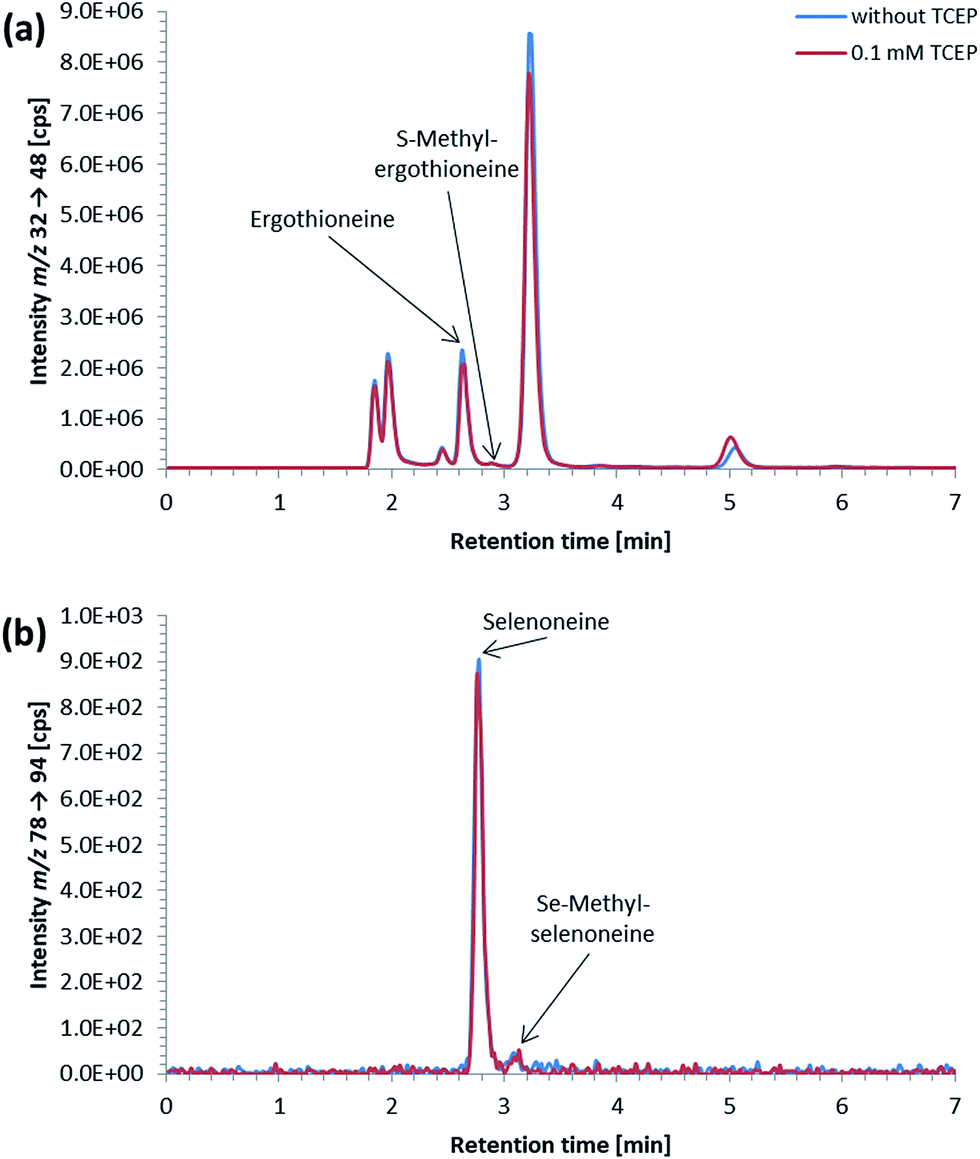 | ||
| Fig. 3 Typical (a) sulfur and (b) selenium elution profile of a lysate of native blood cells (volunteer 3); obtained without TCEP in the mobile phase (blue line) and with 0.1 mM TCEP in the mobile phase (red line); column: Atlantis dC18 4.6 × 150 mm; mobile phase: 20 mM ammonium formate, 3% MeOH, pH 3 (blue line) or 20 mM ammonium formate, 3% MeOH, 0.1 mM TCEP, pH 3 (red line); flow rate: 1.0 mL per minute; injection volume: 10 μL. | ||
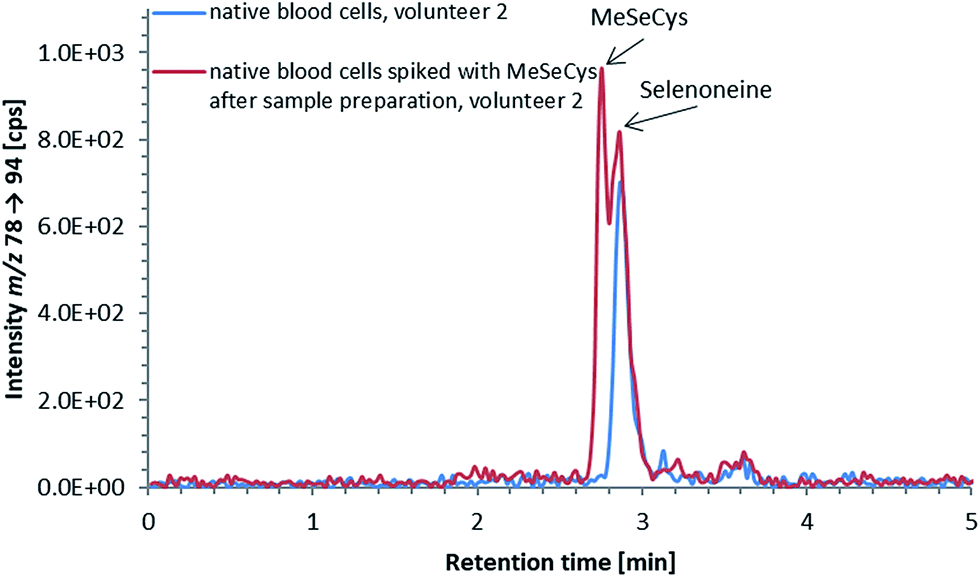 | ||
| Fig. 4 Selenium elution profile of a lysate of native blood cells (volunteer 2); native blood cell lysate (blue line) and native blood cell lysate spiked with 3 μg Se per L MeSeCys after sample preparation (red line); the double peak in the chromatogram of the spiked blood cell lysate indicates, that methylselenocysteine was not present in native blood cell lysates in detectable amounts, and the peak in the selenium elution profile derived from selenoneine; column: Atlantis dC18 4.6 × 150 mm; mobile phase: 20 mM ammonium formate, 3% MeOH, pH 3; flow rate: 1.0 mL per minute; injection volume: 10 μL. | ||
Whereas ergothioneine is present in its thione form under physiological conditions,8 selenoneine can undergo auto-oxidation, resulting in the formation of a dimer,6 which can show different chromatographic behavior compared to its reduced form.24 Both the oxidized and reduced forms can be present at varying concentrations in biological samples, as well as in standard solutions, and can hamper the straight-forward determination of selenoneine. Thus, there is a need to convert selenoneine to a single defined form, either the oxidized or the reduced form, before the HPLC measurements.
An advantage of working with oxidized selenoneine is that it is well retained and separated from other selenium species in reversed-phase chromatography at neutral pH.24 Attempts to oxidize selenoneine in biological samples with hydrogen peroxide, however, resulted in the formation of mixed S–Se-species,24 so this approach was not further explored.
An approach based on reduced selenoneine was then investigated. Although we initially added the reducing agent directly to our samples, we soon found that incorporating the reducing agent into the mobile phase was a more convenient method because it not only saved time during sample preparation but also solved the potential problem of re-oxidation of selenoneine in the period between sample preparation and HPLC injection. Thus, addition of 0.1 mM tris(2-carboxyethyl)phosphine (TCEP) to the mobile phase used for reversed-phase HPLC resulted in the efficient reduction of selenoneine, and, except for selenite, there was no effect on the elution of any of the other selenium species investigated (Fig. 2). The tested sulfur species, including ergothioneine, were also unaffected by the presence of the reducing agent (Fig. 2). Selenite, was not eluted from the column when TCEP was used in the mobile phase, possibly due to its reduction to elemental selenium. Even though the retention of selenoneine on the column was only slightly altered by the addition of TCEP to the mobile phase, better separation from methylselenocysteine was achieved (Fig. 2). Thus, reducing conditions of the mobile phase enabled the conversion of selenoneine to a single (reduced) form, thereby facilitating the quantitative determination of the target species.
Since direct derivatization using a reducing agent in combination with iodoacetamide (IAM) has been shown to convert selenoneine and ergothioneine to a single defined form,14 this approach was also tested. Although derivatization in lysates of blood cells using TCEP/IAM produced quantitative data comparable to that obtained without it (data not shown), the later approach is simpler to use and more time efficient, and was thus used in all subsequent experiments.
The small peak (estimated concentration close to the LoD) eluting shortly after selenoneine in the lysates of blood cell samples (Fig. 3b) was unaffected by treatment with TCEP, which suggested that the selenium atom was “protected”, presumably by a methyl group. Because both selenoneine and its methylated analogue Se-methylselenoneine have been previously identified in human whole blood by Klein et al.,14 we tentatively assigned the small peak in our chromatograms to Se-methylselenoneine. This assignment was supported by co-chromatography with an authentic specimen (extracted from tuna), and then confirmed by high resolution molecular mass spectrometry (ESI, Fig. S3†). Klein et al.14 proposed that selenoneine and its methylated analogue constituted the main pool of non-protein selenium, which was supported by the results from our study (Fig. 3b). The methylated analogue of ergothioneine, namely S-methylergothioneine, was also present in our blood cell lysates (eluting from the column as a small peak after ergothioneine, Fig. 3a; ESI, Fig. S4†), a result consistent with the earlier report of its presence in human whole blood.14 While selenoneine and its methylated analogue were the only detectable small selenium species in the lysates of blood cells, ergothioneine and S-methylergothioneine (approximately 0.2–0.3 mg S per L) constituted only 2–15% of the non-protein sulfur pool in the blood samples from the three volunteers (Fig. 3a).
Sample preparation
Cell lysis is required to access the small target compounds taken up by the blood cells, and several lysis procedures were tested in the initial stages of this study. Although they were all equally effective in releasing selenoneine and ergothioneine from the cells (data not shown), lysis of the blood cells with an equal volume of cold water was the fastest and most convenient, and, therefore, this procedure was chosen for all subsequent experiments.The analysis of small selenium and sulfur species such as selenoneine and ergothioneine in the lysates of blood cells using HPLC/ICPMS is complicated by both the complex matrix and the presence of selenium and sulfur in proteins. Protein precipitation with acetonitrile prior to the analysis of small selenium species has been successfully demonstrated for human serum22 and was, therefore, tested as an alternative to molecular weight cut-off (MWCO) filtration (cut-off 3000 Da)28 in the study at hand. However, acetonitrile precipitation, though cheaper, proved to be time consuming and resulted in losses of 50% of the ergothioneine in the lysates of blood cells compared to cut-off filtration, which was, therefore, used in all experiments.
Stability, spiking recovery and precision
Besides accessing the target compounds for analysis, their stability in the sample matrix during sample preparation and analysis is also important, particularly when large numbers of samples have to be analyzed. During our preliminary studies, we had established that selenoneine and ergothioneine were stable when lysed blood cell samples were kept at room temperature for one hour prior to cut-off filtration. Furthermore, no significant decrease in the target species was observed for lysates of blood cells kept at 4 °C in the autosampler of the HPLC system over a period of two days after sample preparation (data not shown).The sample preparation procedure was tested by spiking ergothioneine and selenoneine to blood cell samples before cell lysis; recoveries for blood cells from three individuals were around 85% and 80% for ergothioneine and selenoneine, respectively (Table 1), demonstrating the applicability for the simultaneous and quantitative determination of ergothioneine and selenoneine in blood cells.
| Analytical figures of merit | Ergothioneine | Selenoneine | ||
|---|---|---|---|---|
| without TCEP | with TCEP | without TCEP | with TCEP | |
| Spiking recovery (n = 5) [%] | 85 ± 3% | 84 ± 3% | 79 ± 1% | 79 ± 2% |
| SD of the method [mg S per L or μg Se per L] | 0.10 | 0.07 | 0.25 | 0.21 |
| RSD of the method [%] for 1 mg S per L or 1 μg Se per L | 10 | 7 | 25 | 21 |
| RSD of the method [%] for 10 mg S per L or 10 μg Se per L | 1 | 1 | 2 | 2 |
The precision of the method was estimated by analysis of two replicates of blood cell lysates for each of the three volunteers prepared fresh on three consecutive days. The standard deviation of the method was around 0.10 mg S per L for ergothioneine corresponding to relative standard deviations (RSD) between 10–1% for ergothioneine concentrations of 1–10 mg S per L, using either one of the chromatographic conditions (Table 1). For selenoneine, RSDs of 2–25% for high (10 μg Se per L) and low (1 μg Se per L) concentrations were obtained (standard deviation of the method around 0.25 μg Se per L, Table 1). Relative standard deviations (RSD) for replicate analysis of a sample from a single volunteer were between 4% and 15% (Table 2). Since selenoneine in contrast to ergothioneine was present in blood cell lysates at much lower concentrations (Table 2), its LoD was of interest. By visual investigation of the signal obtained for the two lowest selenomethionine standard solutions used for the quantification of selenoneine it was estimated to be 0.1–0.5 μg Se per L. Calibration plots for ergothioneine and selenomethionine can be found in Fig. S5 and S6 (ESI†), respectively.
| Sample, (mobile phase) | Total S [g S per L] | Ergothioneine [mg S per L] | |||
|---|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | RSD [%] | ||
| Volunteer 1, (without TCEP) | 1.87, 1.89 | 0.95, 0.96 | 0.72, 0.83 | 0.82, 0.88 | 11 |
| Volunteer 1, (with TCEP) | 0.94, 0.78 | 0.79, 0.91 | 0.77, 0.79 | 9 | |
| Volunteer 2, (without TCEP) | 1.88, 1.95 | 1.8, 1.7 | 1.3, 1.5 | 1.4, 1.5 | 12 |
| Volunteer 2, (with TCEP) | 1.7, 1.6 | 1.4, 1.5 | 1.3, 1.4 | 10 | |
| Volunteer 3, (without TCEP) | 1.98, 1.99 | 6.8, 6.8 | 6.1, 6.1 | 6.3, 6.6 | 5 |
| Volunteer 3, (with TCEP) | 6.8, 6.7 | 6.3, 6.4 | 6.1, 6.0 | 5 | |
| Sample, (mobile phase) | Total Se [μg Se per L] | Selenoneine [μg Se per L] | |||
|---|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | RSD [%] | ||
| Volunteer 1, (without TCEP) | 98.7, 97.0 | 3.4, 3.2 | 3.2, 3.0 | 3.2, 3.4 | 4 |
| Volunteer 1, (with TCEP) | 3.5, 3.6 | 3.1, 3.1 | 3.0, 2.6 | 12 | |
| Volunteer 2, (without TCEP) | 107, 107 | 6.4, 6.2 | 6.4, 5.9 | 6.5, 6.2 | 4 |
| Volunteer 2, (with TCEP) | 7.4, 7.2 | 5.7, 5.5 | 5.4, 5.7 | 15 | |
| Volunteer 3, (without TCEP) | 122, 122 | 7.5, 6.8 | 7.0, 6.9 | 7.9, 8.0 | 7 |
| Volunteer 3, (with TCEP) | 8.4, 8.2 | 6.4, 6.8 | 7.0 6.6 | 12 | |
Ergothioneine and selenoneine in blood cells of three individuals
Ergothioneine and selenoneine were detected in the blood cells of all three individuals (Table 2). Total sulfur concentrations in blood cells of the three individuals ranged around 1.9 g S per L, with ergothioneine concentrations representing about 0.05% to 0.3% of the total sulfur in the blood cells (Table 2). In comparison to ergothioneine and total sulfur, a much larger proportion of total selenium was accounted for by selenoneine, which constituted around 3% to 7% of the total selenium in the blood cells (Table 2).Ergothioneine and selenoneine are not known to be synthesized by humans, therefore, their concentration is dependent on exposure to dietary sources and possibly a polymorphism of the gene encoding the transporter needed for the uptake of ergothioneine11 and selenoneine,30 as suggested by Cheah et al.11 Even though, no information about the dietary habits of the volunteers was gathered, the practicability of our method was demonstrated by this first small study simultaneously quantifying ergothioneine and selenoneine in human blood cells. To gain more insight into the individual variability of selenoneine and ergothioneine in human blood, a larger study including the dietary background of the volunteers should be carried out.
Albeit, selenoneine concentrations determined without or with TCEP in the mobile phase did not show significant differences in the lysates of blood cells (Table 2), adding TCEP to the mobile phase resulted in slightly better separation of methylselenocysteine from selenoneine. Furthermore, comparison of the results obtained with the two chromatographic conditions can give insight into selenoneine possibly bound to thiol-groups present in the sample matrix and freed by the presence of the reducing agent in the mobile phase. Therefore, the method can be of future use to distinguish between total and free selenoneine in the different sample matrices, providing more insight into the role of selenoneine in biological samples.
Conclusion
We report a quantitative HPLC/ICP-QQQ-MS method for the simultaneous determination of the purported antioxidants selenoneine and ergothioneine in human blood cells, and present preliminary results from three individuals. The method might be useful for distinguishing between free and matrix-bound selenoneine, hence, giving more insight into the role of this selenium species and the differences to its sulfur-analogue ergothioneine. Furthermore, it will allow future studies on the relevance of these two related compounds to human health, particularly in terms of dietary sources and individual variability in their uptake and metabolism.Author contributions
The manuscript was written with contributions from all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
We thank the volunteers for participating in this study by donating their blood. We also thank the Austrian Science Fund (FWF, project number I 2262-N28) and the “Deutsche Forschungsgemeinschaft” (DFG, project grant number SCHW 903/9-1) for financially supporting this research and the NAWI Graz Central Lab – Metabolomics for providing the opportunity of recording high resolution mass spectra.References
- K. Schwarz and C. M. Foltz, J. Am. Chem. Soc., 1957, 79, 3292–3293 CrossRef CAS
.
- Y. Ingenbleek and H. Kimura, Nutr. Rev., 2013, 71, 413–432 CrossRef PubMed
-
C. Reilly, Selenium in food and health, Springer Science + Business Media, LCC, New York, 2nd edn, 2006, pp. 1–19 Search PubMed
- L. Johansson, G. Gafvelin and E. S. J. Arnér, Biochim. Biophys. Acta, Gen. Subj., 2005, 1726, 1–13 CrossRef CAS PubMed
- G. N. Schrauzer, J. Nutr., 2000, 130, 1653–1656 CrossRef CAS PubMed
- Y. Yamashita and M. Yamashita, J. Biol. Chem., 2010, 285, 18134–18138 CrossRef CAS PubMed
- I. Erdelmeier, S. Daunay, R. Lebel, L. Farescour and J.-C. Yadan, Green Chem., 2012, 14, 2256–2265 RSC
- I. K. Cheah and B. Halliwell, Biochim. Biophys. Acta, Mol. Basis Dis., 2012, 1822, 784–793 CrossRef CAS PubMed
- P. E. Hartman, Methods Enzymol., 1990, 186, 310–318 CAS
- Y. Yamashita, T. Yabu and M. Yamashita, World J. Biol. Chem., 2010, 1, 144–150 CrossRef PubMed
- I. K. Cheah, R. M. Y. Tang, T. S. Z. Yew, K. H. C. Lim and B. Halliwell, Antioxid. Redox Signaling, 2017, 26, 193–206 CrossRef CAS PubMed
- J. Ey, E. Schömig and D. Taubert, J. Agric. Food Chem., 2007, 55, 6466–6474 CrossRef CAS PubMed
- Y. Yamashita, H. Amlund, T. Suzuki, T. Hara, M. A. Hossain, T. Yabu, K. Touhata and M. Yamashita, Fish. Sci., 2011, 77, 679–686 CrossRef CAS
- M. Klein, L. Ouerdane, M. Bueno and F. Pannier, Metallomics, 2011, 3, 513–520 RSC
- M. Yamashita, Y. Yamashita, T. Ando, J. Wakamiya and S. Akiba, Biol. Trace Elem. Res., 2013, 156, 36–44 CrossRef CAS PubMed
- H. Mitsuyama and J. M. May, Clin. Sci., 1999, 97, 407–411 CrossRef CAS PubMed
- L.-Z. Wang, W.-L. Thuya, D. S.-L. Toh, M. G.-L. Lie, J.-Y. A. Lau, L.-R. Kong, S.-C. Wan, K.-N. Chua, E. J.-D. Lee and B.-C. Goh, J. Mass Spectrom., 2013, 48, 406–412 CrossRef CAS PubMed
- B. Gammelgaard, M. I. Jackson and C. Gabel-Jensen, Anal. Bioanal. Chem., 2011, 399, 1743–1763 CrossRef CAS PubMed
- J. Giner Martínez-Sierra, O. Galilea San Blas, J. M. Marchante Gayón and J. I. García Alonso, Spectrochim. Acta, Part B, 2015, 108, 35–52 CrossRef
- L. Balcaen, G. Woods, M. Resano and F. Vanhaecke, J. Anal. At. Spectrom., 2013, 28, 33–39 RSC
- N. Kroepfl, T. A. Marschall, K. A. Francesconi, T. Schwerdtle and D. Kuehnelt, J. Anal. At. Spectrom., 2017, 32, 1571–1581 RSC
- S. Kokarnig, D. Kuehnelt, M. Stiboller, U. Hartleb and K. A. Francesconi, Anal. Bioanal. Chem., 2011, 400, 2323–2327 CrossRef CAS PubMed
- D. Kuehnelt, N. Kienzl, P. Traar, N. H. Le, K. A. Francesconi and T. Ochi, Anal. Bioanal. Chem., 2005, 383, 235–246 CrossRef CAS PubMed
- N. G. Turrini, N. Kroepfl, K. B. Jensen, T. C. Reiter, K. A. Francesconi, T. Schwerdtle, W. Kroutil and D. Kuehnelt, Metallomics, 2018, 10, 1532–1538 RSC
- N. Kroepfl, K. B. Jensen, K. A. Francesconi and D. Kuehnelt, Anal. Bioanal. Chem., 2015, 407, 7713–7719 CrossRef CAS PubMed
- E. Bianconi, A. Piovesan, F. Facchin, A. Beraudi, R. Casadei, F. Frabetti, L. Vitale, M. C. Pelleri, S. Tassani, F. Piva, S. Perez-Amodio, P. Strippoli and S. Canaider, Ann. Hum. Biol., 2013, 40, 463–471 CrossRef PubMed
- D. Kuehnelt, D. Juresa, N. Kienzl and K. A. Francesconi, Anal. Bioanal. Chem., 2006, 386, 2207–2212 CrossRef CAS PubMed
- K. Flouda, J. M. Dersch, C. Gabel-Jensen, S. Stürup, S. Misra, M. Björnstedt and B. Gammelgaard, Anal. Bioanal. Chem., 2016, 408, 2293–2301 CrossRef CAS PubMed
- D. P. Bishop, D. J. Hare, F. Fryer, R. V. Taudte, B. R. Cardoso, N. Cole and P. A. Doble, Analyst, 2015, 140, 2842–2846 RSC
- M. Yamashita, Y. Yamashita, T. Suzuki, Y. Kani, N. Mizusawa, S. Imamura, K. Takemoto, T. Hara, M. A. Hossain, T. Yabu and K. Touhata, Mar. Biotechnol., 2013, 15, 559–570 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Typical HPLC and ICP-QQQ-MS settings for the quantitative determination of sulfur and selenium in lysates of human blood cells, high resolution mass spectra of an aqueous lysate of blood cells and external calibration plots. See DOI: 10.1039/c8ja00276b |
| This journal is © The Royal Society of Chemistry 2019 |