DOI:
10.1039/C8NA00302E
(Paper)
Nanoscale Adv., 2019,
1, 1331-1336
Hygroscopy-induced nanoparticle reshuffling in ionic-gold-residue-stabilized gold suprananoparticles†
Received
23rd October 2018
, Accepted 19th February 2019
First published on 20th February 2019
Abstract
Polyethyleneimine (PEI)-stabilized gold nanoparticles were used as a model to understand the roles of ionic precursors in the formation of nanoparticles and the impact of their presence on the nanoparticle properties. The low availability of elemental gold and the stabilization of the just-generated gold nanoparticles by the excess gold ions contributed to the production of ultra-small nearly neutral gold nanoparticles, resulting in properties significantly different from those prepared by conventional methods. The cross-linking between gold ions/PEI/nanoparticles further led to the assembly of these small gold nanoparticles into suprananoparticles that were stable in water. The hygroscopic Au(III) residues in the suprananoparticles absorbed moisture to form a micro-water pool and the nanoparticles in the new aqueous solution reshuffled to generate larger nanoparticles, leading to significant changes in their optical properties. Such a phenomenon was formulated into a fast, sensitive and straightforward method for the detection of water content in organic solvents.
Introduction
Stabilization of nanoparticles is usually achieved by either charging the nanoparticle surface (electrostatic stabilization) or capping the nanoparticle surface with ligands (steric stabilization). Because organic capping ligands can also be used to tune the size, morphology, and chemical reactivity of nanoparticles during the nanoparticle formation process, attention has largely focused on how organic molecules influence the growth of nanoparticles.1,2 However, the roles of ionic precursors and the influence of their presence in the reaction mixture on their properties have been mostly overlooked. In the absence of external stabilizers, these ions may appear on the surface of the just-formed nanoparticles, which suppresses further agglomeration of nanoparticles to larger particles. Even though the repulsion among the charged nanoparticles occurs, there is a balance between the electrostatic repulsion and van der Waals attraction among these nanoparticles, resulting in the possible self-assembly of these nanoparticles into supraparticles.3 Gold nanoparticles are excellent species for investigating the impact of the ion residue in the reaction solution in the synthesis and properties of gold nanoparticles owing to their good stability and characteristic photophysical properties.4 Here, we found that gold ions restricted the growth of gold nanoparticles, resulting in the formation of cluster-like small nanoparticle-aggregates (suprananoparticles). Moreover, the presence of gold ions in the solid state of the nanoparticles not only stabilized the gold nanoparticles, but they also acted as desiccants to absorb water molecules. The latter led to the formation of a nanoparticle suspension and induced the reshuffling of the nanoparticles, leading to a significant change in their morphology and photophysical properties.
Results and discussion
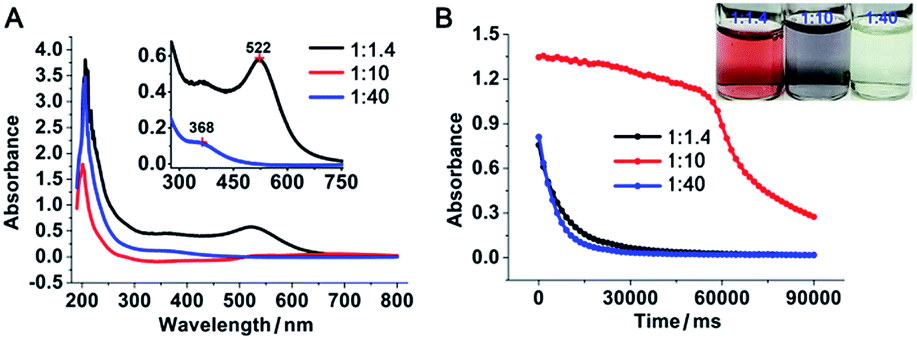
To investigate the role of gold ions during the formation of gold nanoparticles, the newly formed gold nanoparticles were stabilized with a stabilizer that does not neutralize the charge of the gold ions. Because the popular synthetic protocols of gold nanoparticles by citrate reduction in boiled water or borohydride reduction in the presence of alkanethiol likely involve the charged surface of the nanoparticles with a negative zeta potential, we used polyethyleneimine (PEI) as the reducing and stabilizing agent.5–7 PEI has been used as a positive-charge carrier attached to gold nanoparticles for DNA/RNA delivery.8 It has also been applied as a reducing agent for the generation and simultaneous stabilization of gold nanoparticles.7,9 In this report, gold nanoparticles were generated from gold ions (AuCl4−, 0.34 mM) and PEI (average Mn ∼ 1200 Da, 0.24 mM) with a PEI/Au(III) molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.4 in boiling water. The resulting gold nanoparticles were stable, with the characteristic plasmonic absorption of gold nanoparticles at 522 nm (Fig. 1A) and a narrow size distribution centred at 11 nm in diameter (Fig. 2A and B and ESI Fig. S1E†). The properties of the above PEI-stabilized gold nanoparticles were not significantly different from those of gold nanoparticles prepared by other methods. This was likely because all the gold ions were reduced completely and consequently formed nanoparticles. When the above PEI concentration was gradually decreased to a PEI/Au(III) molar ratio of 1:10, 1:17, and 1:30 without altering the gold ion concentration during the reduction process, an increasing number of nanoparticles fused to larger nanoparticles to form gold-wire networks (1:10, Fig. 2C and D) and irregularly shaped plates (1:17, Fig. 2E and F), and further to form polygonal plates (1:30, Fig. 2G and H). The colour of these nanoparticle solutions changed from vivid red to grey with obvious formation of nanoparticle agglomerates as the gold ratio increased (Fig. 1 and photo in Fig. 1B). The higher fusion rate between the nanoparticles in the high gold (low PEI) content solutions suggested that the surfaces of the gold nanoparticles were not capped well with protection groups so that these gold nanoparticles tended to grow larger to form gold nanoparticle networks as well as polygonal plates.10
:1.4 in boiling water. The resulting gold nanoparticles were stable, with the characteristic plasmonic absorption of gold nanoparticles at 522 nm (Fig. 1A) and a narrow size distribution centred at 11 nm in diameter (Fig. 2A and B and ESI Fig. S1E†). The properties of the above PEI-stabilized gold nanoparticles were not significantly different from those of gold nanoparticles prepared by other methods. This was likely because all the gold ions were reduced completely and consequently formed nanoparticles. When the above PEI concentration was gradually decreased to a PEI/Au(III) molar ratio of 1:10, 1:17, and 1:30 without altering the gold ion concentration during the reduction process, an increasing number of nanoparticles fused to larger nanoparticles to form gold-wire networks (1:10, Fig. 2C and D) and irregularly shaped plates (1:17, Fig. 2E and F), and further to form polygonal plates (1:30, Fig. 2G and H). The colour of these nanoparticle solutions changed from vivid red to grey with obvious formation of nanoparticle agglomerates as the gold ratio increased (Fig. 1 and photo in Fig. 1B). The higher fusion rate between the nanoparticles in the high gold (low PEI) content solutions suggested that the surfaces of the gold nanoparticles were not capped well with protection groups so that these gold nanoparticles tended to grow larger to form gold nanoparticle networks as well as polygonal plates.10
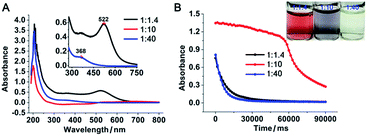 |
| | Fig. 1 Absorption and catalytic activity of gold nanoparticles prepared at a PEI/Au molar ratio of 1:1.4, 1:10 and 1:40. (A) Absorption spectra of the above gold nanoparticles. (B) Catalytic reduction of 4-nitrophenol by these gold nanoparticles. Absorbance at the peak position for 4-nitrophenol (400 nm) as a function of time in the presence of the respective gold nanoparticles. In all cases, the concentrations of 4-nitrophenol and borohydride were 4.3 × 10−3 and 6.3 × 10−2 M, respectively. The concentration of the Au-based catalyst was 3.4 × 10−4 M. Inset: photo of the solutions of the specific gold nanoparticles. | |
 |
| | Fig. 2 Dependence of the gold nanoparticle generation on the PEI/Au(III) molar ratio. TEM images of gold nanoparticles at a PEI/Au(III) ratio of 1:1.4 (A, B), 1:10 (C, D), 1:17 (E, F), 1:30 (G, H) and 1:40 (I, J); SEM image (K) and size distribution (L) of nanoparticles at a PEI/Au(III) ratio of 1:40. The TEM images in the lower row were obtained at a higher resolution compared to those in the upper row. The gold concentration was kept at 3.4 × 10−4 M. Scale bar: 100 nm (A, C, E, G, I, and K) and 20 nm (B, D, F, H, and J). | |
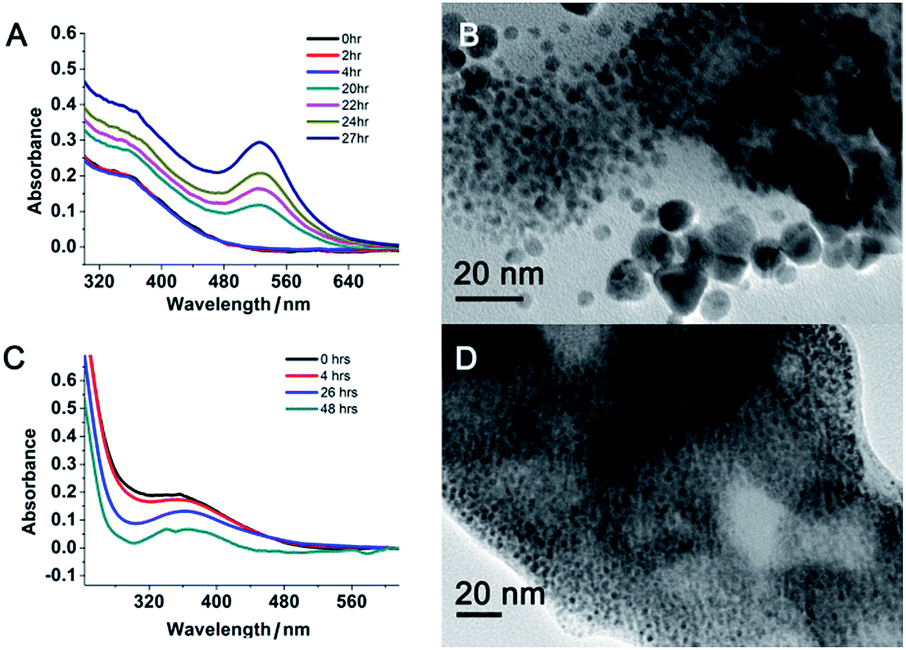
However, when the gold concentration was further increased to a PEI/Au(III) molar ratio of 1:40, the gold nanoparticle solution became light yellow (Fig. 1B inset) and the characteristic plasmonic absorption of gold nanoparticles disappeared (Fig. 1A). Such an abrupt transition in their optical properties was due to a change in the nature of the nanoparticles. The TEM and SEM images of the high-gold-content gold nanoparticles at a lower resolution indicated that the gold nanoparticles were large with an average diameter of 40.8 nm (Fig. 2I, K and L). However, these “large nanoparticles” were spherical clusters (suprananoparticles) composed of many ultra-small gold nanoparticles that had an average diameter of 2.4 nm (Fig. 2J and L). This was in line with the fact that the plasmonic absorption (∼520 nm) of nanoparticles less than 3 nm in diameter vanishes.11 The 368 nm absorption in the absorption spectra was quite rarely assigned to the absorption of gold nanoparticles, but could be more likely ascribed to the absorption of the oxidized PEI12 or Aumn+ clusters (m > n) that either stayed on the surface of the gold nanoparticles or to the nanoparticle per se.13 Further increasing the gold content to 1:50 or 1:60 in the above gold nanoparticle production protocol resulted in similar suprananoparticles to those observed at the 1:40 molar ratio (ESI Fig. S1†). Because the formation of the suprananoparticles might be due to the dewetting process during the TEM sample preparation,14 we examined their hydrodynamic radii with dynamic light scattering (DLS). The DLS results showed that there were four major components with a hydrodynamic diameter of 12 ± 2, 107 ± 16, 1010 ± 340, and 24100 ± 3600 nm (ESI Fig. S2†); they were assigned to the free tiny gold nanoparticles, the suprananoparticles, larger aggregates of these suprananoparticles or polygonal plates, and agglomerates of these nanoparticles, respectively. This indicates that the suprananoparticles already formed before they were cast onto the TEM grids. The zeta potential of these suprananoparticles was only 0.4 mV, whereas the small gold nanoparticles had a zeta potential of 13 mV. Due to the nearly neutral surface of the nanoparticles that resulted in weaker repulsion but stronger van der Waals attraction among these nanoparticles, these nanoparticles tended to form aggregates. Moreover, the hydrogen-bonding between the PEI networks and the coordinate bonding between the amino groups on the polymer chain and the gold ions also contributed to the formation of suprananoparticles.15,16 These gold suprananoparticles showed a similar catalytic activity for the reduction of 4-nitrophenol to 4-aminophenol to the small nanoparticles (1:1.4), but much better than the large nanoparticles (1:10).17,18 As shown in Fig. 1B, the characteristic absorption of 4-nitrophenol (400 nm) decreased faster upon the catalysis by the small gold nanoparticles (1:1.4) and suprananoparticles (1:40), compared to that by the large nanoparticles (1:10). This suggests that the networks inside the suprananoparticles were quite permeable so that small molecules could diffuse into the core. Given the nanoparticle size of 2.4 nm in diameter, the pore in the spherical cluster (suprananoparticles) is about 2 nm in diameter, which allows the entry of small molecules into the inner part of the suprananoparticles. However, these suprananoparticles only illustrated moderate colloidal stability, fusing into larger nanoparticles within a day and then exhibiting the characteristic 520 nm absorption of gold nanoparticles (Fig. 3A and B). This was likely due to the lower charge density on the surface of these suprananoparticles. It has been reported that a higher sodium chloride concentration decreases the intra-chain repulsion of the PEI owing to the salt-screening effect and subsequently induces the contraction of the PEI chain.19 This may lower the levels of chain extension as well as the cross-linking between inter-PEI chains through hydrogen-bonding, and then causes the breakdown of the inter-PEI networks. Consequently, the suprananoparticles may disassemble in the presence of sodium chloride. As expected, no suprananoparticles were detected by TEM after the addition of sodium chloride (150 mM) to the suprananoparticle solution, but no change in its absorption spectrum was observed (Fig. 3C and D). Instead, the sodium chloride significantly delayed the transformation of the small nanoparticles into larger gold nanoparticles, as seen in the absorption spectra (Fig. 3C).
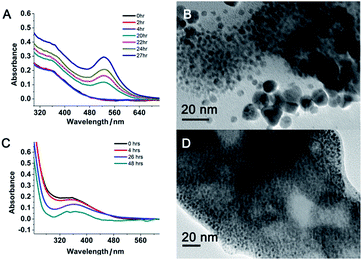 |
| | Fig. 3 Colloidal stability of the gold suprananoparticles prepared at a PEI/Au(III) molar ratio of 1:40. Time-dependent absorption spectra (A) and TEM image of the suprananoparticles after 27 h in water (B). Same sample but in the presence of sodium chloride (150 mM). Time-dependent absorption spectra (C) did not show the occurrence of the plasmonic absorption of the large gold nanoparticles at 520 nm. However, the TEM image of the solution after 48 h in water (D) did not show any suprananoparticles but ultra-small nanoparticles. Scale bar, 20 nm. | |
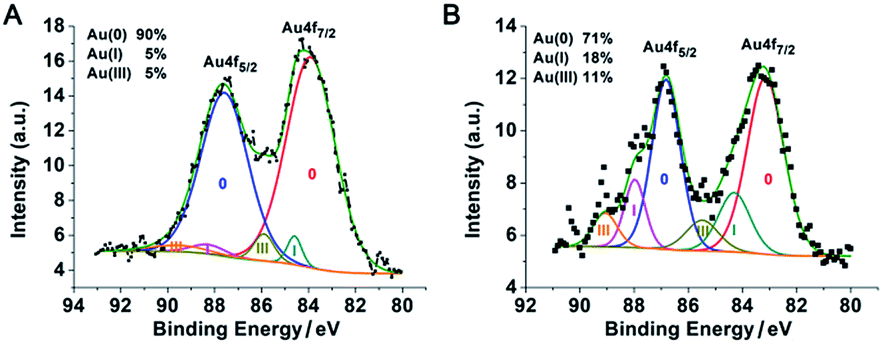
The PEI networks may contribute to the formation of nanoparticle clusters, but did not control the size of an individual gold nanoparticle. With a PEI/HAuCl4 mass ratio of 1:1 in DMF, larger PEI molecules (Mn ∼ 25 kDa) stabilized and encapsulated multiple gold nanoparticles in PEI polymer shells.20 That reaction was equivalent to our protocol with a PEI/Au molar ratio of 1:1.4. The sizes of the nanoparticles in these two reactions were similar but there were two major differences. (1) No obvious polymer shells were formed in our experiment because the polymer chain used in our protocol was too short.20 (2) The resulting PEI suprastructure of gold nanoparticles in the previous report was not stable in water. In our subsequent experiments, with less PEI and more gold content, we successfully obtained stable suprananoparticles in aqueous solution. The key to such an improvement was the excess of gold ions. As the number of PEI molecules decreased, the reducing power of PEI depleted, leading to the accumulation of Au(III) or Au(I) in the solution. Usually, a metal cluster can be further stabilized by capping the cluster with metal ions.21 Partially charged Auδ+ species have also been found on the surface of citrate-anion-stabilized gold nanoparticles.22 Therefore, in our case, it was more likely that these gold ions stayed on or even bonded to the surface of the newly formed gold nanoparticles, which were further stabilized by coordination with the amino groups on the PEI chains,23 resulting in thermodynamically stable nanoparticles. Both the low level of reduced elemental gold and the outstanding stability of these ion-capping nanoparticles contributed to the generation of these ultra-small nanoparticles, as shown in Fig. 2J. To verify the above hypothesis, the XPS spectrum of the suprananoparticles was obtained. It clearly showed that the suprananoparticles consisted of 90% elemental gold, 5% Au(I), and 5% Au(III) (Fig. 4A). Meanwhile, the excess gold ions formed complexes with PEI chains to build cross-linked networks between these nanoparticles, resulting in gold suprananoparticles with excellent stability in water.
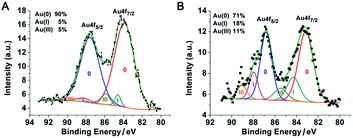 |
| | Fig. 4 XPS spectra of the suprananoparticles prepared at a PEI/Au(III) molar ratio of 1:40. More elemental gold was oxidized after the colour of the lyophilised gold suprananoparticles turned from yellow (A) to black (B). | |
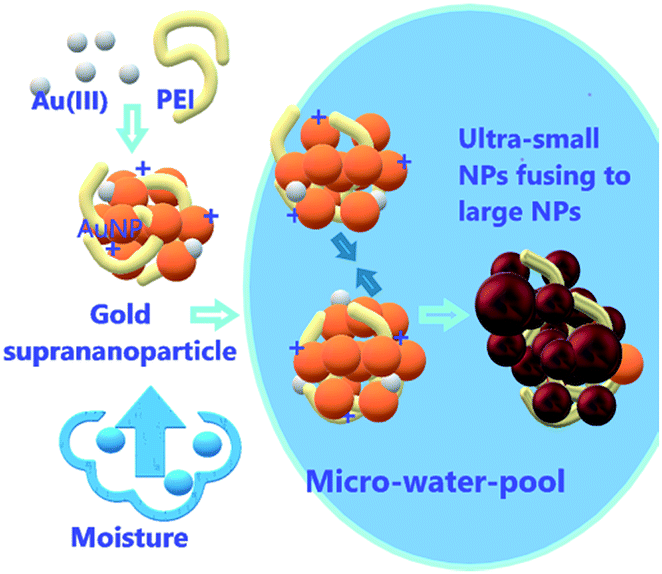
Interestingly, the freeze-dried gold suprananoparticles for the XPS test were light yellow powders. Once the powders were re-suspended in water, the suprananoparticles were restored without changes in their optical properties. However, such powders were extremely sensitive to moisture in the air. When exposed to air at a relative humidity of 50% at 25 °C, the yellow powders changed to black within an hour but retained the yellow colour in a desiccator for a month. It was highly possible that gold nanoparticles consisting of the suprananoparticles changed to larger nanoparticles because the freeze-dried gold nanoparticles (PEI/Au(III) molar ratio of 1:1.4, 11 nm in diameter) were also black, but it may be difficult for the solid gold suprananoparticles to proceed through such a dramatic change in the solid phase. However, the hygroscopic Au(III) residues in the suprananoparticles may absorb moisture to form a micro-water pool and subsequently the nanoparticles freely defused to meet their counterparts (Scheme 1) and then fused to larger nanoparticles. Such a phenomenon was recorded with a camera on an optical microscope. Right after the yellow powders were exposed to air on a coverslip, the yellow powders had started to absorb moisture; the granules of gold powders on the coverslip looked like they were surrounded by a thin layer of liquid (Fig. 5A). Within 40 min, these gold granules had merged into a liquid film (Fig. 5B) and then the film became dark brown in 80 min (Fig. 5C). A detailed video (Fig. 5E and ESI Video-1†) showed that the granules absorbed water, grew larger, and finally merged into a larger film in 300 s. Meanwhile, the colour of the water pools became darker. Such excellent hygroscopic capability was only exhibited by the suprananoparticles. The black powder of the freeze-dried gold nanoparticles (PEI/Au(III) molar ratio of 1:1.4, 11 nm in diameter) did not change its morphology at all in two hours when exposed to the above conditions (Fig. 5D and ESI Video-2†). These images/videos were consistent with our previous claim that the suprananoparticle powders absorbed water to form a micro-water pool and then nanoparticle-reshuffling occurred in the aqueous solution. Such reshuffling was concentration-dependent. It was much slower in dilute aqueous solution (taking place in 24 h) but much faster in the above water pools. Moreover, the colour change of the gold species was simply a consequence of the fusion of smaller nanoparticles to form larger ones (ESI Fig. S3†), but was not because of the generation of more nanoparticles. XPS of these black gold powders suggests that more elemental gold was oxidized to Au(I) (18%) and Au(III) (11%), as shown in Fig. 4B. This shows that the ultra-small gold nanoparticles are relatively less thermodynamically stable.24 While they were oxidized, these gold nanoparticles merged into larger gold nanoparticles that displayed different photophysical properties. The Au 4f7/2 binding energy for the Au(0) component in the suprananoparticles was 83.9 eV, 0.1 eV shifted from that of gold foil (84.0 eV).25 Upon aggregation, the gold suprananoparticles grew larger and their binding energy decreased to 83.2 eV, in line with the fact that the larger the gold nanoparticles, the lower the binding energy.26 It is possible that the Au+ binds to the surface of the gold nanoparticle with a binding energy of 84.5 eV for Au(I), lower than the expected value of 85.2 eV for Au+ ions.22,27 The Au 4f7/2 binding energy for the Au(I) component in the suprananoparticles was 84.6 eV, suggesting the possible bonding between Au+ and the gold nanoparticles. However, we cannot exclude the possible presence of organometallic Au(I) in the gold suprananoparticles.
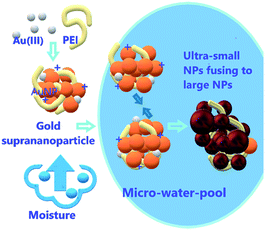 |
| | Scheme 1 Schematic showing hygroscopy-induced nanoparticle reshuffling. Gold ions in the suprananoparticles absorb moisture to form a micro-water-pool in which suprananoparticles reshuffle to form larger nanoparticles. | |
 |
| | Fig. 5 Hygroscopic effect of the suprananoparticles. Photos of lyophilised suprananoparticles on a glass coverslip taken by using a colour camera in situ (A), after 40 min (B) and after 80 min (C) from an eyepiece of a microscope with a 20× objective under white light irradiation. They showed that the gold granules immersed in the newly formed water pool and the yellow gold species become dark brown. Such a process was recorded with a CCD camera on the same microscope, as shown in the screenshots of the video clip (E). The numbers in (E) stand for the time in seconds the lyophilised suprananoparticles were exposed to air. A photo of the lyophilised large gold nanoparticles (PEI/Au(III) molar ratio of 1:1.4), taken 2 h after exposure to air showing no hygroscopic effect (D). Scale bar: 35 μm. | |
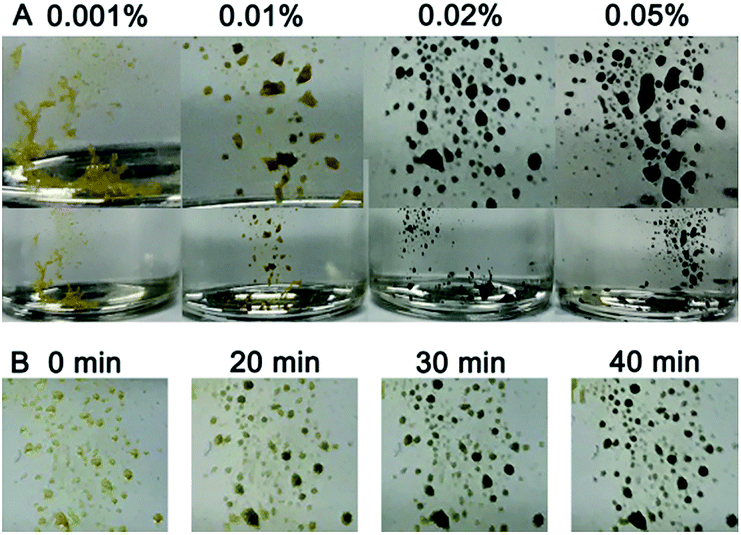
The sensitivity of the gold suprananoparticles to moisture inspired us to apply the suprananoparticle as a water indicator. The main methods for water content detection in organic solvents have been based on an impedance tester,28 spectrometry,29,30 chromatography,31 and electrochemistry.32 Our approach may be an alternative to the above methods and presented a fast and straightforward detection of water content in organic solvents. Alcohols in which the suprananoparticles can dissolve should be avoided. The yellow suprananoparticle powders can be immersed in most organic solvents such as toluene, chloroform, acetonitrile and cyclohexane, in which the suprananoparticles were insoluble. The water in the solvent was extracted to the suprananoparticles, which formed water pools and subsequently the suprananoparticles reshuffled to generate large and black gold nanoparticles. As shown in Fig. 6A, the gold suprananoparticles detected as low as 0.01% water content in toluene within an hour, which is among the best detection limits reported. The higher the water concentration, the darker the gold suprananoparticles became. This method could be fast as well. The change in the colour of the gold suprananoparticles could be observed 20 min after the immersion of the gold suprananoparticles in the solvent (Fig. 6B). Moreover, the gold suprananoparticles can be a sensitive marker in solvents: no water in the solvent, no change in the colour. Once water dissolves in the solvent, the colour of the gold suprananoparticles will change to black. We are still working on minimizing the gold used for water detection while maximizing the signal from the optical change of the nanoparticles.
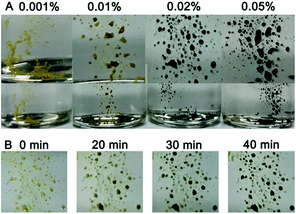 |
| | Fig. 6 Detection of water content in toluene with lyophilised gold suprananoparticles. (A) Photos of the suprananoparticles 60 min after they were immersed in toluene in the presence of 0.001%, 0.01%, 0.02% and 0.05% of water. The upper row shows magnified images of the samples in the lower row. (B) Photos of the sample in (A) (0.02%) taken in situ, 20 min, 30 min and 40 min after immersion in toluene. | |
Experimental
Synthesis of PEI–AuNPs at a PEI/Au(III) molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :x
:x
PEI solution (2.4 mM, 840/x μL) was added dropwise into pre-heated gold(III) chloride trihydrate solution (0.34 mM, 6 mL) with stirring in the dark for 5 min at 75 °C. After cooling down for 60 min in the dark, (600–840/x) μL PEI stock solution (2.4 mM) was added, followed by stirring for 1 h in the dark.
Comparison of catalytic ability
The concentrations of each component were maintained throughout except where indicated. In a quartz cuvette (10 mm), DI water (1 mL), catalyst (0.3 mM, 1 mL) and 4-nitrophenol stock solution (30 μL) were mixed. Sodium borohydride stock solution (50 μL) for 4-nitrophenol reduction was then added to the mixture, followed by a quick shake of the cuvette. The absorption spectra were then recorded on a SCINCO S-4100 Scan UV-visible spectrophotometer at room temperature.
The reduction was visualized by the disappearance of the 400 nm peak of 4-nitrophenol and the appearance of a new peak at 300 nm for 4-aminophenol. We observed the kinetics of the reaction by recording the change in the absorbance of the 4-nitrophenolate ions at 400 nm.
Size distribution of nanoparticles
The diameter of the nanoparticles in the TEM/SEM images was measured with ImageJ, in which a line was drawn across the distance of interest and Analyze/Measure was applied. The “length” in the result report was the diameter of the gold nanoparticles. The diameters of the gold nanoparticles and suprananoparticles in the TEM/SEM images were respectively measured randomly. At least 500 suprananoparticles were processed.
Detection of water content in organic solvents
A glass vial containing the freeze-dried gold nanoparticles was filled with organic solvents with known water contents. The cap of the vial was then closed tightly.
Conclusions
In summary, nanoparticles fused to form larger nanoparticles to form gold-wire networks and irregularly shaped plates, and further to form polygonal plates as the reducing capability of PEI decreased for the production of PEI-stabilized gold nanoparticles. However, as the reducing power of PEI was depleted at a PEI/Au molar ratio of 1:40, suprananoparticles consisting of ultra-small gold nanoparticles were generated. The low level of elemental gold and the stabilization of gold nanoparticles by the excess gold ions contributed to the production of ultra-small nearly neutral gold nanoparticles whereas the cross-linking between gold ions/PEI/nanoparticles further led to the assembly of these small gold nanoparticles into suprananoparticles that were stable in water. The suprananoparticles showed excellent catalytic activity for the reduction of 4-nitrophenol, suggesting that substrates could permeate through the suprananoparticle network. Such suprananoparticles could be stored stably in a dry place after lyophilisation. The hygroscopic Au(III) residues in the suprananoparticle solid absorbed moisture to form a micro-water pool and subsequently the nanoparticles in the new aqueous solution reshuffled to larger nanoparticles, leading to a colour change of the solid from yellow to black. We formulated the material into a fast, sensitive and straightforward tool to detect the water content in organic solvents. Our results indicated that ionic precursors may play an important role in the formation and stabilization of nanoparticles. Moreover, the ionic residues, regardless of whether they are a part of the nanostructure or impurities, may introduce further characteristics and significantly change the properties of the nanostructure.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Korean National Research Foundation (2015R1A2A1A15055721 and 2018R1D1A1B07050171). S. Choi thanks NRF (2015R1D1A1A01057710 and 2018R1D1A1B07046393).
Notes and references
- M. L. Personick and C. A. Mirkin, J. Am. Chem. Soc., 2013, 135, 18238–18247 CrossRef CAS PubMed.
- M. Rycenga, C. M. Cobley, J. Zeng, W. Li, C. H. Moran, Q. Zhang, D. Qin and Y. Xia, Chem. Rev., 2011, 111, 3669–3712 CrossRef CAS PubMed.
- Y. Xia, T. D. Nguyen, M. Yang, B. Lee, A. Santos, P. Podsiadlo, Z. Tang, S. C. Glotzer and N. A. Kotov, Nat. Nanotechnol., 2011, 6, 580 CrossRef CAS PubMed.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802, 10.1039/c39940000801.
- X. Sun, S. Dong and E. Wang, J. Colloid Interface Sci., 2005, 288, 301–303 CrossRef CAS PubMed.
- M. Thomas and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9138–9143 CrossRef CAS PubMed.
- F. S. Mohammed, S. R. Cole and C. L. Kitchens, ACS Sustainable Chem. Eng., 2013, 1, 826–832 CrossRef CAS.
- Y. Liu, K. B. Male, P. Bouvrette and J. H. T. Luong, Chem. Mater., 2003, 15, 4172–4180 CrossRef CAS.
- V. Subramanian, E. E. Wolf and P. V. Kamat, J. Am. Chem. Soc., 2004, 126, 4943–4950 CrossRef CAS PubMed.
- H. C. Haas, N. W. Schuler and R. L. Macdonald, J. Polym. Sci., Part A: Polym. Chem., 1972, 10, 3143–3158 CrossRef CAS.
- C. M. Aikens, J. Phys. Chem. C, 2008, 112, 19797–19800 CrossRef CAS.
- E. Rabani, D. R. Reichman, P. L. Geissler and L. E. Brus, Nature, 2003, 426, 271–274 CrossRef CAS PubMed.
- Y. Yao, M. Xue, Z. Zhang, M. Zhang, Y. Wang and F. Huang, Chem. Sci., 2013, 4, 3667–3672 RSC.
- C.-L. Chen, P. Zhang and N. L. Rosi, J. Am. Chem. Soc., 2008, 130, 13555–13557 CrossRef CAS PubMed.
- S. Choi, Y. Jeong and J. Yu, RSC Adv., 2016, 6, 73805–73809 RSC.
- S. Choi, Y. Jeong and J. Yu, Catal. Commun., 2016, 84, 80–84 CrossRef CAS.
- K. A. Curtis, D. Miller, P. Millard, S. Basu, F. Horkay and P. L. Chandran, PLoS One, 2016, 11, e0158147 CrossRef PubMed.
- F. Kretschmer, U. Mansfeld, S. Hoeppener, M. D. Hager and U. S. Schubert, Chem. Commun., 2014, 50, 88–90 RSC.
- A. Henglein, Chem. Rev., 1989, 89, 1861–1873 CrossRef CAS.
- H. Al-Johani, E. Abou-Hamad, A. Jedidi, C. M. Widdifield, J. Viger-Gravel, S. S. Sangaru, D. Gajan, D. H. Anjum, S. Ould-Chikh, M. N. Hedhili, A. Gurinov, M. J. Kelly, M. El Eter, L. Cavallo, L. Emsley and J.-M. Basset, Nat. Chem., 2017, 9, 890 CrossRef CAS PubMed.
- T. Rajský and M. Urban, J. Phys. Chem. A, 2016, 120, 3938–3949 CrossRef PubMed.
- H. G. Boyen, G. Kastle, F. Weigl, B. Koslowski, C. Dietrich, P. Ziemann, J. P. Spatz, S. Riethmuller, C. Hartmann, M. Moller, G. Schmid, M. G. Garnier and P. Oelhafen, Science, 2002, 297, 1533–1536 CrossRef CAS PubMed.
- A. Villa, N. Dimitratos, C. E. Chan-Thaw, C. Hammond, G. M. Veith, D. Wang, M. Manzoli, L. Prati and G. J. Hutchings, Chem. Soc. Rev., 2016, 45, 4953–4994 RSC.
- S. Peters, S. Peredkov, M. Neeb, W. Eberhardt and M. Al-Hada, Surf. Sci., 2013, 608, 129–134 CrossRef CAS.
- G. Ertas, U. Korcan Demirok and S. Suzer, Appl. Surf. Sci., 2005, 249, 12–15 CrossRef CAS.
- F. Opekar and P. Tůma, Sens. Actuators, B, 2015, 220, 485–490 CrossRef CAS.
- H. Sun, B. Wang and S. G. DiMagno, Org. Lett., 2008, 10, 4413–4416 CrossRef CAS PubMed.
- L. Ding, Z. Zhang, X. Li and J. Su, Chem. Commun., 2013, 49, 7319–7321 RSC.
- C. Dumas and C. C. Hsu, J. Chromatogr. A, 1982, 240, 508–514 CrossRef CAS.
- Y. Hui, E. L. K. Chng, C. Y. L. Chng, H. L. Poh and R. D. Webster, J. Am. Chem. Soc., 2009, 131, 1523–1534 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and TEM images. See DOI: 10.1039/c8na00302e |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Minyoung
Lim‡
,
Yanlu
Zhao
,
Minyoung
Lim‡
,
Yanlu
Zhao