Repertoire Builder: high-throughput structural modeling of B and T cell receptors†
Received
12th February 2019
, Accepted 21st May 2019
First published on 21st June 2019
Abstract
Repertoire Builder (https://sysimm.org/rep_builder/) is a method for generating atomic-resolution, three-dimensional models of B cell receptors (BCRs) or T cell receptors (TCRs) from their amino acid sequences. It is currently capable of handling batches of up to 104 sequences in approximately 30 minutes. This performance was achieved by applying a multiple sequence alignment extension technique originally developed for phylogenetic analysis to the template selection problem of complementarity determining region (CDR) loops. Under comparable conditions, average all-atom root-mean square deviations (RMSDs) from experimentally-determined structures of CDRH3 loops in BCRs were significantly lower than tested third-party high-throughput modeling methods, including ABodyBuilder, PigsPro, and LYRA. For TCRs, similar trends were observed when Repertoire Builder was compared with TCRmodel and LYRA. We also found that Repertoire Builder model errors were, in general, lower than those produced by our earlier Kotai Antibody Builder, even when CDRH3 loop refinement was used. However, in a subset of cases, which could be distinguished by poor Repertoire Builder scores, refinement by Kotai Antibody Builder or Rosetta Antibody, both of which utilize extensive structural sampling, improved the third heavy chain CDR (CDRH3) RMSD on average. Taken together, these results indicate that the MSA extension approach used by Repertoire Builder resulted in a favorable balance between speed and accuracy when compared to alternative methods. Furthermore, we conclude that more sensitive scoring, rather than extended structural sampling, is needed to further improve the accuracy of BCR and TCR modeling.
Design, System, Application
Repertoire Builder is a tool for building 3D models of B cell receptors (BCRs) or T cell receptors (TCRs) to atomic resolution. The strategy used by Repertoire Builder is an application of the multiple sequence alignment (MSA) extension feature of the MAFFT software. The particular application here is to represent structural templates for each complementarity-determining region (CDR) of a given length by a single MSA. By repeatedly applying the MSA extension method, a complete set of templates that covers the 3 CDRs and 1 framework for each chain can be obtained. The input must be either paired (heavy and light) or unpaired (heavy or light) chain amino acid sequence of the variable region for the receptor in question (BCR or TCR). The immediate application of Repertoire Builder is to render 3D models in a high-throughput and accurate manner, in order to allow structure-based analyses for BCR or TCR sequence data. Because the volume of such data is currently growing exponentially, Repertoire Builder represents a unique approach to large-scale BCR or TCR repertoire data analysis.
|
Introduction
Recent single-cell resolution sequencing technologies can elucidate the natively paired (heavy–light) B cell receptor (BCR) and T cell receptor (TCR) sequences in a high-throughput manner.1 Although the coverage has not yet reached that of bulk sequencing methods, state-of-the-art paired sequencing platforms can yield thousands of unique receptor sequences in a single experiment. Because of the critical role of B and T cells in the prevention or progression of disease, new methods to functionally analyze emerging sequence data are needed. Structural modeling can, in principle, contribute to such functional analysis, since structures allow physical and chemical concepts to be applied to the prediction of receptor-antigen molecular recognition2,3 or antibody developability.4 In practice, however, a major limitation with available BCR and TCR structural modeling methods is the tradeoff between speed and accuracy. In the most recent single-blind antibody modeling assessment (AMA-II),4 for example, all of the most accurate methods were inherently low throughput, requiring thousands of CPU hours, or more, to build a single atomic-resolution model.
Most of the computational cost in current high-resolution BCR or TCR modeling methods is due to extended structural sampling of loop conformations in complementarity-determining regions (CDRs). While CDRs do not make up the majority of BCR or TCR residues, they are the most important in terms of antigen recognition, and thus can be considered the most functionally important part of the structure. The problem of CDR modeling has been intensively studied for many years and recently a number of groups, including our own, have developed extended structural sampling methods to tackle the problem.5,6 While accuracy of CDRs is critical to a structural understanding of antigen and epitope specificity, the recent emergence of high-throughput TCR and BCR sequencing methods demands that high-throughput structural modeling methods also be developed if such methods are to keep up with the growth in data in the coming years. To date several high-throughput BCR modeling tools have been described, including PigsPro,18 ABodyBuilder17 and LYRA.19 Here, we sought to develop an efficient approach that would require only minimal structural sampling in the modeling process and would work for both BCRs and TCRs. To this end, we utilized a multiple sequence alignment (MSA) extension technique implemented in the software MAFFT,7 wherein a query sequence is added to a pre-aligned MSA.8 The original motivation of the MAFFT extension procedure was accurate phylogenetic inference. However, we show here that this approach also has an important application in BCR and TCR structural modeling. The individual steps of this approach are described in more detail in Methods.
Methods
Overview
The overall scheme used by Repertoire Builder is illustrated in Fig. 1. Our application of the MAFFT MSA extension utilizes known structures (templates) in a pre-aligned MSA. We combine all known structures containing a given CDR loop of a given length into a single MSA, which we refer to here as a template MSA. We also align templates for all non-CDR (i.e. “framework”) regions into a single MSA. Finally, we merge together heavy (beta) and light (alpha) chains into a single MSA, which is used as a template for heavy–light chain orientation (Fig. 1A). We separate CDR MSAs by loop length because, when we use them to predict the structure of a query sequence with unknown structure, we can first assign the lengths of the query CDRs then align these to a CDR template MSA such that there will be no gaps in the CDR part of the alignment (Fig. 1B). When a template MSA is extended by addition of a query sequence, the relationships between the pre-aligned templates remain unchanged (Fig. 1C). The extension procedure thus produces a set of query-template alignments with a common index (the MSA column) in a single step. In order to rank the templates within each MSA, each query-template alignment is expressed as a feature-vector, indexed by the MSA column, containing the pairwise alignment scores of each aligned residue pair. A scalar query-template score is computed by taking the dot-product of each feature vector with a weight vector where the weight vector represents the importance of each MSA column for the region of interest (Fig. 1D). We emphasize that, although each CDR template MSA corresponds to a given loop with a specified length, the MSA includes all the residues of the BCR or TCR variable region. This allows residues outside of the region of interest (e.g., CDR) to contribute to the score. The complete backbone structure is constructed by assembling the aligned CDR, framework and orientation templates using conserved anchor residues (Fig. 1E). Where needed (i.e., where query and template amino acids differ), side-chains are then replaced with those of the query using SCWRL4,9 which constitutes the only explicit structural sampling step in the procedure. Details of these steps are given below.
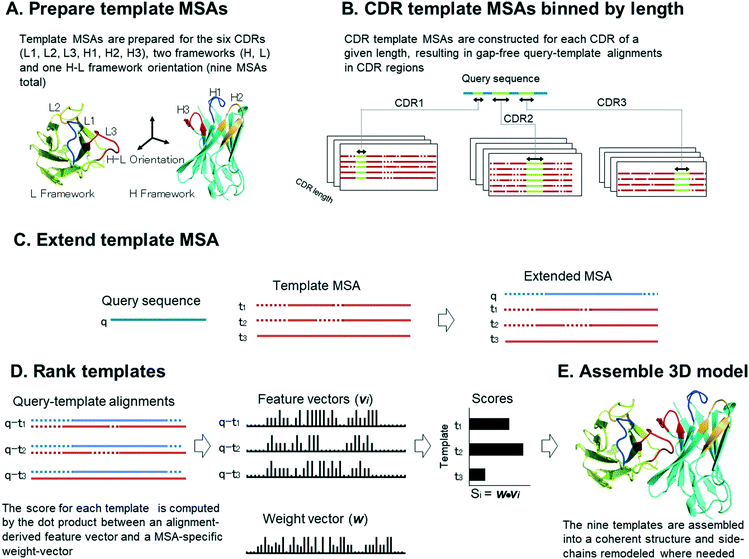 |
| | Fig. 1 Overview of Repertoire Builder template selection and modeling pipeline. | |
Structure data preparation
Crystal structures of each human, mouse or rat BCR or TCR receptor with resolution no worse than 3.0 Å were collected from PDB (April 25th, 2017). Receptor constant regions within these structures were removed. The PISCES program was then used to select a non-redundant set of variable domains at 99% sequence identity. For entries to be used as modelling templates, no missing C-alpha atoms were allowed. In addition, at least 4 residues (anchors) before CDR1 and after CDR3 were required. From such templates, those including no modified residues were used to optimize weight vectors for template selection.
Post-flu vaccination BCR sequences
9313 natively paired, full variable region antibody sequences were generated by applying Immune Repertoire Capture® technology10 to healthy donor peripheral blood mononuclear cells isolated and cryopreserved 1 week after administration of seasonal flu vaccine. 5743 paired sequences represented plasmablasts sorted as described previously11 and 3570 represented CD19 + B cells, which were cultured for 4 days in IMDM medium (Invitrogen) in the presence of FBS, Normocin, IL-2 (PeproTech), IL-21 (PeproTech), rCD40 ligand (R&D Systems), and His-Tag antibodies (R&D Systems), prior to single cell sorting. Sequence generation and annotation was as described previously.11
Template MSA construction
For each receptor type (BCR/TCR), variable domain MSAs were constructed for each heavy or light chain and for each region (CDRs 1–3 and framework), as well as for the heavy/light orientation (HL orientation). The sequences were multiply aligned by MAFFT using constraints derived from ASH12 pairwise structural alignments as described previously.7 For a given CDR, templates were binned according to the length of the CDR and only templates with a given CDR length were aligned. For the framework MSAs, a non-redundant set of PDB variable domains was clustered at 99% sequence identity. Orientation MSAs were constructed by concatenating the individual H and L alignments for a given template, as if they constituted a single chain. The definition of each CDR boundary was taken from Honegger and Pluckthun wherein the BCR CDR2 definition is approximately twice as long as the standard (e.g. IMGT13) definition, and includes the beta strand and loop following the CDR2 loop.7 This definition reflects the fact that the beta strand and loop are not structurally well-conserved and thus are not suitable as anchor points in the structural assembly step, as described below. Note that these non-standard definitions were only used for modeling; in all RMSD calculations standard IMGT definitions were used.
Query-template scoring
Given an MSA m(i,k), where i is an aligned sequence (row) and k is the alignment position (column), we defined the feature vector 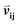 as vij(k) = B(m(i,k), m(j,k)) where B(a,b) is the BLOSUM62 matrix element for amino acids a and b after extension to include a constant gap penalty. We then defined the sequence similarity between sequences i and j as
as vij(k) = B(m(i,k), m(j,k)) where B(a,b) is the BLOSUM62 matrix element for amino acids a and b after extension to include a constant gap penalty. We then defined the sequence similarity between sequences i and j as 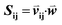 , where
, where 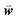 is a weight vector to be optimized to achieve the best agreement between Sij and the structural similarity of template sequences i and j for each MSA, as described below.
is a weight vector to be optimized to achieve the best agreement between Sij and the structural similarity of template sequences i and j for each MSA, as described below.
Weight vector optimization
First, the structural deviation between all templates for a given region was defined. For CDR templates within the same length bin, all pairs of CDR templates were superimposed by their anchor residues (4 framework residues before and after CDR); RMSDs were then computed from the corresponding C-alpha atoms of CDR pairs. For framework templates, structure pairs were superimposed using conserved framework residues (Table S1†); RMSDs were computed based on MSA-aligned C-alpha atoms of conserved framework residues. For weight vector training, structural similarity-based ranking was expressed in terms of ordered template triplets [Tr, Ti, Tj] according to the RMSDs above. Tr is a template designated as the reference, and templates (Ti, Tj) satisfy the requirement RMSD (Tr, Tj)–RMSD (Tr, Ti) > 0.1 Å. For template regions that had a large number of entries, down-sampling was applied to reduce the number of triplets to no more than 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000. The goal was to maximize the number of triplets for which Sri − Srj > 0. The input features
000. The goal was to maximize the number of triplets for which Sri − Srj > 0. The input features 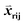 consisted of the differences between two feature vectors for each template triplet. That is, for [Tr, Ti, Tj],
consisted of the differences between two feature vectors for each template triplet. That is, for [Tr, Ti, Tj], 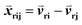 . Outputs were the hyperbolic tangent values of the dot product between inputs and the weight-vector,
. Outputs were the hyperbolic tangent values of the dot product between inputs and the weight-vector, 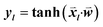 , where l indicates a particular triplet. The optimization was performed using the single-layer neural network implementation in the Lasagne Python library.14
, where l indicates a particular triplet. The optimization was performed using the single-layer neural network implementation in the Lasagne Python library.14
Model rendering
We can efficiently align a query sequence q whose structure we wish to predict, to a pre-aligned MSA m without changing the relationship between the pre-aligned templates.8 In order to render a model of a given query, we first inferred the CDR lengths by alignment to the framework MSAs. We next selected the templates from each of the 9 MSAs (2 frameworks, 6 CDRs and 1 orientation) using the feature vector-based score. To construct a coherent structure, the two framework templates were first superimposed on the orientation template using structurally conserved residues. Next, each top-scoring CDR template was superimposed onto the appropriate framework template using the four anchor residues before and after the CDR. Where needed, side-chain replacement was then carried out using SCWRL4.9
Quality assessment of modeled structures
For each receptor type (BCR/TCR), PDB crystal structures with resolution no worse than 3.0 Å were clustered by Cd-hit at 95% sequence identity using their variable domain sequences. Representative entries were used as a benchmark set. For each benchmark entry, a template blacklist was built by querying all homologs in the PDB with ≥80% sequence identity in either chain. All such templates were then masked from the alignment step. The accuracy of models was measured by all-residue all-atom and CDRH3 all-atom RMSDs between each model and its native structure after superimposing structurally conserved framework residues. In this way, 637 BCR and 66 TCR benchmark entries were prepared. Models built by reference methods were assessed in the same way. The statistical significance of the difference in the resulting all-atom RMSDs for Repertoire Builder and each reference method was computed using the Wilcoxon signed-rank test for the common set of successful models. For Kotai Antibody Builder, a customized environment was constructed using the subset of templates released before 2013, and tested on 246 queries released since 2013.
Web server
The Repertoire Builder public web server allows batches of up to 10000 paired or unpaired BCR or TCR queries to be run, with support for a separate blacklist for each query (Fig. 2A). The web server assumes paired (light–heavy or alpha–beta) chains if both chain types are input. It will not sample multiple pairings. If single chains are input, single chain models are built. The web server was written as a lightweight service in Go with Supervisor being used for front-end scaling and load-balancing. The job manager was also written in Go, but with a modular structure in order to be able to support different server environments. The web server backend utilizes 150 cores per job. These cores are a mixture of Intel Xeon and AMD Opteron processors. The specific core type and number fluctuates depending on core availability at the time the job is submitted. In a test of the BCR benchmark set on an Intel Xeon-E5-2660v3 the average CPU time usage was 10.4 seconds per model. No differences were observed in the average time taken for TCR models. We wish to emphasize that the per processor speed of Lyra, PigsPro, ABodyBuilder and Repertoire Builder are all fast enough to handle large numbers of sequences efficiently if distributed over a large number of processors. In our tests, Repertoire Builder will return results for 10000 paired sequences in approximately 30 minutes (Fig. 2B).
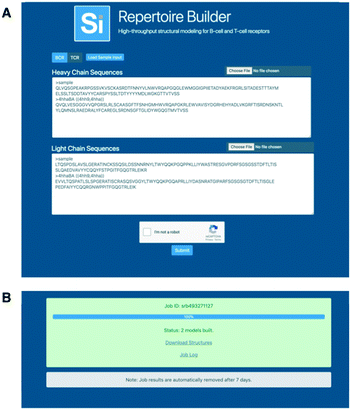 |
| | Fig. 2 Web Server. A) The web server accepts paired or unpaired BCR or TCR sequences. B) Output is returned as a zip file containing all models and a log file containing any errors. | |
Conserved framework residues for RMSD calculation
MSA profiles were constructed for each chain type (BCR light/heavy, TCR alpha/beta). MSA column index boundaries between frameworks and CDRs were determined according to the IMGT annotation of CDRs. Structurally equivalent residues averaged over each column in framework regions computed as defined on a 0–9 scale using ASH,15 and positions with equivalence score of 8 or higher were annotated as “conserved” framework positions. The sequences of benchmark sequences were then aligned onto the above-mentioned reference MSAs, and residues that were mapped to conserved framework columns were used as reference residues to superimpose models on native structures for RMSD calculations. The corresponding indices of these structurally conserved framework residues in the IMGT numbering scheme can be found in Table S1.†
Third-party BCR and TCR modeling tools
ABodyBuilder,17 PigsPro,18 and TCRmodel20 were run via web interface using default parameters other than blacklists. The latest version of LYRA19 was downloaded (October 18th, 2017) and run locally with blacklists and with the refinement step disabled, as recommended by the authors.
Results
Benchmark design
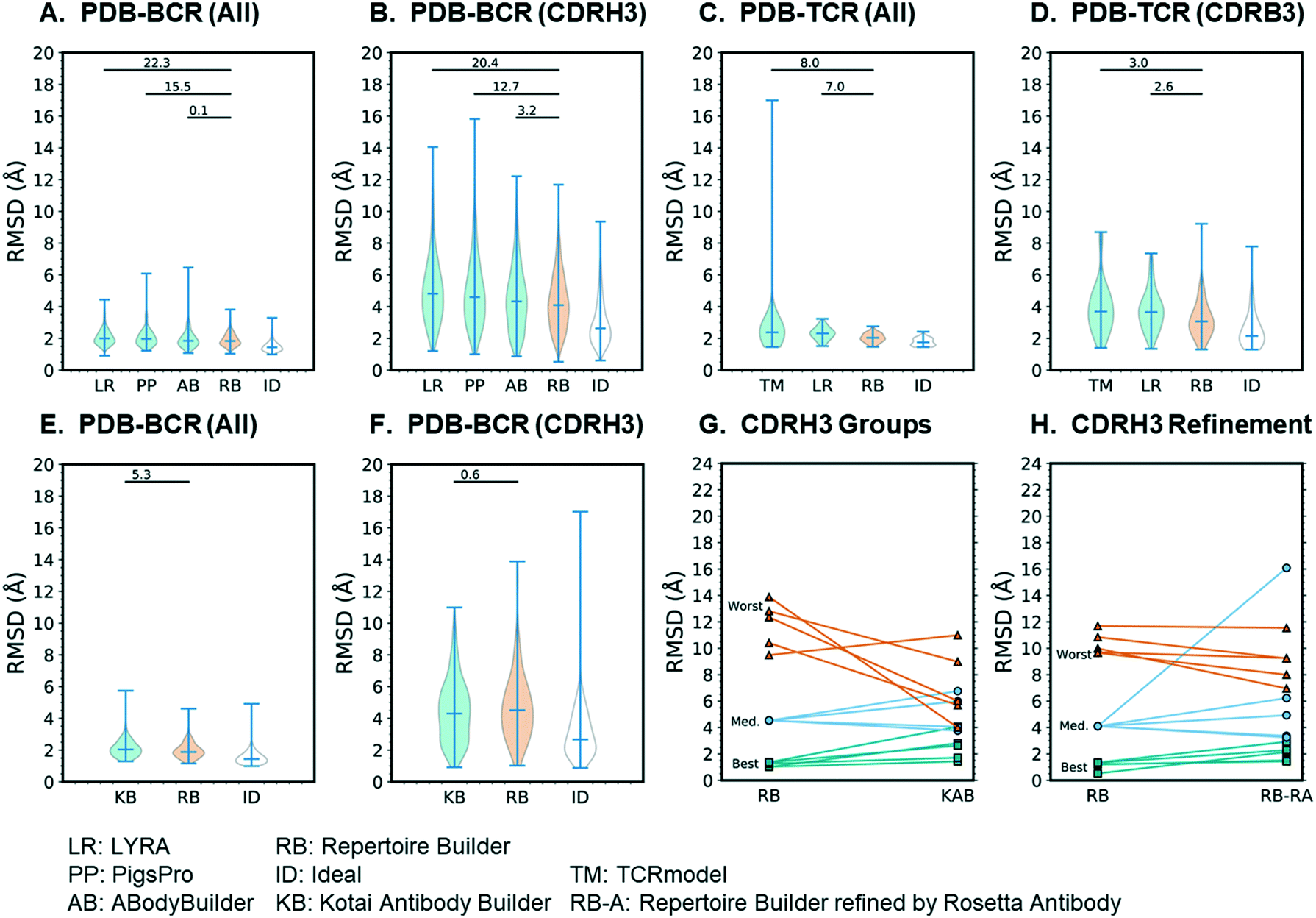
In order to quantitatively compare Repertoire Builder with other modeling methods, we utilized a representative set of 637 BCR and 66 TCR sequences with known structures extracted from the Protein Data Bank (PDB).16 The difference in the number of BCR and TCR sequences reflects the number of PDB entries: the number of BCRs in the PDB exceeds that of TCRs by roughly a factor of 10. To simulate a realistic use-case scenario where the structures of the queries are unknown, we blacklisted any templates from the modeling procedure that had a sequence identity above a given threshold across the variable domain of each query. We employed two thresholds, 80% and 90% (Table S2†). Tested modeling methods included ABodyBuilder,17 PigsPro,18 and LYRA19 for BCRs; LYRA and TCRmodel20 for TCRs. Each of these methods can be considered “high-throughput” in the sense that extensive structural sampling is not performed and the computational cost is typically no more than several minutes per model. In addition, for the 80% blacklist benchmark, we constructed an “Ideal” method where the template with lowest-RMSD from the query was selected for each region (CDR, framework, orientation). By definition the ideal method represents the lower bound of the RMSD for Repertoire Builder (i.e. to decouple “scoring” from “sampling” under the condition where the score is optimal). The top-scoring model of each method, including ideal, was assessed by all-residue RMSD and CDRH3 RMSD from the native (PDB) structure after superimposing structurally conserved residues in the heavy and light frameworks.
Performance of high-throughput methods
The results of the 80% blacklist benchmark are summarized in Fig. 3. As shown in Fig. 3A, the all-residue all-atom RMSDs of BCR models followed the trend: ideal (1.51) < Repertoire Builder (1.92) < ABodyBuilder (1.96) < PigsPro (2.09) < LYRA (2.10) and the differences between Repertoire Builder and two of the other (non-ideal) methods (PIGSPro and LYRA) were statistically significant (p < 0.05). The number of successful cases (i.e. for which a model could be built) was comparable between Repertoire Builder (629) and ABodyBuilder (631), and higher than PigsPro (587) or LYRA (587). The ideal method was, however, significantly better than Repertoire Builder (p < 0.001), indicating that there is still room for improvement in the Repertoire Builder score. For CDRH3, the all-atom RMSD trend was the same and all differences were statistically significant (p < 0.001; Fig. 3B). For TCRs, the trend was again ideal (1.77) < Repertoire Builder (2.04) < LYRA (2.31) < TCRmodel (2.64) for all residues (p < 0.001), and ideal (2.53) < Repertoire Builder (3.23) < LYRA (3.77) < TCRmodel (3.93) for CDRB3 (p < 0.001) despite the smaller number of queries and structural templates available for TCRs (Fig. 3C and D). The number of successful TCR runs was comparable between all methods with LYRA (63) > Repertoire Builder (60) > TCRmodel (57).
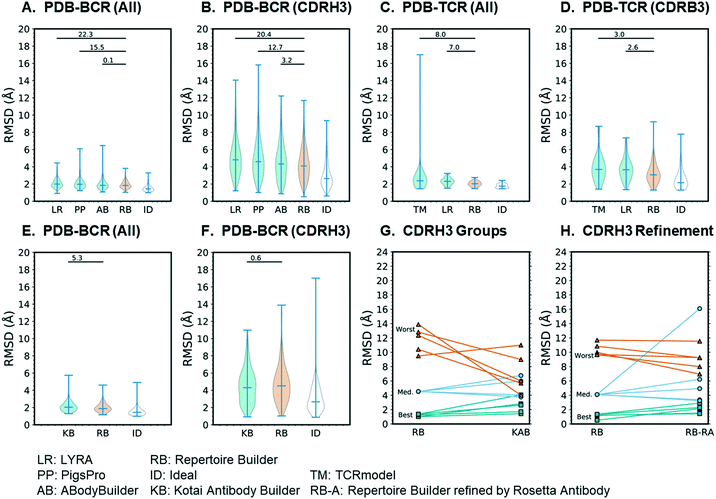 |
| | Fig. 3 Benchmark results. In all cases the Y-axis shows the all-atom RMSD. Significance of difference from Repertoire Builder was measured by the Wilcoxon signed-rank test and expressed as −log(p). A–D) high-throughput methods; E–F) Kotai Antibody Builder; G) comparison of CDRH3 RMSDs between Kotai Repertoire Builder and Antibody Builder for three representative groups of models; H) refinement of three representative groups of models Repertoire Builder models by Rosetta Antibody. | |
Analysis of extensive CDRH3 structural sampling
We next examined our earlier low-throughput BCR modeling method, Kotai Antibody Builder.21 Kotai Antibody Builder uses extensive structural sampling and performed well in the AMA-II assessment, but does not allow explicit template blacklisting. We took advantage of the fact that the template library had not been updated since January 2013 and used a representative set of 237 PDB entries released after this date as queries. We then constructed a customized Repertoire Builder environment that utilized only templates released before January 2013. We used the “refine” option in Kotai Antibody Builder, which achieved the highest accuracy at the time of its publication,21 but consumes ∼3 × 103-fold more CPU time than Repertoire Builder. We found that the mean all-residue all-atom RMSD of Repertoire Builder models (2.00) was significantly lower than that of Kotai Antibody Builder (2.10; p < 0.001; Fig. 3E). The number of successful Repertoire Builder cases (242) was also 16% higher than for Kotai Antibody Builder (202). Although there was no significant difference in CDRH3 all-atom RMSD (p = 0.28), the mean value for Kotai Antibody Builder (4.4) was lower than that of Repertoire Builder (4.7; Fig. 3F).
Analysis of CDRH3 loops
Since both Kotai Antibody Builder and Repertoire Builder CDRH3 all-atom RMSDs were significantly worse than the ideal method, the source of the error is not only due to sampling but also to scoring. We next examined the outliers for which the Repertoire Builder query-template CDRH3 score was unable to select the best templates. We hypothesized that the Repertoire Builder CDRH3 outliers would have poor CDRH3 query-template scores. To test this hypothesis, we selected three groups of five representative models based on CDRH3 all-atom RMSD (best-5: median-5; worst-5) and examined their query-template scores. As expected, the mean scores of each group followed the trend best-5 (0.69 ± 0.28) > median-5 (0.09 ± 0.20) > worst-5 (−0.05 ± 0.10), where the values in brackets indicate the mean and standard deviation from the mean. Consistent with mean CDRH3 all-atom RMSDs (Fig. 3F), Kotai Antibody Builder modeled the worst-5 group much more accurately than Repertoire Builder (Fig. 3G; Table S3†).
CDRH3 Refinement
The relatively small Kotai Antibody Builder benchmark results suggest that poor-quality Repertoire Builder CDRH3 models can be identified by their query-template CDRH3 scores, and, potentially improved by refinement. We next assessed the best-5, median-5 and worst-5 models from the larger high-throughput benchmark. Again, Repertoire Builder CDRH3 query-template scores followed the trend: best-5 (0.56 ± 0.40) > median-5 (−0.02 ± 0.05) > worst-5 (−0.08 ± 0.12), indicating that high CDRH3 RMSD outliers have lower scores on average. We next subjected these three groups to CDRH3 refinement using Rosetta Antibody, an extended structural sampling protocol that typically requires ∼1000 CPU hours per query.6 Again, the worst-5 models improved, on average, while the median-5 and best-5 either did not change significantly or became worse upon refinement (Fig. 3H; Table S4†). Taken together, these results suggest that it may be possible to improve Repertoire Builder CDRH3 models by applying loop refinement when the scores are low. However, given the high computational cost, along with the overall increase in the all-residue RMSD upon refinement (Fig. 3E), this approach should be used with caution. Moreover, the significant gap between ideal and actual Repertoire Builder CDRH3 RMSDs (Fig. 3F) suggests the core problem is scoring and not sampling.
Tests using high-throughput sequence data
We next assessed the performance of Repertoire Builder using sequences with unknown structure. Here we used 9313 paired human BCR sequences acquired post flu vaccination and a set of 1079 paired TCR entries downloaded from VDJdb.22 The average pairwise sequence identities within the PDB and flu vaccination datasets were similar: 0.53 ± 0.08 and 0.52 ± 0.08, respectively. The success rate of the flu vaccine set (93.2%) was consistent with that of the PDB-based benchmark, while that for the VDJdb set (85%) was lower, due to the poorer coverage of some TCR loops in the PDB. The Repertoire Builder CDRH3 query-template scores in the PDB-BCR and flu vaccination runs were 0.098 ± 0.25 and 0.021 ± 0.17, respectively. The CDRB3 scores in the PDB-TCR and VDJdb runs were 0.086 ± 0.16 and 0.28 ± 0.40, respectively. The VDJdb set contained both a number of sequences that could not be modeled (lowering the success rate) as well as number of sequences that were highly similar to known PDB entries (increasing mean CDRB3 score). When comparing PDB sequences to sequences with unknown structures, the differences between the mean scores of the sets (i.e., PDB-BCR vs. flu vaccination or PDB-TCR vs. VDJdb) were smaller than the standard deviations of the scores within each set, suggesting that the results on PDB sets are representative of what may be expected with novel sequence sets. The accuracy and efficiency of Repertoire Builder will enable immune receptors to be structurally analyzed in a high-throughput fashion. This, in turn, will open the possibility of identifying functional BCR or TCR relationships in unrelated donors.
Conclusions
In this study we showed that Repertoire Builder can process large numbers of BCR or TCR sequences efficiently and accurately. In our tests, the mean errors in the Repertoire Builder models were significantly lower than those of all tested methods. X-ray crystallographic studies have shown that BCRs targeting common antigens in unrelated donors can share structural features.23,24 However, X-ray crystallography cannot scale with the current growth of BCR sequence data. For TCRs, the situation is much worse: sequencing technology has grown faster, while the number of crystal structures is an order of magnitude lower than for BCRs. To our knowledge, only two third-party structural modeling tools (LYRA, TCRmodel) can accept TCR data. For these reasons we believe Repertoire Builder can play an important role in the analysis of large-scale BCR and TCR sequence datasets. Current limitations include a lack of structural coverage for some CDR lengths, especially in the case of TCRs. There is a potential for refinement of CDR3 loops in such cases, but expensive structural sampling methods should probably be applied on smaller subsets of sequences of interest and only in cases where Repertoire Builder scores are low. Another concern is the simplicity of the scoring function, which, although favorable in our benchmark over existing methods, has room for improvement, as indicated by the difference between the ideal and actual scores.
Conflicts of interest
D. S, S. L and D. M. S own equity in KOTAI Biotechnologies Inc. K. Y is an employee of and owns equity in KOTAI Biotechnologies Inc. G. C and W. V. are employees of and own equity in Atreca, Inc.
Acknowledgements
We would like to thank all members of the Systems Immunology Lab for helpful discussions. This research was supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number 17am0101108j0001. This study has received grants from the JSPS KAKENHI (Grants-in-Aid for Scientific Research) 18H02430.
References
- S. Friedensohn, T. A. Khan and S. T. Reddy, Trends Biotechnol., 2017, 35, 203–214 CrossRef CAS PubMed.
- A. C. Martin, J. C. Cheetham and A. R. Rees, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 9268–9272 CrossRef CAS PubMed.
- V. Morea, A. M. Lesk and A. Tramontano, Methods, 2000, 20, 267–279 CrossRef CAS PubMed.
- J. C. Almagro, A. Teplyakov, J. Luo, R. W. Sweet, S. Kodangattil, F. Hernandez-Guzman and G. L. Gilliland, Proteins, 2014, 82, 1553–1562 CrossRef CAS PubMed.
- H. Shirai, K. Ikeda, K. Yamashita, Y. Tsuchiya, J. Sarmiento, S. Liang, T. Morokata, K. Mizuguchi, J. Higo, D. M. Standley and H. Nakamura, Proteins, 2014, 82, 1624–1635 CrossRef CAS PubMed.
- B. D. Weitzner, J. R. Jeliazkov, S. Lyskov, N. Marze, D. Kuroda, R. Frick, J. Adolf-Bryfogle, N. Biswas, R. L. Dunbrack, Jr. and J. J. Gray, Nat. Protoc., 2017, 12, 401–416 CrossRef CAS PubMed.
- K. Katoh and D. M. Standley, Mol. Biol. Evol., 2013, 30, 772–780 CrossRef CAS PubMed.
- K. Katoh and M. C. Frith, Bioinformatics, 2012, 28, 3144–3146 CrossRef CAS PubMed.
- G. G. Krivov, M. V. Shapovalov and R. L. Dunbrack, Jr., Proteins, 2009, 77, 778–795 CrossRef CAS PubMed.
- Y. C. Tan, L. K. Blum, S. Kongpachith, C. H. Ju, X. Cai, T. M. Lindstrom, J. Sokolove and W. H. Robinson, Clin. Immunol., 2014, 151, 55–65 CrossRef CAS PubMed.
- J. DeFalco, M. Harbell, A. Manning-Bog, G. Baia, A. Scholz, B. Millare, M. Sumi, D. Zhang, F. Chu, C. Dowd, P. Zuno-Mitchell, D. Kim, Y. Leung, S. Jiang, X. Tang, K. S. Williamson, X. Chen, S. M. Carroll, G. Espiritu Santo, N. Haaser, N. Nguyen, E. Giladi, D. Minor, Y. C. Tan, J. B. Sokolove, L. Steinman, T. A. Serafini, G. Cavet, N. M. Greenberg, J. Glanville, W. Volkmuth, D. E. Emerling and W. H. Robinson, Clin. Immunol., 2018, 187, 37–45 CrossRef CAS PubMed.
- D. M. Standley, H. Toh and H. Nakamura, BMC Bioinf., 2007, 8, 116 CrossRef PubMed.
- M. P. Lefranc, C. Pommie, M. Ruiz, V. Giudicelli, E. Foulquier, L. Truong, V. Thouvenin-Contet and G. Lefranc, Dev. Comp. Immunol., 2003, 27, 55–77 CrossRef CAS PubMed.
-
S. Dieleman, J. Schlüter, C. Raffel, E. Olson, S. K. Sønderby, D. Nouri, D. Maturana, M. Thoma, E. Battenberg, J. Kelly, J. D. Fauw, M. Heilman, D. M. de Almeida, B. McFee, H. Weideman, G. Takács, P. de Rivaz, J. Crall, G. Sanders, K. Rasul, C. Liu, G. French and J. Degrave, Lasagne: First release, 2015, http://dx.doi.org/10.5281/zenodo.27878 Search PubMed.
- D. M. Standley, H. Toh and H. Nakamura, Proteins, 2004, 57, 381–391 CrossRef CAS PubMed.
- H. Berman, K. Henrick and H. Nakamura, Nat. Struct. Biol., 2003, 10, 980 CrossRef CAS PubMed.
- J. Leem, J. Dunbar, G. Georges, J. Shi and C. M. Deane, mAbs, 2016, 8, 1259–1268 CrossRef CAS PubMed.
- R. Lepore, P. P. Olimpieri, M. A. Messih and A. Tramontano, Nucleic Acids Res., 2017, 45(W1), W17–W23 CrossRef CAS PubMed.
- M. S. Klausen, M. V. Anderson, M. C. Jespersen, M. Nielsen and P. Marcatili, Nucleic Acids Res., 2015, 43, W349–355 CrossRef CAS PubMed.
- R. Gowthaman and B. G. Pierce, Nucleic Acids Res., 2018, 46, W396–W401 CrossRef CAS PubMed.
- K. Yamashita, K. Ikeda, K. Amada, S. Liang, Y. Tsuchiya, H. Nakamura, H. Shirai and D. M. Standley, Bioinformatics, 2014, 30, 3279–3280 CrossRef CAS PubMed.
- M. Shugay, D. V. Bagaev, I. V. Zvyagin, R. M. Vroomans, J. C. Crawford, G. Dolton, E. A. Komech, A. L. Sycheva, A. E. Koneva, E. S. Egorov, A. V. Eliseev, E. Van Dyk, P. Dash, M. Attaf, C. Rius, K. Ladell, J. E. McLaren, K. K. Matthews, E. B. Clemens, D. C. Douek, F. Luciani, D. van Baarle, K. Kedzierska, C. Kesmir, P. G. Thomas, D. A. Price, A. K. Sewell and D. M. Chudakov, Nucleic Acids Res., 2018, 46, D419–D427 CrossRef CAS PubMed.
- M. G. Joyce, A. K. Wheatley, P. V. Thomas, G. Y. Chuang, C. Soto, R. T. Bailer, A. Druz, I. S. Georgiev, R. A. Gillespie, M. Kanekiyo, W. P. Kong, K. Leung, S. N. Narpala, M. S. Prabhakaran, E. S. Yang, B. Zhang, Y. Zhang, M. Asokan, J. C. Boyington, T. Bylund, S. Darko, C. R. Lees, A. Ransier, C. H. Shen, L. Wang, J. R. Whittle, X. Wu, H. M. Yassine, C. Santos, Y. Matsuoka, Y. Tsybovsky, U. Baxa, N. C. S. Program, J. C. Mullikin, K. Subbarao, D. C. Douek, B. S. Graham, R. A. Koup, J. E. Ledgerwood, M. Roederer, L. Shapiro, P. D. Kwong, J. R. Mascola and A. B. McDermott, Cell, 2016, 166, 609–623 CrossRef CAS PubMed.
- J. F. Scheid, H. Mouquet, B. Ueberheide, R. Diskin, F. Klein, T. Y. Oliveira, J. Pietzsch, D. Fenyo, A. Abadir, K. Velinzon, A. Hurley, S. Myung, F. Boulad, P. Poignard, D. R. Burton, F. Pereyra, D. D. Ho, B. D. Walker, M. S. Seaman, P. J. Bjorkman, B. T. Chait and M. C. Nussenzweig, Science, 2011, 333, 1633–1637 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9me00020h |
| ‡ Current address: Department of Biosciences and Nutrition, Karolinska Institutet. |
| § Equal contribution. |
| ¶ Current address: KOTAI Biotechnologies Inc, 3-1 Yamadaoka, Suita, Osaka 565-0871. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab

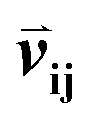 as vij(k) = B(m(i,k), m(j,k)) where B(a,b) is the BLOSUM62 matrix element for amino acids a and b after extension to include a constant gap penalty. We then defined the sequence similarity between sequences i and j as
as vij(k) = B(m(i,k), m(j,k)) where B(a,b) is the BLOSUM62 matrix element for amino acids a and b after extension to include a constant gap penalty. We then defined the sequence similarity between sequences i and j as 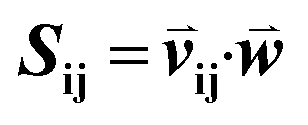 , where
, where 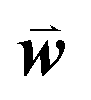 is a weight vector to be optimized to achieve the best agreement between Sij and the structural similarity of template sequences i and j for each MSA, as described below.
is a weight vector to be optimized to achieve the best agreement between Sij and the structural similarity of template sequences i and j for each MSA, as described below.
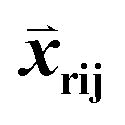 consisted of the differences between two feature vectors for each template triplet. That is, for [Tr, Ti, Tj],
consisted of the differences between two feature vectors for each template triplet. That is, for [Tr, Ti, Tj], 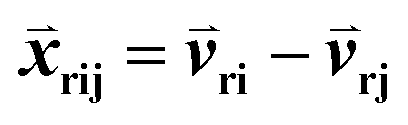 . Outputs were the hyperbolic tangent values of the dot product between inputs and the weight-vector,
. Outputs were the hyperbolic tangent values of the dot product between inputs and the weight-vector, 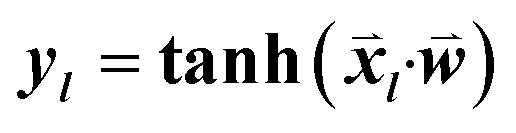 , where l indicates a particular triplet. The optimization was performed using the single-layer neural network implementation in the Lasagne Python library.14
, where l indicates a particular triplet. The optimization was performed using the single-layer neural network implementation in the Lasagne Python library.14