Fluorescence switching with subphthalocyanine-dihydroazulene dyads†
Received
12th October 2018
, Accepted 8th January 2019
First published on 14th January 2019
Abstract
One convenient way of reversibly modulating the emission properties of a fluorophore is by introducing a photochromic unit that upon irradiation is converted into an isomeric unit that can quench the fluorescence of the fluorophore. Here we present the synthesis of two dyads containing a boron subphthalocyanine (SubPc) fluorophore and the dihydroazulene (DHA) photoswitch. Upon photoisomerization, the DHA unit is converted to a vinylheptafulvene (VHF) unit that was found to efficiently quench the SubPc fluorescence when placed at the axial boron position of the SubPc. The original fluorescence was restored by thermal conversion of the VHF to DHA. Fluorescence was thereby turned off and on by successive light-heat cycles. The other dyad has the DHA unit placed at the periphery of the SubPc, and this dyad showed instead limited photostability.
Design, System, Application
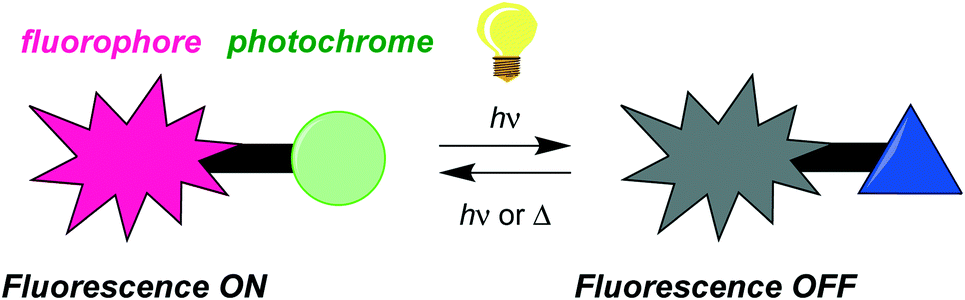
The molecular design covered in this work is based on the ability to modulate the fluorescence of a fluorophore by the reversible switching of a photochromic unit. The photochromic unit can exist in two isomeric forms, and for the concept to work, one form should not alter the emission, while the other should efficiently quench it. This may be achieved if the electronic properties of the two forms are very different, and if one of them has an absorption overlapping with the emission of the fluorophore. Specifically, the boron subphthalocyanine fluorophore was here functionalized with a dihydroazulene photoswitch. This functionalization leaves the fluorescence of the subphthalocyanine unaltered, but upon photoisomerization of the dihydroazulene into a vinylheptafulvene, the fluorescence is almost completely quenched. The vinylheptafulvene will in the dark thermally return to the more stable dihydroazulene isomer, thereby restoring the fluorescence. For these isomerizations to occur reversibly, there are some constraints on the system – the dihydroazulene needs to be placed at the axial position at the boron atom of the subphthalocyanine unit rather than at the peripheral position. Future application potentials of such fluorophore–photochrome dyads are as optical memory devices, systems for bioimaging, or light-controllable biological markers.
|
Introduction
Design of fluorophores for which fluorescence can be turned on and off via the state of a covalently attached photochromic unit has attracted significant interest in recent years as they have potential applications in optical memory devices, bioimaging, and as light-controllable biological markers.1 In such systems, schematically presented in Scheme 1, the emission of the fluorophore unit is usually quenched by one of the isomeric forms of the photochromic unit. When there is overlap between the fluorophore emission and the generated photoisomer absorption, quenching may occur by energy transfer. If the generated photoisomer is a strong electron acceptor, quenching may occur instead by an electron transfer mechanism.
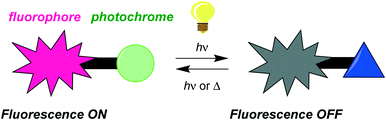 |
| | Scheme 1 Schematic illustration of fluorescence modulation by photoisomerization of a photochromic unit linked to a fluorophore. | |
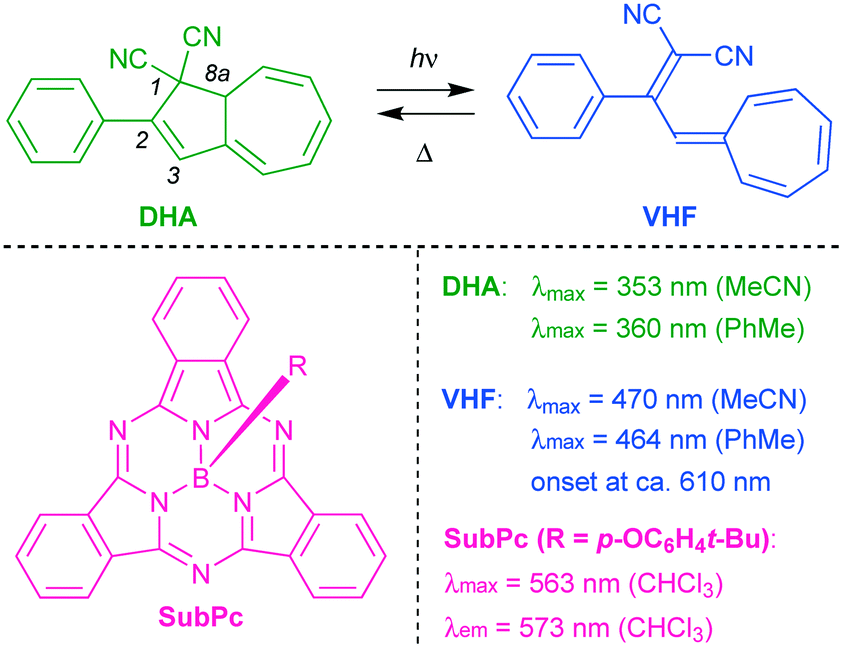
To mention a few examples, dithienylethene photoswitches have been used to modulate the fluorescence of naphthalimide,2 of perylene bisimide,3 of tetraphenylethylene,4 of Zn(II) based coordination polymers consisting of fluorescent ligands,5 and of a nanoassembly incorporating lanthanide-doped nanoparticles;6 spiropyrans the fluorescence of coumarin7 and of dipyrromethene;8 an imidazole dimer the fluorescence of fluorescein;9 and a dihydroazulene the fluorescence of boron dipyrromethene.10 The dihydroazulene (DHA) photoswitch mentioned in the last example undergoes a photoisomerization to a vinylheptafulvene (VHF) as shown in Scheme 2, which thermally reverts to DHA.11 The VHF exhibits a redshifted longest-wavelength absorption maximum, which overlaps with the boron dipyrromethene emission and thereby causes quenching of the fluorescence by energy transfer.10
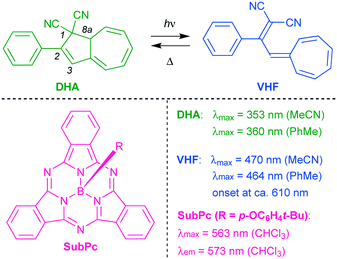 |
| | Scheme 2 Dihydroazulene (DHA)/vinylheptafulvene (VHF) photo-/thermoswitch and general structure of boron subphthalocyanine (SubPc) with an axial substituent R. Absorption and emission data are listed (taken from ref. 13 and 14). | |
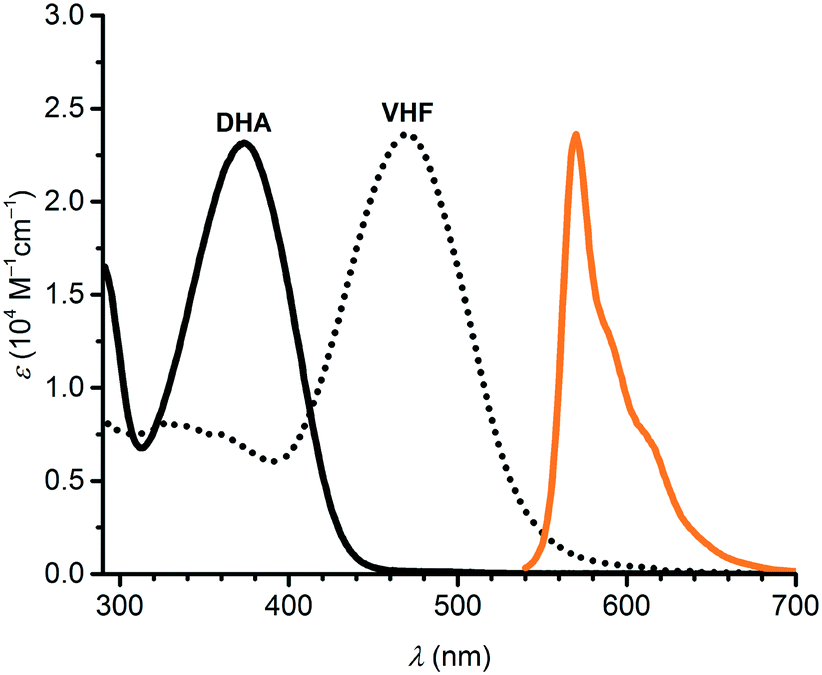
Boron subphthalocyanines (SubPcs; Scheme 2) present a class of fluorophores with properties being attractive for photovoltaics, organic light-emitting diodes, optical imaging, and photodynamic therapy applications.12 SubPcs are synthetically easily accessible12 and can be functionalized at the peripheral positions or at the axial boron position, which allows, as we shall show in this work, for the introduction of a photochrome at various locations in space relative to the SubPc. SubPcs with no peripheral substituents usually emit with a maximum around 570–575 nm12b (for R = p-OC6H4t-Bu: 573 nm; fluorescence quantum yield of 30% in CHCl3 (ref. 13)). While DHA absorbs at a maximum of 360 nm in PhMe, VHF exhibits a broad absorption with maximum at 464 nm and onset around 610 nm,14 and its absorption profile would thereby overlap with the emission of SubPc. For this reason, we imagined that the DHA-to-VHF photoisomerization could be employed to control the SubPc fluorescence. Moreover, DHA–VHF isomerizations can conveniently be performed quantitatively in both directions, with no photostationary state involved in the photoisomerization (characterized by a quantum yield of 60% in PhMe15). In regard to the attractive use of SubPc as fluorophore, it is worth mentioning that the cone structure of the SubPc unit provides a potential host site for fullerene electron acceptors in the solid or solution states,16 thereby offering an alternative “input” for reversibly quenching the fluorescence and hence for constructing advanced devices. Moreover, synthetic methods are available for constructing oligomeric SubPc systems with novel optical properties12b that could be interesting as well to modulate by one or more photochromes. Here we present new SubPc–DHA conjugates and fluorescence modulation via DHA–VHF conversions.
Results and discussion
Synthesis
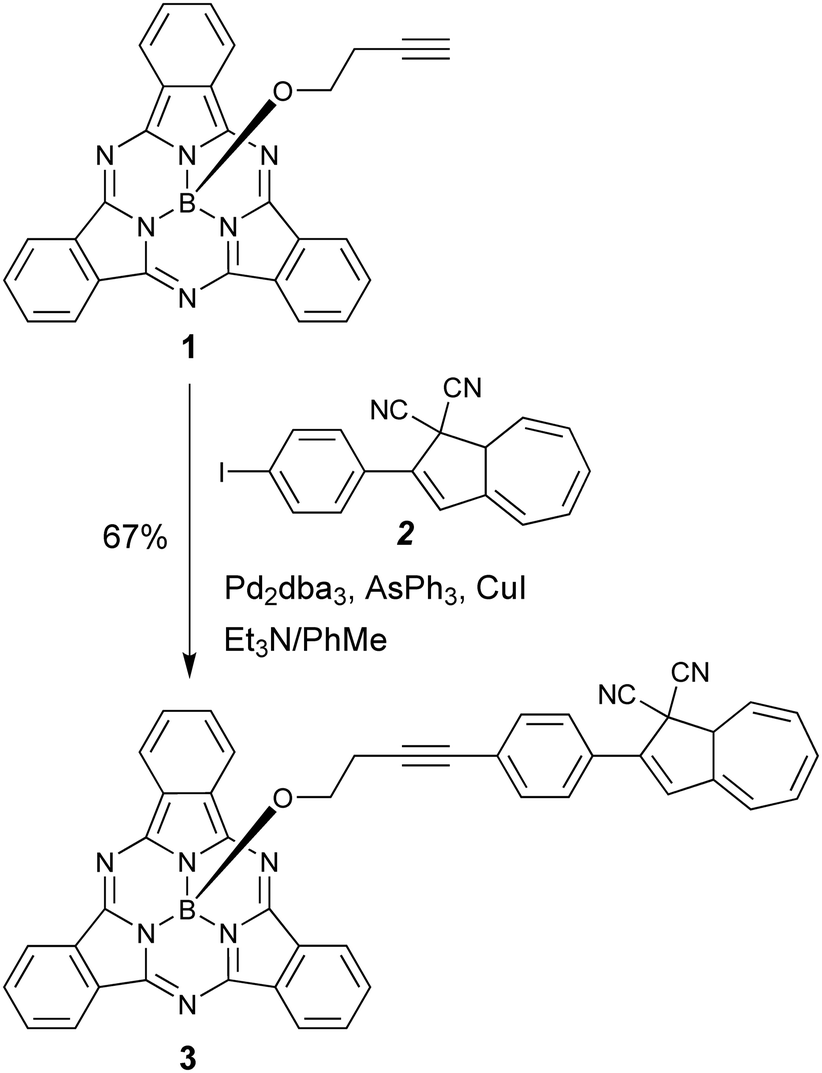
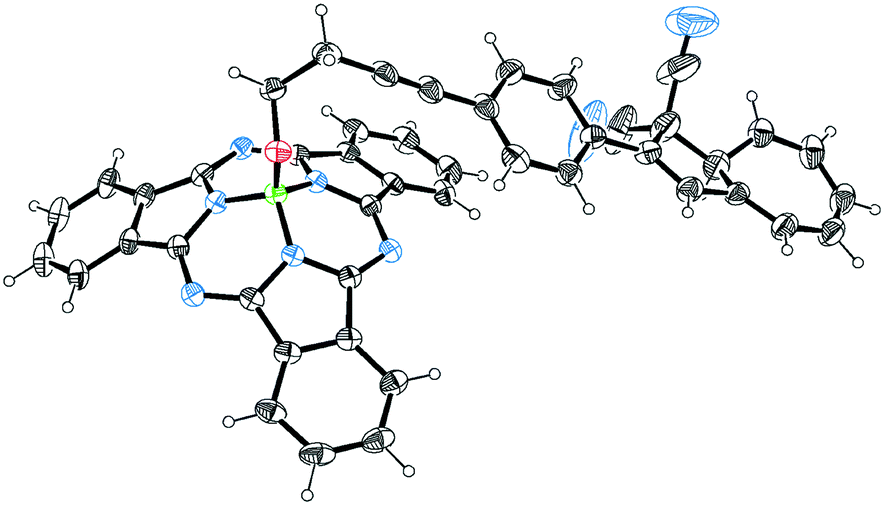
Our first objective was to introduce a DHA at the axial position of SubPc. We have recently reported the SubPc building block 1 (Scheme 3) with a terminal alkyne at the axial position and shown that it can be used as a coupling partner in Sonogashira reactions.17 Treatment of this compound with the known18 iodo-functionalized DHA 2 using Pd2dba3/AsPh3 and CuI as catalyst system in triethylamine and toluene gave the axial SubPc–DHA 3 in good yield. The structure of this compound was confirmed by X-ray crystallographic analysis (Fig. 1). The 1H-NMR spectrum of 3 reveals that the OCH2 protons are significantly shielded (resonating around 1.7 ppm), which can be explained by the diatropic ring current of the 14 π-aromatic SubPc and the location of the OCH2 protons close to the center of the ring.
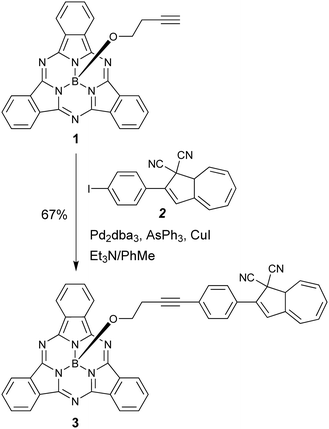 |
| | Scheme 3 Synthesis of axial SubPc–DHA dyad. dba = dibenzylidene acetone. | |
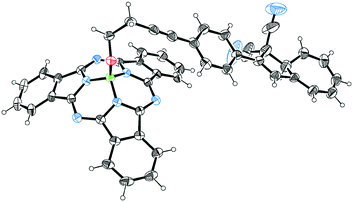 |
| | Fig. 1 Molecular structure of 3 obtained by X-ray crystallographic analysis. Crystals grown from a bilayer of CH2Cl2/heptane. Ellipsoids displayed at 30% probability. CCDC 1861766. | |
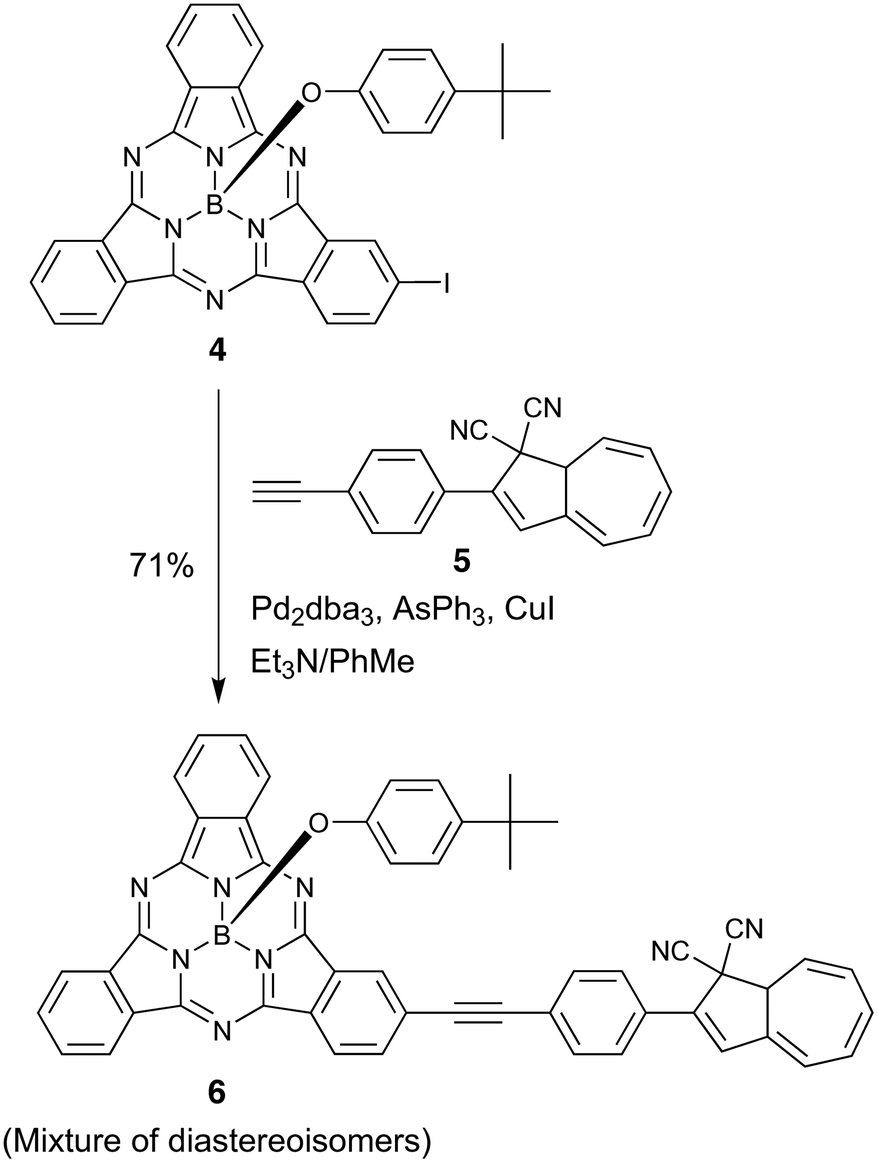
Next, we incorporated DHA at the peripheral position of SubPc by starting out with the known19 iodo-SubPc 4 (Scheme 4). A Sonogashira coupling with the known20 ethynylated DHA 5 gave the peripheral SubPc–DHA 6 as a mixture of diastereoisomers – owing to the presence of a stereocenter in both starting materials (C-8a of DHA 5 and the tetrahedral boron atom of SubPc 4).
 |
| | Scheme 4 Synthesis of peripheral SubPc–DHA dyad. | |
Optical properties and switching studies
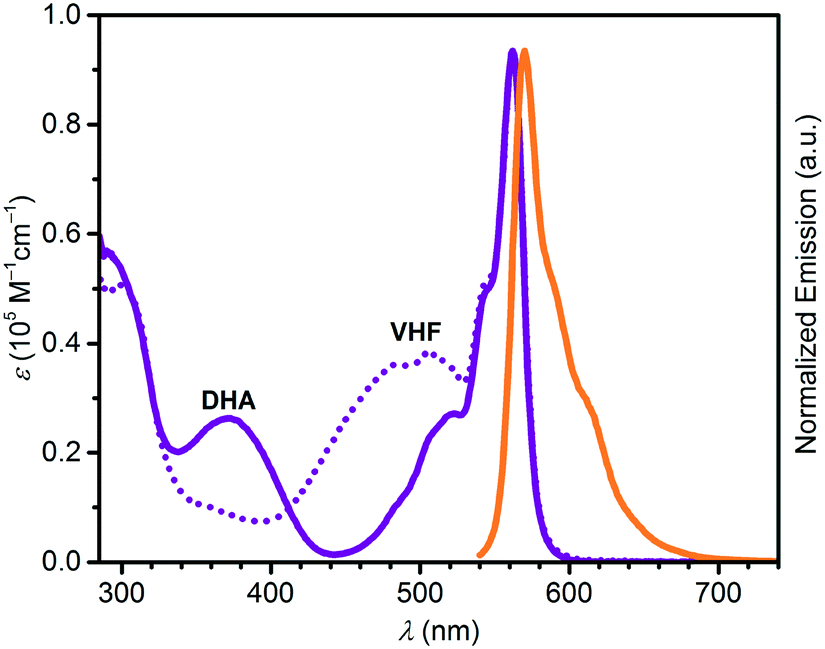
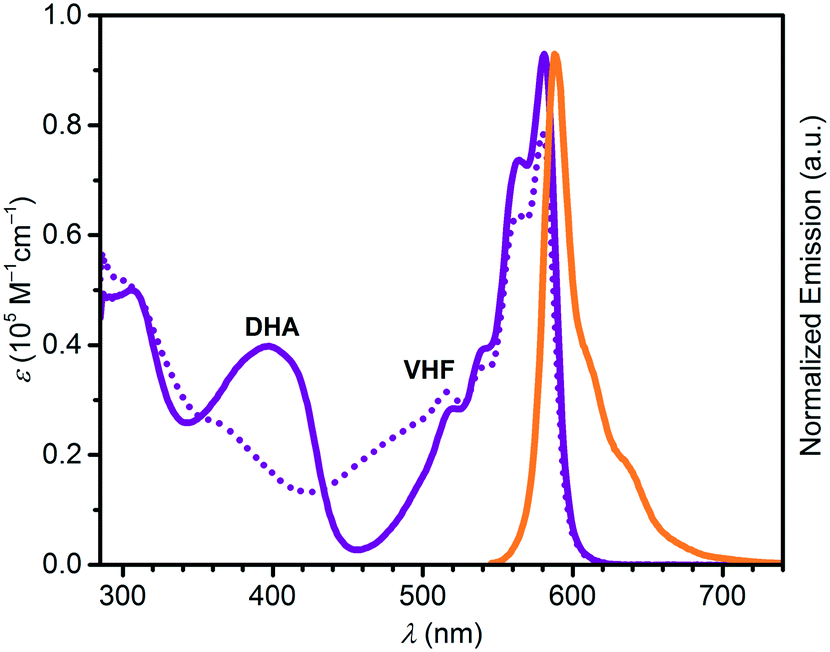
Absorption maxima of SubPc–DHA conjugates 3 and 6 in toluene and of their isomeric VHF forms (generated upon irradiation, vide infra) are compiled in Table 1. The absorption spectra are shown in Fig. 2 and 3 together with the emission spectra of the SubPc–DHA isomers. The two conjugates exhibit the characteristic SubPc absorption maxima at 562 nm (3) and 581 nm (6) and emission maxima at 570 nm (3) and 588 nm (6); they thus exhibit Stokes shifts of 7–8 nm. The maxima are redshifted for 6 on account of the extended π-conjugation at the SubPc periphery. A fluorescence quantum yield of 0.30 in toluene was determined for dyad 3 (see ESI†), which is identical to that obtained for the simple SubPc shown in Scheme 2 (with R = p-OC6H4t-Bu) in chloroform.13 Thus, the DHA unit has no significant influence on the SubPc fluorescence. The characteristic DHA absorptions are observed at 371 nm for 3 and at 398 nm for the larger conjugated π-system 6. The characteristic VHF absorptions are observed at 485 nm for 3 and as a shoulder around 482 nm for 6; in both cases these bands overlap with absorptions of the SubPc chromophore.
Table 1 UV-vis absorption data in PhMe. sh = shoulder
| Compound |
λ
max (nm) (ε (103 M−1 cm−1)) |
|
3-DHA
|
562 (93.5) |
545sh (49.8) |
523 (27.1) |
371 (26.3) |
294sh (55.6) |
— |
|
3-VHF
|
562 (92.4) |
545sh (51.6) |
505 (38.3) |
485 (36.0) |
299 (50.7) |
— |
|
6-DHA
|
581 (93.0) |
564 (73.7) |
539sh (39.0) |
522sh (28.4) |
398 (39.8) |
307 (50.0) |
|
6-VHF
|
580 (78.6) |
564 (63.9) |
539sh (35.9) |
516 (31.5) |
363sh (25.9) |
306sh (50.6) |
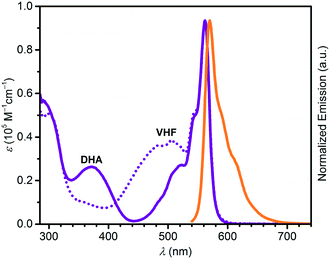 |
| | Fig. 2 UV-vis absorption spectra of 3-DHA (purple) and 3-VHF (purple, dotted), and normalized emission spectrum of 3-DHA (orange) after excitation at 530 nm. All spectra recorded in toluene. | |
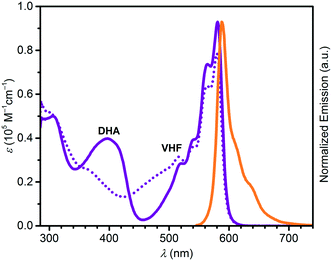 |
| | Fig. 3 UV-vis absorption spectra of 6-DHA (purple) and 6-VHF (purple, dotted), and normalized emission spectrum of 6-DHA (orange) after excitation at 530 nm. All spectra recorded in toluene. Decomposition seems to occur of the SubPc unit during ring-opening as indicated by the decrease in the SubPc absorption at 580 nm. | |
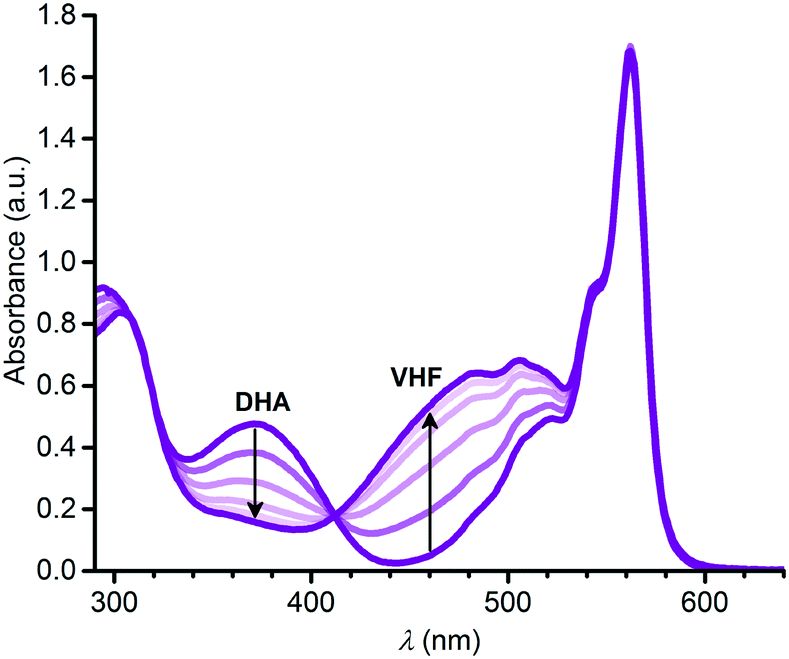
The VHF form of 3, 3-VHF, was formed by irradiation of 3 in toluene with 420 nm light; this wavelength was chosen as lower wavelengths closer to the DHA absorption maximum seemed to cause degradation. The conversion was observed by a decrease in the characteristic DHA absorption and an increase in the characteristic VHF absorption as shown in Fig. 4. The VHF returned to DHA in the dark, in this case also with isosbestic points in the absorption spectra (see ESI,† Fig. S10). By fitting the VHF decay in absorbance at 460 nm over time at 25 °C by an exponential function (ESI,† Fig. S12), we find a half-life for the VHF-to-DHA conversion of 1503 min, close to that obtained previously for the “parent VHF” shown in Fig. 2 (1474 min in toluene).14
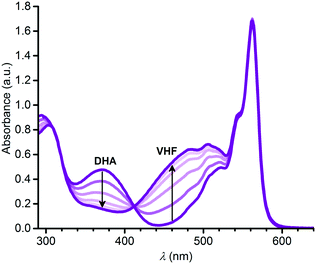 |
| | Fig. 4 Change in absorption spectra upon converting 3-DHA to 3-VHF in toluene by irradiation at 420 nm. | |
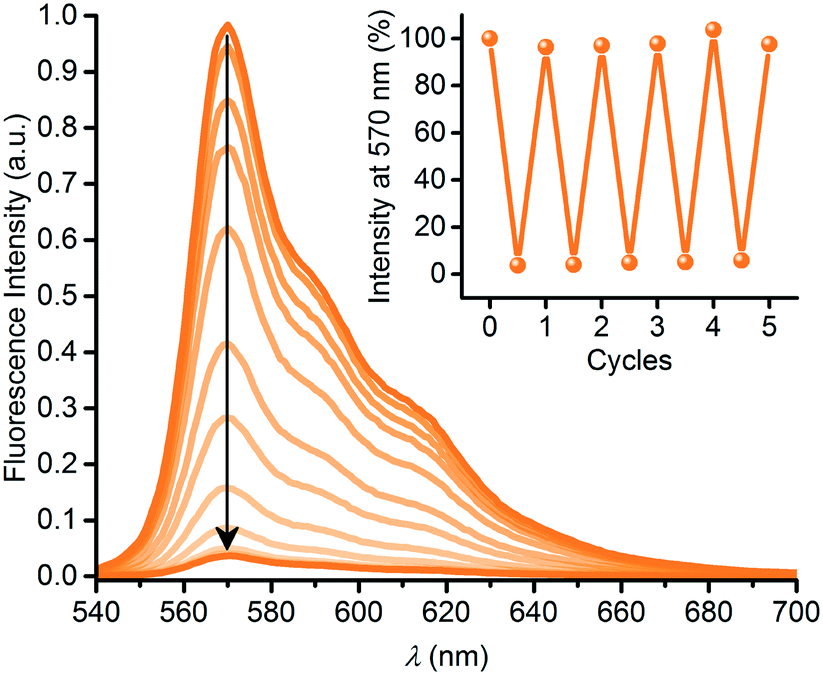
The DHA–VHF conversions of 3 had significant impact on the fluorescence properties. Thus, the SubPc emission decreased in intensity as shown in Fig. 5 during conversion of 3-DHA to 3-VHF, and the final product is almost non-fluorescent (while the initial 3-DHA has a fluorescence quantum yield of 0.30 in toluene). Heating the sample in toluene at 60 °C for a day, causing VHF-to-DHA isomerization, fully restored the fluorescence. As shown in the inset of Fig. 5, the fluorescence could reversibly be turned on and off over several successive irradiation-heating cycles.
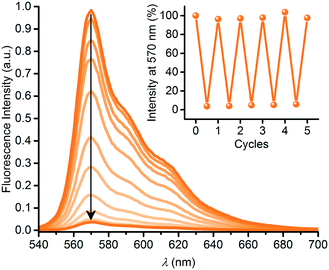 |
| | Fig. 5 Fluorescence quenching upon 3-DHA-to-3-VHF conversion by irradiation at 420 nm for 0 to 14 minutes. Inset: Fluorescence switching over five cycles. For each cycle, fluorescence was restored by heating (60 °C) the sample in the dark for 14–26 hours. | |
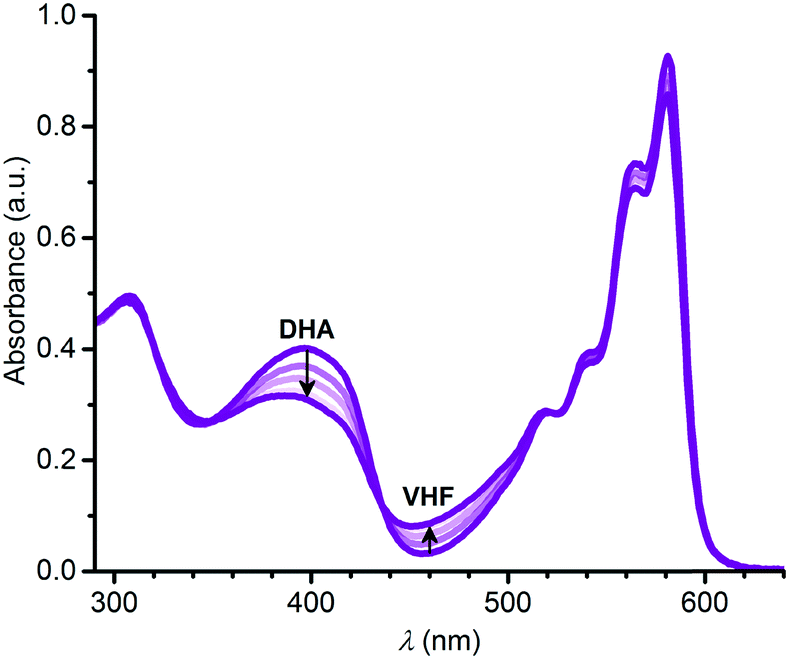
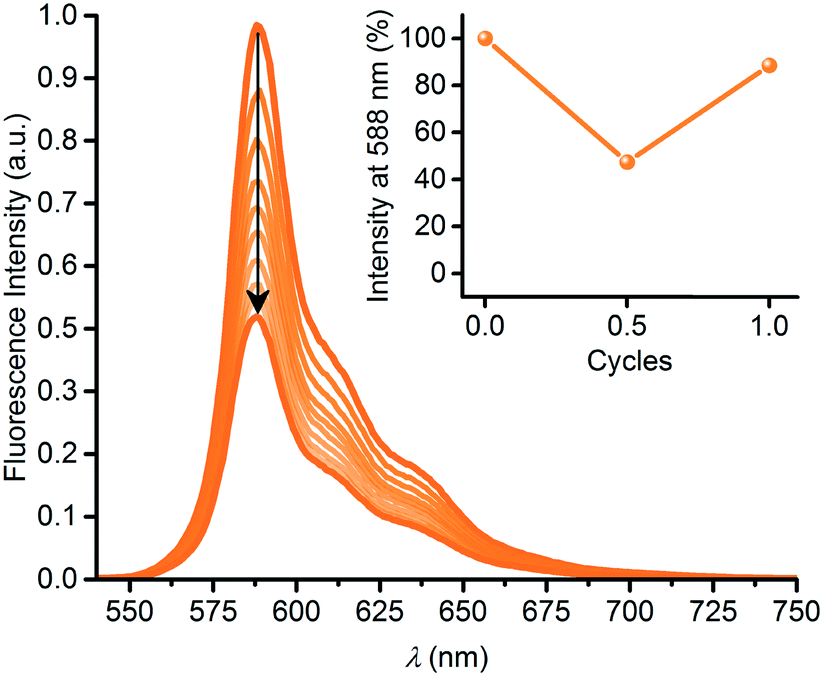
In a similar manner, we subjected the dyad 6 to DHA–VHF isomerizations, but, unfortunately, the photoisomerization occurred in this case with some degradation as clean isosbestic points were not observed (Fig. 6). Ignoring this issue, we find for the thermal back-reaction of 6-VHF (plus degradation products) a half-life of 1226 min at 25 °C (see ESI,† Fig. S11 and S13). This smaller half-life may be explained by the electron-withdrawing character of the SubPc-ethynyl substituent, as electron-withdrawing groups at this position of the VHF are known to enhance the ring-closure reaction.21 Similarly, we observe degradation when following the change in fluorescence of 6 upon irradiation. Moreover, the DHA-to-VHF required irradiation for a longer time (almost full conversion after 525 min, while it only took 14 min for 3-DHA under similar conditions). As was the case for 3, formation of the 6-VHF isomer resulted in reduced SubPc emission (although not as significantly as for the other dyad), but the original fluorescence was not fully restored after one cycle (Fig. 7).
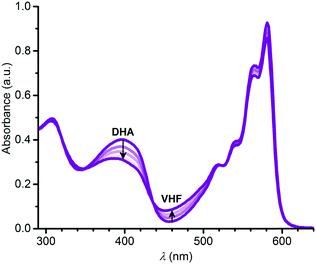 |
| | Fig. 6 Change in absorption spectra upon converting 6-DHA to 6-VHF in toluene by irradiation at 420 nm. | |
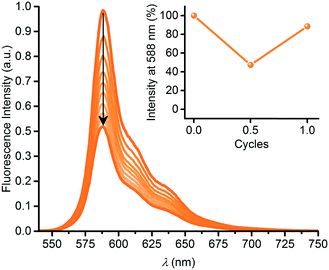 |
| | Fig. 7 Fluorescence quenching upon 6-DHA-to-6-VHF conversion by irradiation at 420 nm for 0 to 525 minutes. Inset: fluorescence switching over 1 cycle. Fluorescence was restored by heating (60 °C) the sample in the dark for 13 hours. Decomposition to 88% based on fluorescence after just one cycle. | |
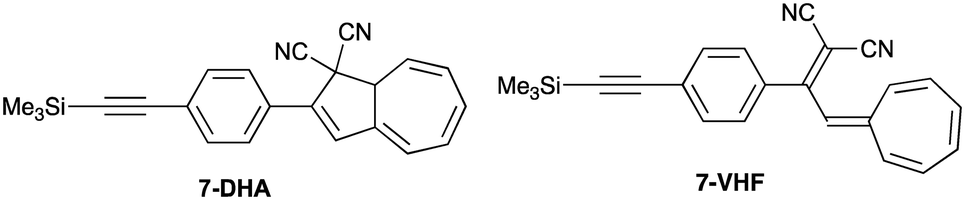
The photostability of the axial SubPc–DHA 3 provides superior properties in comparison to the peripheral SubPc–DHA 6. The efficient quenching of 3's fluorescence by conversion to the SubPc–VHF is likely due to an energy transfer mechanism (either Dexter or Förster). The distance between the two units is below 10 Å and there is a small overlap between the SubPc emission and VHF absorption spectra; the absorption spectrum of the related 7-VHF (generated from the known207-DHA; Fig. 8), being structurally similar to the VHF chromophore unit of 3-VHF, and the emission spectrum of 3 are shown in Fig. 9. A competing quenching by electron transfer cannot, however, be outruled, but the energetics is not trivial to calculate due to irreversible or partly reversible redox properties of the entities13,22 and an unknown distance between charges after electron transfer.
 |
| | Fig. 8 Reference compounds. | |
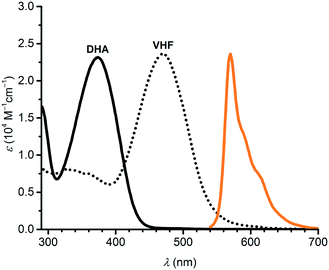 |
| | Fig. 9 Absorption spectra of 7-DHA and 7-VHF in toluene. The emission spectrum of 3 is shown in orange color (arbitrary units). | |
Experimental
General methods
All reagents and solvents were obtained from commercial suppliers and used as received unless otherwise stated. Purification by column chromatography was carried out on silica gel (SiO2, 60 Å, 40–63 μm). Thin-layer chromatography (TLC) was carried out using commercially available aluminum sheets precoated with silica gel with fluorescence indicator and visualized under UV light at 254 or 360 nm. 1H and 13C NMR spectra were recorded on a 500 MHz instrument equipped with a noninverse cryoprobe at 500 MHz and 126 MHz, respectively. 11B NMR spectra were recorded on a 500 MHz instrument equipped with a broad-band probe. Chemical shift values are quoted in ppm and coupling constants (J) in Hz. 1H and 13C NMR spectra are referenced against the residual solvent peak (CDCl3δH = 7.26 ppm, δC = 77.16 ppm). 11B NMR spectra are referenced against an external standard of BF3 diethyl etherate (BF3·(OC2H5)2; δB = 0 ppm). HRMS MALDI spectra were recorded on a FT-ICR instrument equipped with a 7 T magnet (prior to the experiments, the instrument was calibrated using NaTFA cluster ions). All melting points are uncorrected. UV-vis absorption measurements were performed in a 1 cm path-length cuvette, and the neat solvent was used as baseline; sh = shoulder. For irradiation studies, the lamp used was an ozon free 450 W Xe-lamp connected to a monochromator. Fluorescence measurements were carried out in a 1 cm path-length cuvette on samples with absorbances below 0.1 units. For details on fluorescence quantum yield measurements, see ESI.†
SubPc–DHA 3
Degassed Et3N (0.10 mL) was added to an argon-purged suspension of 1 (62 mg, 0.13 mmol), 2 (77 mg, 0.20 mmol), Pd2dba3 (12 mg, 0.013 mmol), CuI (2.5 mg, 0.013 mmol) and AsPh3 (33 mg, 0.11 mmol) in PhMe (7 mL). The reaction mixture was stirred at room temperature in the dark overnight, filtered through a plug of SiO2 (EtOAc) and concentrated under reduced pressure. Purification by flash column chromatography (SiO2, 30% EtOAc/heptane) afforded the title compound (64 mg, 67%) as a golden-brown solid. Rf (30% EtOAc/heptane) = 0.24. M.p. > 230 °C. UV-vis (toluene) λmax (ε) 290 (57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), 371 (26300), 523 (27100), 546sh (50100), 562 (93500) nm (M−1 cm−1). 1H NMR (500 MHz, CDCl3) δ 8.82 (dd, J = 5.9, 3.0 Hz, 6H), 7.88 (dd, J = 5.9, 3.0 Hz, 6H), 7.58 (d, J = 8.5 Hz, 2H), 7.22 (d, J = 8.5 Hz, 2H), 6.88 (s, 1H), 6.57 (dd, J = 11.3, 6.3 Hz, 1H), 6.48 (dd, J = 11.3, 6.1 Hz, 1H), 6.35 (br d, J = 6.3 Hz, 1H), 6.32 (ddd, J = 10.1, 6.1, 2.0 Hz, 1H), 5.84 (dd, J = 10.1, 3.8 Hz, 1H), 3.81 (dt, J = 3.8, 2.0 Hz, 1H), 1.74–1.67 (m, 2H), 1.67–1.62 (m, 2H) ppm. 13C NMR (126 MHz, CDCl3) δ 151.7, 139.8, 138.8, 132.6, 132.4, 131.1, 131.1, 129.9, 129.3, 127.8, 126.0, 125.6, 122.2, 121.3, 119.7, 115.3, 112.8, 90.0, 80.5, 58.0, 51.3, 45.2, 22.2 ppm (one signal coincident or not observed). 11B NMR (160 MHz, CDCl3) δ −15.1 ppm. HRMS (MALDI): m/z = 719.2474 [M + H+], calc. for (C46H28BN8O+) m/z = 719.2489. Crystals suitable for X-ray crystallographic analysis were grown from a bilayer of CH2Cl2 and heptane.
000), 371 (26300), 523 (27100), 546sh (50100), 562 (93500) nm (M−1 cm−1). 1H NMR (500 MHz, CDCl3) δ 8.82 (dd, J = 5.9, 3.0 Hz, 6H), 7.88 (dd, J = 5.9, 3.0 Hz, 6H), 7.58 (d, J = 8.5 Hz, 2H), 7.22 (d, J = 8.5 Hz, 2H), 6.88 (s, 1H), 6.57 (dd, J = 11.3, 6.3 Hz, 1H), 6.48 (dd, J = 11.3, 6.1 Hz, 1H), 6.35 (br d, J = 6.3 Hz, 1H), 6.32 (ddd, J = 10.1, 6.1, 2.0 Hz, 1H), 5.84 (dd, J = 10.1, 3.8 Hz, 1H), 3.81 (dt, J = 3.8, 2.0 Hz, 1H), 1.74–1.67 (m, 2H), 1.67–1.62 (m, 2H) ppm. 13C NMR (126 MHz, CDCl3) δ 151.7, 139.8, 138.8, 132.6, 132.4, 131.1, 131.1, 129.9, 129.3, 127.8, 126.0, 125.6, 122.2, 121.3, 119.7, 115.3, 112.8, 90.0, 80.5, 58.0, 51.3, 45.2, 22.2 ppm (one signal coincident or not observed). 11B NMR (160 MHz, CDCl3) δ −15.1 ppm. HRMS (MALDI): m/z = 719.2474 [M + H+], calc. for (C46H28BN8O+) m/z = 719.2489. Crystals suitable for X-ray crystallographic analysis were grown from a bilayer of CH2Cl2 and heptane.
DHA–SubPc 6 (mixture of diastereoisomers)
A degassed mixture of Et3N/toluene 1:3 (4 mL) was added to an argon-purged reaction vessel charged with 4 (52 mg, 0.078 mmol), 5 (28 mg, 0.10 mmol), Pd2dba3 (18 mg, 0.019 mmol), AsPh3 (49 mg, 0.16 mmol) and CuI (4.0 mg, 0.021 mmol). The reaction mixture was stirred at room temperature for 4 hours in the dark, diluted with EtOAc (4 mL), filtered through a plug of SiO2 (EtOAc/toluene 1:1) and concentrated in vacuo. The residue was dissolved in a small volume of toluene and subjected to flash column chromatography (SiO2, 35% EtOAc/heptane) followed by size-exclusion chromatography (S-X3 Biobeads, CH2Cl2) to afford the title compound (45 mg, 71%) as a golden-brown solid. Rf (35% EtOAc/heptane) = 0.35. M.p. > 230 °C. UV-vis (toluene) λmax (ε) 306 (50000), 398 (39800), 522sh (28400), 543sh (39400), 564 (73700), 581 (93000) nm (M−1 cm−1). 1H NMR (500 MHz, CDCl3) δ 9.05 (s, 1H), 8.90–8.77 (m, 5H), 8.02 (dd, J = 8.2, 1.4 Hz, 1H), 7.94–7.87 (m, 4H), 7.78 (d, J = 8.6 Hz, 2H), 7.71 (d, J = 8.6 Hz, 2H), 6.95 (s, 1H), 6.77 (d, J = 8.8 Hz, 2H), 6.54 (ddd, J = 10.7, 6.3, 3.7 Hz, 1H), 6.46 (ddd, J = 10.7, 6.0, 3.9 Hz, 1H), 6.36 (d, J = 6.3 Hz, 1H), 6.33–6.28 (m, 1H), 5.83 (dd, J = 10.1, 3.8 Hz, 1H), 5.32 (d, J = 8.8 Hz, 2H), 3.81 (dt, J = 3.8, 1.9 Hz, 1H), 1.08 (s, 9H) ppm. 13C NMR (126 MHz, CDCl3) δ 152.39, 151.82, 151.81, 150.66, 150.65, 150.14, 143.81, 139.45, 138.62, 133.24, 132.63, 132.58, 131.34, 131.30, 131.17, 131.13, 130.99, 130.98, 130.94, 130.58, 130.21, 130.19, 130.09, 130.00, 127.87, 126.37, 125.91, 125.86, 124.74, 124.31, 122.45, 122.38, 122.29, 121.71, 119.64, 117.85, 115.17, 112.75, 92.42, 91.50, 51.27, 45.19, 33.98, 31.46 (four signals coincident or not observed) ppm. 11B NMR (160 MHz, CDCl3) δ −15.0 ppm. HRMS (MALDI+): m/z = 823.3093 [M + H+], calc. for (C54H36BN8O+) m/z = 823.3100.
Conclusions
In conclusion, two SubPc–DHA conjugates were prepared with the DHA unit placed at the axial and peripheral position of SubPc, respectively. While the photostability of the peripherally substituted SubPc was poor, the axially substituted one exhibited good photostability above 420 nm, allowing for reversible DHA–VHF isomerizations. The photoisomerization of DHA into VHF was found to quench almost completely the fluorescence of the SubPc fluorophore, while the DHA isomer had a fluorescence similar to that of a simple SubPc with a tert-butylphenoxy as axial substituent. Thus, this SubPc–DHA presents a convenient system for reversibly modulating the fluorescence of SubPc by consecutive light and heat stimuli. In future work it could be interesting to combine the fluorescence modulation with binding of a guest molecule with specific optical and redox properties in the SubPc cavity by virtue of π–π interactions. Such complex formation may offer an alternative way of altering the fluorescence or it may instead influence the photoactivity of the DHA unit.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
University of Copenhagen is acknowledged for support.
Notes and references
-
(a) F. M. Raymo and M. Tomasulo, J. Phys. Chem. A, 2005, 109, 7343–7352 CrossRef CAS PubMed;
(b) I. Yildiz, E. Deniz and F. M. Raymo, Chem. Soc. Rev., 2009, 38, 1859–1867 RSC;
(c) T. Fukaminato, J. Photochem. Photobiol., C, 2011, 12, 177–208 CrossRef CAS.
- G. Jiang, S. Wang, W. Yuan, L. Jiang, Y. Song, H. Tian and D. Zhu, Chem. Mater., 2006, 18, 235–237 CrossRef CAS.
- T. Fukaminato, T. Doi, N. Tamaoki, K. Okuno, Y. Ishibashi, H. Miyasaka and M. Irie, J. Am. Chem. Soc., 2011, 133, 4984–4990 CrossRef CAS PubMed.
- J. Qi, C. Chen, X. Zhang, X. Hu, S. Ji, R. T. K. Kwok, J. W. Y. Lam, D. Ding and B. Z. Tang, Nat. Commun., 2018, 9, 1848 CrossRef.
- J. Kim, Y. You, S.-J. Yoon, J. H. Kim, B. Kang, S. K. Park, D. R. Whang, J. Seo, K. Cho and S. Y. Park, Chem. – Eur. J., 2017, 23, 10017–10022 CrossRef CAS PubMed.
- T. Wu, D. Wilson and N. R. Branda, Chem. Mater., 2014, 26, 4313–4320 CrossRef CAS.
- E. Deniz, S. Sortino and F. M. Raymo, J. Phys. Chem. Lett., 2010, 1, 3506–3509 CrossRef CAS.
- M. Tomasulo, E. Deniz, R. J. Alvarado and F. M. Raymo, J. Phys. Chem. C, 2008, 112, 8038–8045 CrossRef CAS.
- K. Mutoh, M. Sliwa and J. Abe, J. Phys. Chem. C, 2013, 117, 4808–4814 CrossRef CAS.
- C. Trieflinger, H. Röhr, K. Rurack and J. Daub, Angew. Chem., Int. Ed., 2005, 44, 6943–6947 CrossRef CAS PubMed.
- J. Daub, T. Knöchel and A. Mannschreck, Angew. Chem., Int. Ed. Engl., 1984, 23, 960–961 CrossRef.
-
(a) G. E. Morse, M. G. Helander, J. F. Maka, Z.-H. Lu and T. P. Bender, ACS Appl. Mater. Interfaces, 2010, 2, 1934–1944 CrossRef CAS;
(b) C. G. Claessens, D. González-Rodríguez, M. S. Rodríguez-Morgade, A. Medina and T. Torres, Chem. Rev., 2014, 114, 2192–2277 CrossRef CAS PubMed;
(c) M. E. El-Khouly, A. El-Refaey, W. Nam, S. Fukuzumi, Ö. Göktug and M. Durmus, Photochem. Photobiol. Sci., 2017, 16, 1512–1518 RSC;
(d) E. van de Winckel, M. Mascaraque, A. Zamarrón, Á. J. de la Fuente, T. Torres and A. de la Escosura, Adv. Funct. Mater., 2018, 28, 1705938 CrossRef;
(e) R. K. Garner, D. S. Josey, S. R. Nyikos, A. Dovijarski, J. M. Wang, G. J. Evans and T. P. Bender, Sol. Energy Mater. Sol. Cells, 2018, 176, 331–335 CrossRef CAS;
(f) H. Gotfredsen, T. Neumann, F. E. Storm, A. V. Muñoz, M. Jevric, O. Hammerich, K. V. Mikkelsen, M. Freitag, G. Boschloo and M. B. Nielsen, ChemPhotoChem, 2018, 2, 976–985 CrossRef CAS.
- H. Gotfredsen, F. E. Storm, A. V. Muñoz, M. Santella, A. Kadziola, O. Hammerich, K. V. Mikkelsen and M. B. Nielsen, Chem. – Eur. J., 2017, 23, 16194–16198 CrossRef CAS PubMed.
- S. L. Broman, S. L. Brand, C. R. Parker, M. Å. Petersen, C. G. Tortzen, A. Kadziola, K. Kilså and M. B. Nielsen, ARKIVOC, 2011, ix, 51–67 Search PubMed.
- H. Görner, C. Fischer, S. Gierisch and J. Daub, J. Phys. Chem., 1993, 97, 4110–4117 CrossRef.
- H. Gotfredsen, T. Holmstrøm, A. V. Muñoz, F. E. Storm, C. G. Tortzen, A. Kadziola, K. V. Mikkelsen, O. Hammerich and M. B. Nielsen, Org. Lett., 2018, 20, 5821–5825 CrossRef CAS PubMed , and references cited therein.
- H. Gotfredsen, M. Jevric, A. Kadziola and M. B. Nielsen, Eur. J. Org. Chem., 2016, 17–21 CrossRef CAS.
-
(a) L. Gobbi, P. Seiler and F. Diederich, Angew. Chem., Int. Ed., 1999, 38, 674–678 CrossRef CAS;
(b) L. Gobbi, P. Seiler, F. Diederich, V. Gramlich, C. Boudon, J.-P. Gisselbrecht and M. Gross, Helv. Chim. Acta, 2001, 84, 743–777 CrossRef CAS.
- A. V. Muñoz, H. Gotfredsen, M. Jevric, A. Kadziola, O. Hammerich and M. B. Nielsen, J. Org. Chem., 2018, 83, 2227–2234 CrossRef PubMed.
- M. Santella, V. Mazzanti, M. Jevric, C. R. Parker, S. L. Broman, A. D. Bond and M. B. Nielsen, J. Org. Chem., 2012, 77, 8922–8932 CrossRef CAS PubMed.
- S. L. Broman, M. Jevric and M. B. Nielsen, Chem. – Eur. J., 2013, 19, 9542–9548 CrossRef CAS PubMed.
- S. Lara-Avila, A. V. Danilov, S. E. Kubatkin, S. L. Broman, C. R. Parker and M. B. Nielsen, J. Phys. Chem. C, 2011, 115, 18372–18377 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and UV-vis absorption spectra, fluorescence quantum yield determination, and kinetics studies. CCDC 1861766. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8me00075a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ,
Martin Drøhse
Kilde
,
Martin Drøhse
Kilde