
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceParameters affecting the synthesis of carbon dots for quantitation of copper ions†
Yu-Syuan
Lin‡
a,
Yaling
Lin‡
a,
Arun Prakash
Periasamy
a,
Jinshun
Cang
b and
Huan-Tsung
Chang
 *ac
*ac
aDepartment of Chemistry, National Taiwan University, Taipei 10617, Taiwan. E-mail: changht@ntu.edu.tw
bDepartment of Chemical Engineering, Yancheng Institute of Industry Technology, Yancheng, Jiangsu 224005, China
cDepartment of Chemistry, Chung Yuan Christian University, Taoyuan City 32023, Taiwan
First published on 14th May 2019
Abstract
A simple, eco-friendly, and low-cost electrochemical approach has been applied to the synthesis of carbon dots (C dots) from histidine hydrochloride in the absence or presence of halides (Cl, Br, and I) at various potentials up to 10 V. The as-formed C dots refer to C dots, Cl–C, Br–C, and I–C dots. The time-evolution UV-vis absorption and photoluminescence (PL) spectra provide more detailed information about the formation of C dots. Upon increasing the reaction time from 1 to 120 min, more and more C dots are formed, leading to increased PL intensity. The halides play two important roles in determining the formation of C dots; controlling the reaction rate and surface states. When compared to chloride and bromide, iodide has a greater effect on varying surface states and inducing PL quenching through intersystem crossing. The PL intensities of the four types of C dots all decrease upon increasing Cu2+, Hg2+, and Ag+ concentrations. In the presence of 0.8 mM I−, I–C dots compared to C dots, Cl–C dots, and Br–C dots are slightly better for quantitation of Cu2+. Fourier transform infrared spectroscopy, cyclic voltammetry, electrochemical impedance spectroscopy, and X-ray photoelectron spectroscopy results of I–C dots reveal the interactions of Cu2+ with the surface ligands (imidazole and histidine). The I–C dot probe in the presence of 0.8 mM I− is selective toward Cu2+ over the tested metal ions such as Hg2+ and Ag+. The assay provides a limit of detection of 0.22 μM for Cu2+ at a signal-to-noise ratio of 3. Practicality of this probe has been validated by the analyses of tap, lake, and sea water samples, with negligible matrix effects.
1. Introduction
Photoluminescent nanomaterials such as carbon dots (C dots), quantum dots, nanoclusters, and gold nanodots have become interesting optical sensing materials for detecting various analytes.1–5 Among them, C dots are more biocompatible and stable against salt and photoirradiation induced photoluminescence (PL) quenching. Most C dots possess interesting excitation dependent PL properties; their emission undergoes a red shift upon increasing the excitation wavelength.6 Usually, their PL intensity decreases upon increasing the emission wavelength.7 The interesting PL properties of C dots are related to their core size, surface defects and ligands, oxidation state, and the number of conjugated π electrons.1,8–14Many approaches, including laser ablation, ultrasonication, direct heating, microwave-assisted heating, plasma treatment, hydrothermal routes, have been applied for the preparation of C dots from different precursors.15–21 Among them, the hydrothermal approach is the most popular, mainly because of its simplicity.6,21 It has been reported that C dots are formed through four steps i.e. condensation, polymerization, carbonization, and passivation.22–24 Various sources, including coffee powder, tea, and apple juice, have been used for the preparation of C dots through hydrothermal approaches.6,25–30 However, hydrothermal approaches require high energy and are usually limited to the preparation of few grams of C dots per batch.
Although the formation processes and optical properties of C dots have been suggested,22–24 strong evidence to support the suggestion/hypothesis is still needed. In this study, we applied our previously developed electrochemical approach to the preparation of C dots from histidine.31 We previously showed that control of the solution pH value is important for the electrochemical preparation of C dots from glycine. However, detailed information about the formation of C dots is missing. We in this study further tested several important factors for controlling the preparation of C dots from histidine, including electrolysis time, concentration of histidine, and species and concentration of salts (electrolytes). We measured the UV-vis absorption and PL spectra of C dots that had been prepared in alkaline solution at 10 V for different periods of time up to 120 min. Through the obtained optical data, we provided more detailed information about the effect of halides on the formation of C dots. The C dots prepared in the presence of iodide (I−) are selective and sensitive for quantitation of copper ions (Cu2+) in the presence of 0.8 mM NaI.
2. Experimental
2.1 Materials
L-Histidine hydrochloride monohydrate was purchased from TCI (Tokyo, Japan). Potassium nitrate (>99.0%), silver nitrate (>99.0%), and sodium chloride (>99.0%) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Copper nitrate trihydrate (>99.0%), lead nitrate (>99.0%), and sodium bromide (99.5%) were purchased from Acros (Morris Plains, NJ, USA). Magnesium nitrate hexahydrate (>98.0%) and sodium iodide (>99.0%) were procured from SHOWA (Tokyo, Japan). Cobalt chloride (97.0%), iron chloride (98.0%), and mercury chloride (>98.0%) were obtained from Alfa Aesar (Heysham, England). Calcium nitrate tetrahydrate (>99.0%) and manganese chloride dehydrate (>99.0%) were purchased from Merck (Kenilworth, NJ, USA). Monobasic, dibasic, and tribasic sodium salts of phosphate, phosphoric acid (85.0%), sodium hydroxide (98.0%), and zinc nitrate hexahydrate (>99.0%) were obtained from J.T. Baker (Center Valley, PA, USA). Ultrapure water (18.2 MΩ cm) from a Milli-Q system (Millipore, Billerica, MA, USA) was used to prepare all solutions.2.2 Preparation of C dots
A two-platinum-electrode electrochemical system was used for the synthesis of C dots. The length and diameter of the Pt electrode are 6 and 0.05 cm, respectively. A single output direct current (DC) power supply (TP-1303C, Taipei, Taiwan) was used for the synthesis of C dots under a static potential (1 to 10 V) between the two electrodes. Known amounts (0.38–3.83 g) of histidine hydrochloride were dissolved separately in NaOH solution (1 M) to obtain final concentrations of 0.09–0.90 M, with a final pH value of 9.0 that was adjusted with 10 M HCl or NaOH. To study the effect of halides on the formation of C dots from histidine, aqueous solutions of NaCl (0–2.34 g), NaBr (2.06 g), or NaI (3.00 g) were added to the histidine solutions (20 mL). Aliquots (10 μL) of the solutions after being subjected to electrolysis for various periods of time (1–120 min) were taken out for spectrochemical measurement. For quantitation of Cu2+ ions, the C dots that had been prepared at 10 V for 120 min and had been left undisturbed at ambient temperature (25 °C) for two days were used. This solution was then filtered through a 0.22 μm membrane to remove large particles, and was then subjected to dialysis against pure water through a membrane (MWCO = 100–500D) for 24 h. For simplicity, C dots prepared in the presence of Cl−, Br−, and I− are denoted as Cl–C, Br–C, and I–C dots, respectively. Their concentrations are presented as 1× for simplicity.2.3 Characterization
Aqueous solutions of four different types of C dots were subjected to PL measurements using a microplate fluorometer (Synergy 4 Multi-Mode Microplate Reader) from BioTek instruments (Winooski, VT, USA). Their PL decay curves were recorded using a FluoTime 300 system from PicoQuant (Berlin, Germany) with a diode laser emitting at 375 nm as the light source. A Synergy H1 multi-mode monochromatic spectrophotometer from BioTek instruments (Winooski, VT, USA) was used to measure their UV-vis absorption spectra. The Fourier transform infrared (FTIR) spectra of histidine and C dots were recorded using a Varian 640 FTIR spectrophotometer from Varian (Palo Alto, CA, USA). The transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images of the C dots were acquired using an H-7100 TEM from Hitachi (Tokyo, Japan) and a JSM-1200EX II HRTEM from JEOL (Tokyo, Japan), respectively. The X-ray photoelectron spectroscopy (XPS) measurements were conducted using a K-Alpha X-ray photo electron spectrometer system from Thermo (Waltham, MA, USA). The binding energy values of C dots were calibrated using the C1s signal at 284.6 eV. Their Raman spectra were acquired using an Olympus BX51 microscope and a Nicolet Almega XR dispersive Raman spectrometer from Thermo (Waltham, MA, USA) with a 10 mW laser emitting at a 780 nm peak wavelength, using the Raman peak of Si wafer (520 cm−1) as a standard. Cyclic voltammetry (CV) curves of phosphate buffer (20 mM, pH 3.0) without and with C dots (200 μL) and metal ions (200 μM) were recorded using a CHI 760D electrochemical work station from CH instruments (Austin, TX, USA). A glassy carbon electrode, saturated Ag/AgCl, and Pt wire were used as the working, reference, and counter electrodes, respectively. Electrochemical impedance spectra (EIS) and the Nyquist plots of the glassy carbon electrode in the frequency range of 0.01 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz at an amplitude of 5 mV were recorded using the CHI 760D electrochemical work station in N2 saturated phosphate buffer (20 mM, pH 3.0) without and with C dots (200 μL) and Cu2+ ions (200 μM).
000 Hz at an amplitude of 5 mV were recorded using the CHI 760D electrochemical work station in N2 saturated phosphate buffer (20 mM, pH 3.0) without and with C dots (200 μL) and Cu2+ ions (200 μM).
2.4 Quantitation of Cu2+ ions
Quantitation of Cu2+ ions (0–50 μM) using C-dots (0.001×, final concentration) was conducted in phosphate buffer solutions (20 mM) at pH values of 3.0 and 7.0. Linearity, limit of detection, and selectivity were investigated at pH 3.0. To obtain linearity for quantitation of Cu2+ ions, aliquots of C dots (0.01×, 100 μL) were added to phosphate buffer solution (25 mM, pH 3.0, 800 μL) prior to the addition of Cu2+ ions (0–500 μM, 100 μL). The mixtures were equilibrated by shaking at ambient temperature for 60 min, and their PL spectra were recorded when excited at 420 nm. To test the selectivity, C dots (0.1×, 100 μL) were added to phosphate buffer solution (25 mM, 800 μL). Various metal ions (K+, Mg2+, Ca2+, Mn2+, Fe3+, Co2+, Cu2+, Zn2+, Pb2+, Hg2+, and Ag+; 1 mM, 100 μL) were then added into the resulting solution. The mixtures were equilibrated by shaking at ambient temperature for 60 min, and their PL spectra were recorded with an excitation wavelength of 420 nm. This assay was validated by the analyses of lake and tap water samples that had been collected from the National Taiwan University campus, and the sea water sample that had been collected from Keelung coast (East China Sea; Taiwan). Each of the water samples was filtered through a 0.22 μm membrane. Aliquots (100 μL) of the real samples were spiked with various concentrations (0.3–3 μM; final concentrations) of Cu2+ ions. Then, phosphate buffer (200 mM, pH 3.0, 100 μL), C dots (0.01×, 100 μL), and ultrapure water were added subsequently to the spiked solutions, each with a final volume of 1.0 mL. The mixtures were equilibrated by shaking at ambient temperature for 60 min, and their PL spectra were recorded under excitation at 420 nm.3. Result and discussion
3.1 Formation of C dots
When applying a voltage greater than 1.31 V, oxidation of histidine occurred on the Pt anode, in which the oxidized histidine was adsorbed onto the electrode through its terminal carboxyl group and secondary amino group. Most of the adsorbed molecules were then converted to form imidazoles.31,32 During the course of electrolysis, the solution pH value increased slightly as a result of the formation of NH3. The adsorbed imidazoles were converted to form C dots through a process of polymerization, carbonization and passivation.32 The production yield of C dots prepared from amino acids through a simple electrochemical approach under alkaline conditions is usually higher than that under acidic conditions.31 The yields of C dots, Cl–C dots, Br–C dots, and I–C dots are 5.1, 12.3, 15.6 and 33.8%, respectively. The yield of C dots is lower than that of the other three dots, mainly because they were prepared in the solution containing lower concentration of electrolyte. The yield of I–C dots is the highest, mainly because I− is a stronger leaving group than Br− and Cl−, leading to higher reaction efficiency.3.2 Effect of applied voltage
Applied voltage affects the electrolysis rate and thus the formation of C dots. At an applied voltage <1.31 V,33 C-dots were not formed, mainly because the oxidation of histidine did not occur. At a constant electrolysis time (3 min), the PL intensity of the solution increases upon increasing the applied voltage from 1 to 10 V as shown in Fig. 1A. Interestingly, the PL profiles are all similar, revealing that the applied voltage is unlikely to affect the structure of C-dots. The result reveals that more C dots were formed when applying a higher voltage for the same reaction time. Considering reaction speed and PL intensity, we selected 10 V for the preparation of C dots hereafter. Since the amount of heat and bubbles generated increased upon increasing the voltage, a voltage greater than 10 V is not suggested unless a cooling system is applied. | ||
| Fig. 1 Effects of (A) applied voltage, (B) histidine concentration, (C) pH, and (D) NaCl concentration on the formation of C dots at 10 V for 2 h. Excitation and emission wavelengths are 420 nm and 505 nm, respectively. (A) 0.63 M histidine at pH 9.0; (B) 1 M NaCl at pH 9.0; (C) 0.63 M histidine and 1 M NaCl; (D) 0.63 M histidine at pH 9.0. | ||
3.3 Effect of histidine concentration and pH value
Fig. 1B shows the PL spectra of C dots prepared from different concentrations of histidine (0.09–0.90 M) at pH 9.0 and 10 V for 120 min. The PL intensity at 505 nm increases upon increasing the concentration of histidine up to 0.63 M. Upon further increasing the histidine concentration, the PL intensity decreases, mainly because through intermolecular attractions some unreacted histidine molecules formed aggregates and then adsorbed onto the surfaces of C dots, leading to aggregation of C dots. At the optimal histidine concentration (0.63 M), we tested the effect of pH (3.0–9.0) on the formation of C dots. Fig. 1C shows that the PL intensity increases upon increasing the pH value from 3.0 to 9.0. Under acidic conditions (pH ∼3.0), the protonated imidazole ring interacts weakly with the Pt anode surface, leading to poor efficiency of oxidation, polymerization, and carbonization. Upon increasing the pH value (e.g. pH ∼5.0), the interaction of histidine with the Pt electrode increases, thus C dots are formed quickly, leading to higher PL than that when synthesized at pH 3.0. More C dots are formed and PL further increases upon increasing the pH value from 5.0–7.0, simply because the interaction of the adsorbed molecule with the Pt electrode is stronger as a result of increased dissociation degree of its carboxylic acid group.34 At pH 9.0, the PL intensity reaches its maximum. The anodic generation of oxygen/or hydroxyl ions accelerates the electrochemical oxidation/or polymerization of the adsorbed molecules, leading to the formation of C dots with more emissive traps and higher PL intensity.35 Upon further increasing the pH value, the amount of C dots formed increases slightly. Since it is difficult to prepare a solution with a similar composition to that at lower pH values, synthesis of C dots at pH >9.0 was not conducted.3.4 Effect of sodium halides
It has been reported that the addition of sodium halides into amino acid solution enhances its electrochemical oxidation rates due to increased conductivity.36 We thus investigated the effects of NaCl (0–2.0 M) on the formation of C dots under optimal conditions (0.63 M histidine and pH 9.0). Fig. 1D shows that the PL intensity of Cl–C dots increases upon increasing the NaCl concentration up to 1 M, mainly because of increased reaction rates. At NaCl concentrations greater than 1 M, histidine molecules aggregated as evidenced by the formation of a white solid product. Thus 1 M NaCl was found to be optimal for the preparation of C dots. Under similar synthesis conditions, Br–C dots and I–C dots were prepared for comparison of their physicochemical and spectrochemical properties with those of Cl–C dots and C dots. Fig. S1† shows the TEM images of C dots, Cl–C dots, Br–C dots and, I–C dots. All of the X–C dots were prepared under similar conditions (similar ionic strengths). By counting 100 of the four types of C dots, their average sizes were determined to be 3.2 ± 0.6, 3.9 ± 0.9, 3.8 ± 0.6, and 4.9 ± 0.7 nm, respectively. The HRTEM images displayed in the insets show that each of them has lattices with a d-spacing of 0.31 nm which are consistent with the (002) diffraction planes of sp2 graphitic carbon. The FTIR spectra (Fig. S2†) were recorded to identify the functional groups on the surfaces of the four types of C dots. The peaks at 2962, 1640, 1462 and 1150 cm−1 were assigned to –CH2–, amide I/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, amide III/C–N, and C–O stretching vibrations, respectively. The broad bands at around 3320 cm−1 suggest the existence of functional groups such as –COOH, –OH, –NH2, and –NH3+ on the surface of all four types of C dots.37 The suggestion is supported by their high water dispersibility. The XPS spectra displayed in Fig. S3† show their C1s spectra, revealing the presence of C–C (284.6 eV), C–I (285.2 eV), C–Br (285.6 eV), C–N/or C–O (286.0 eV), C–Cl (286.6 eV), CO (286.8 eV), and O–CO (287.8 eV) bonds.38,39 Each of their N1s spectra shows four peaks at 398.1 eV, 399.1 eV, 399.8 eV and 400.6 eV, which are attributed to pyridinic N, pyrrolic N, graphitic N, and N–H, respectively.40 The deconvoluted C1s and N1s spectra support the presence of ring structures and the formation of C dots. Table S1† lists the elemental compositions of the as-prepared C dots. Note that the Br–C dots and I–C dots have higher oxygen content than the rest, indicating their greater surface passivation. The Raman spectra of the four types of C dots displayed in Fig. S3† show the characteristic D- and G-bands at 1311 cm−1 and 1531 cm−1, which are attributed to the first-order scattering of the E2g vibration mode in the graphite sheets and the structural defects, respectively.41 The peak at 1420 cm−1 is attributed to the carboxylate groups (COO−).31 The D-band to G-band ratio (ID/IG) of the four types of C dots decreases in the order of I–C dots (0.98) > Br–C dots (0.96) > C dots (0.94) > Cl–C dots (0.90), showing that I–C dots possess the highest density of defects. The result suggests that the halides affect the surface reconstruction processes and defects differently.42
O, amide III/C–N, and C–O stretching vibrations, respectively. The broad bands at around 3320 cm−1 suggest the existence of functional groups such as –COOH, –OH, –NH2, and –NH3+ on the surface of all four types of C dots.37 The suggestion is supported by their high water dispersibility. The XPS spectra displayed in Fig. S3† show their C1s spectra, revealing the presence of C–C (284.6 eV), C–I (285.2 eV), C–Br (285.6 eV), C–N/or C–O (286.0 eV), C–Cl (286.6 eV), CO (286.8 eV), and O–CO (287.8 eV) bonds.38,39 Each of their N1s spectra shows four peaks at 398.1 eV, 399.1 eV, 399.8 eV and 400.6 eV, which are attributed to pyridinic N, pyrrolic N, graphitic N, and N–H, respectively.40 The deconvoluted C1s and N1s spectra support the presence of ring structures and the formation of C dots. Table S1† lists the elemental compositions of the as-prepared C dots. Note that the Br–C dots and I–C dots have higher oxygen content than the rest, indicating their greater surface passivation. The Raman spectra of the four types of C dots displayed in Fig. S3† show the characteristic D- and G-bands at 1311 cm−1 and 1531 cm−1, which are attributed to the first-order scattering of the E2g vibration mode in the graphite sheets and the structural defects, respectively.41 The peak at 1420 cm−1 is attributed to the carboxylate groups (COO−).31 The D-band to G-band ratio (ID/IG) of the four types of C dots decreases in the order of I–C dots (0.98) > Br–C dots (0.96) > C dots (0.94) > Cl–C dots (0.90), showing that I–C dots possess the highest density of defects. The result suggests that the halides affect the surface reconstruction processes and defects differently.42
Fig. 2A–C show the effect of sodium halides on the PL of C dots at different excitation wavelengths. As shown in Fig. 2A, C dots prepared in the absence and presence of 1 M NaCl upon excitation at 280 nm all exhibit one sharp peak at 330 nm and two broad peaks at 445 and 505 nm. The sharp peak at 330 nm is attributed to strong emission from the π–π* transitions in the core.43 The two broad peaks are attributed mainly to strong emissive contribution from the surface states. The PL intensities of Cl–C dots at 445 and 505 nm are higher than the intensities of those prepared in the absence of NaCl. Increased NaCl concentration greatly enhanced the oxidation rate of histidine, leading to the formation of more strong emissive surface states and thus high PL intensity. Unlike for Cl–C dots, the peak at 330 nm for Br–C dots or I–C dots was quenched significantly due to the inner effect, greater specific adsorption capacity and greater covalent character of Br−/I− than Cl−.44 Br–C and I–C dots both show two peaks at 445 and 505 nm, revealing the existence of emissive surface states. Although I− has a greater effect on the surface state, the PL intensities of I–C dots at the two peaks are weaker than those of Cl–C and Br–C dots, mainly because it induces greater PL quenching through intersystem crossing. The quenching efficiency of C dots at 505 nm induced by 1 mM NaI is 43.9%, which supports our reasoning. As shown in Fig. 2B, upon excitation at 360 nm, two emission peaks at 445 and 490 nm were observed for all the four types of C dots. Their PL intensities decrease in the order of Cl–C dots > Br–C dots > C dots > I–C dots because of strong intersystem crossing (heavy-atom effect). As shown in Fig. 2C, upon excitation at 420 nm, an emission peak at 505 nm was observed, with a decreasing order of PL intensity: Cl–C dots > C dots > Br–C dots > I–C dots. The differential PL increasing/decreasing orders at various excitation wavelengths are mainly because the three halides have stronger effects on the surface emission than on the core emission. Fig. 2D shows the absorption spectra of all four tested C dot solutions and histidine hydrochloride solution. A sharp peak at 220 nm for histidine hydrochloride solution is assigned to its imidazole ring.45 The peak at 220 nm for Br–C dots and I–C dots undergoes a red shift, mainly due to the absorption of the two halides. The broad absorption over the wavelength range from 300 to 400 nm is attributed to the n–π* transition of the CO bond and the π–π* transition of the conjugated CC bond.24
 | ||
| Fig. 2 Effect of sodium halides on the formation of C dots from 0.63 M histidine solution at pH 9.0 with/without 1 M NaX at 10 V for 2 h. PL spectra (A–C) and UV-vis absorption spectra (D). PL spectra of C dots (a), Cl–C dots (b), Br–C dots (c), and I–C dots (d) at excitation wavelengths of (A) 280, (B) 360, and (C) 420 nm. UV-vis absorption spectra of C dots (a), Cl–C dots (b), Br–C dots (c), I–C dots (d) and histidine (e). | ||
3.5 Effect of reaction time
To provide more detailed information about the formation of C dots, we investigated the effect of reaction time on the formation of the four types of C dots. Fig. 3 shows the time-dependent PL intensities of all four types of C dots when excited at different wavelengths. As shown in Fig. 3A, upon excitation at 280 nm, the PL of C dots at 330 nm increases gradually and reaches a plateau at around 50 min. Meanwhile the PL of Cl–C dots does not change significantly for the first 20 min period. Instead, it increases gradually and reaches a plateau at 50 min. In both cases, the PL decreases slightly after 50 min and becomes stable after 120 min. The results suggest that Cl− suppresses the core emission when its content in the Cl–C dots is high. Upon increasing the reaction time, the amount of C dots increases, leading to lower density of adsorbed Cl− on the surface of each C dot. Because the emission at 330 nm is small in the Br–C and I–C dots, the effects of Br− and I− on their time-evolution PL are hard to observe. Fig. 3B shows the comparison of the PL intensities of the four types of C dots at 445 nm. Besides the PL of I–C dots, the PL intensities from the other three all increased gradually upon increasing the reaction time, revealing that more surface states are formed. For I–C dots, the PL increases gradually and reaches a plateau at around 8 min, and then decreases before reaching a constant at around 30 min, mainly because of the formation of I3− which has strong absorption bands centered separately near 280 and 350 nm, leading to PL quenching of the C dots due to the inner filter effect. The PL intensities of Cl–C dots, Br–C dots and I–C dots are all higher than those of C dots, supporting that the halides affect the surface emission and formation rate of C dots. Besides I–C dots, gradually increased PL intensities were found for the other C dots when excited at 360 or 420 nm (Fig. 3C–F). Because of stronger effects of iodide on the surface defects and intersystem crossing, the PL intensities of I–C dots decrease once they reach their maxima. Interestingly, all the three types of C dots prepared in the presence of sodium halides show detectable PL within 1 min electrolysis when excited at 360 or 420 nm (Fig. 3C–F). Also displayed in the insets of Fig. 3C–F are their time-dependent UV-vis absorption spectra. The sharp peak at 220 nm for each of the four types of C dots is assigned to that of the imidazole ring.45 Upon increasing the electrolysis time, the peak of Br–C dots or I–C dots at 220 nm undergoes a red shift due to increased absorption of Br2 or I2 as a result of increases in their formation amounts. The absorbance values over 300 to 400 nm increase upon increasing the reaction time, mainly because of gradual increases in the formation of C dots. Table 1 reveals negligible differences in the PL lifetimes (excitation wavelength/emission wavelength: 375/440 nm) of C dots, Cl–C dots, Br–C dots and I–C dots regardless of the reaction times. The lifetime data support that iodide induced greater intersystem crossing. Besides I–C dots, the other three types of C dots obtained after 60 min have slightly longer lifetimes, mainly because of increases in their surface oxidation with oxygen. | ||
| Fig. 3 Effect of reaction time on the PL intensity and UV-vis absorption spectra of C dots (a), Cl–C dots (b), Br–C dots (c), and I–C dots (d). The emission wavelengths are 330 (A), 445 (B), and 505 nm (C), respectively, when excited at 280 nm. The emission wavelengths are 445 (D) and 490 nm (E), respectively, when excited at 360 nm. The emission wavelength is 505 nm (F) when excited at 420 nm. Insets in (C)–(F) are their corresponding UV-vis absorption spectra. All C dots were prepared at 10 V from 0.63 M histidine solutions at pH 9.0 with/without 1 M NaX. | ||
| Nanomaterials | τ 1 min (ns) | τ 30 min (ns) | τ 60 min (ns) | τ 120 min (ns) |
|---|---|---|---|---|
| C dots | 5.57 | 4.92 | 4.83 | 5.05 |
| Cl–C dots | 4.58 | 4.71 | 4.45 | 4.54 |
| Br–C dots | 4.51 | 4.89 | 4.72 | 4.84 |
| I–C dots | 4.29 | 4.40 | 4.01 | 3.91 |
3.6 Formation routes
Scheme 1 summarizes the formation of C dots from histidine at pH 9.0. After adsorption of histidine onto the anodic Pt electrode, histidine undergoes electrochemical oxidation to form imidazole. Subsequently, the adsorbed imidazole goes through a process of polymerization, carbonization, and passivation to form C dots. As shown in Scheme 1 the solution color changes can be easily observed by the naked eye. The solution becomes darker upon increasing the reaction time, mainly because greater amounts of C-dots and larger aggregates (particles) of C dots are formed. Oxidation also occurs, leading to changes in their surface states. Addition of the halides to the reaction solution increases the conductivity and thus the electrolysis rate. In addition, the halides also induce the formation of greater surface defects. Their different nucleophilic substitution rates, specific adsorption capacity, and migration/ diffusion rates are also responsible for the formation of different C dots.46 | ||
| Scheme 1 Schematic representation of preparation of C dots from histidine hydrochloride through an electrochemical route. | ||
3.7 Sensitivity and selectivity
To test the analytical application of the four types of C-dots, their stability in aqueous solutions at different pH values (3.0–11.0) was tested. As displayed in Fig. S5A,† they are all stable in the tested pH region. The main reason for choosing the excitation/emission wavelengths of 420/505 nm is because the optical properties are related to the surface defects of the C dots and are expected to be more sensitive to an environmental change. Fig. S5B† shows their high salt tolerance, being stable up to 3 M NaCl. The results reveal their potential in analytical applications. Because histidine has high specificity towards Cu2+ (formation constant K = 2.34 × 1010 at pH 4.0),47 we used the as-prepared C dots to detect Cu2+ ions. Cu2+ plays important roles in many biological functions, while at high concentrations also potentially causes some diseases such as gastrointestinal disorders and liver/kidney damage.48–50 Although the sensitivity for the detection of Cu2+ is slightly higher at pH 7.0 than at 3.0, mainly because Cu2+ ions interact more strongly with the adsorbed histidine and the surface functional groups such as amino and carboxylate, interference from Zn2+ ions is severe at pH >3.0. In addition, formation of metal oxides is another disadvantage. On the other hand, poor sensitivity and lower stability of C dots at pH < 3.0 are disadvantageous.To evaluate the selectivity of the four types of C dots toward Cu2+ at pH 3.0, we determined their relative PL intensity [(IF − IFo)/IFo] at 505 nm in the presence of different metal ions when excited at a wavelength of 420 nm. Fig. S6† shows the selectivity of all four types of tested C dots toward various metal ions at a concentration of 100 μM. Cu2+, Hg2+, and Ag+ all induced significant PL quenching of the four types of C dots. To mask the interferences from Hg2+ and Ag+ ions, NaI was added. The solubility product (Ksp) values of Hg2I2 and AgI are 4.6 × 10−29 and 8.3 × 10−17, respectively, which are much smaller than that (1.0 × 10−12) of CuI. To minimize interference from Ni2+, in addition to NaI, citric acid that can form stronger complexes with Ni2+ than with Cu2+ was added.51 In the presence of 0.8 mM NaI and 1 mM citric acid, the I–C dot probe provides high selectivity toward Cu2+ ions (100 μM) over the potential interfering metal ions (100 μM) as shown in Fig. 4A. We note that the other three types of C dots also can be used for selective detection of Cu2+ ions, with slightly higher interference from some potential interfering species such as Fe3+. Hereafter, we only discuss quantitation of Cu2+ using I–C dots.
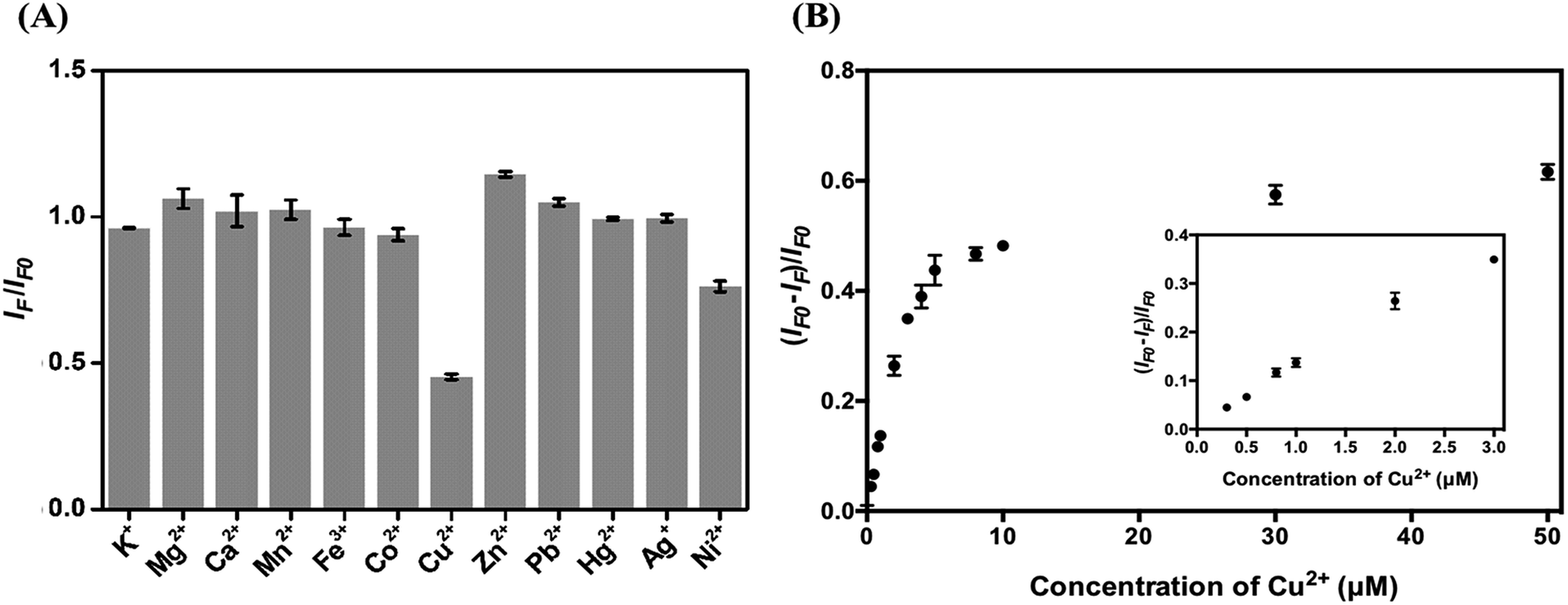 | ||
| Fig. 4 Selectivity (A) and sensitivity (B) of I–C dots towards Cu2+ ions (100 μM) in phosphate buffer (20 mM, pH 3.0) containing 0.8 mM NaI and 1 mM citric acid. The concentrations of I–C dots in (A) and (B) are 0.01× and 0.001×, respectively. IF0 and IF are the PL intensities of I–C dots at 505 nm in the absence and presence of Cu2+, respectively, when excited at 420 nm. The concentrations for the other metal ions are all 100 μM. I–C dots were prepared from 0.63 M histidine solution at pH 9.0 containing 1 M NaI at 10 V for 2 h. | ||
Fig. 4B shows that the relative PL intensity of I–C dots decreases upon increasing Cu2+ ion concentration in the range of 0–50 μM. The Cu2+ induced PL quenching is mainly through an electron transfer process.52 The quenching efficiencies of C dots and I–C dots induced by 100 μM Cu2+ are 65.2% and 65.6%, respectively, ruling out strong interaction of the surface iodides with Cu2+. The I–C dot probe exhibits linearity toward Cu2+ ions over the concentration range of 0.3–3 μM (R2 = 0.98), with a limit of detection (LOD) of 0.22 μM at a signal-to-noise ratio of 3. The LOD is lower than the tolerance level (20 μM) of Cu2+ ions in drinking water defined by the US Environmental Protection Agency (EPA).4 Table S2† lists the linear ranges and the LODs of different nanomaterial based probes toward the quantitation of Cu2+ ions, showing that a lower LOD is provided by the I–C dots than that by the rest.53–60 In addition, organic solvent is not required when applying this simple assay. Having high selectivity and a low LOD, the I–C dot probe holds great potential for the quantification of Cu2+ ions in real samples containing complex matrixes.
3.8 Sensing mechanism
To provide more information about the specificity of I–C dots toward Cu2+ ions, a simple and sensitive electrochemical tool (CV) was applied. A bare GCE exhibits a featureless CV curve over the potential range from −0.6 to 0.8 V in the presence of I–C dots (1 mL) as shown in Fig. S7A.† Faradaic or non-faradaic reactions did not occur even when CVs were recorded over a wide potential window (−0.6–1.3 V), revealing that the active binding sites did not contribute to the electron transfer or double layer capacitance. In a solution containing I–C dots and Fe3+ ions, oxidation or reduction peaks of Fe3+ ions were not detected. After spiking Cu2+ ions (100 μM) into the solution, two reduction peaks at ca. −0.1 and −0.31 V and one oxidation peak at ca. 0.1 V appeared.61 Peaks at −0.31 and 0.1 V are attributed to the reduction and oxidation of Cu–histidine complexes, respectively. Upon increasing Cu2+ concentration, the reduction peaks shifted to more negative potentials, revealing that Cu2+ ions were reduced to metallic copper (Cu0) at ca. −0.1 V. To identify the interfacial changes during the formation of the complex, EIS measurements were conducted. The Nyquist plot of a bare GCE recorded in the presence of I–C dots with N2 purging is displayed in Fig. S7B.† The bare GCE exhibits a small arc in the high frequency region due to strong adsorption of I–C dots on the electrode surface. The arc diameter becomes smaller in the presence of Cu2+ as a result of improved charge transfer. CV and EIS results revealed the plausible binding sites of Cu2+ ions in I–C dots and the reduction of Cu2+ ions to form metallic copper, supporting our proposed sensing mechanism. As shown in Table S1,† the oxygen content of I–C dots increased significantly after reaction with Cu2+ ions. As depicted in Fig. S2,† the characteristic absorption peaks at 1640 cm−1 and 1596 cm−1 disappeared after reaction with Cu2+ ions, revealing the oxidation of amide I groups on the surface of I–C dots.62 Two new bands at 1289 and 2383 cm−1 are ascribed to the C–O and CO2 stretching vibrations, which revealed that the surface functional groups of I–C dots were oxidized. The 2p3/2 and 2p1/2 peaks at 932.3 and 952.1 eV in the Cu 2p spectrum are attributed to the metallic copper, respectively (Fig. S3†).63 There is a small characteristic satellite peak around 942.0 eV for Cu 2p3/2, which indicates that some unreacted copper ions were bound onto the surface of I–C dots. All these results confirm that the I–C dots were oxidized by the surface Cu2+ ions/complexes and metallic copper was formed on the surface of I–C dots.3.9 Real sample analysis
Practicality of the I–C dot probe was validated through the determination of the concentrations of Cu2+ ions in various real samples, including tap water, lake water and sea water, using a standard addition method. Fig. S8† shows good linearity (R2 = 0.98) of relative PL intensity [(IF0 – IF)/IF0] responses at 505 nm toward spiked Cu2+ ions over the concentration range from 0.3 to 3 μM. There was no obvious difference in the linearity and the slope among the three samples, revealing negligible matrix effects. Having high sensitivity and selectivity, this probe is useful for quantitation of Cu2+ ions in water samples.4. Conclusions
A simple and cost-effective electrochemical approach was applied to prepare C dots from histidine under alkaline conditions, which allows one to gain more information about the formation of C dots. Upon increasing the reaction time, more C dots were formed. Because the formation speed of C dots increases upon increasing the electrolysis rate, larger amounts of C-dots can be prepared in a short period of time from precursors in high-conductivity media at a high voltage using a large electrode. When compared to hydrothermal routes, this electrochemical approach produces a much lower amount of large particles. Halides induce surface defects, leading to enhanced surface emission intensity, but they also cause PL quenching through intersystem crossing. Having high sensitivity and selectivity, the I–C dot probe holds great potential for quantitation of Cu2+ in complicated samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by the Ministry of Science and Technology of Taiwan (MOST) under contract 107-2113-M-002-015-MY3. A. P. Periasamy is grateful to MOST for a postdoctoral fellowship under the contract number MOST 106-2811-M-002-083. We thank Ms. S.-J. Ji and C.-Y. Chien of the Precious Instrument Center of NTU for their assistance in HRTEM analyses.References
- Z. X. Liu, B. B. Chen, M. L. Liu, H. Y. Zou and C. Z. Huang, Green Chem., 2017, 19, 1494–1498 RSC.
- H. Jin, R. Gui, J. Gong and W. Huang, Biosens. Bioelectron., 2017, 92, 378–384 CrossRef CAS PubMed.
- W. Y. Chen, C. C. Huang, L. Y. Chen and H. T. Chang, Nanoscale, 2014, 6, 11078–11083 RSC.
- G. Y. Lan, C. C. Huang and H. T. Chang, Chem. Commun., 2010, 46, 1257–1259 RSC.
- J. Feng, W. J. Wang, X. Hai, Y. L. Yu and J. H. Wang, J. Mater. Chem. B, 2016, 4, 387–393 RSC.
- P. C. Hsu, Z. Y. Shih, C. H. Lee and H. T. Chang, Green Chem., 2012, 14, 917–920 RSC.
- H. Nie, M. Li, Q. Li, S. Liang, Y. Tan, L. Sheng, W. Shi and S. X. A. Zhang, Chem. Mater., 2014, 26, 3104–3112 CrossRef CAS.
- S. Liu, J. Tian, L. Wang, Y. Zhang, X. Qin, Y. Luo, A. M. Asiri, A. M. A. O. Al-Youbi and X. Sun, Adv. Mater., 2012, 24, 2037–2041 CrossRef CAS PubMed.
- X. Wang, K. Qu, B. Xu, J. Ren and X. Qu, J. Mater. Chem., 2011, 21, 2445–2450 RSC.
- L. Ma, W. D. Xiang, H. H. Gao, J. Wang, Y. Ni and X. Liang, Dyes Pigm., 2016, 128, 1–7 CrossRef CAS.
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. A. Tsang, X. Yang and S. T. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef CAS PubMed.
- D. Pan, J. Zhang, Z. Li and M. Wu, Adv. Mater., 2010, 22, 734–738 CrossRef CAS PubMed.
- G. E. LeCroy, F. Messina, A. Sciortino, C. E. Bunker, P. Wang, S. K. A. Fernando and Y. P. Sun, J. Phys. Chem. C, 2017, 121, 28180–28186 CrossRef CAS.
- V. Strauss, J. T. Margraf, C. Dolle, B. Butz, T. J. Nacken, J. Walter, W. Bauer, W. Peukert, E. Spiecker, T. Clark and D. M. Guldi, J. Am. Chem. Soc., 2014, 136, 17308–17316 CrossRef CAS PubMed.
- Y. P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S. Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed.
- Q. L. Zhao, Z. L. Zhang, B. H. Huang, J. Peng, M. Zhang and D. W. Pang, Chem. Commun., 2008, 5116–5118 RSC.
- J. Niu and H. Gao, J. Lumin., 2009, 149, 159–162 CrossRef.
- Z. Ma, H. Ming, H. Huang, Y. Liu and Z. Kang, New J. Chem., 2012, 36, 861–864 RSC.
- V. N. Mehta, S. Jha, R. K. Singhal and S. K. Kailasa, New J. Chem., 2014, 38, 6152–6160 RSC.
- S. Y. Park, C. Y. Lee, H. R. An, H. Kim, Y. C. Lee, E. C. Park, H. S. Chun, H. Y. Yang, S. H. Choi, H. S. Kim, K. S. Kang, H. G. Park, J. P. Kim, Y. Choi, J. Lee and H. U. Lee, Nanoscale, 2017, 9, 9210–9217 RSC.
- P. C. Hsu and H. T. Chang, Chem. Commun., 2012, 48, 3984–3986 RSC.
- A. B. Bourlinos, A. Stassinopoulos, D. Anglos, R. Zboril, V. Georgakilas and E. P. Giannelis, Chem. Mater., 2008, 20, 4539–4541 CrossRef CAS.
- S. Mitra, S. Chandra, T. Kundu, R. Banerjee, P. Pramanik and A. Goswami, RSC Adv., 2012, 2, 12129–12131 RSC.
- X. Jia, J. Li and E. Wang, Nanoscale, 2012, 4, 5572–5575 RSC.
- P. C. Hsu, P. C. Chen, C. M. Ou, H. Y. Chang and H. T. Chang, J. Mater. Chem. B, 2013, 1, 1774–1781 RSC.
- C. L. Li, C. M. Ou, C. C. Huang, W. C. Wu, Y. P. Chen, T. E. Lin, L. C. Ho, C. W. Wang, C. C. Shih, H. C. Zhou, Y. C. Lee, W. F. Tzeng, T. J. Chiou, S. T. Chu, J. Cangm and H. T. Chang, J. Mater. Chem. B, 2014, 2, 4564–4571 RSC.
- Y. S. Liu, Y. N. Zhao and Y. Y. Zhang, Sens. Actuators, B, 2014, 196, 647–652 CrossRef CAS.
- B. S. B. Kasibabu, S. L. D'souza, S. Jha, R. K. Singhal, H. Basu and S. K. Kailasa, Anal. Methods, 2015, 7, 2373–2378 RSC.
- S. Sahu, B. Behera, T. K. Maiti and S. Mohapatra, Chem. Commun., 2012, 48, 8835–8837 RSC.
- V. N. Mehta, S. Jha, H. Basu, R. K. Singhal and S. K. Kailasa, Sens. Actuators, B, 2015, 213, 434–443 CrossRef CAS.
- C. I. Wang, W. C. Wu, A. P. Periasamy and H. T. Chang, Green Chem., 2014, 16, 2509–2514 RSC.
- K. Ogura, M. Kobayashi, M. Nakayama and Y. Miho, J. Electroanal. Chem., 1999, 463, 218–223 CrossRef CAS.
- D. M. Close and P. Wardman, J. Phys. Chem. A, 2018, 122, 439–445 CrossRef CAS.
- S. Li and M. Hong, J. Am. Chem. Soc., 2011, 133, 1534–1544 CrossRef CAS PubMed.
- N. L. Weinberg and H. R. Weinberg, Chem. Rev., 1968, 68, 449–523 CrossRef CAS.
- M. R. Thombare and G. S. Gokavi, J. Braz. Chem. Soc., 2014, 25, 1545–1551 CAS.
- J. Jiang, Y. He, S. Li and H. Cui, Chem. Commun., 2012, 48, 9634–9636 RSC.
- M. Patel, W. Feng, K. Savaram, M. R. Khoshi, R. Huang, J. Sun, E. Rabie, C. Flach, R. Mendelsohn, E. Garfunkel and H. He, Small, 2015, 11, 3358–3368 CrossRef CAS PubMed.
- X. L. Zhou, F. Solymosi, P. M. Blass, K. C. Cannon and J. M. White, Surf. Sci., 1989, 219, 294–316 CrossRef CAS.
- Z. Liu, Y. Yang, Z. Du, W. Xing, S. Komarneni, Z. Zhang, X. Gao and Z. Yan, Nanoscale Res. Lett., 2015, 10, 333–343 CrossRef.
- Y. Q. Dong, L. S. Wan, J. H. Cai, Q. Q. Fang, Y. W. Chi and G. N. Chen, Sci. Rep., 2015, 5, 10037 CrossRef CAS PubMed.
- J. C. Charlier, Acc. Chem. Res., 2002, 35, 1063–1069 CrossRef CAS.
- J. Schneider, C. J. Reckmeier, Y. Xiong, M. V. Seckendorff, A. S. Andrei, S. Susha, P. Kasák and A. L. Rogach, J. Phys. Chem. C, 2017, 121, 2014–2022 CrossRef CAS.
- K. Ogura, J. CO2 Util., 2013, 1, 43–49 CrossRef CAS.
- L. J. Saidel, A. R. Goldfarb and S. Waldman, J. Biol. Chem., 1952, 197, 285–291 CAS.
- H. Wang, B. C. W. Gu, D. W. Lee and J. S. Lee, Sci. Rep., 2017, 7, 43794 CrossRef PubMed.
- R. Leberman and B. R. Rabin, Trans. Faraday Soc., 1959, 55, 1660–1670 RSC.
- D. G. Barceloux and D. Barceloux, J. Toxicol., Clin. Toxicol., 1999, 37(2), 217–230 CrossRef CAS PubMed.
- M. Olivares and R. Uauy, Am. J. Clin. Nutr., 1996, 63, 791S–796S CrossRef CAS PubMed.
- P. G. Georgopoulos, A. Roy, M. J. Yonone-Lioy, R. E. Opiekun and P. J. Lioy, J. Toxicol. Environ. Health, Part B, 2001, 4(4), 341–394 CAS.
- O. Y. Zelenin, Russ. J. Coord. Chem., 2007, 33, 346–350 CrossRef CAS.
- J. Q. Tian, Q. Liu, A. M. Asiri, A. O. Al-Yubi and X. P. Sun, Anal. Chem., 2013, 85, 5595–5599 CrossRef CAS.
- A. Zhu, Q. Qu, X. Shao, B. Kong and Y. Tian, Angew. Chem., Int. Ed., 2012, 124, 7297–7301 CrossRef.
- M. Koneswaran and R. Narayanaswamy, Sens. Actuators, B, 2009, 139, 104–109 CrossRef CAS.
- G. He, C. Liu, X. Liu, Q. Wang, A. Fan, S. Wang and X. Qian, PLoS One, 2017, 12, e0186994 CrossRef.
- M. C. Rong, K. X. Zhang, Y. R. Wang and X. Chen, Chin. Chem. Lett., 2017, 28, 1119–1124 CrossRef CAS.
- C. Boonme, T. Noipa, T. Tuntulani and W. Ngeontae, Spectrochim. Acta, Part A, 2013, 169, 161–168 CrossRef.
- X. F. Hou, F. Zeng, F. K. Du and S. Z. Wu, Nanotechnology, 2013, 24, 335502 CrossRef PubMed.
- Y. Wang, W.-T. Wu, M.-B. Wu, H.-D. Sun, H. Xie, C. Hu, X.-Y. Wu and J.-H. Qiu, New Carbon Mater, 2015, 30, 550–559 CrossRef.
- L. Zhao, H. Li, Y. Xu, H. Liu, T. Zhou, N. Huang, Y. Li and L. Ding, Anal. Bioanal. Chem., 2018, 410, 4301–4309 CrossRef CAS PubMed.
- Y. C. Weng and T. H. Cheng, Z. Naturforsch., 2011, 66, 279–288 CAS.
- G. Oza, K. Oza, S. Pandey, S. Shinde, A. Mewada, M. Thakur, M. Sharon and M. Sharon, J. Fluoresc., 2015, 25, 9–14 CrossRef CAS PubMed.
- Y. J. Lin, P. C. Chen, Z. Yuan, J. Y. Ma and H. T. Chang, Chem. Commun., 2015, 51, 11983–11986 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00137a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |