
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDye-sensitized TiO2 nanotube membranes act as a visible-light switchable diffusion gate†
Imgon
Hwang
a,
Francesca
Riboni
 ab,
Ekaterina
Gongadze
c,
Aleš
Iglič
cd,
JeongEun
Yoo
a,
Seulgi
So‡
a,
Anca
Mazare
a and
Patrik
Schmuki
*abe
ab,
Ekaterina
Gongadze
c,
Aleš
Iglič
cd,
JeongEun
Yoo
a,
Seulgi
So‡
a,
Anca
Mazare
a and
Patrik
Schmuki
*abe
aDepartment of Materials Science WW4-LKO, University of Erlangen-Nuremberg, Martensstrasse 7, 91058 Erlangen, Germany. E-mail: schmuki@ww.uni-erlangen.de
bRegional Centre of Advanced Technologies and Materials, Šlechtitelů 27, 78371 Olomouc, Czech Republic
cLaboratory of Biophysics, Faculty of Electrical Engineering, University of Ljubljana, Tržaška 25, SI-1000 Ljubljana, Slovenia
dLaboratory of Clinical Biophysics, Faculty of Medicine, University of Ljubljana, Zaloška 9, SI-1000 Ljubljana, Slovenia
eDepartment of Chemistry, Faculty of Science, King Abdulaziz University, P. O. Box 80203, Jeddah 21569, Saudi Arabia
First published on 11th November 2019
Abstract
Here we report that both-end open anodic TiO2 nanotube membranes, after sensitization with a Ru(II)-based dye, exhibit visible-light switching properties for flow-through the nanotube channels. Under illumination, the gate is in an open state providing ∼four-times faster permeation of small molecules through the membrane compared to a dark state. Switching is reversible with no apparent dye degradation being observed. Gating is possible not only of permeating dye molecules but also of nanoprobes such as polystyrene nanospheres. Supported by quantitative modelling, we attribute the switching mechanism to light-induced changes of the charge distribution at the dye/TiO2 interface which in turn alters the hydrodynamics within the anodic tube membranes. This demonstrates that these simple dye-sensitized nanotube membranes can be used as an optically addressable flow-through gate in nanofluidics.
Introduction
Gating of nanoscale flow-through devices has received major attention in the field of system engineering for controlled “diffusion” applications, for example in drug-delivery, bio-separation, and sensing applications.1–5 It is important in such devices that nanochannels are reversibly gated, i.e., they can be switched between an open (“on”) and closed (“off”) state in response to an external stimulus (e.g. voltage, pH, or temperature variation).6–10Stimuli-responsive membranes (SRMs) are based on hydrogels,11 ion-track polymers,8,10,12 and inorganic solid-state nanochannels such as anodic alumina and silica7,9,13,14 and the latter can be conveniently modified to exhibit an adaptive gating behavior based on surface termination, reconstruction, and/or local molecular rearrangement.1 Oxide based nanochannels show remarkable intrinsic robustness and stability, along with an easy chemistry governing channel functionalization for implementing diffusion-gating characteristics i.e., self-assembly or anchoring of (mono)layers of a stimuli-responsive material on the oxide nanochannel surface.1 Using this approach, diffusion gates based e.g. on charge exclusion, hydrophobicity/hydrophilicity switching, and size-selectivity have been explored for ionic current control, molecular separation, and protein fractioning.7,9,13–16
The most convenient “wireless” approach to trigger switching in flow-through membranes is the use of light as an on–off switching input. Up to now this is mostly achieved by grafting photo-responsive organic moieties onto the walls of the nanochannels, and the switching is induced by light induced conformational change of the molecule attached to the wall, which in turn affects the permeability properties of the membrane (e.g., wettability, hydrophilicity/hydrophobicity, hydrodynamic diameter, etc.).13,15,17,18
Here we show that one of the most explored dye sensitized oxides – TiO2 – can effectively be used for visible light flux gating if fabricated as both-end open sensitized membranes. TiO2 nanotube (NT) membranes can be fabricated by combining (i) self-organizing electrochemical anodization to produce one-dimensional TiO2 nanotubes (NTs) on a Ti substrate,19–21 with (ii) a lift-off process, and this results in self-standing, both-end-open and highly aligned nanochannel/nanotube arrays.22–25
Such TiO2 nanotube layers have been recently used as flow-through membranes for the size-selective separation of different solutes, including e.g. biologically relevant molecular substrates and micro- and nano-spheres.26–30
In the present work, we dye-sensitized TiO2 NT membranes with N719 molecular dye ([RuII(2,2′-bipyridyl-4,4′-dicarboxylate)-(NCS)2]TBA2, TBA = tetra-n-butylammonium), a dye that is most commonly used in dye sensitized solar cells (DSSCs). We find that such membranes show visible light-gating effects for trans-membrane transport. Supported by quantitative modelling, we show that light-induced perturbation of the electric properties and surface charge of N719/TiO2 tubes affects the tube hydrodynamic properties, and thus can alter the permeation of molecules or nanoscale objects such as polystyrene beads (20 nm). These dye sensitized/TiO2 NT membranes exhibit periodically reversible, visible light-modulated gating characteristics in repetitive light ON–OFF cycles.
Experimental
Fabrication of TiO2 nanotube membranes
For anodic nanotube growth, Ti sheets with a thickness of 0.125 mm (99.6% purity, Advent Materials, UK) were used. Prior to anodization, Ti foil was cleaned in acetone, ethanol, and distilled water in an ultrasonic bath and then dried in a N2 stream.Anodization was carried out in a two-electrode electrochemical cell, with Ti and Pt foils as working and counter electrodes, respectively. An ethylene glycol (EG, 99.5% Sigma-Aldrich) based electrolyte was used that also contained 1.5 M lactic acid (LA, 89% Sigma-Aldrich), 0.1 M ammonium fluoride (NH4F, 98% Sigma-Aldrich) and 5 wt% distilled water.31 Anodization was carried out at 120 V (Jaissle IMP88 PC) for 10 min. The nanotube layers were then annealed in air at 250 °C for 1 h (heating/cooling rate, 30 °C min−1) using a rapid thermal annealer (Jipelec JETFIRST 100 RTA). A second anodization was carried out at 120 V and 60 °C (the temperature was fixed using a thermostat, HAAKE F3) for 3 min to produce a thin underlayer of amorphous TiO2 NTs. This amorphous layer could then selectively be dissolved by dipping the NT films in a 30 wt% H2O2 aqueous solution for 90 min at room temperature, resulting in the detachment of the upper layer (that is, the tube layer annealed at 250 °C) in the form of a free-standing TiO2 nanotube membrane.
In order to convert the TiO2 nanotubes to anatase, membranes were annealed for 1 h in air at 450 °C (heating/cooling rate, 30 °C min−1) using a rapid thermal annealer (Jipelec JETFIRST 100 RTA).
Materials physico-chemical characterization
Morphological characterization of the nanotubes was carried out with a field-emission scanning electron microscope (FE-SEM, Hitachi S4800). In particular, the NT thickness was measured from the cross sectional SEM images.The crystallographic properties of the materials were analyzed by X-ray diffraction (XRD) performed with an X'pert Philips MPD (equipped with a Panalytical X'celerator detector) using graphite monochromatic Cu Kα radiation (λ = 1.54056 Å). XRD patterns were collected by placing the membranes on a quartz glass slide. XPS measurements were performed by using X-ray photoelectron spectroscopy (XPS, PHI 5600, US) and peaks were shifted to Ti 2p 458.5 eV.
Light-modulated gating tests
The functionality of the tube membranes was investigated for the (photo-mediated) transport of the N719 Ru(II) dye ([RuII(2,2′-bipyridyl-4,4′-dicarboxylate)-(NCS)2]TBA2). For this, TiO2 NT membranes were mounted in a PVC holder (with a 0.53 cm-diameter opening) that was used to separate the two compartments (A and B) of a cell. One cell (B) wall was equipped with a quartz glass window.24,29Each compartment was filled with a fixed amount (6 mL) of two different solutions. In detail, a 1.5 mM solution of N719 was prepared in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetonitrile/t-BuOH mixture; chamber A was then filled with 20 v/v% of the dye solution in EtOH, in order to attain an initial dye concentration of ∼300 μM. Chamber B was filled with a 1:1 t-BuOH/EtOH mixture. The (light-modulated) diffusion of N719 through the NT membrane was monitored on both sides of the cell by measuring the decrease (in chamber A) and increase (in chamber B) of the intensity of the dye main absorption band (λ = 534 nm) by means of a spectrophotometer (Lambda Bio XLS, PerkinElmer); the dye concentration was afterwards calculated by applying the Beer–Lambert law.32 A 532 nm LED light (150 mW cm−2) was used as the irradiation source, and both the dark and visible light-modulated experiments lasted 8 h.
:1 acetonitrile/t-BuOH mixture; chamber A was then filled with 20 v/v% of the dye solution in EtOH, in order to attain an initial dye concentration of ∼300 μM. Chamber B was filled with a 1:1 t-BuOH/EtOH mixture. The (light-modulated) diffusion of N719 through the NT membrane was monitored on both sides of the cell by measuring the decrease (in chamber A) and increase (in chamber B) of the intensity of the dye main absorption band (λ = 534 nm) by means of a spectrophotometer (Lambda Bio XLS, PerkinElmer); the dye concentration was afterwards calculated by applying the Beer–Lambert law.32 A 532 nm LED light (150 mW cm−2) was used as the irradiation source, and both the dark and visible light-modulated experiments lasted 8 h.
AgNO3 (1 mM) was added to the dye solution (chamber A) to evaluate the effect of an electron scavenger on the (light-modulated) diffusion of N719 through the tubes. Finally, the membrane functionality was also evaluated for the (light-modulated) permeability of 20 nm polystyrene beads, functionalized with sulfate groups (FluoSpheres® Sulfate Microspheres, 505/515, 2% solids), thus being negatively charged.33 For this, a specific amount of sulfate microspheres was added to the N719 solution in chamber A to achieve an initial microsphere concentration of ∼0.2 mM. The diffusion of the nanospheres through the NT membrane was monitored in both cell compartments by measuring the decrease (A) and increase (B) in the intensity of the microsphere main absorption band (λ = ca. 500 nm) by means of a spectrophotometer (Lambda Bio XLS, PerkinElmer).
In order to exclude all the possible contributions of dye and/or nanosphere diffusion to the overall kinetics of the different experiments, two sets of control experiments were also performed using TiO2 NT arrays (that is, anchored on Ti foil) and in a more conventional (photo)reactor configuration.34 In particular, a first set of experiments was performed in the presence of the dye (t-BuOH/acetonitrile/ethanol) solution, with and without Ag+, and in the dark or under 532 nm illumination. For a second set of experiments, Ag ions were replaced by the most classic I−/I2 redox couple for DSSCs.
Calculations of the spatial dependence of electric potential and electric field strength in the N719 electrolyte solution in contact with a TiO2 surface
The spatial distributions of electric potential and electric field strength were calculated for an ethanol solution of the dissociated Ru(II)-based N719 dye in contact with a TiO2 surface.35In bulk ethanol solution, N719 dissociates into its anionic part (N7192− – for the sake of ease, N719) with a net −2e0 charge (e0 being the unit charge) and two cations [N(n-Bu)4]+ (TBA+ – for the sake of ease, TBA), each with a net +e0 charge. The charge of the TiO2 surface is described by its surface charge density, σ0.
It is typically reported that N719 anions anchor on the TiO2 surface through carboxylate functional groups. Therefore, owing to a dN719 of ∼ 1.5 nm,36 we assume a distance of closest approach, for both free N719 anions and TBA cations (dTBA = ca. 1 nm) from the TiO2 surface, equal to b2 = 1.5 nm. Under these conditions, b1 represents the distance between the center of the chemisorbed N719 dye and the TiO2 surface, i.e., b1 = 0.75 nm. Finally, the surface charge density σ1 describes the plane charge distribution at a distance b1 from the TiO2 surface.
Taking into account the finite and asymmetric size of ions, the expressions for the spatial distribution of TBA cations (n+(x)), N719 anions (n−(x)) and ethanol (ne(x)) in the electric double layer near a charged TiO2 surface37 can be derived by using the method of lattice statistics with Boltzmann correction factors30 which was proven to be equivalent to the method of minimization of the free energy of the system.37 In the following, α+ and α− are the number of lattice sites occupied by a single positive and negative ion, respectively, while a single solvent (ethanol) molecule occupies just one lattice site. Considering dTBA ∼ 0.95 nm,30dN719 ∼ 1.5 nm,36 and dethanol ∼ 0.46 nm,37 we assume α+ ≈ 3 and α− ≈ 13 and the bulk number density of lattice sites ns/NA = 17.36 mol L−1.
The number densities n+(x), n−(x) and ne(x) can be derived by calculating the corresponding probabilities that a single lattice site in the bulk solution is occupied by one of the three particles (i.e. cations, anions or ethanol molecules):
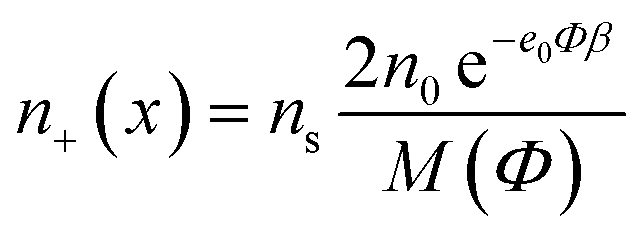 | (1) |
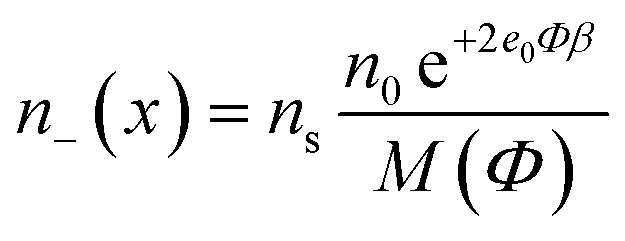 | (2) |
| M(Φ) = α+2n0e−e0Φβ + α−n0e+2e0Φβ + n0e | (3) |
| ns = α+2n0 + α−n0 + n0e | (4) |
| n0e = ns − α+2n0 − α−n0. |
The macroscopic volume charge density in the solution for x ≥ b2 is:
| ρ(x) = e0n+(x) − 2e0n−(x) = −2e0nsn0[e2e0ϕβ − e−e0ϕβ]/M(Φ) | (5) |
The corresponding Poisson's equation (i.e. modified Eigen–Wicke45 equation) is therefore:
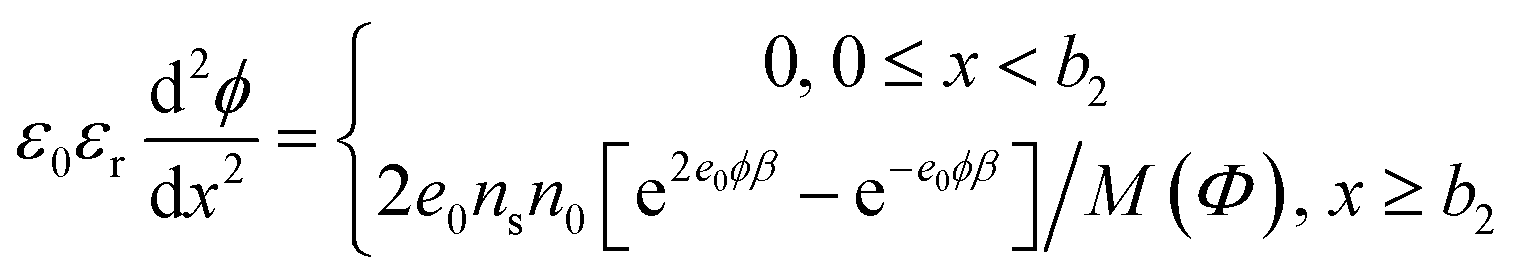 | (6) |
 at x = 0, x = b1 and x = b2:
at x = 0, x = b1 and x = b2: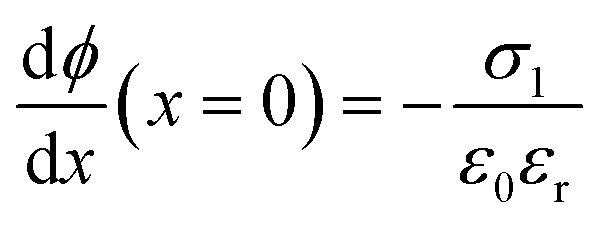 | (7) |
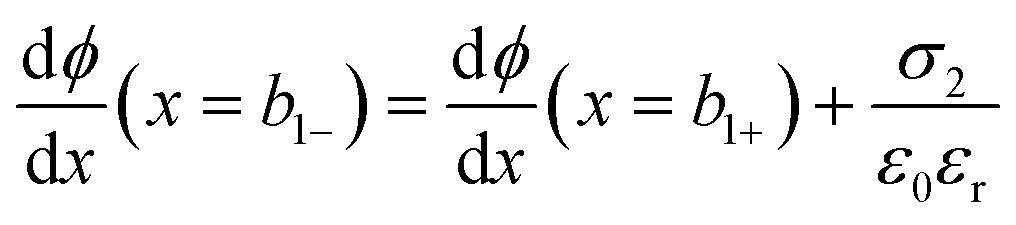 | (8) |
| ϕ(x = b1−) = ϕ(x = b1+) | (9) |
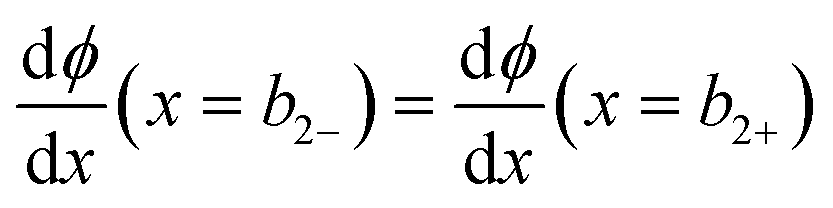 | (10) |
| ϕ(x = b2−) = ϕ(x = b2+) | (11) |
All the above equations are based on the electroneutrality in the bulk solution, where 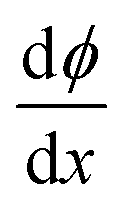 = 0 and ϕ assumes a constant value (zero, in our case). The electroneutrality of the system is described by the boundary condition in eqn (7), that is, for
= 0 and ϕ assumes a constant value (zero, in our case). The electroneutrality of the system is described by the boundary condition in eqn (7), that is, for 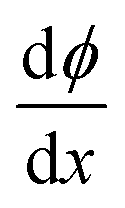 at x = 0. Other parameter values are σ0 = −0.05 A s m−2,39 relative permittivity εr = 25,40 and absolute temperature T = 293 K.
at x = 0. Other parameter values are σ0 = −0.05 A s m−2,39 relative permittivity εr = 25,40 and absolute temperature T = 293 K.
The differential eqn (2), subjected to the boundary conditions (eqn (7)–(11)), was solved numerically using COMSOL 5.3.
Results and discussion
TiO2 nanotube membranes
Fig. 1(a) illustrates the experimental procedure for the fabrication of self-standing membranes of vertically aligned TiO2 nanotubes. The as-fabricated membranes (1 cm diameter) are robust, mechanically stable (Fig. 1(b)), and can easily be handled with tweezers; in addition, no serious cracking or damage was observed prior to or after dipping in the N719 dye-based solution or after extended permeation experiments. The membranes (∼80–100 nm nanotube diameter) were fabricated using an optimized sequence of double-anodization and mild annealing steps, followed by lift-off from the Ti metal substrate by a chemical etching step (see the ESI for details†).24,25,41,42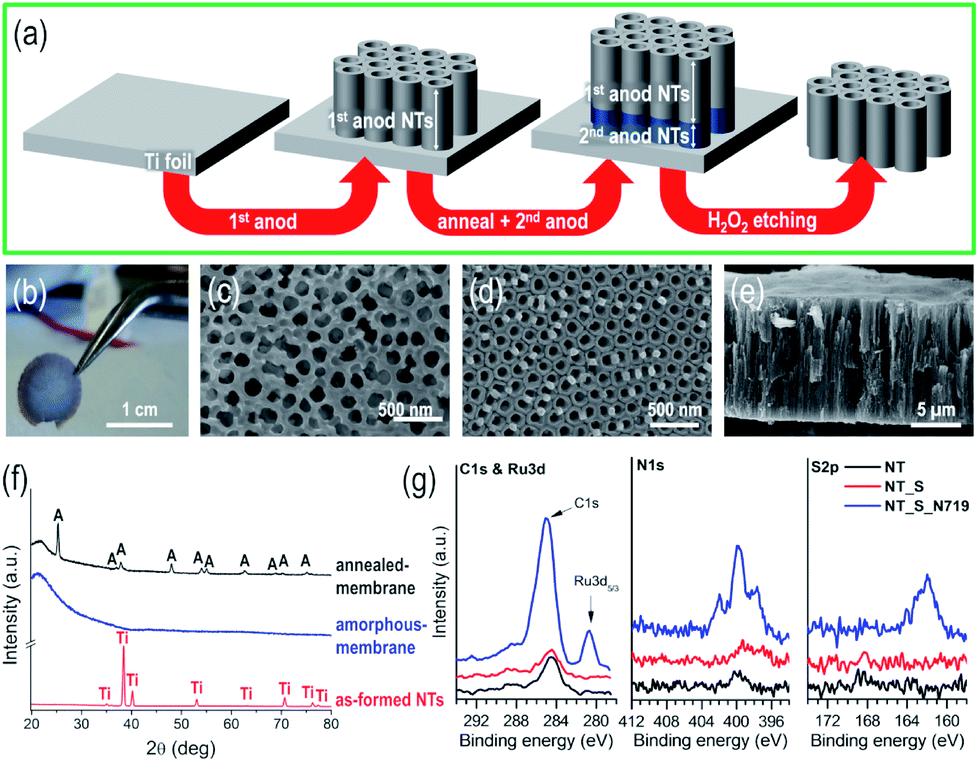 | ||
| Fig. 1 (a) Schematic of the fabrication of self-standing TiO2 nanotube membranes through a sequence of anodization (anod) steps, annealing and etching. The optical image in (b) shows a robust TiO2 nanotube membrane that could be held with tweezers. (c–e) Top, bottom, and cross-sectional SEM views of the TiO2 NT layer after H2O2 treatment for the formation of self-standing nanochannel membranes. (f) XRD patterns of (i) TiO2 NTs before detachment from the Ti substrate (red plot), (ii) amorphous self-standing TiO2 tube membrane (blue plot) and (iii) air-annealed (450 °C – 1 h) self-standing TiO2 tube membrane (black plot). XRD patterns of tube membranes were collected by transferring the layer onto a quartz glass. (g) High resolution XPS graphs of C 1s and Ru 3d, and N 1s and O 1s for bare nanotubes (NTs), nanotubes immersed in solution without the dye (NT_S) and nanotubes immersed in the dye solution (NT_S_N719). | ||
The etching step not only leads to the complete dissolution of the lower anodic tube layer, but also to that of the bottom of the upper layer, thus resulting in the flow-through nanochannel morphology. Despite the number of steps involved, this sequence leads to self-standing and crack-free tubular layers (Fig. 1(c–e)). The as-formed anodic TiO2 nanotube layers are usually amorphous, and as expected, for the as-synthesized NT layer before chemical etching, only reflections from the underneath Ti foil could be detected, while the tube membrane (that is, detached from the metal substrate and transferred onto a quartz glass) exhibits no specific features, i.e., neither crystalline TiO2 nor Ti reflections could be observed (in line with the amorphous nature of the original tube layer), as shown in Fig. 1(f). Annealing the membrane in air at 450 °C converted TiO2 from amorphous to crystalline anatase; to note, in the absence of the Ti foil underneath, the formation of the undesired rutile phase at the metal/oxide interface is also prevented (this occurs due to the thermal oxidation of metallic Ti).24,43
To evaluate the binding of the N719 molecular dye, we immersed the as-synthesized NT layers in the N719 dye solution used during the flow through experiments, i.e. a solution containing N719, acetonitrile, t-BuOH and EtOH – see Experimental. As a reference, we used a bare NT layer and additionally a NT layer immersed in a solvent solution without N719, NT_S (to account for the possible organics adsorption). The high resolution spectra of the three samples are shown in Fig. 1(g) for C 1s and Ru 3d peaks, N 1s and S 2p; we clearly observe no adsorption from the solvents used in the solution and a clear indication of N719 adsorption due to the increase in C 1s signals, and the appearance of peaks corresponding to Ru 3d5/3, N 1s and S 2p. First, the C 1s and Ru 3d peaks have a strong overlap; however the Ru 3d5/3 peak is visible at 280.8 eV (corresponding to RuII),44,45 while only the Ru 3d3/2 peak is hidden in the C 1s peaks (peak splitting of Ru 3d is at 4.2 eV and that of Ru 3d3/2 is at ∼285 eV). Thus the C 1s peak evident for the N719 nanotube sample (NT_S_N719) actually shows contributions from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–C, C–O, CN (from the NCS groups), TBA groups and Ru 3d3/2, with small contributions from C–C, C–O and COOH adsorbed on the TiO2 nanotubes38 (as seen from the small C 1s peaks of NT and NT_S). The N 1s and S 2p spectra confirm dye adsorption due to the presence of N 1s (very low N pickup from the environment for bare nanotubes) and S 2p peaks and these are attributed to the NCS groups (plus the CN bonds in the pyridine rings, in the case of N1s). The Ti 2p and O 1s peaks are listed in Fig. S1,† and while we observe no difference for the Ti 2p peaks, in the case of O 1s peaks for the N719 nanotube sample, we observe a broadening of the O peak towards higher binding energies which is correlated with the CO and COOH groups of the dye.
C, C–C, C–O, CN (from the NCS groups), TBA groups and Ru 3d3/2, with small contributions from C–C, C–O and COOH adsorbed on the TiO2 nanotubes38 (as seen from the small C 1s peaks of NT and NT_S). The N 1s and S 2p spectra confirm dye adsorption due to the presence of N 1s (very low N pickup from the environment for bare nanotubes) and S 2p peaks and these are attributed to the NCS groups (plus the CN bonds in the pyridine rings, in the case of N1s). The Ti 2p and O 1s peaks are listed in Fig. S1,† and while we observe no difference for the Ti 2p peaks, in the case of O 1s peaks for the N719 nanotube sample, we observe a broadening of the O peak towards higher binding energies which is correlated with the CO and COOH groups of the dye.
Visible light-responsive diffusion gate
The as-fabricated N719/TiO2 NT membranes are active in the visible spectral region where TiO2 is transparent and photo-inert (Eg,TiO2 ∼ 3.2 eV).46 The functionality of these highly defined, robust and crystalline TiO2 NT membranes was tested in a flow-through configuration for gated ion-diffusion, namely for (photo-assisted) diffusion experiments with the membranes sandwiched as a separator in the two-compartment cell, as shown in Fig. 2(a). The two sides of the cell were filled with a 20 v/v% N719 solution in EtOH (see details in Experimental), and with an EtOH based “reference solution”; before diffusion, the solution in compartment A exhibits a dark orange color owing to the Ru(II)-based dye, while the dye-free solution in compartment B is transparent and colorless. The temperature increase in both cell compartments during the illumination experiments is small (e.g. for 8 h illumination, the temperature increase was 3 °C, from 23.8 to 27 °C).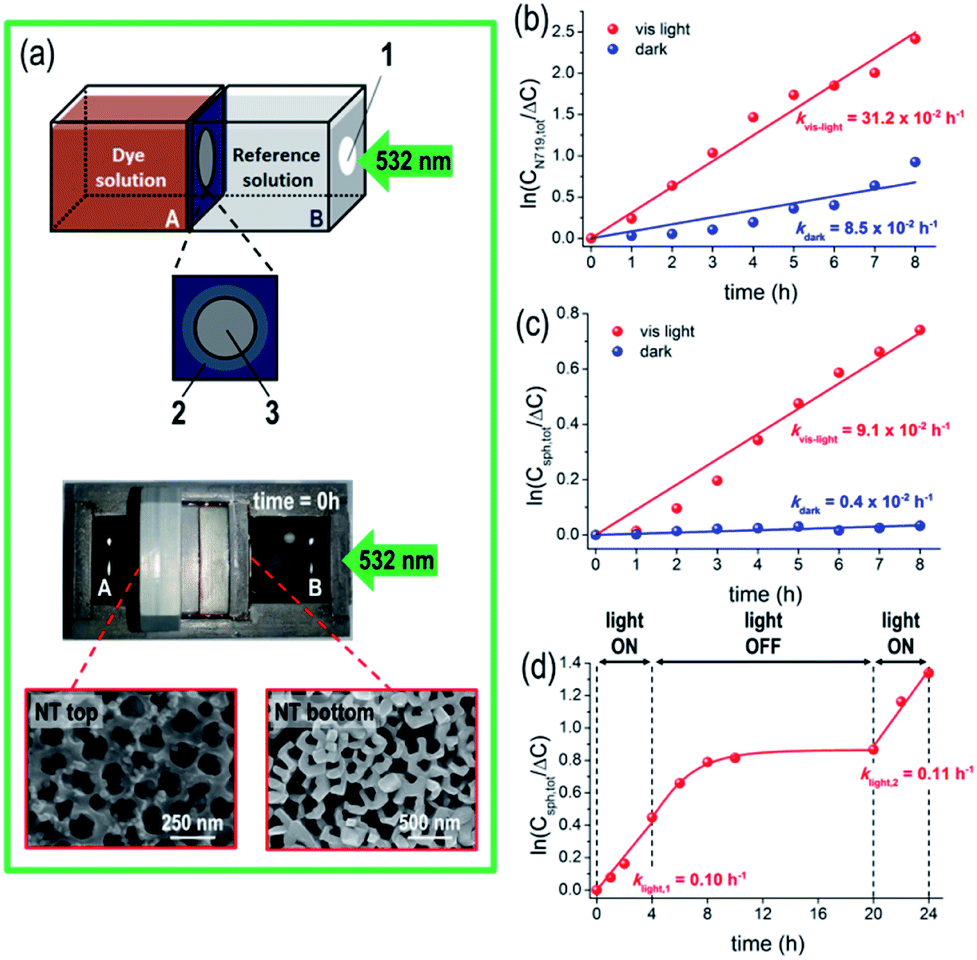 | ||
| Fig. 2 (a) Upper panel shows the sketch of the setup used for flow-through photocatalytic experiments. Chamber A contains 20 v/v% dye solution in ethanol (dye solution: 300 μM N719 in 1:1 acetonitrile/tert-butanol mixture) – for the polystyrene nanosphere experiments, the required amount of nanoparticles was also added in order to attain 200 μM initial concentration; chamber B contains the blank reference solution (1:1 tert-butanol/ethanol mixture). 1 represents the quartz glass window for 532 nm illumination, 2 indicates the entire tube membrane (d = 1 cm) and 3 is the actual membrane surface exposed to solutions and light (d = 0.53 cm). Lower panel shows the optical image of the two-compartment reactor before the diffusion experiment starts (t = 0 h), filled-in with the dye (A) and blank background (B) solutions. The SEM images show the top and bottom of a tube membrane. The membrane was always mounted with the tube top facing the dye solution and the tube bottom facing the reference solution. Kinetics of the dye (b) and nanosphere (c) flow across a tube membrane, in the dark and under 532 nm illumination. A first order rate law, that is, linear dependence of ln(Ctot/ΔC) vs. time, is assumed. (d) Kinetics of nanosphere separation through a dye pre-sensitized tube membrane, in light ON–OFF cycles. | ||
We first tested the permeability of the NT membrane to N719 molecules in the dark by measuring the dye concentration in both cell compartments and as anticipated, the N719 molecules diffuse through the membrane from the higher dye concentration side (A) to the lower one (B), until an equilibrium state is achieved, after ca. 7 hours, as discussed in the ESI and Fig. S2(a–d).† The concentration gradient (ΔC) across the tube membrane is the driving force and assuming a first-order kinetics for dye diffusion, a linear dependence of ln(ΔC/Ctot) vs. time is observed (Fig. 2(b)), with kdark = 8.5 × 10−2 h−1. A N719 diffusion coefficient D of ∼10−7 cm2 s−1 was extracted using a classic Fick approach47 – this is in line with previous reports on dye permeability through an anodic flow-through tube membrane.24,27 The value therefore confirms the absence of cracks or leaks within the membrane.
To note, the dye becomes partially adsorbed on the TiO2 tube membrane during flow through, and even if a significant change of the membrane color is also observed after 8 h of immersion in the N719 solution; however the total amount of free N719 in solution was not significantly affected by the amount adsorbed on the tubes. This, as the total dye concentration (measured in both cells), remains constant over the whole time range of the flow through experiments (middle panel, Fig. S2(a)).†
When the system is irradiated with a 532 nm-LED light, the diffusion of N719 molecules through the TiO2 nanotube membrane becomes significantly affected, as under illumination, the decrease/increase of the N719 amount on both sides of the cell is faster than that in the absence of light (red plots in Fig. S3(a)).† From the quantitative data, a ca. 4-times larger kinetic constant was calculated for the light-driven diffusion (i.e., klight = 31.2 × 10−2 h−1, see Fig. 2(b)), confirming the first-order kinetics of the reaction, resulting in a higher N719 diffusion coefficient D of ∼4 × 10−7 cm2 s−1.
Regarding the photo-induced degradation of the dye, when Ag+ ions are present in the diffusing electrolyte no significant dye degradation is observed (that is, the total N719 concentration remains constant over time, lower panel in Fig. S2(a)).† We attribute this effect of Ag+ ions to their electron-scavenging properties and their ability of coordinating (free) –NCS ligands of the Ru(II)-based dye. In general, dye photo-degradation can be prevented if the composition of the diffusing medium contains a suitable electron scavenging species (see also Fig. S3 and the ESI for discussion†).
Based on the above data, the dye/TiO2 NT membranes behave as smart nanofluidic systems with visible light-tunable hydrodynamic and permeability properties29 (i.e., klight/kdark = ca. 4). Supported by theoretical modeling (see below), we show that this effect can be ascribed to a light-promoted perturbation of the dye/tube electric properties upon electron injection from N719 (adsorbed on the tube walls) to TiO2 (i.e., in line with the principles reported for DSSCs48,49).
The full visible light-responsive on/off gating functionality of a dye-sensitized TiO2 flow-through membrane was then explored for the diffusion of an inert nanoprobe, that is, 20 nm diameter spherical, fluorescence labeled polystyrene nanoparticles (NPs or nanospheres).33 Nanosphere diffusion through the tube membrane was followed by UV-vis absorption spectroscopy in both chambers of the reactor, by measuring the change in the intensity of the nanosphere label with a maximum absorption band at λ = ca. 500 nm. The N719/TiO2 NT membrane behaves also in this case as a photo-switchable diffusion gate (Fig. S4(a)),† that is, NP diffusion is almost fully prevented in the dark, but it occurs when the system is under 532 nm illumination. In other words, the membrane switches from a (apparent) closed state in the absence of light to a gate-open state under 532 nm light.
Assuming a first-order kinetic dependence of the nanosphere concentration vs. time (Fig. 2(c)), the rate constants for the dark and light processes indicate a significantly faster nanosphere flux across the tube membrane under light than in the dark (klight/kdark = ca. 23). Similarly, we observed that nanospheres adsorb to a small extent on the NT tops, as seen in the SEM images of the tube top and bottom of a membrane after the experiment (Fig. 2(a)). However, the total amount of nanospheres (determined on both sides of the reactor) remains constant over the investigated time (middle and lower panels in Fig. S4(a));† thus no (photo-)degradation and/or significant adsorption on the tube wall occurs that would affect the experiment.
Excluding any clogging of the tube membrane by nanospheres (the tube diameter is ∼80–100 nm vs. the NPs diameter ∼20 nm), the slower NP trans-membrane permeation observed in the dark compared with the N719 dye is approximately in line with the Stokes–Einstein relation, where D is inversely proportional to the radius (r) of the diffusing species, that is, D ∝ r−1.50 Therefore, the diffusion coefficient of 20 nm diameter nanospheres (r = 10 nm) is expected to be more than 10 times smaller than that of N719 molecules (rN719 ∼ 0.8 nm36).27
However, it is evident from Fig. 2(c) (red plot) that the flux of NPs through the NT membrane becomes significantly enhanced when the system is irradiated with the 532 nm LED. In line with the results of dye ion-mobility, we attribute this effect to the light-mediated nanofluidics of the dye/TiO2 flow-through membrane due to the perturbation of the electrical and hydrodynamic properties which follows electron injection from the photo-excited dye into TiO2. Furthermore, in the absence of a light-responsive molecule (i.e. no dye adsorbed on the tube walls) the TiO2 tube membrane maintains a “closed” state that prevents polystyrene nanosphere diffusion (see below and the ESI, Fig. S5†).
These experiments show that the modulation of the nanofluidic gating properties of flow-through sensitized TiO2 tube membranes is valid regardless of the nature (i.e., light-responsive vs. light-inert) and size (i.e., 1.5 nm vs. 20 nm) of the diffusing species, indicating the underlying mechanism to be the same and to originate from the dye-sensitized tube walls.
Fig. 2(d) shows the results for the nanosphere diffusion using a dye-sensitized tube membrane in repetitive light ON–OFF cycles. During the initial light-ON period, diffusion of nanospheres through the membrane is observed, namely the membrane is in an open state providing fast NP diffusion. A kinetic constant klight,1 = 0.10 h−1 in line with that measured for the system with free dye molecules in the diffusing medium (Fig. 2(c)) confirms that a plain dye-sensitized membrane can deliver a similar light-regulated diffusion efficiency – i.e., no excess of free dye molecules in the diffusing medium is required. When the light is turned off, a slow decay of NP diffusion is observed (that is, switching of the sensitized tube membrane diffusion properties occurs on a longer time scale), followed by a significant drop in the kinetics of NP flow-through, i.e., the membrane is switched to a “gate-off” state. The permeability characteristics could be fully restored when the 532 nm LED light was turned on again, namely when the photo-promoted electron (from N719) mediated process was re-activated and the membrane switched back to a “gate-on” state for NP diffusion. We did not observe a decay of the activity of the membranes during several days and in addition, switching is reversible with no apparent dye degradation (to note, there is no photo-induced degradation of the N719 dye in the solution in the presence of Ag+ ions). This, as a comparison of the kinetic constants of the light-ON periods, with klight,1 ∼ klight,2, is in line with the reversible gate on/off characteristics of a dye-sensitized tube membrane, and also confirms the long-term stability of this approach for fabricating light-responsive flow-through systems.
Modelling
In order to assess the causes of the effect, we modelled the investigated dye/TiO2 NT system according to the schemes in Fig. 3. A key consideration is to simulate the effects produced under irradiation on the tube electric potential and electric field as a function of the distance from the tube surface (x).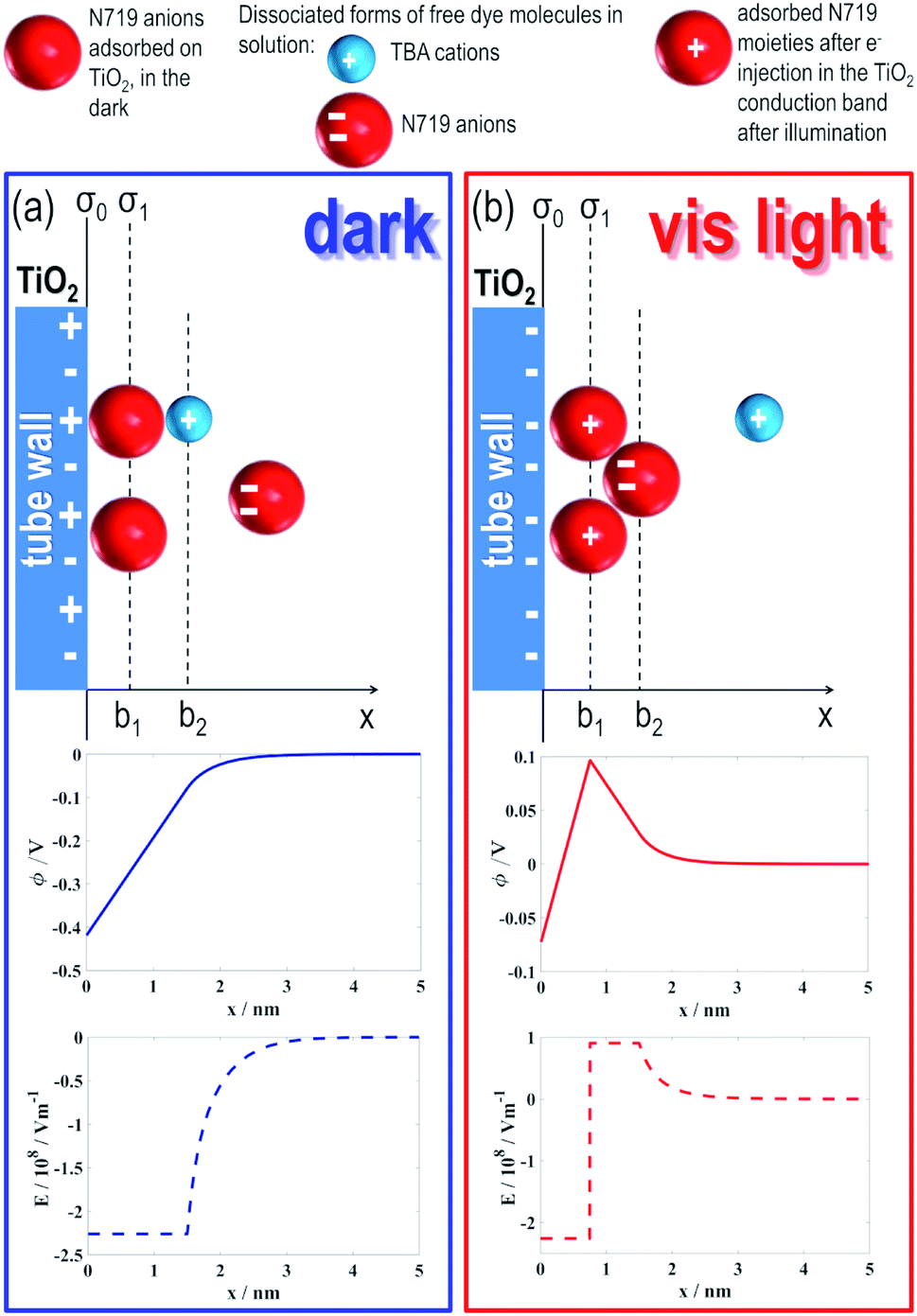 | ||
| Fig. 3 (a and b) represent the dye/TiO2 tube system at equilibrium (under dark) and under 532 nm illumination, respectively. In particular, the upper panels represent the model used for calculating the spatial distributions of the electric potential (ϕ, middle panels) and the electric field strength (E, lower panels) as a function of the distance from the surface of a TiO2 tube (x). Positive charges on the TiO2 surface in the dark represent under-coordinated Ti surface sites where N719 anions likely adsorb, thus becoming electrically neutral. Red spheres, with two negative charges (−2e0), and light blue spheres, with a positive charge (+e0), represent the dissociated form of free dye molecules in the diffusing medium, that is, N719 anions and TBA cations, respectively. Red spheres, with a positive charge (+e0), represent adsorbed N719 moieties after electron injection into the TiO2 conduction band upon illumination (i.e., Ru(III)). ϕ(x) and E(x) were calculated by quantitatively solving the modified Wicke–Eigen equation, with α+ = 3, α− = 13, ns/NA = 17.36 mol L−1 and n0/NA = 0.06 mol L−1 and room temperature, subjected to appropriate boundary conditions (for details, see Experimental). | ||
In solution, N719 typically dissociates into two tetrabutylammonium cations (TBA), each holding a positive charge +e0, and a Ru(II)-based molecular complex, the “dye”, that features two negative charges (–2e0) localized on –COO− groups.
N719 anions adsorb on the TiO2 surface through carboxylate binding/interaction e.g., with under-coordinated Ti surface sites, via hydrogen bonding or van der Waals interactions51,52 – typically, the adsorption mode of carboxylic groups on TiO2 is crucial to enable efficient charge transfer from the dye donor group to the semiconductor.53
For TiO2 nanotubes, we recently showed that the adsorption/binding efficiency of a molecule is surface curvature-dependent and that at highly curved rims (i.e., tube tops) is maximized due to a maximum of the TiO2 surface charge density (σ).37,38,54 Based on these and previous findings that showed that the surface of 20 nm diameter TiO2 nanoparticles (i.e., NPs with a highly curved surface) is uniformly covered by a N719-based monolayer,55 we assume that the (sharp edges of) tube walls (either at NT tops or bottoms) and the NT surfaces are decorated uniformly with N719 molecules (see models in Fig. 3).
To estimate the space dependence of the electric potential (ϕ) and the corresponding electric field strength (E) in the vicinity of the dye-decorated TiO2 surface (before and after light irradiation), we quantitatively solved the modified Wicke–Eigen equation (for derivation and definitions of the symbols, see Experimental and the two upper panels in Fig. 3):
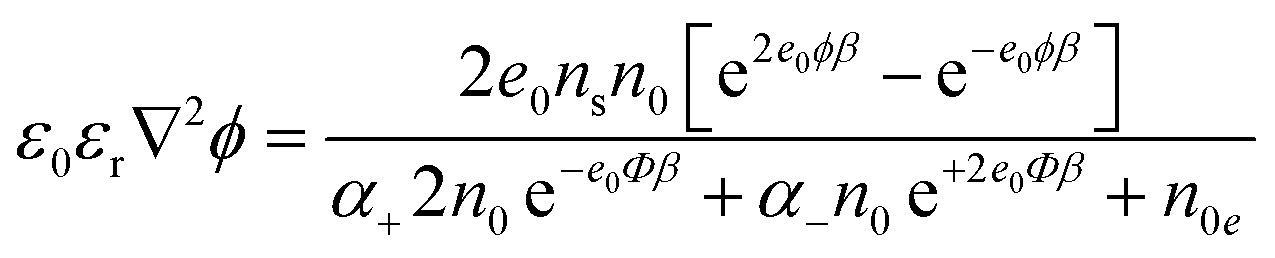 | (12) |
At equilibrium, i.e. when no light is provided (Fig. 3(a)), N719 adsorbed on the TiO2 surface “shares” (a fraction of) the initial −2e0 charge with TiO2 and, in line with this, a negative electric potential (Φ < 0) region (middle panel, Fig. 3(a)) and a corresponding negative electric field (E < 0) region (lower panel, Fig. 3(a)) were found to extend to a minimum of 1.5 nm (i.e., the diameter of a N719 molecule36) from the TiO2 surface (see Experimental for calculation details). This leads to a (electrostatic) repulsion (see the first column in Table 1) between the TiO2 surface covered by adsorbed dye molecules (Fig. 3(a)) and free anions in solution and therefore to hindered access to the free anions in the tube membrane, resulting consequently in a slow flow-through diffusion kinetics. These simulated findings are in accordance with our experimental results (Fig. 2(b)).
However, under light irradiation (Fig. 3(b)), that is, when N719 is photo-excited and photo-promoted electrons are injected from the LUMO of the dye into the CB of TiO2, the electric charge situation of the dye/TiO2 system dramatically changes: at a specific distance, x > b1 where b1 corresponds to the radius of N719 (i.e., b1 = 0.75 nm), a positive electric potential and electric field strength occur, as shown in the middle and lower panel, respectively, Fig. 3(b). Namely, the light-promoted e− injection from the dye creates a negative-charge depleted region in the vicinity of the TiO2 nanotube surface. In addition, a direct consequence of the e− injection on TiO2 from the photo-excited dye is in fact the oxidation of Ru(II) to Ru(III), that is, the “dye” becomes positively charged (which is well-known for the N719 dye used in dye sensitized solar-cells based on nanostructured TiO2).
Therefore, the plane charge density σ1 = e0/(dN719)2 at b1 describing the charge of a photo-oxidized dye layer on TiO2 becomes positive, ∼0.07 A s m−2 (calculated for n0/NA = 0.06 mol L−1), in contrast to σ1 = 0 A s m−2 before light irradiation (in the dark, the dye/TiO2 system is indeed considered under an electroneutrality regime).
The positively charged layer of irradiated N719 molecules produces a positive electric field (lower panel, Fig. 3(b)) and a consequent short-range attractive force on the free negatively charged dye molecules in solution (see right column in Table 1). Hence, the access of free dye molecules in the tube membrane is promoted with a direct consequence on the dye trans-membrane flow, in accordance with our experimental results (Fig. 2(b)).
In the calculations of attractive and repulsive forces presented in Table 1, a much smaller bulk concentration of ions (n0/NA) was assumed than that in Fig. 3; therefore the electric double layer also protrudes much deeper into the solution than as shown in Fig. 3. More importantly, the n0/NA value of 0.2 mmol L−1 used in the calculations presented in Table 1 is in the range of the used experimental value (see for example Fig. S2(a) in the ESI†). Table 1 reveals that the effect of light-induced charge switching extends more than 10 nm away from the inner surface of the TiO2 nanotube. Thus, a 10 nm thick ring on the inner surface of the nanotube presents around 40% of the inner cross sectional area of an 80–100 nm inner diameter nanotube. This affects in this range the hydrodynamic flow of charge (the flux of charged particles) through the tubes. In more detail, the positive charges accumulated on the surface of the tube wall (created by adsorbed N719 moieties after electron injection into the TiO2 conduction band upon illumination) produce a positive electric field and a short-range attractive force that can affect the free N719 anion movement in the solution (diffusion). Therefore, it can be concluded that light-induced charge switching can considerably change the transport through nanotubes, in accordance with experimental observations presented in this work.
We have recently shown that a modification of the electrical properties of a TiO2 tube membrane, induced e.g. by adjusting the electrolyte pH and/or composition, affects its surface electrokinetic properties.29 Based on these findings, we now assume that a similar effect on the hydrodynamics of a dye/tube membrane can also be observed due to a change of its charge surface density promoted by visible light illumination: positive charges accumulated at x = b1 may indeed attract free (negatively charged) N719 ions in solution and boost their diffusion. In line with this, our experimental results show that a ∼4-times faster flow-through migration of the dye occurs across the NT membrane under 532 nm light, with respect to diffusion in the dark (i.e., concentration gradient-driven).
This visible-light promoted effect could be exploited and, most remarkably, can be modulated reversibly under light ON–OFF conditions for transporting inert spectator molecules, that is, molecular substrates that are not directly involved in the photopromoted charge distribution switching, as we show e.g. for the negatively charged polystyrene nanospheres that undergo a similar gating mechanism.
Conclusions
In the present work, we show that dye sensitized anodic TiO2 nanotube membranes can be exploited as visible light switchable gates. TiO2 NT membranes sensitized with N719 can be stimulated with 532 nm illumination and show an altered flux (diffusion) through the tube membrane of not only dye molecules but also of inert polystyrene nanospheres. This effect can be regulated reversibly in light ON–OFF cycles. We attribute these visible light-adjustable diffusion features to the modification of the dye/TiO2 interface electrical properties which occurs under illumination. The re-arrangement of the electric charge along the walls of the NT membrane is key and affects the hydrodynamic properties within a tube channel and thus the permeability characteristics. These N719/TiO2 tube gated membranes, with a visible-light driven trigger, can be regarded as a new platform to optically control flow-through in nanofluidic devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge ERC (project ID: 340511), DFG and the DFG “cluster of excellence” EAM (Grant no. EXC 315), and ARRS grant No. P2-0232 for financial support.Notes and references
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, E. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101 CrossRef PubMed.
- A. K. Fard, G. McKay, A. Buenkenhoudt, H. Al Sulaiti, F. Motmans, M. Khraisheh and M. Atieh, Materials, 2018, 11, 74 CrossRef PubMed.
- E. M. Hoek, V. V. Tarabara, K.-H. Wee and R. Bai, Stimuli-Responsive Membranes, in Encyclopedia of Membrane Science and Technology – Membrane Materials, Characterization and Module Design, ed. E. M. Hoek and V.V. Tarabara, 2013 Search PubMed.
- R. van den Hurk and S. Evoy, Sensors, 2015, 15, 14045 CrossRef CAS PubMed.
- M. E. Idrissi, C. E. Meyer, L. Zarntner and W. Meier, J. Nanobiotechnol., 2018, 16, 63 CrossRef PubMed.
- O. Beckstein, P. C. Biggin and M. S. P. Sansom, J. Phys. Chem. B, 2000, 105, 12902 CrossRef.
- S. Kim, E. I. Ozalp, M. Darwish and J. A. Weldon, Nanoscale, 2018, 10, 20740 RSC.
- M. Ali, P. Ramirez, S. Mafé, R. Neumann and W. Ensinger, ACS Nano, 2009, 3, 603 CrossRef CAS PubMed.
- S. N. Smirnov, I. V. Vlassiouk and N. V. Lavrik, ACS Nano, 2011, 5, 7453 CrossRef CAS PubMed.
- L. J. Small, D. R. Wheeler and E. D. Spoerke, Nanoscale, 2015, 7, 16909 RSC.
- D. J. Beebe, J. S. Moore, J. M. Bauer, Q. Yu, R. H. Liu, C. Devadoss and B.-H. Jo, Nature, 2000, 404, 588 CrossRef CAS PubMed.
- B. Yameen, M. Ali, R. Neumann, W. Ensinger, W. Knoll and O. Azzaroni, Nano Lett., 2009, 9, 2788 CrossRef CAS PubMed.
- I. Vlassiouk, C.-D. Park, S. A. Vail, D. Gust and S. Smirnov, Nano Lett., 2006, 6, 1013 CrossRef CAS PubMed.
- Q. Zhang, Z. Zhang, H. Zhou, Z. Xie, L. Wen, Z. Liu, J. Zhai and X. Diao, Nano Res., 2017, 10, 3715 CrossRef CAS.
- Q. Zhang, J. Kang, Z. Xie, X. Diao, Z. Liu and J. Zhai, Adv. Mater., 2018, 30, 1703323 CrossRef PubMed.
- D. G. Haywood, A. Saha-Shah, L. A. Baker and S. C. Jacobson, Anal. Chem., 2015, 87, 172 CrossRef CAS PubMed.
- N. Liu, D. R. Dunphy, P. Atanassov, S. D. Bunge, Z. Chen, G. P. López, T. J. Boyle and C. Jeffrey Brinker, Nano Lett., 2004, 4, 551 CrossRef CAS.
- Q. Q. Zhang, Z. Y. Liu, K. F. Wang and J. Zhai, Adv. Funct. Mater., 2015, 25, 2091 CrossRef CAS.
- K. Lee, A. Mazare and P. Schmuki, Chem. Rev., 2014, 114, 9385 CrossRef CAS PubMed.
- F. Riboni, N. T. Nguyen, S. So and P. Schmuki, Nanoscale Horiz., 2016, 1, 445 RSC.
- S. Ozkan, A. Mazare and P. Schmuki, Electrochim. Acta, 2018, 268, 435 CrossRef CAS.
- F. Mohammadpour, M. Moradi, K. Lee, G. Cha, S. So, A. Kahnt, D. Guldi, M. Altomare and P. Schmuki, Chem. Commun., 2015, 51, 1631 RSC.
- F. Mohammadpour, M. Moradi, G. Cha, S. So, K. Lee, M. Altomare and P. Schmuki, ChemElectroChem, 2015, 2, 204 CrossRef CAS.
- S. So, I. Hwang, F. Riboni, J. E. Yoo and P. Schmuki, Electrochem. Commun., 2016, 71, 73 CrossRef CAS.
- S. So, I. Hwang, S. Mohajernia, M. Mackovic, E. Spiecker, G. Cha, A. Mazare and P. Schmuki, Adv. Energy Mater., 2018, 8, 1800981 CrossRef.
- P. Roy, T. Dey, K. Lee, D. Kim, B. Fabry and P. Schmuki, J. Am. Chem. Soc., 2010, 132, 7893 CrossRef CAS PubMed.
- S. P. Albu, A. Ghicov, S. Berger, H. Jha and P. Schmuki, Electrochem. Commun., 2010, 12, 1352 CrossRef CAS.
- J. Xu, L. Yang, Y. Han, Y. Wang, X. Zhou, Z. Gao, Y.-Y. Song and P. Schmuki, ACS Appl. Mater. Interfaces, 2016, 8, 34 Search PubMed.
- S. Mohajernia, A. Mazare, E. Gongadze, V. Kralj-Iglič, A. Iglič and P. Schmuki, Electrochim. Acta, 2017, 245, 25 CrossRef CAS.
- Z. Dai, L. Yang, Y. Li, C. Zhao, J. Guo, Z. Gao and Y.-Y. Song, Chem. Commun., 2019, 55, 10571 RSC.
- S. So, K. Lee and P. Schmuki, J. Am. Chem. Soc., 2012, 134, 11316 CrossRef CAS PubMed.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Gratzel, J. Am. Chem. Soc., 1993, 115, 6382 CrossRef CAS.
- S. Coertjens, R. De Dier, P. Moldenaers, L. Isa and J. Vermant, Langmuir, 2017, 33, 2689 CrossRef CAS PubMed.
- I. Paramasivam, J. M. Macak and P. Schmuki, Electrochem. Commun., 2008, 10, 71 CrossRef CAS.
- S. McLaughlin, Annu. Rev. Biophys. Biophys. Chem., 1989, 18, 113 CrossRef CAS PubMed.
- P. Marquet, G. Andersson, A. Snedden, L. Kloo and R. Atkin, Langmuir, 2010, 26, 9612 CrossRef CAS PubMed.
- M. Kulkarni, A. Mazare, E. Gongadze, S. Perutkova, V. Kralj-Iglič, I. Milošev, P. Schmuki, A. Iglič and M. Mozetič, Nanotechnology, 2015, 26, 62002 CrossRef CAS PubMed.
- M. Kulkarni, A. Mazare, J. Park, E. Gongadze, M. S. Killian, S. Kralj, K. von der Mark, A. Iglič and P. Schmuki, Acta Biomater., 2016, 45, 357 CrossRef CAS PubMed.
- M. Lorenzetti, E. Gongadze, M. Kulkarni, I. Junkar and A. Iglič, Nanoscale Res. Lett., 2016, 11, 378 CrossRef PubMed.
- M. Mohsen-Nia, H. Amiri and B. Jazi, J. Solution Chem., 2010, 39, 701 CrossRef CAS.
- J. Choi, S.-H. Park, Y. S. Kwon, J. Lim, I. Y. Song and T. Park, Chem. Commun., 2012, 48, 8748 RSC.
- G. Cha, P. Schmuki and M. Altomare, Chem.–Asian J., 2016, 11, 789 CrossRef CAS PubMed.
- K. Zhu, N. R. Neale, A. F. Halverson, J. Y. Kim and A. J. Frank, J. Phys. Chem. C, 2010, 114, 13433 CrossRef CAS.
- J. Singh, A. Gusain, V. Saxena, A. K. Chauhan, P. Veerender, S. P. Koiry, P. Jha, A. Jain, D. K. Aswal and S. K. Gupta, J. Phys. Chem. C, 2013, 117, 21096 CrossRef CAS.
- L. M. Martinez-Prieto, S. Carenco, C. H. Wu, E. Bonnefille, S. Axnanda, Z. Liu, P. F. Fazzini, K. Philippot, M. Salmeron and B. Chaudret, ACS Catal., 2014, 4, 3160 CrossRef CAS.
- M. Grätzel, Nature, 2001, 414, 338 CrossRef PubMed.
- K. L. Kostka, M. D. Radcliffe and E. J. von Meerwall, Phys. Chem., 1992, 96, 2289 CrossRef CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef.
- M. Grätzel, Inorg. Chem., 2005, 44, 6841 CrossRef PubMed.
- A. Einstein, Ann. Phys., 1905, 322, 549 CrossRef.
- K. Kalyanasundaram and M. Grätzel, Coord. Chem. Rev., 1998, 177, 347 CrossRef CAS.
- F. De Angelis, S. Fantacci, A. Selloni, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. C, 2010, 114, 6054 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- E. Gongadze, D. Kabaso, S. Bauer, T. Slivnik, P. Schmuki, U. van Rienen and A. Iglič, Int. J. Nanomed., 2011, 6, 1801 Search PubMed.
- R. Katoh, A. Huijser, K. Hara, T. J. Savenije and L. D. A. Siebbeles, J. Phys. Chem. C, 2007, 111, 10741 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00480g |
| ‡ Current address: POSCO Technical Research Laboratories, Automotive Steel Surface Research Group. |
| This journal is © The Royal Society of Chemistry 2019 |