
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric bifunctional protein nanoparticles through redesign of self-assembly†
Santiago
Sosa
ab,
Andrés H.
Rossi
a,
Alan M.
Szalai
b,
Sebastián
Klinke
 ac,
Jimena
Rinaldi
a,
Ana
Farias
a,
Paula M.
Berguer
a,
Alejandro D.
Nadra
d,
Fernando D.
Stefani
be,
Fernando A.
Goldbaum
ac and
Hernán R.
Bonomi‡
*a
ac,
Jimena
Rinaldi
a,
Ana
Farias
a,
Paula M.
Berguer
a,
Alejandro D.
Nadra
d,
Fernando D.
Stefani
be,
Fernando A.
Goldbaum
ac and
Hernán R.
Bonomi‡
*a
aFundación Instituto Leloir, IIBBA-CONICET, Av. Patricias Argentinas 435, (C1405BWE) Ciudad Autónoma de Buenos Aires, Argentina. E-mail: hbonomi@leloir.org.ar
bCentro de Investigaciones en Bionanociencias (CIBION)-CONICET, Godoy Cruz 2390 (C1425FQD), Ciudad Autónoma de Buenos Aires, Argentina
cPlataforma Argentina de Biología Estructural y Metabolómica PLABEM, Av. Patricias Argentinas 435 (C1405BWE) Ciudad Autónoma de Buenos Aires, Argentina
dDepartamento de Fisiología, Biología Molecular y Celular, Departamento de Química Biológica and IQUIBICEN-CONICET, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Ciudad Universitaria, Pabellón 2 (C1428EHA), Ciudad Autónoma de Buenos Aires, Argentina
eDepartamento de Física, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Pabellón 1 Ciudad Universitaria (C1428EHA), Ciudad Autónoma de Buenos Aires, Argentina
First published on 21st February 2019
Abstract
Engineering oligomeric protein self-assembly is an attractive approach to fabricate nanostructures with well-defined geometries, stoichiometry and functions. The homodecamer Brucella Lumazine Synthase (BLS) is a highly stable and immunogenic protein nanoparticle (PNP). Here, we engineered the BLS protein scaffold to display two functions in spatially opposite regions of its structure yielding a Janus-like nanoparticle. An in silico analysis of the BLS head-to-head dimer of homopentamers shows major inter-pentameric interactions located in the equatorial interface. Based on this analysis, two BLS protomer variants were designed to interrupt pentamer self-dimerization and promote heteropentameric dimers. This strategy enabled us to generate a decameric particle with two distinct sides formed by two independent pentamers. The versatility of this new self-assembly nanofabrication strategy is illustrated with two example applications. First, a bifunctional BLS bearing Alexa Fluor 488 fluorophores on one side and sialic acid binding domains on the other side was used for labelling murine and human cells and analyzed by flow cytometry and confocal microscopy. Second, multichromophoric FRET nanoparticles were fabricated and characterized at the single molecule level, showing discrete energy transfer events. The engineered BLS variants constitute a general platform for displaying two functions in a controlled manner within the same PNP with potential applications in various areas such as biomedicine, biotechnology and nanotechnology.
Introduction
Self-assembly of biological macromolecules is a key aspect of life. Nucleic acids, lipids and proteins organize into supramolecular arrangements that contribute to the existence of larger structures such as genomes, membranes, cytoskeletons and tissues. Recently, molecular biology knowledge is being applied to obtain bottom-up nanofabrication strategies. DNA nanotechnology, and in particular the DNA origami strategy, has been developed intensely in recent years. In spite of the incredible versatility of DNA self-assembly, the structures formed by nucleic acids tend to be mechanically flexible. Obtaining rigid nanostructures requires a careful design of cross-linked hybridization between key parts of the DNA structures. In fact, in nature, rigid biological structures are abundant in proteins. In comparison to nucleic acids, engineering and controlling protein self-assembly is considerably more complex, but holds the potential to generate more rigid and larger structures.Proteins are highly versatile and present diverse self-organized arrangements. Protein oligomerization depends on the primary peptide sequence as well as external conditions such as pH, temperature, ionic strength and the presence of ligands. The quaternary structure is intimately related to protein functions with a huge diversity in nature. An analysis of the quaternary arrangements of the Protein Data Bank (PDB) entries (https://www.rcsb.org/) classified by the OLIGAMI database1 reveals that approximately 47% of the catalogued proteins are monomeric and that most of the remaining form low copy number oligomers, mainly dimers (30.9%), trimers (4.6%), tetramers (8.7%) and hexamers (2.9%). In some special cases proteins form bigger complexes of, for example, 10, 12, 24 or 60 subunits. Interestingly, most of the latter structures are homopolymeric and present a high degree of symmetry.
Protein nanoparticles (PNPs) are a special subset of supramolecular protein assemblies. The PNP hallmark is a symmetric, either oligomeric or multimeric repetitive structure, sometimes conferring enhanced stability (e.g. chemical, mechanical or thermal)2,3 and high immunogenicity.4 Most of these particles derive from viral scaffold proteins such as virus-like proteins (VLPs), but some non-viral proteins, such as ferritins or small heat shock proteins, also belong to this particular protein class.5,6 In some cases, these arrangements form molecular cages able to entrap, for example, small molecules, nucleic acids7 or multiple proteins8 within their structure. These characteristics make PNPs very attractive models for bio- and nanotechnological developments. By combining protein engineering and chemical manipulation of PNPs, it has been possible to develop novel systems with potential applications in a wide range of fields including vaccine platforms,5,9 nanomedicine,10 cargo delivery11 and nanomaterials.6
The oligomeric state of polypeptides can be altered by manipulating either the external physicochemical conditions, its primary sequence or by producing chemical modifications to the scaffold. Engineering protein modules to control molecular self-assembly is a current new challenging and technologically relevant nanofabrication approach.12,13 Mutations may be predicted in a target sequence in order to produce a desired effect in an oligomer. Due to the complex nature of the protomer–protomer interaction networks and their difficulty to be accurately predicted, this strategy is particularly well suited for protein ensembles for which their molecular structure is available.
The lumazine synthase (LS, EC 2.5.1.78) enzyme, also known as RibH, belongs to the riboflavin biosynthetic pathway found in plants and microorganisms.14 The LS tertiary structure is highly conserved across taxa, even in very distant primary sequences. Strikingly, very subtle changes in the tertiary structure give rise to radically different quaternary assemblies (Fig. S1†): the Type-I LS or RibH/RibH1 group is composed of pentamers and icosahedrons, while the Type-II LS or RibH2 group encompasses only decamers.15,16 In all known LS variants, pentamers act always as the basic building blocks of these molecules. Interestingly, the icosahedral Type-I LSs have been extensively used as a model of protein cages for multiple purposes, recently reviewed by Azuma et al.17
The Brucella spp. Lumazine Synthase (BLS) is a homodecamer (Type-II LS) formed by the head-to head association of two homopentamers.15,18 BLS plays a key role in the Brucella infectious process,19 shows high thermal and chemical stability20 and elicits a strong immunogenic response in different animal models.21–27 Its disordered N-termini (first 8–10 residues of all chains) allow to genetically fuse polypeptides without affecting its structural properties, giving rise to chimeric constructs. BLS has been successfully used as a structural scaffold for vaccine design displaying a multiple variety of epitopes,28–31 ranging from linear peptides to protein domains and to entire proteins.32–34
Two strategies have been previously applied to link two different functions into the same BLS particle. One of them consists of the chemical dissociation of two different homodecameric chimeras of BLS into monomers using a chaotropic agent (for example guanidinium chloride), followed by a mixture and re-association of the monomers by dialysis.28 The second strategy is based on the genetic fusion of a set of leucine zipper peptide adaptors (leu1 and leu2), one to the BLS N-termini and the other to the proteins of interest.34 Then, by mixing leu1-BLS with the two target proteins each fused to leu2, the leucine zippers link both target proteins to BLS. In both cases, the target proteins are stochastically distributed within the scaffold, obtaining a heterogeneous population of heterodecamers.
The next step in tailoring functions of PNPs consists of organizing two or more functional moieties in exact spatial configurations within the same particle. Here, we demonstrate a redesign of the BLS into a Janus-like nanoparticle35 by generating a decameric structure built from two different pentamers. Based on structural analyses and rational design, we engineered the BLS pentamer–pentamer interface in order to (i) disrupt major interactions between homopentamers and (ii) enable the interaction between heteropentamers. Finally, we performed proof-of-concept functional assays in which we show that this novel protein scaffold presents potential uses in nanobiotechnology such as vaccine design, antigen production and imaging tool development.
Results
In silico analysis of Type-II LS pentamer–pentamer interfaces
As a first step, we sought to understand the nature of the Type-II LS pentamer–pentamer interaction by analyzing in detail its dimeric equatorial interface. To date, besides BLS, only one Type-II LS crystallographic structure has been solved: RibH2 from the soil bacterium Mesorhizobium loti (MLS) (Fig. 1A).16 The protein sequence alignment between both LSs shows 61% identical and 77% similar residues. Secondary structural elements and interface contacts between pentamers were identified in both structures and indicated in the primary sequences (Fig. 1B and Table 1). Here, we assume interface contacts when any of the atoms from two residues from different pentamers are at a shorter distance than 4.0 Å. The locations of the α-helices and β-sheets in the primary structure are found to be almost identical in both proteins. Table 1 lists all pairs of pentamer–pentamer contacts found in BLS and MLS. As it can be observed, MLS presents additional contacts compared to BLS, two of these interactions (Y120-H121 and R128-E132) being of electrostatic nature. These findings indicate that the matrix of interactions in the pentamer–pentamer interface is variable among decameric Type-II LS.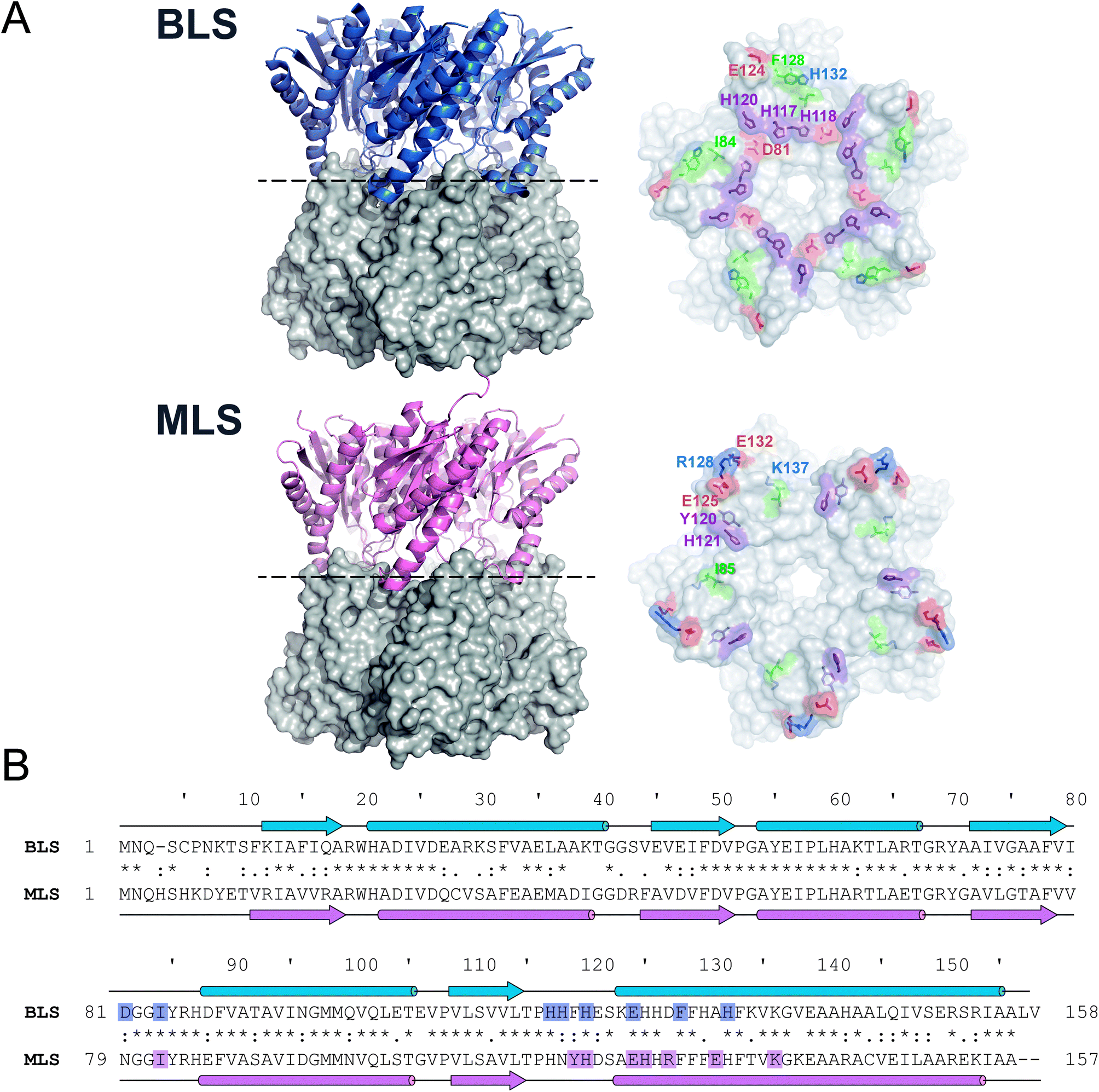 | ||
| Fig. 1 Type-II LS comparative analysis. (A) B. abortus and M. loti Type-II LS structures; PDB entries 1XN1 and 2OBX, respectively. Left panels: side view of the decameric particles. Each pentamer is differently shown in cartoon and surface representations. Right panels: top view of pentamer homodimeric interfaces. Pentamer–pentamer interacting residues are colored by type: positive (blue), negative (red), hydrophobic (green) and polar (violet). Interface interacting residues from one protomer are labeled. (B) Structural alignments of BLS (UniProt Q2YKV1, blue) and MLS (UniProt Q986N2, pink). Arrows and cylinders represent α-helices and β-sheets, respectively; interacting residues from the pentamer–pentamer interfaces (see Table 1) are highlighted. | ||
| Pair of residues | Type of interaction | Distance (Å) |
|---|---|---|
| BLS | ||
| D81-H120 | Electrostatic | 3.9 ± 0.3 |
| D81-H117 | Electrostatic | 3.5 ± 0.7 |
| I84-H118 | Hydrophobic | 3.2 ± 0.2 |
| H117-H118 | π–π stacking | 3.3 ± 0.2 |
| H120-H120 | π–π stacking | 3.2 ± 0.2 |
| E124-H132 | Electrostatic | 3.4 ± 0.6 |
| F128-F128 | Hydrophobic | 3.9 ± 0.3 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| MLS | ||
| I85-H126 | Hydrophobic | 3.6 ± 0.3 |
| Y120-H121 | Electrostatic | 3.8 ± 0.2 |
| H121-H121 | π–π stacking | 3.1 ± 0.2 |
| E125-K137 | Electrostatic | 3.1 ± 0.5 |
| R128-E132 | Electrostatic | 3.2 ± 0.4 |
In order to evaluate the significance of the role of each interface interaction, we decided to further characterize the BLS interface. For that aim, we performed thermodynamic calculations in silico by an alanine scanning strategy on all residues identified above as part of the interactions between the two pentamers, called A and B. We used the software FoldX36 to calculate Gibbs free energy between pentamers (ΔGpp) by finding local minima using a library of rotamers for each mutant (Table S2†). The ΔGpp relative changes of the alanine mutants, taking the wild-type protein (BLSWT) as the reference, are shown in Fig. 2A. Interestingly, two important sets of interactions between pentamers A and B were identified, exhibiting ΔGpp reductions of 42% and 90%, which were designated “velcro 1” (H117A and H118A) and “velcro 2” (E124A and F128A/H132A), respectively (Fig. 2A). These patches of interactions are highlighted in the BLS structure shown in Fig. 2B in more detail as two insets.
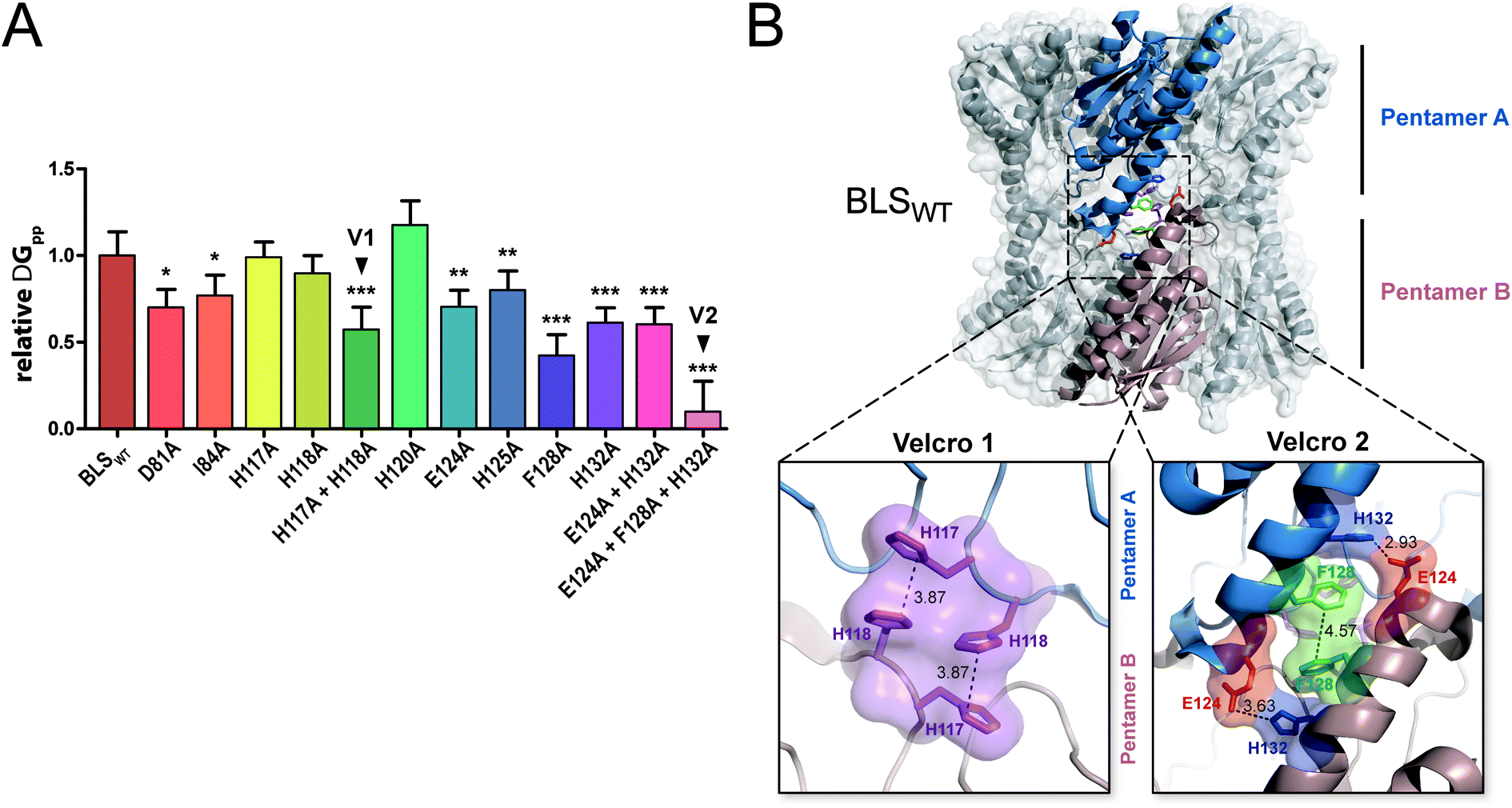 | ||
| Fig. 2 In silico analysis of the pentamer–pentamer interface contacts in BLS. (A) Relative pentamer–pentamer interaction energy calculations between BLSWT and BLS alanine interface mutants using FoldX and PDB 1XN1 as templates. Residues from “velcro 1” and “velcro 2” are indicated as V1 and V2, respectively. Values are expressed as mean ± SD. Statistical analysis was performed by non-parametric analysis of variance (Kruskal–Wallis) followed by paired comparisons (Mann–Whitney) (*P < 0.05, **P < 0.01, ***P < 0.001). Data derive from five independent calculations. (B) The identified interactions from velcros 1 and 2 are shown in the structure of BLS. A side view of the BLS superimposing surface and the cartoon representation highlights two monomers from different pentamers with different colors. Velcros side chains are represented as sticks within the dashed box; insets show in detail the velcro 1 and velcro 2 from opposing pentamers. Residues are indicated and color coded by type: positive (blue), negative (red), hydrophobic (green) and polar (violet). | ||
Redesign of the BLS pentamer–pentamer interface
In order to guide the self-assembly of BLS towards the formation of a Janus-like nanoparticle we decided (i) to disrupt the BLS homodecamer assembly by the mutagenesis of some residues at the pentamer–pentamer interface and (ii) to reconstitute a heterodecameric particle by the association of two different mutant pentameric species. Residues within velcros 1 and 2 were initially contemplated as potential candidates to be mutated in order to disable the pentamer–pentamer association. The alanine substitutions of velcro 2 residues (Fig. 2A) indicate that this patch of interactions is a major contributor in cementing together the two pentamers. A closer look at velcro 2 in the protein structure also suggests its relevant role for maintaining both pentamers bound together by one hydrophobic (F128-F128) and two electrostatic interactions (E124-H132) per pair of opposing protomers (Fig. 2B). Hence, we decided to leave velcro 2, including F128, unchanged and focus on modifying the interactions of velcro 1. The rationale behind this decision is that a modification of this key interaction patch may result in a high destabilization of the dimeric interface, probably making it more difficult to reconstitute it with complementary mutations for achieving a stable decamer. The interaction between H117 (from pentamer A) and H118 (from pentamer B), in velcro 1, is mainly due to a π–π stacking between the imidazole moieties from both residues (Fig. 2B).37 We opted to disrupt this π–π interaction by introducing charged residues at the positions 117 and 118. Taking into account their distances and volumes available from the crystallographic model, we designed two different mutants: H117D/H118D and H117K/H118K. Due to the electrostatic forces generated by the charged residues, these mutations should generate the desired dual effect. Electrostatic repulsion hinders homopentamer self-dimerization due to charges of the same sign at the interface (pentamer A: H117D/H118D, and pentamer B: H117K/H118K) while heterodimer formation is favored by electrostatic attraction (H117D-H118K and H117K-H118D). Based on the same rationale, we decided to introduce two additional salt-bridges in order to strengthen the interaction between mutant pentamers A and B, while further weakening their self-association. The D127-A131 BLS positions correspond in sequence and space to the R128-D132 salt bridge found at the MLS interface (Fig. 1). Therefore, a set of mutations was formulated (pentamer A: D127R/A131R; pentamer B: D127E/A131E) to emulate MLS in this respect. Furthermore, the introduction of two positively and two negatively charged residues in each mutant maintains the net charge null, preserving their theoretical isoelectric points similar to BLSWT. In total, four point mutations per monomer were conceived, which we named BLSDR and BLSKE for the introduction of Asp, Arg, Lys and Glu residues, respectively (Table 2). The introduced mutations at the interface are represented for both mutants in Fig. 3, after modeling their conformation in FoldX. The BLSDR and BLSKE association in a decameric particle, referred to as BLSDRKE, was also modeled. The mutant side chains were able to form the proposed salt bridges, suggesting that these contacts may indeed exist in BLSDRKE.| Mutant | Interface mutations | |||
|---|---|---|---|---|
| BLSDR | H117D | H118D | D127R | A131R |
| BLSKE | H117K | H118K | D127E | A131E |
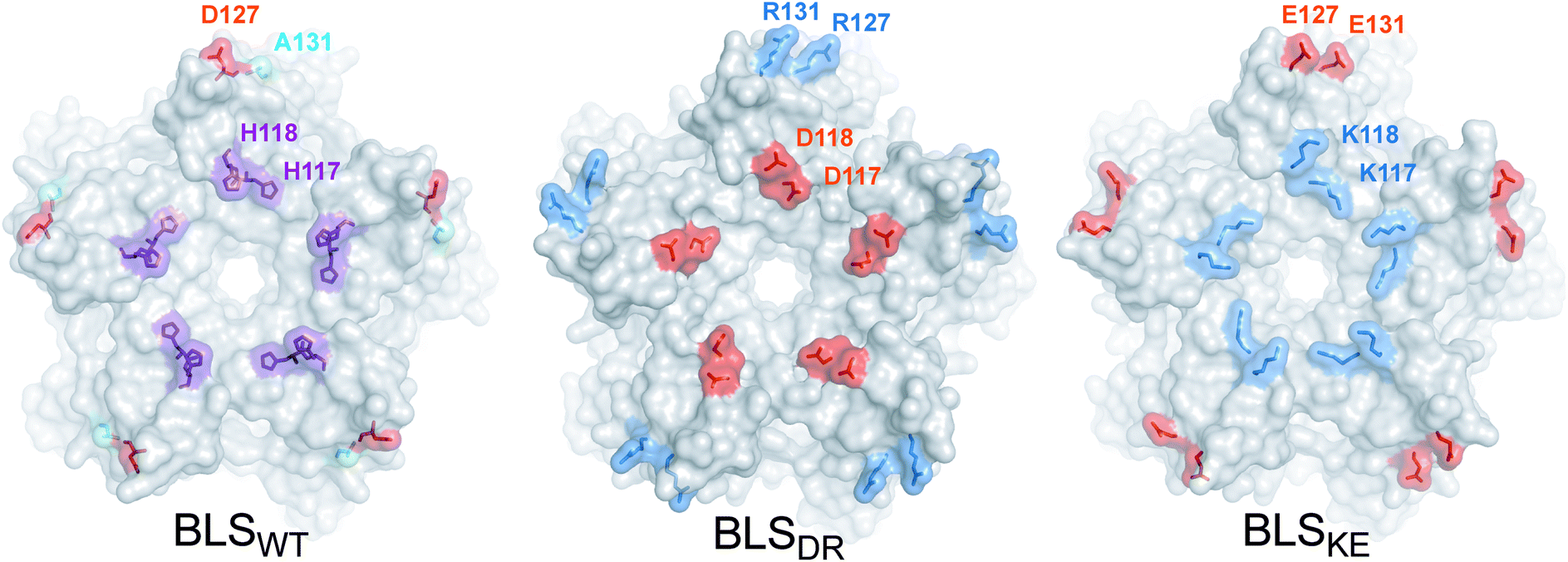 | ||
| Fig. 3 BLSDRKE pentameric interface structural model. Top view of the pentameric interfaces of BLS proteins. The wild-type residues to be substituted from BLSWT are shown in the structure (PDB 1XN1). The mutant residues from BLSDR and BLSKE were modeled using FoldX. The proteins are depicted in surface representation and the wild-type and mutant residues are represented as surfaces and sticks. Alanine, histidine, positive and negative residues are colored in cyan, violet, blue and red, respectively. Residues from one protomer are indicated. | ||
Pentameric BLSKE and BLSDR associate to form a stable heterodecamer
Recombinant BLSKE and BLSDR mutants were successfully generated, expressed and purified in high yields (Fig. S2,† see![[M with combining low line]](https://www.rsc.org/images/entities/char_004d_0332.gif) aterials and methods for details). The quaternary structure of these new proteins was first characterized by performing size exclusion chromatography (SEC) coupled to static light scattering (SLS) measurements for each mutant separately and mixed together, from which their molecular weight (MW) was determined. Fig. 4A shows the SEC elution profiles, indicating the mean MW calculated for each sample. Both BLSDR and BLSKE present higher elution volumes (smaller particles) than BLSWT, and the calculated MWs correspond to pentameric species. In accordance with our design, pre-incubation of an equimolar mix of BLSDR and BLSKE produces a new species in the SEC-SLS experiment that matches the elution volume of BLSWT with a MW of 176 ± 2 kDa, which is in agreement with a decameric structure (Fig. 4A). The BLSDRKE decameric peak was assayed again by SEC-SLS immediately after mixing (0 h) and after 24 h. In both cases, a single decameric peak was detected of approximately the same MW (Fig. 4B). This rules out the existence of a pentamer–decamer dynamic equilibrium, and allows us to conclude that the BLSDRKE heterodimer assembly is stable under these experimental conditions. Due to the electrostatic nature of the novel pentamer–pentamer interactions introduced in BLSDRKE, we wondered whether ionic strength could alter the heteropentamer dimer stability. Therefore, we pre-incubated BLSDRKE in buffers with different NaCl concentrations ranging from 0 to 2 M, and performed SEC-SLS measurements of the samples using the same buffers. All NaCl concentrations assayed yielded decameric species (Fig. 4C) similar to BLSWT, indicating a comparably high stability in both decamers under elevated ionic strength conditions.
aterials and methods for details). The quaternary structure of these new proteins was first characterized by performing size exclusion chromatography (SEC) coupled to static light scattering (SLS) measurements for each mutant separately and mixed together, from which their molecular weight (MW) was determined. Fig. 4A shows the SEC elution profiles, indicating the mean MW calculated for each sample. Both BLSDR and BLSKE present higher elution volumes (smaller particles) than BLSWT, and the calculated MWs correspond to pentameric species. In accordance with our design, pre-incubation of an equimolar mix of BLSDR and BLSKE produces a new species in the SEC-SLS experiment that matches the elution volume of BLSWT with a MW of 176 ± 2 kDa, which is in agreement with a decameric structure (Fig. 4A). The BLSDRKE decameric peak was assayed again by SEC-SLS immediately after mixing (0 h) and after 24 h. In both cases, a single decameric peak was detected of approximately the same MW (Fig. 4B). This rules out the existence of a pentamer–decamer dynamic equilibrium, and allows us to conclude that the BLSDRKE heterodimer assembly is stable under these experimental conditions. Due to the electrostatic nature of the novel pentamer–pentamer interactions introduced in BLSDRKE, we wondered whether ionic strength could alter the heteropentamer dimer stability. Therefore, we pre-incubated BLSDRKE in buffers with different NaCl concentrations ranging from 0 to 2 M, and performed SEC-SLS measurements of the samples using the same buffers. All NaCl concentrations assayed yielded decameric species (Fig. 4C) similar to BLSWT, indicating a comparably high stability in both decamers under elevated ionic strength conditions.
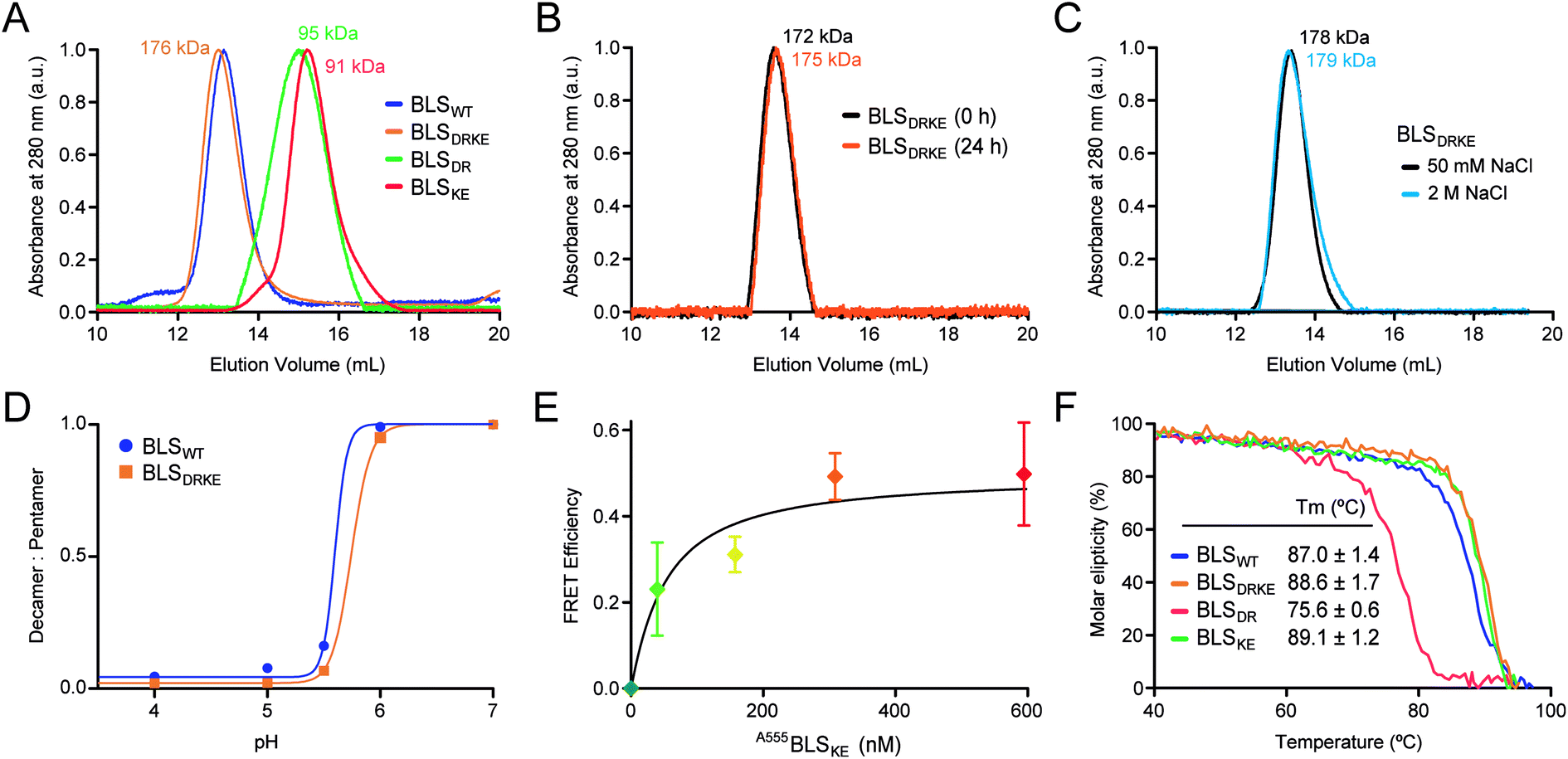 | ||
| Fig. 4 BLSDRKE heteropentamer dimer assembly and stability. (A) The MWs of BLSWT, BLSDR, BLSKE or a 30 min co-incubation of both species (BLSDRKE) were calculated from SEC-SLS measurement elution profiles. (B) The BLSDRKE peak from (A) was collected and re-injected immediately (0 h) or after 24 h. (C) SEC-SLS BLSDRKE in 50 mM or 2 M NaCl. (A–C) Calculated MWs are indicated. (D) BLSWT and BLSDRKE decamer dissociation at pH values 4–7. The data presented correspond to the decamer:pentamer ratio calculated from SEC-SLS measurements at each pH value (see Fig. S3†). (E) FRET efficiency calculated from 520 and 570 nm signals obtained from A488BLSDR fluorescence spectra (λexc: 470 nm) incubated with increasing A555BLSKE concentrations (Fig. S4†) using eqn (1), see Materials and methods. Values are expressed as mean ± SD (n = 3). A fitting curve using eqn (2) is shown. (F) Apparent melting temperature of BLS proteins. Temperature dependence of circular dichroism molar ellipticity signal at 222 nm of BLS variants relative to 25 °C values. The graph corresponds to a representative experiment of three independent experiments. Values in the inset table are expressed as mean ± SD (n = 3). | ||
BLSWT presents a pH-dependent pentamer–pentamer dimer dissociation, with a 50% dissociation at a pH of ∼5.7 (Fig. 4D and S3†). Histidine residues are natural candidates for explaining this phenomenon. Due to the elevated amount of histidine residues participating in pentamer–pentamer contacts (25 from a total of 55 residues involved per decamer) and considering that BLSDRKE has 10 histidine residues replaced, we next evaluated whether the pH dependent stability remained unchanged. The results show that, BLSDRKE exhibits a nearly identical pH dissociation curve compared to BLSWT, with a 50% dissociation at a pH value of ∼5.7 (Fig. 4D). Thus, the replaced histidine residues H117 and H118 do not play a significant role in pentamer–decamer equilibrium driven by pH.
To further verify the interpentameric interaction, we performed a Förster resonance energy transfer (FRET) assay by conjugating donor and acceptor fluorophores on each pentamer. A point mutation was introduced at position 123 to replace a lysine residue by a cysteine residue, which was subsequently used to chemically link dyes containing maleimide groups in positions near the interface. According to the structure of BLS, the expected separation distance between donor and acceptor fluorophores ranges from 19 to 23 Å, being suitable for efficient FRET. The new cysteine mutant variants for BLSDR and BLSKE were expressed, purified and derivatized with Alexa Fluor 488 and Alexa Fluor 555, named A488BLSDR and A555BLSKE, respectively. The fluorescence spectrum of A488BLSDR incubated at increasing A555BLSKE amounts was determined under excitation at 470 nm (Fig. S4†). At higher A555BLSKE concentrations, the emission peak corresponding to the acceptor (570 nm) increases while the emission peak corresponding to the donor (520 nm) decreases. A FRET efficiency plot was derived from these values (Fig. 4E). The presence of a clear FRET effect indicates the close spatial proximity of both pentamers in the BLSDRKE particles.
Additionally, the structure and stability of BLSDR, BLSKE and BLSDRKE were assessed by temperature curves measured by circular dichroism (CD). First, we compared the CD spectra of BLS pentameric and decameric variants at room temperature, presenting similar curves with two main negative peaks at 208 and 222 nm, and a positive peak at 190–195 nm (Fig. S5A†), evidencing a combination of α/β structures with a major α-helical component. These results indicate that the mutations do not introduce significant changes in the secondary structures of BLSDR, BLSKE or BLSDRKE. Next, in order to interrogate the thermal stability of the BLS variants, their molar ellipticity signal at 222 nm was recorded as a function of temperature. The results show similar unfolding curves for BLSDRKE and BLSWT with melting temperatures (Tm) of 86.7 and 85.4 °C, respectively (Fig. 4F). BLSDR displays a thermal unfolding curve shifted towards lower temperatures and a lower Tm (75.6 °C). This result is in concordance with previous work where it was shown that the pentameric form of BLS has lower thermal stability.20 Surprisingly, BLSKE appeared to be as stable as the decameric structures. The mutational effects in BLSDR in the decameric context of the heteropentamer dimer BLSDRKE are counteracted by the interaction with BLSKE even showing a slightly enhanced Tm of ca. 2 °C compared to BLSWT (Fig. 4F).
BLSDRKE retains immunological properties of BLSWT
BLSWT is a powerful immunogen,21–27 and hence we decided to evaluate the capacity of BLSDRKE and the pentameric variants to elicit humoral immune responses, taking BLSWT as a reference. BALB/cJ mice were injected intraperitoneally with 10 μg of each of the proteins. After 14, 28 and 42 days post-immunization (d.p.i.) sera samples were collected and the antibody production was assessed by an Enzyme-linked immunosorbent assay (ELISA). Fig. 5 shows the ELISA results from a 1:200 serum dilution. The response curves from BLSDRKE and BLSKE recapitulate the BLSWT curve, while BLSDR does not induce a significant humoral response. Noteworthy, these results are reminiscent of the thermal stabilities obtained in Fig. 4F, where the heteropentamer decamer and the BLSKE pentamer both share comparable Tm values and humoral responses to BLSWT; while on the other hand, BLSDR presents both significantly lower Tm and capacity to elicit antibodies in mice.
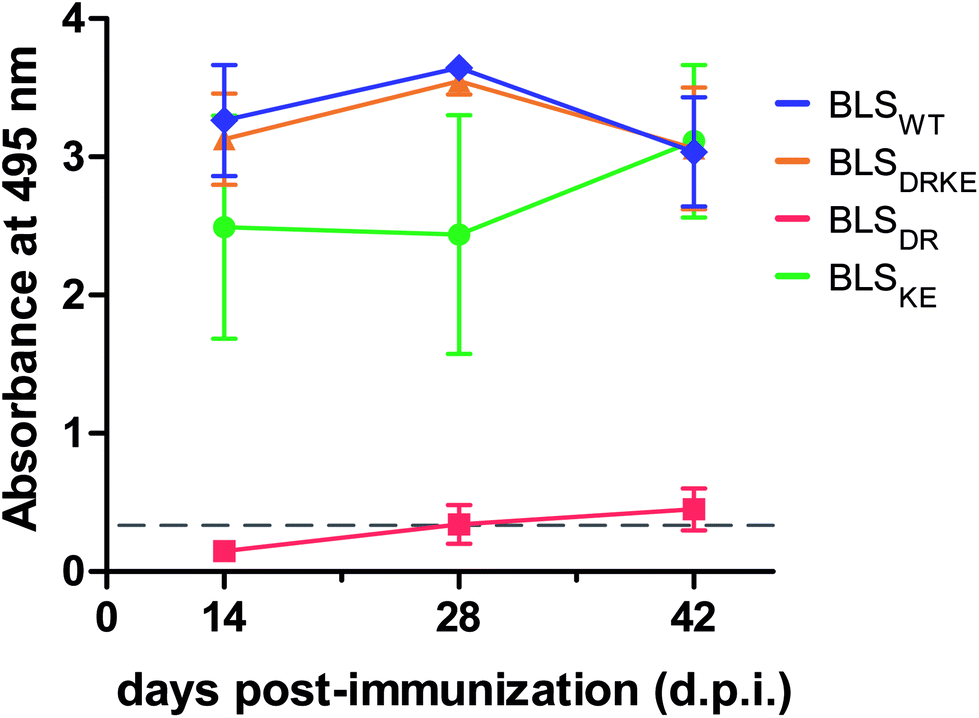 | ||
| Fig. 5 Humoral response in immunized mice with BLS variants. BALB/cJ mice were immunized intraperitoneally using BLS decameric (BLSWT, BLSDRKE) or pentameric variants (BLSDR, BLSKE). After 14, 28 or 42 d.p.i. mice sera reactivity was quantified by ELISA (1:200 dilutions). Values are expressed as mean ± SD (n = 4). | ||
Fluorescent cell-labeling
We next explored different applications for the tool we generated. As a first application example we decided to build a bifunctional PNP to fluorescently label mammalian cells. The rationale and the design are as follows: a BLSKE pentamer fused to the VP8 rotavirus sialic acid binding domain (VP8-BLSKE)29 would bind to the sialic acid molecules on the surface of cellular membranes and the A488BLSDR pentamer would assemble forming a decameric fluorescent complex. Hence, we first generated, expressed and purified VP8-BLSKE and corroborated by CD, SEC-SLS, and SDS-PAGE analyses that: (i) the secondary and tertiary structures of both domains are not affected when fused into a chimera (Fig. S5B†), (ii) it is in fact a pentamer in solution, and (iii) it has the capacity to bind to BLSDR giving rise to a particle with a MW corresponding to a decamer (Fig. 6).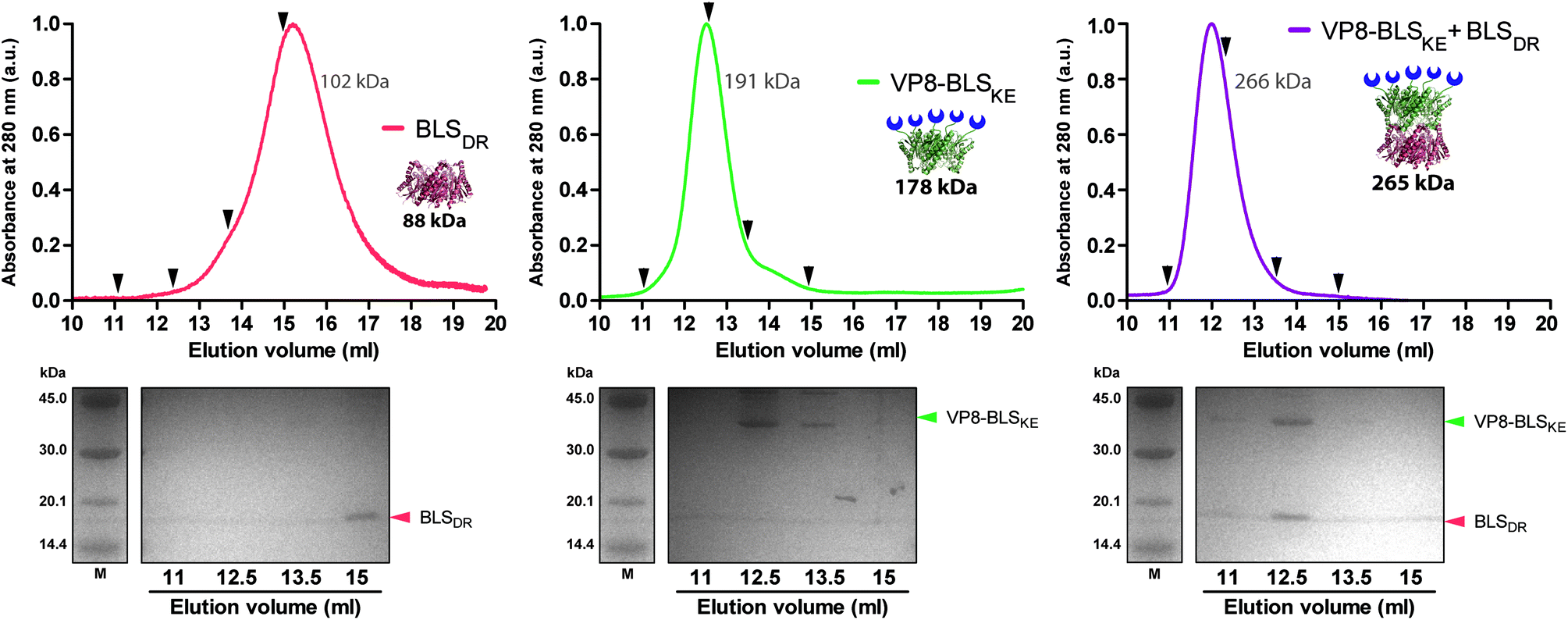 | ||
| Fig. 6 Quaternary structure determinations of VP8-BLSKE in solution. Upper panels: SEC-SLS chromatograms of BLSDR, VP8-BLSKE and VP8-BLSKE-BLSDR. The UV absorbance signal at 280 nm was recorded and normalized. The theoretical and SLS-derived MWs for each oligomeric molecule are indicated in black bold and grey types, respectively. Samples were taken at the marked elution volumes (black arrows) for SDS-PAGE analysis. Lower panels: 15% SDS-PAGE of the samples. The monomer identities matching their MWs are indicated at the right of each gel. | ||
Then, we performed two different labelling assays to demonstrate the potential, versatility and specificity of this platform. The first assay consisted of incubating a monolayer of adhered HeLa cells with VP8-BLSKE and A488BLSDR, followed by several washing steps and then visualization by confocal microscopy (Fig. 7A). Cells treated in this way display a clear fluorescent signal. Control treatment using BLSKE and A488BLSDR showed no substantial signal, indicating that the labeling is specific due to the presence of VP8 (Fig. 7A). The second labeling assay was accomplished by incubating NS0 cells in suspension with a VP8-BLSKE and A488BLSDR equimolar mixture. The cells were then washed and analyzed by flow cytometry. NS0 cells exhibited an ∼10-fold higher fluorescence signal compared to control cells treated with an A488BLSDR and BLSKE mixture (Fig. S6†). Altogether, these results indicate that the presence of the VP8 domain in the fluorescent decamer confers binding specificity to molecules of sialic acid present on the cell surface. We then assayed how the heterodecamer concentration affects the labeling. For that purpose, cell suspensions were incubated with increasing amounts of VP8-BLSKE and A488BLSDR. This experiment showed that there is a clear dependency of the signal distribution on the amount of VP8-BLSKE and A488BLSDR used in each treatment, also indicating that the system did not reach its saturation limit in the range tested (Fig. 7B).
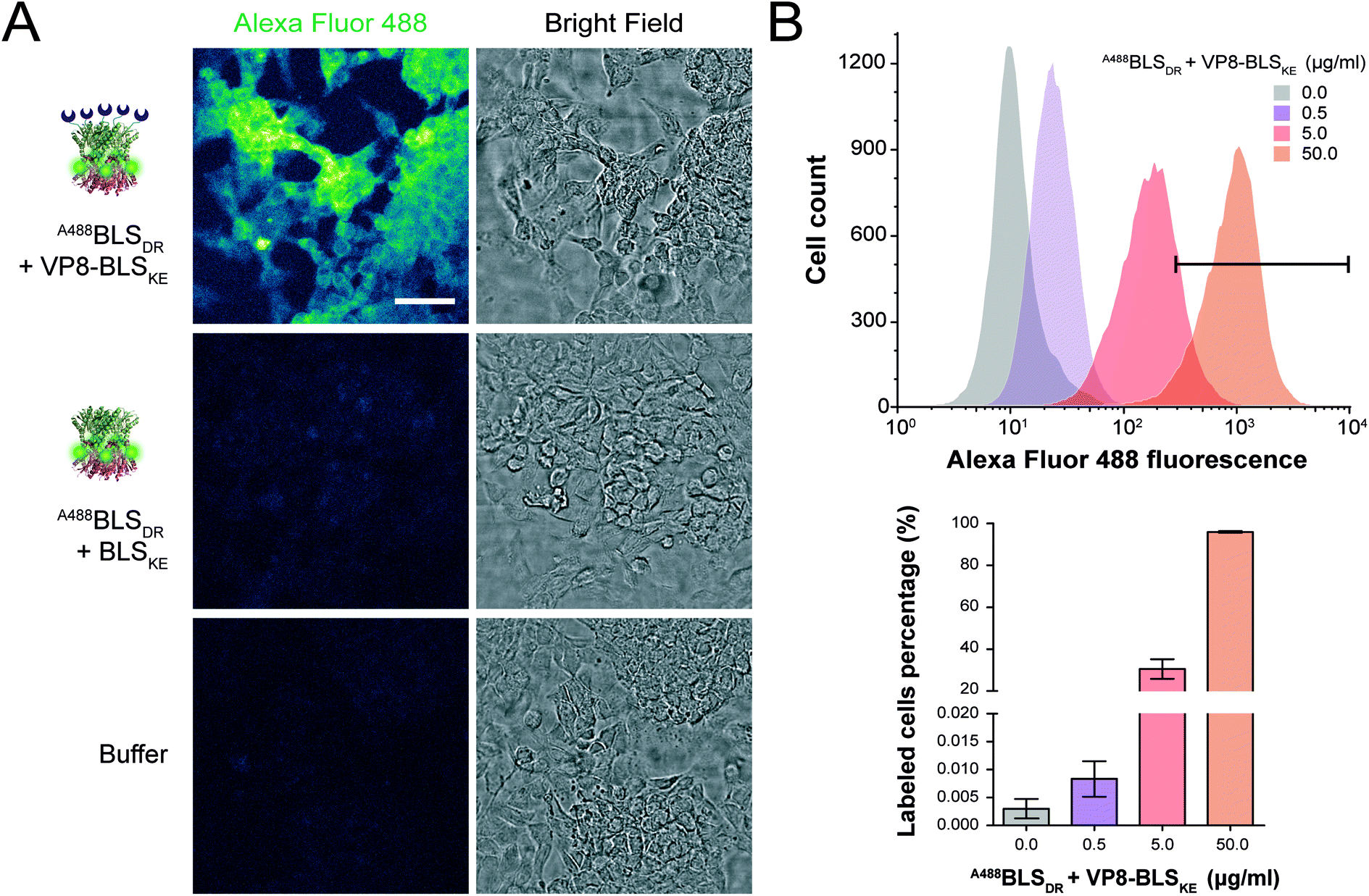 | ||
| Fig. 7 Fluorescence cell labeling assays. (A) Confocal microscopy images of HeLa cell monolayers incubated with BLSDR labeled with Alexa Fluor 488 (A488BLSDR), with or without sialic acid-binding VP8-BLSKE. Control cells without fluorescent labelling were treated with PBS buffer. Scale bar represents 50 μm. All fluorescence images were equalized using ImageJ software (color scale: “green fire blue” lookup table). (B) Upper panel: flow cytometry fluorescence intensity histograms incubating NS0 cells with increasing amounts of A488BLSDR + VP8-BLSKE. The areas under the fluorescence intensity values > 5 × 102 are indicated with the black bar. Lower panel: percentage of labelled cells presenting fluorescence intensity values ≥5 × 102 from the upper panel. | ||
Building a multichromophoric FRET model
FRET is a mechanism of energy transfer based on the dipole–dipole interaction that is active between two fluorophores in close proximity. Typically, it occurs when the separation distance is below 5–10 nm. Multichromophoric energy transfer plays a key role in natural systems such as light harvesting complexes.38 Fabrication of artificial multichromophoric systems is highly challenging, since it requires the organization of fluorophores with regular separation distances in a well-defined geometry with a precision of just a few nanometers.39–42 Our BLS model is a promising platform to achieve this goal taking advantage of protein self-assembly, analogously to natural complexes.We began to explore the use of the bifunctional BLS PNPs as a synthetic model to study multichromophoric FRET at the single molecule level (smFRET). We labeled positions 123 of the monomers of BLSDR and BLSKE with the Cy3 and Cy5 fluorescent dyes, respectively, as suitable donor and acceptor dyes for smFRET. Individual Cy3BLSDR and Cy5BLSKE pentamers were readily detected on a two-color wide-field epifluorescence microscope in total internal reflection illumination mode (TIRF), upon excitation with laser light of 532 and 642 nm, respectively (Fig. S7†). Although 10–12% of cross-talk was found in the Cy5 channel when Cy3BLSDR was excited at 532 nm, this is clearly differentiated from direct signals (Fig. S8†). The isolated Cy3BLSDR and Cy5BLSKE particles display multiple discrete fluorescence intensity steps. Approximately 10% of the PNPs showed 5 intensity steps (Fig. 8A and B), while the rest of the PNPs displayed 2, 3 or 4 fluorescence steps, indicating that the protein complexes were labeled with varying amounts of fluorophores.
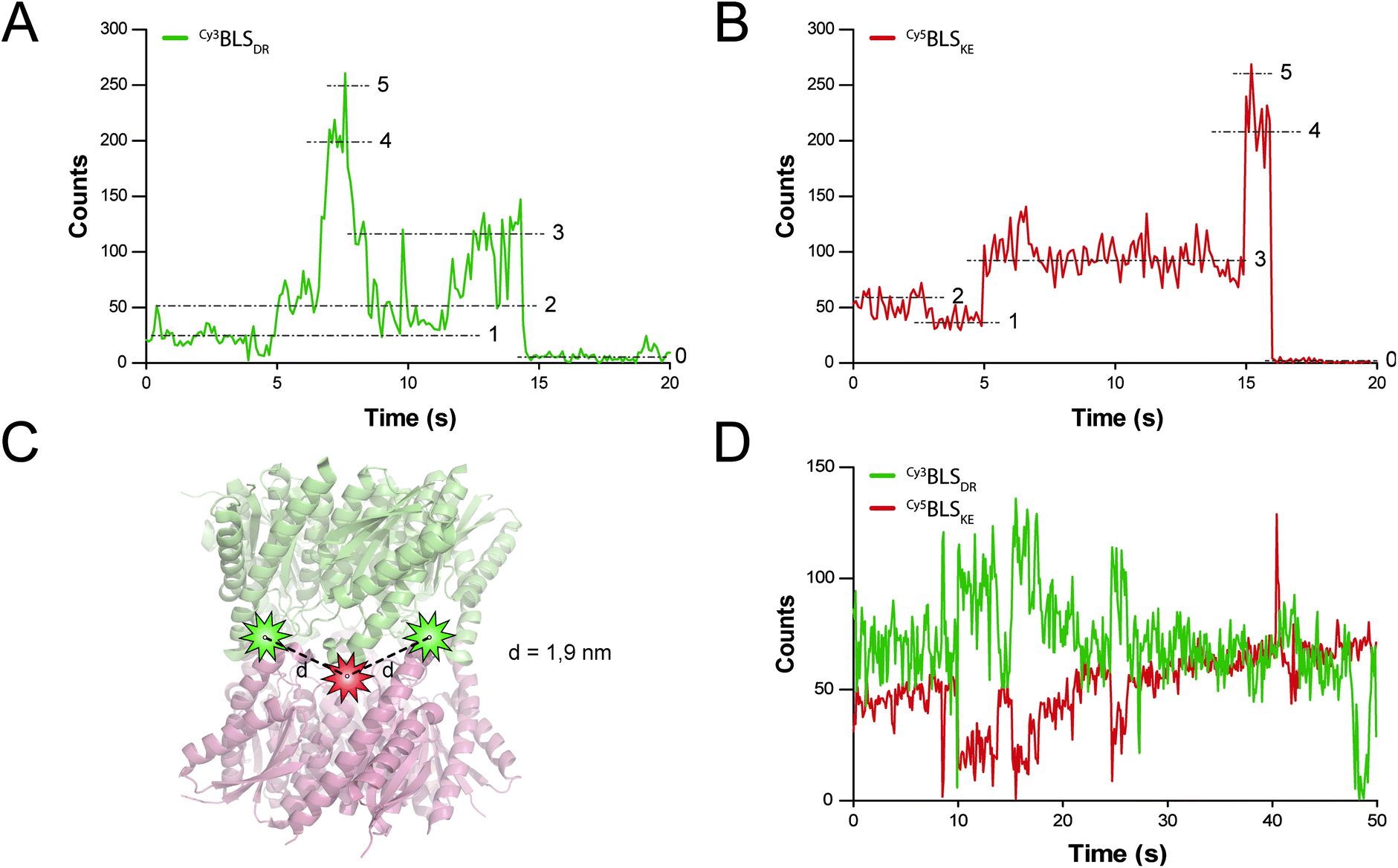 | ||
| Fig. 8 smFRET assays in BLS particles. (A and B) Fluorescence time traces of a representative single spot of Cy3BLSDR and Cy5BLSKE excited with 532 and 642 nm lasers, respectively. The five fluorescence intensity steps identified are indicated with dashed lines and their corresponding number. (B) Scheme of the spatial location of Cy3 (green stars) and Cy5 (red star) fluorescent dyes bound to the cysteine residues at positions 123 in a modified BLSDRKE. The calculated distances d between one Cy3 dye to its flanking Cy5 molecules are indicated. (D) Fluorescence time traces of a single spot of co-incubated Cy3BLSDR and Cy5BLSKE, excited with a 532 nm laser. | ||
The next step was to assemble the heterodecamer, positioning multiple fluorophores in the equatorial part of the molecules (Fig. 8C). For that aim, equimolar mixtures of labeled pentamers or pentamers alone were pre-incubated at room temperature prior to their deposition onto microscope coverslips. Co-incubation of Cy3BLSDR and Cy5BLSKE produced fluorescent particles with signals corresponding to Cy5BLSKE that could be detected in the Cy5 emission channel when excited with both 532 and 642 nm. These fluorescent spots also overlapped with fluorescent signals from the Cy3 emission channel under excitation at 532 nm (Fig. S8†). These results indicate that FRET indeed occurs within those doubly labeled fluorescent PNPs. The two-color emission time traces of the multichromophoric particles present complex anticorrelated signals (Fig. 8D), revealing a variety of smFRET events probably due to the different number and orientation of active fluorophores (e.g. caused by chemical bond rotations and protein backbone movements) at each given time within individual particles.
Discussion and conclusions
In the present work we have achieved the engineering of protein self-assembly in order to obtain a Janus-like functional particle scaffold. Our strategy consisted of generating two mutant pentameric variants of the BLS homodecameric protein, introducing charged residues in their pentamer–pentamer interfaces which prevented self-dimerization and simultaneously promoted heterodecamer formation through charge complementation. By this means, a precise control of the BLS multimer spatial configuration was achieved. The variants of BLS pentamers may be fused to other proteins or to chemical groups that could be employed to build novel nano-objects and nanomaterials, creating PNPs that can hold up to five copies of each function ordered in both sides of the scaffold. The pentavalent display of certain molecules such as fluorophores, enzymes or binding domains, with high local concentration, may enable for example, enhanced fluorescent signals, higher enzymatic reaction speeds and increased binding avidities. Moreover, following this scheme it is now possible to develop a pentameric library with different kinds of functions, which may be combined as desired in the same PNP. The addition of each new function could increase the combinatorial, and thus the possible technological applications.It is important to note that these new pentameric proteins conserve many of the structural properties of BLSWT and bring new characteristics that may be interesting for nanotechnological developments. When BLSKE and BLSDR are assembled into a decameric particle, it shows high temperature stability, and quaternary structure stability in a wide range of ionic strengths and at pH values over 5.5. Decameric BLSDRKE also conserves the intrinsic immunogenicity of BLSWT, which makes this system useful for vaccine engineering. In this sense, the BLS scaffold has been demonstrated to be successful for displaying foreign antigens, which has led to the patenting of this technology.43,44 In this work, we have expanded its capabilities, bringing the possibility to display two types of antigens (i.e. for multivalent vaccines) or an antigen and an immunomodulator (i.e. for immunomodulatory antigen delivery systems) within the same particle. On the other hand, the cellular labeling assays performed in this work indicate that the engineered versions of BLS could eventually be used as diagnostic or therapeutic tools.
DNA origami technology has recently become a powerful nanofabrication tool. However, achieving 3D or rigid structures using DNA is challenging due to the intrinsic flexibility of DNA. Proteins, on the other hand, can present naturally rigid and thermally stable tertiary and quaternary structures. Here, we have demonstrated that BLS and its variants can be used as versatile nanometric scaffolds to organize various molecular entities in space. In addition to the multivalent fluorescent labeling, we have used bifunctional PNPs to construct a multichromophoric energy transfer system, a highly challenging task as it requires the organization of multiple molecules at separation distances of 1–3 nm. Model systems in which the location and distances between chromophores are well controlled at this scale are powerful tools to understand in deeper detail photosynthetic processes and/or to engineer artificial photosynthetic structures.
In summary we have presented a new method for devising asymmetric, bifunctional PNPs with clear potential applications in (bio)nanotechnology. As we gain detailed knowledge of protein structures and interactions, new nanofabrication avenues through biomolecular self-assembly analogous to the one here presented will be opened.
Materials and methods
In silico structural analysis
The crystallographic structures of BLS and MLS (PDB entries 1XN1 and 2OBX, respectively) were analyzed to identify the residues involved in the pentamer–pentamer interactions.16,37 Interatomic distances between all residue-atoms from the different pentamers were measured using the web server CoCoMaps (https://www.molnac.unisa.it/BioTools/cocomaps/). Contacts were considered for residue atoms involved in their interactions (i.e. electrostatic, hydrophobic or π–π stacking) at distances shorter than 4.0 Å and are detailed in Table 1.In silico thermodynamic stability studies and complex formation were carried out using the FoldX program (http://foldxsuite.crg.eu/). The PDB 1XN1 model was used as a template for all BLS in silico mutations. An alanine scanning was performed in all residues identified in the pentamer dimer interface using CoCoMaps. For each residue, mutations were made in all monomers at the same time. Each mutation was performed 5 times and the contribution to the total interaction energy between pentamers was calculated as the average. The set of selected mutations to simultaneously disrupt the self-interactions between mutant pentamers and to favor heteropentamer interactions are shown in Table 2. BLSDR, BLSKE and BLSDRKE mutant decamers were analyzed using FoldX Stability and AnalyzeComplex functions.
Site-directed mutagenesis and VP8-BLS fusion
The pBLSWT plasmid carries the BLS gene (UniProt Q2YKV1) cloned in a pET11a plasmid (Novagen) between NdeI/BamHI sites, and possesses a wild-type interphase and an N-terminal C5S substitution to avoid the use of reducing agents to impair aggregation. All BLS mutant versions were generated by whole plasmid amplification PCR using the Q5 High-Fidelity DNA Polymerase (New England Biolabs), specific primers (Table S1†) and pET11a derived vectors as templates. Briefly, inverted and overlapping forward and reverse primers containing the mutations were used for 14–18 cycles PCRs using methylated plasmids as templates. Then, the reactions were treated with DpnI and transformed into E. coli DH5α cells. Clones were screened and confirmed by Sanger sequencing. By this means we generated the pBLSWT, pBLSD, pBLSDR, pBLSK, pBLSKE, pBLSDR-C and pBLSKE-C plasmids, as detailed in Table S1.†The VP8-BLS fusion construct was achieved first by PCR amplification of the 18 kDa sialic acid binding domain product from the C486 bovine rotavirus (BRV) VP8 core protein (VP8d), including the pentapeptide linker GSGSG, using Pfx DNA polymerase (Invitrogen), specific primers with homologous ends to the target plasmid and the pVP8-BLS plasmid as templates. The pVP8-BLS plasmid was kindly provided by Dr Patricio Craig.29 Then, the 0.5 kbp VP8d amplicon was used as the primer in a whole plasmid amplification PCR using the Q5 High-Fidelity DNA Polymerase and the pBLSKE plasmid as templates. Similarly as explained above, the reaction was treated with DpnI and transformed into E. coli DH5α cells. Clones carrying the 1.0 kbp fragment from the VP8d-linker fused to the BLSKE N-terminus at residue number 7 were screened using PCR and confirmed by Sanger sequencing, resulting in the pVP8-BLSKE plasmid (Table S1†).
For full details on the protein sequences used in this work see section “BLS protein sequences” in the ESI.†
Protein expression and purification
Vectors coding for BLSWT and its variants were transformed into E. coli BL21(DE3) cells. Protein expression was induced during exponential growth (OD600 = 0.7) with 0.5 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG) for 4 h at 37 °C. Then, bacterial cultures were pelleted at 9000 g for 15 min, sonicated at 10 W for 10 min and ultracentrifuged at 150000 g for 30 min. The soluble fraction was purified using a Q-Sepharose column (GE Healthcare Life Sciences) in a fast performance liquid chromatography apparatus (Gilson) with a 0–1 M NaCl linear gradient in 50 mM Tris pH 8.0. The samples enriched in BLS proteins were further purified on a Superdex-200 column (GE Healthcare Life Sciences) in 50 mM Tris–HCl pH 8.0, 0.25 M NaCl buffer. Each step was monitored by 15% SDS-PAGE. BLS proteins were concentrated up to 1–10 mg ml−1, flash-frozen in liquid N2, and stored at −80 °C for future use.
Static light scattering measurements
The molecular weight (MW) of proteins in solution was determined using a Precision Detectors PD2010 light scattering instrument connected in tandem to a Gilson high-performance liquid chromatography (HPLC) system, a Waters 486 UV detector and a LKB 2142 differential refractometer. BLS proteins (0.2–1 mg ml−1) were analyzed in a Superdex 200 HR-10/30 column (24 ml; GE Healthcare Life Sciences) and using an isocratic elution buffer (50 mM Tris–HCl pH 8.0, 0.25 M NaCl). The effect of ionic strength on dimerization was evaluated by only varying the total salt concentration (0–2 M NaCl). The effect of pH on dimerization was studied using a citrate-phosphate buffer (pH 4.0–8.0, 0.25 M NaCl). Scattered light at 90°, UV absorbance at 280 nm, and the refractive index (RI) signals were measured. The data were analyzed with Discovery32 software (Precision Detectors). The MW for each sample was calculated by using the SLS and RI signals, with bovine serum albumin (BSA) as a calibration standard (66.5 kDa).Far-UV circular dichroism
Circular dichroism (CD) spectra were monitored in the far-UV region using a J810 spectropolarimeter (Jasco). Ten scans were averaged for each measurement at 25 °C. Data were collected with a scan rate of 100 nm min−1 in a 0.1 cm path length cuvette. Data were then converted to molar ellipticity [θ] per dmol of protein [mdeg cm2 dmol−1]. Thermal unfolding of BLSWT, BLSDRKE, BLSDR and BLSKE was performed in 50 mM Tris–HCl pH 7.0, 0.1 M NaCl buffer by recording CD signals at 222 nm as a function of temperature. The samples were heated by increasing the temperature at 4 °C min−1 with a Peltier system (Jasco) in the range of 25–95 °C and measurements were performed at 0.5 °C intervals. Fast cooling back to 25 °C showed no recovery of the ellipticity, indicative of the irreversibility of the process. Thus, the Tm of the thermal transition in this work is reported as apparent Tm and is the temperature at which half of the difference between the initial and final signals is calculated from a sigmoidal fit to the experimental data.Protein labeling
BLSDR-C and BLSKE-C were labeled with (i) Alexa Fluor 488C5 Maleimide and Alexa Fluor 555C5 Maleimide (Thermo Fisher Scientific), and (ii) Cy3 Maleimide and Cy5 Maleimide Mono-Reactive Dye (GE Healthcare), following the manufacturers' instructions. Briefly, proteins at 100 μM concentration in a Tris–HCl 50 mM buffer pH 7.3, supplemented with 1 mM of dithiothreitol (DTT) or 0.7 mM Tris(2-carboxyethyl)phosphine (TCEP), were incubated overnight at 4 °C in the presence of 2 M of the maleimide fluorophores. The reactions were stopped with the addition of β-mercaptoethanol. Then, the unbound dyes were removed using a G25 minitrap column (GE Healthcare). Finally, the labeled proteins were concentrated to ∼50 mg ml−1 stocks and stored at −80 °C for further use. The resulting labeled proteins were named A488BLSDR, A555BLSKE, Cy3BLSDR and Cy5BLSKE, respectively.Förster resonance energy transfer (FRET)
The A488BLSDR ⇔ A555BLSKE association was assayed using steady-state FRET measurements. Briefly, a solution of 15 nM A488BLSDR in PBS, pH 8 and 600 μl total volume was mixed with increasing concentrations (0–600 nM) of A555BLSKE. The excitation and emission spectra were measured using a Jasco FP-500 spectrofluorometer. The 520 and 570 nm signals were determined from the emission spectra of the protein mixture by exciting the donor at 470 nm. The emission at 570 nm was then determined by exciting the acceptor at 540 nm. The raw fluorescence emission signals were corrected for background and averaged over three experiments at each specific condition. FRET efficiency was calculated using the following equation: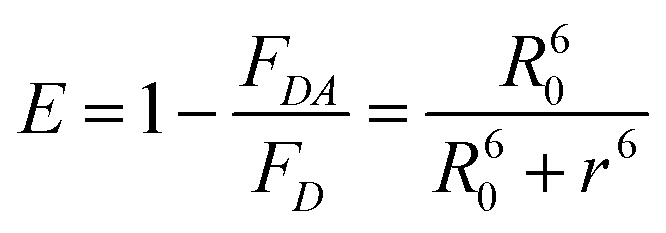 | (1) |
A fitting to FRET efficiency was performed assuming a simplified model of one-to-one interaction between A488BLSDR and A555BLSKE using the following equation:45
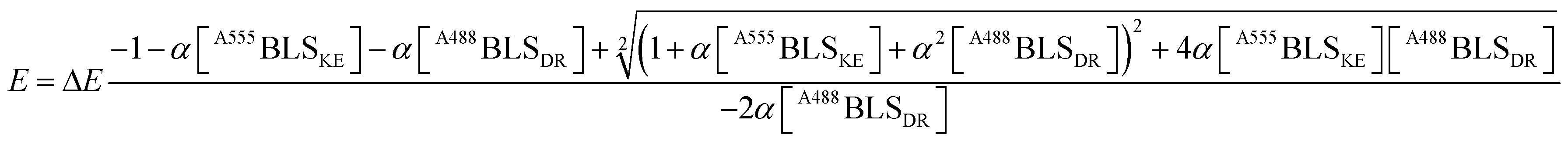 | (2) |
Immunization assays in mice
BALB/cJ mice were obtained from The Jackson Laboratory (Bar Harbor, Maine, USA), bred under specific pathogen-free conditions, housed and treated according to the policies of the protocol approved by the Committee on the Ethics of Animal Experiments of the Leloir Institute following the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The immunizations were performed in groups of 4–5 mice 8 to 10 weeks old, intraperitoneally, with 100 μl of phosphate saline buffer (PBS) containing 10 μg of BLSWT, BLSDRKE, BLSDR or BLSKE, previously incubated twice with polymyxin B-agarose (Sigma-Aldrich) to eliminate LPS, as previously described.22 Mice sera were collected at 14, 28 and 42 d.p.i. and stored at −20 °C for future use.Enzyme-linked immunosorbent assay (ELISA)
Standard ELISA procedures were followed to measure antibody production against the BLS protein variants in mice sera. Briefly, Nunc Maxisorp plate wells were coated or not (for non-specific signal) with 50 μl of BLS proteins (0.2 μg per well), and then blocked with 1% skim milk in PBS. Then, serial dilutions for each serum were incubated, washed and treated with peroxidase-conjugated polyclonal antibodies against mice IgG (Sigma, St. Louis, MO). The serum reactivity was developed by adding 50 μl of 2 μg μl−1o-phenylenediamine (OPD) and 0.03% H2O2 in 0.1 M citrate phosphate buffer. The reaction was stopped with 50 μl of 4 N H2SO4 and the absorbance at 492 nm was recorded using a plate reader (SLT Lab Instruments). The reported values for each serum dilution correspond to the mean absorbance at 492 nm for each group of animals.Monolayer cell culture labeling assay
HeLa cells were cultured at 37 °C, 5% CO2 in R10 medium consisting of RPMI 1640 endotoxin-free medium (Gibco) supplemented with 10% (v/v) Fetal Bovine Serum (FBS, Gibco), 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 1 mM pyruvate and 4 mM L-glutamine. Then, 2.5 × 105 cells were transferred to a multiwell culture plate containing microscope glass coverslips and incubated for 30 min at 37 °C in 5% CO2. Afterwards, the cells were washed with R10 medium and fixed in 8% paraformaldehyde (PFA) and 4% sucrose in water for 30 min at 37 °C. The coverslips were then incubated with 2% BSA and 2% (v/v) donkey serum (Sigma-Aldrich) in PBS for 4 h and then washed with PBS. Washed coverslips were incubated for 30 min at room temperature with PBS, A488BLSDR + BLSKE or A488BLSDR + VP8-BLSKE at a final protein concentration of 10 μg ml−1 in PBS. Finally, after washing with water, the coverslips were mounted on microscope slides and visualized using an LSM 510 confocal microscope (Zeiss). Images were processed using ImageJ 1.51u software (https://imagej.nih.gov/ij/).Suspension cell culture labeling assay
NS0 murine myeloma cells (ATCC PTA-4174) were grown at 37 °C 5% CO2 in DMEM medium containing FBS 10% (Gibco), 200 U ml−1 penicillin, 100 μg ml−1 streptomycin, 1 mM pyruvate and 4 mM L-glutamine. Then, 0.25 × 105 cells were transferred to a cell culture plate with PBS buffer containing 0.1% sodium azide and 3% (v/v) SFB in a 200 μl final volume. Afterwards, cells were incubated for 30 min at 4 °C in PBS, A488BLSDR + BLSKE or A488BLSDR + VP8-BLSKE at a final protein concentration of 50 μg ml−1 in PBS. Later, the samples were washed twice with PBS, centrifuged at 400 g and resuspended in 200 μl PBS. Finally, cellular bound fluorescence was quantified by flow cytometry using a fluorescence-activated cell sorting system (FACScan cytometer, BD Biosciences). Data were analyzed with the CellQuest software (BD Immunocytometry Systems) and FlowJo v7.6.2 software (https://www.flowjo.com/).Single-molecule FRET (smFRET)
Samples were prepared in Lab-Tek II chambered coverglasses (Nunc), previously treated with KOH 1 M, a 0.48% (v/v) aqueous solution of Poly(diallyldimethylammonium) chloride (Sigma Aldrich) and blocked BSA 1%. Cy3BLSDR and Cy5BLSKE PBS buffer solutions were pre-incubated for 30 min at room temperature, separated or in equimolar mixtures, at 0.5 μM for each labeled pentamer. Then, coverglasses were treated for 1 s with each protein solution and washed 10 times with PBS buffer. Each sample was incubated with imaging buffer (to stabilize blinking and avoid photobleaching) containing 50 mM Tris pH 8, 10 mM NaCl, 10% (w/v) glucose, 100 mM mercaptoethylamine, 1 mg ml−1 glucose oxidase, 0.5 mg ml−1 catalase and 2 mM Trolox (Acros Organics, Fisher Scientific).smFRET was carried out using a microscope home-built around an Olympus IX-73 body, operating in wide-field epifluorescence mode. Two color images were acquired with an oil immersion objective Olympus PlanApo 60x NA 1.42. Total internal reflection illumination mode (TIRF) was enabled by moving a linear stage (Thorlabs) so that the focus of the lasers translated laterally within the back focal plane of the objective. Fluorescence emission from Cy3 and Cy5 was separated with a dichroic mirror (Chroma ZT647rdc), filtered with bandpass emission filters (Semrock 582/75 BrightLine HC and Chroma ET700/75m) and imaged onto adjacent areas of the EMCCD camera operated at −50 °C (Andor iXon3 897). The camera and the lasers were controlled with custom software previously described.46 A 642 nm 1.5 W laser (MPB Communications, 2RU/VFL/P/1500/642) and a 532 nm 1.5 W laser (Laser Quantum, ventus 532) were used for fluorescence excitation of Cy5 and Cy3, respectively. The lasers were combined with dichroic mirrors (Semrock LM01-427, LM01-552 and Di03-R405/488/532/635-t1) and then focused to the back focal plane of the objective. Further blocking of the illumination lasers was performed with a multi-edge notch filter (Semrock NF03-05/488/532/635E). Laser power densities used during acquisition were 555 W cm−2 and 610 W cm−2 for 532 and 642 nm, respectively. A Pre-Amp Gain of 5.1 and an EM Gain of 50 were used in the CCD camera. Different regions of 128 × 128 pixels (∼290 μm2) were observed using 100 ms exposure times. Single molecule time traces were achieved after background subtraction, using a region of interest of 5 × 5 pixels (∼0.44 μm2). Cross-talk was characterized in samples containing either Cy3 or Cy5, and was then used to correct Cy3 and Cy5 traces. Differences between the two channels were corrected by imaging isolated fluorescent markers visible on both channels (Life Technologies Tetraspeck 0.1 μm) and then finding the affine transformation that minimizes the distance between the same markers as detected in each detection channel as previously described.46
Statistical analysis
Statistical analysis was performed with the InfoStat 2014 software (https://www.infostat.com.ar/) and GraphPad Prism version 6.00 software (https://www.graphpad.com/). One-way ANOVA followed by non-parametric analysis of variance (Kruskal–Wallis) and paired comparisons (Mann–Whitney) were performed.Conflicts of interest
The authors declare no conflicts of interestAcknowledgements
This work was supported by the Argentinian Research Council (CONICET), the Argentinian Agency for Science and Technology Promotion (ANPCyT) (grant PICT-2014-0959 and PICT-2014-3729), and the Argentinian Ministry of Science, Technology and Productive Innovation (MINCyT). Santiago Sosa was partially supported by a fellowship from Consejo Interuniversitario Nacional (CIN), Argentina. We thank Dr Patricio O. Craig for fruitful discussions.References
- K. Fujiwara and M. Ikeguchi, Open Bioinf. J., 2008, 2, 50–53 CrossRef CAS.
- A. Llauró, B. Schwarz, R. Koliyatt, P. J. de Pablo and T. Douglas, ACS Nano, 2016, 10, 8465–8473 CrossRef PubMed.
- Y. Tanaka, K. Tsumoto, Y. Yasutake, M. Umetsu, M. Yao, H. Fukada, I. Tanaka and I. Kumagai, J. Biol. Chem., 2004, 279, 32957–32967 CrossRef CAS PubMed.
- C. Ludwig and R. Wagner, Curr. Opin. Biotechnol., 2007, 18, 537–545 CrossRef CAS PubMed.
- J. López-Sagaseta, E. Malito, R. Rappuoli and M. J. Bottomley, Comput. Struct. Biotechnol. J., 2016, 14, 58–68 CrossRef PubMed.
- B. Schwarz, M. Uchida and T. Douglas, Adv. Virus Res., 2017, 97, 1–60 CAS.
- P.-Y. Fang, L. M. Gómez Ramos, S. Y. Holguin, C. Hsiao, J. C. Bowman, H.-W. Yang and L. D. Williams, Nucleic Acids Res., 2017, 45, 3519–3527 CrossRef CAS PubMed.
- M. Uchida, K. McCoy, M. Fukuto, L. Yang, H. Yoshimura, H. M. Miettinen, B. LaFrance, D. P. Patterson, B. Schwarz, J. A. Karty, P. E. Prevelige Jr, B. Lee and T. Douglas, ACS Nano, 2017, 12, 942–953 CrossRef PubMed.
- K. M. Frietze, D. S. Peabody and B. Chackerian, Curr. Opin. Virol., 2016, 18, 44–49 CrossRef CAS PubMed.
- H. Masarapu, B. K. Patel, P. L. Chariou, H. Hu, N. M. Gulati, B. L. Carpenter, R. A. Ghiladi, S. Shukla and N. F. Steinmetz, Biomacromolecules, 2017, 18, 4141–4153 CrossRef CAS PubMed.
- T. G. W. Edwardson, T. Mori and D. Hilvert, J. Am. Chem. Soc., 2018, 140, 10439–10442 CrossRef CAS PubMed.
- N. Kobayashi and R. Arai, Curr. Opin. Biotechnol., 2017, 46, 57–65 CrossRef CAS PubMed.
- L. Yang, A. Liu, S. Cao, R. M. Putri, P. Jonkheijm and J. J. L. M. Cornelissen, Chemistry, 2016, 22, 15570–15582 CrossRef CAS PubMed.
- A. Bacher, S. Eberhardt, M. Fischer, K. Kis and G. Richter, Annu. Rev. Nutr., 2000, 20, 153–167 CrossRef CAS PubMed.
- V. Zylberman, S. Klinke, I. Haase, A. Bacher, M. Fischer and F. A. Goldbaum, J. Bacteriol., 2006, 188, 6135–6142 CrossRef CAS PubMed.
- S. Klinke, V. Zylberman, H. R. Bonomi, I. Haase, B. G. Guimarães, B. C. Braden, A. Bacher, M. Fischer and F. A. Goldbaum, J. Mol. Biol., 2007, 373, 664–680 CrossRef CAS PubMed.
- Y. Azuma, T. G. W. Edwardson and D. Hilvert, Chem. Soc. Rev., 2018, 47, 3543–3557 RSC.
- N. Ainciart, V. Zylberman, P. O. Craig, D. Nygaard, H. R. Bonomi, A. A. Cauerhff and F. A. Goldbaum, Proteins, 2011, 79, 1079–1088 CrossRef CAS PubMed.
- H. R. Bonomi, M. I. Marchesini, S. Klinke, J. E. Ugalde, V. Zylberman, R. A. Ugalde, D. J. Comerci and F. A. Goldbaum, PLoS One, 2010, 5, e9435 CrossRef PubMed.
- V. Zylberman, P. O. Craig, S. Klinke, B. C. Braden, A. Cauerhff and F. A. Goldbaum, J. Biol. Chem., 2004, 279, 8093–8101 CrossRef CAS PubMed.
- F. A. Goldbaum, C. A. Velikovsky, P. C. Baldi, S. Mörtl, A. Bacher and C. A. Fossati, J. Med. Microbiol., 1999, 48, 833–839 CrossRef CAS PubMed.
- P. M. Berguer, J. Mundiñano, I. Piazzon and F. A. Goldbaum, J. Immunol., 2006, 176, 2366–2372 CrossRef CAS.
- S. M. Estein, M. A. Fiorentino, F. A. Paolicchi, M. Clausse, J. Manazza, J. Cassataro, G. H. Giambartolomei, L. M. Coria, V. Zylberman, C. A. Fossati, R. Kjeken and F. A. Goldbaum, Vaccine, 2009, 27, 6704–6711 CrossRef CAS PubMed.
- P. M. Berguer, V. A. Alzogaray, A. H. Rossi, J. Mundiñano, I. Piazzon and F. A. Goldbaum, PLoS One, 2012, 7, e45705 CrossRef CAS PubMed.
- A. G. Díaz, M. Clausse, F. A. Paolicchi, M. A. Fiorentino, G. Ghersi, V. Zylberman, F. A. Goldbaum and S. M. Estein, Vet. Immunol. Immunopathol., 2013, 154, 36–41 CrossRef PubMed.
- A. H. Rossi, A. Farias, J. E. Fernández, H. R. Bonomi, F. A. Goldbaum and P. M. Berguer, PLoS One, 2015, 10, e0126827 CrossRef PubMed.
- M. Clausse, A. G. Díaz, R. P. Pardo, V. Zylberman, F. A. Goldbaum and S. M. Estein, Vet. Immunol. Immunopathol., 2017, 184, 36–41 CrossRef CAS PubMed.
- D. A. Laplagne, V. Zylberman, N. Ainciart, M. W. Steward, E. Sciutto, C. A. Fossati and F. A. Goldbaum, Proteins, 2004, 57, 820–828 CrossRef CAS PubMed.
- D. Bellido, P. O. Craig, M. V. Mozgovoj, D. D. Gonzalez, A. Wigdorovitz, F. A. Goldbaum and M. J. Dus Santos, Vaccine, 2009, 27, 136–145 CrossRef CAS PubMed.
- G. Fragoso, F. Esquivel-Guadarrama, M. A. Santana, R. J. Bobes, B. Hernández, J. Cervantes, R. Segura, F. A. Goldbaum, E. Sciutto and G. Rosas, Clin. Vaccine Immunol., 2011, 18, 1067–1076 CrossRef CAS PubMed.
- M. P. Mejias, G. Ghersi, P. O. Craig, C. A. Panek, L. V. Bentancor, A. Baschkier, F. A. Goldbaum, V. Zylberman and M. S. Palermo, J. Immunol., 2013, 191, 2403–2411 CrossRef CAS PubMed.
- P. O. Craig, P. M. Berguer, N. Ainciart, V. Zylberman, M. G. Thomas, L. J. Martinez Tosar, A. Bulloj, G. L. Boccaccio and F. A. Goldbaum, Proteins, 2005, 61, 1089–1100 CrossRef CAS PubMed.
- L. Ratier, M. Urrutia, G. Paris, L. Zarebski, A. C. Frasch and F. A. Goldbaum, PLoS One, 2008, 3, e3524 CrossRef PubMed.
- P. O. Craig, V. Alzogaray and F. A. Goldbaum, Biomacromolecules, 2012, 13, 1112–1121 CrossRef CAS PubMed.
- L.-T.-C. Tran, S. Lesieur and V. Faivre, Expert Opin. Drug Delivery, 2014, 11, 1061–1074 CrossRef CAS PubMed.
- R. Guerois, J. E. Nielsen and L. Serrano, J. Mol. Biol., 2002, 320, 369–387 CrossRef CAS.
- S. Klinke, V. Zylberman, D. R. Vega, B. G. Guimarães, B. C. Braden and F. A. Goldbaum, J. Mol. Biol., 2005, 353, 124–137 CrossRef CAS PubMed.
- T. Mirkovic, E. E. Ostroumov, J. M. Anna, R. van Grondelle, Govindjee and G. D. Scholes, Chem. Rev., 2017, 117, 249–293 CrossRef CAS PubMed.
- R. A. Miller, A. D. Presley and M. B. Francis, J. Am. Chem. Soc., 2007, 129, 3104–3109 CrossRef CAS PubMed.
- J. Iehl, J.-F. Nierengarten, A. Harriman, T. Bura and R. Ziessel, J. Am. Chem. Soc., 2012, 134, 988–998 CrossRef CAS PubMed.
- P. K. Dutta, S. Levenberg, A. Loskutov, D. Jun, R. Saer, J. T. Beatty, S. Lin, Y. Liu, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2014, 136, 16618–16625 CrossRef CAS PubMed.
- E. A. Hemmig, C. Creatore, B. Wünsch, L. Hecker, P. Mair, M. A. Parker, S. Emmott, P. Tinnefeld, U. F. Keyser and A. W. Chin, Nano Lett., 2016, 16, 2369–2374 CrossRef CAS PubMed.
- USPTO, 8685670, US Patent, 2014 Search PubMed.
- USPTO, 20160115459:A1, US Patent, 2016 Search PubMed.
- Y. Song, V. Madahar and J. Liao, Ann. Biomed. Eng., 2011, 39, 1224–1234 CrossRef PubMed.
- F. M. Barabas, L. A. Masullo and F. D. Stefani, Rev. Sci. Instrum., 2016, 87, 126103 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8na00375k |
| ‡ Current address: Centre de Biochimie Structurale, CNRS UMR5048, INSERM U1054, Université de Montpellier, 29 rue de Navacelles (34090), Montpellier, France. |
| This journal is © The Royal Society of Chemistry 2019 |