
Hemi-core@frame AuCu@IrNi nanocrystals as active and durable bifunctional catalysts for the water splitting reaction in acidic media†
Jongsik
Park‡
 a,
Songa
Choi‡
a,
Aram
Oh
b,
Haneul
Jin
a,
Jinwhan
Joo
a,
Hionsuck
Baik
b and
Kwangyeol
Lee
*a
a,
Songa
Choi‡
a,
Aram
Oh
b,
Haneul
Jin
a,
Jinwhan
Joo
a,
Hionsuck
Baik
b and
Kwangyeol
Lee
*a
aDepartment of Chemistry, Korea University, Seoul 02841, Republic of Korea. E-mail: kylee1@korea.ac.kr
bKorea Basic Science Institute (KBSI), Seoul 02841, Republic of Korea
First published on 12th February 2019
Abstract
Highly efficient and economically sustainable hydrogen production via electrocatalytic water splitting can be realized by the advent of active and durable electrocatalysts toward the oxygen evolution reaction (OER) and hydrogen evolution reaction (HER). Multimetallic nanoframe structures have received great attention as promising electrocatalysts for these reactions, because their inherent high surface area and tunable surface energy states are beneficial to the electrocatalyst performance. We envisaged that the stability and activity of multimetallic nanoframe catalysts could be simultaneously augmented by introducing an additional structural feature of activity-enhancing lattice mismatch via formation of a structure-fortifying core–shell structure. Herein, we successfully demonstrate that hemi-core@frame AuCu@IrNi nanocrystals, possessing structural features of both nanoframe and core–shell, are active and durable bifunctional catalysts toward both the OER and HER under acidic conditions. The hemicore@frame AuCu@IrNi nanocrystals exhibit superior efficient electrocatalytic performance toward the overall water splitting reaction, and show 355 mV overpotential at the current density of 10 mA cm−2 in a 0.5 M H2SO4 electrolyte. The robustness of the catalysts was also verified through the long-term stability test.
Conceptual insightsIr-Based multimetallic nanomaterials with large surface area are well-known active electrocatalysts for the oxygen evolution reaction (OER) due to their high activity. From a structural point of view, both nanoframeworks with a high surface area and core@shell nanoparticles with a strain effect driven from the lattice mismatch are regarded as promising electrocatalysts. However, it is difficult to combine these advantages into one single nanoparticle. In this work, we demonstrate the synthetic method of hemi-core@nanoframes, namely AuCu@IrNi alloy nanoframes which show enhanced electrocatalytic activity arising from the large surface area and the surface strain via lattice mismatch between the core and shell. The hemi-core@frame AuCu@IrNi nanocrystals exhibit excellent electrocatalytic activity and durability for both the OER and hydrogen evolution reaction (HER) under acidic conditions. We expect that this novel synthetic methodology will pave the way for further development of multicomponent hemi-core@nanoframes as bifunctional electrocatalysts toward important electrocatalytic processes. |
Introduction
Bifunctional electrocatalysts for the oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) of water splitting in acid electrolytes can greatly impact the renewable and sustainable production of hydrogen energy.1–7 Among various nanocatalyst structural motifs, multimetallic nanoframes are particularly promising in realization of economically viable bifunctional electrocatalysts because the inherent high surface area and the tunable surface energy states of multimetallic nanoframes are beneficial to the electrocatalyst performance.8–13 Furthermore, the prudent choice of constituent metal atoms can result in the much desired bifunctionality toward the OER and HER. Thus far, Ir-based oxide catalysts via oxidation of Ir metal have led in their catalytic performance toward the OER in terms of both activity and durability under acidic electrolytes.14–19 Efforts to further enhance the structural integrity of Ir oxide catalysts have been intensively exerted in recent years and the most notable advances have been accomplished by introducing secondary elements such as Sr and Co to the Ir matrix.20,21 On the other hand, Ir metal itself can be employed for the HER, while the best HER performances have been found with Pt-based or Pt alloy catalysts.22,23 It has also been demonstrated that Ir-based alloy nanoparticles exhibit a good catalytic activity toward the HER, which is comparable to those of Pt-based catalysts.24–27 Therefore, Ir-based alloy nanoparticles can be potentially employed as bifunctional catalysts toward the OER and HER.On the other hand, lattice-mismatch found at the interface of a core–shell nanoparticle has been advantageously exploited to boost the catalytic activity, particularly in the development of Pt-based electrocatalysts toward the oxygen reduction reaction (ORR).28–34 Since we have recently demonstrated that Ir-based nanoframe structures obtained by using leachable Cu or Ni nanotemplates are excellent OER catalysts after surface oxidation,14,20 we have been particularly intrigued by the possibility of further improving the catalytic activity of the Ir-based nanoframe structure via implementation of the additional core–shell structural feature. The structural robustness of flimsy nanoframes might be further fortified by the presence of a structure-supporting core.14,35,36 Specifically, we envisioned a structure of an Ir-based multimetallic nanoframe encapsulating an Au-based core as a bifunctional catalyst toward the OER and HER, in which the Au-based core should not be completely detached from the Ir-based nanoframe lest the core–shell mismatch effect be lost. Herein, we demonstrate that the hybrid hetero nanostructure of IrNi nanoframe/AuCu@IrNi core–shell, possessing both the nanoframe morphology and core–shell structural motif, exhibits excellent bifunctional catalytic performance toward overall water splitting. The fabrication of the complex hemicore@shell structural feature could be possible by understanding the mixing degree and decomposition kinetics among the multiple components.37–39 The hemicore@frame AuCu@IrNi nanocrystals deliver highly efficient catalytic activity by achieving the current density of 10 mA cm−2 at a cell voltage of only 1.585 V for overall water splitting in 0.5 M H2SO4 electrolyte. Furthermore, only 9 mV of overpotential increase was observed for hemicore@frame AuCu@IrNi nanoparticles after 24 h of continuous long-term stability test, indicating their excellent structural robustness during the cycling test.
Results and discussion
In a typical synthesis of a hybrid hetero nanostructure of IrNi nanoframe/AuCu@IrNi hemi core–shell (ACIN-HF), a slurry of Ir(acac)3 (0.02 mmol), Ni(acac)2 (0.04 mmol), CTAC (0.08 mmol), 1,2-HDD (0.04 mmol), oleylamine (98%) (12 mmol), and AuCu seed solution (0.01 mmol in 1 mL in oleylamine; see the ESI,† for preparation of AuCu seeds, Fig. S1) was prepared in a 100 mL Schlenk tube with magnetic stirring. After the solution was placed at 60 °C for 5 min, the Schlenk tube was directly placed in a hot oil bath, which was preheated to 260 °C. After being heated at the same temperature for 40 min, the reaction mixture was cooled to room temperature with magnetic stirring. The reaction mixture, after being cooled to room temperature and with added 15 mL of toluene and 25 mL of ethanol, was centrifuged at 4000 rpm for 5 min. The resulting precipitates (precursor to ACIN-HF; PACIN-HF) were dispersed in 2 mL of toluene and 2 mL of ethanol and then mixed with 2 mL of 3 M HCl solution. The mixture was placed at 60 °C for 1 h. Finally, the precipitated ACIN-HF was centrifuged and washed with ethanol (10 mL) two times and then dried under vacuum.The characterization data of PACIN-HF and ACIN-HF are shown in Fig. 1. A representative transmission electron microscopy (TEM) image of PACIN-HF is shown in Fig. 1a. The inset in Fig. 1a shows the overall distribution of different elements within PACIN-HF. The corresponding elemental mapping images for Au (orange), Ir (green), Ni (red), and Cu (blue) in Fig. 1b clearly show the formation of hetero structural features. The line profile analysis of PACIN-HF clearly demonstrates the hetero-structural morphology (Fig. 1c). The core is composed of totally segregated AuCu and Ni phase. The energy-dispersive X-ray spectra (EDS) data reveals that the atomic compositions of PACIN-HF are Au 9.3%, Ir 16.6%, Ni 64.3%, and Cu 9.7% (Fig. S2a, ESI†). In order to visualize the formation mechanism of the hemi-core hetero structure, we also analyzed TEM, corresponding elemental mapping data and line profile analysis of ACIN-HF as shown in Fig. 1d–f. The bright hemispherical core of the HAADF-STEM image is composed of an AuCu rich phase and the outer frame is composed of an IrNi phase. The EDS data reveals that the atomic compositions of ACIN-HF are Au 19.8%, Ir 35.1%, Ni 27.9%, and Cu 17.2% (Fig. S2b, ESI†). The line profile analysis of ACIN-HF also clearly demonstrates the hemi-core@frame morphology (Fig. 1f). The size distribution data of PACIN-HF and ACIN-HF are shown in Fig. S3 (ESI†). The diameter and length of PACIN-HF are 12.5 ± 1.2 nm and 16.9 ± 1.5 nm. These values are similar to those of ACIN-HF (diameter: 12.9 ± 0.7 nm, length: 16.3 ± 1.1 nm) which indicates that only the core Ni phase is removed during an etching process, without affecting the overall crystal size. To further investigate the compositions of both unetched and etched nanocrystals, we analyzed high-resolution TEM (HRTEM) images of PACIN-HF and ACIN-HF (Fig. S4, ESI†). We found that three kinds of alloy compositions are observed through the FFT patterns. The (i) rectangle area in Fig. S4a (ESI†) indicates the twinned AuCu phase. The lattice spacing of {110}AuCu is 0.196 nm, which is close to lattice spacing (d = 0.199 nm) of a bulk AuCu phase. At the edge of PACIN-HF ((ii) rectangle), the measured lattice spacing is 0.214 nm, which is shorter than that of pure {111}Ir (d = 0.221 nm) but larger than that of pure {111}Ni (d = 0.203 nm). The incorporation of Ni phase would induce the lattice contraction in the Ir-based alloy phase of PACIN-HF. The lattice spacing of the core phase in the (iii) rectangle is measured to be 0.179 nm, consistent with the presence of {200}Ni. While both AuCu phase and IrNi phase are clearly shown in the FFT patterns in Fig. S4b (ESI†), the Ni phase has been removed through chemical etching. In addition, it is clearly visible that the Ir-based frame of ACIN-HF is polycrystalline because they formed through the epitaxial growth on the pre-synthesized twinned AuCu seeds. The powder X-ray diffraction patterns (PXRDs) of PACIN-HF and ACIN-HF have broad major peaks, which are due to combination of AuCu, IrNi, and Ni-rich phases (Fig. S5a, ESI†). After the chemical etching process, the peak intensity of the Ni-rich phase (red arrow) was drastically decreased due to Ni phase dissolution, while the IrNi phase (green arrow) was still visible (Fig. S5b, ESI†). Interestingly, the XRD peak of AuCu measured at near 40° became broader (yellow arrow). This peak broadness would have been driven from the segregation of the Cu phase, resulting in the formation of several AuxCu1−x alloy phases. The gradient AuxCu1−x composition also could be detected through the line profile analysis in Fig. 1f.
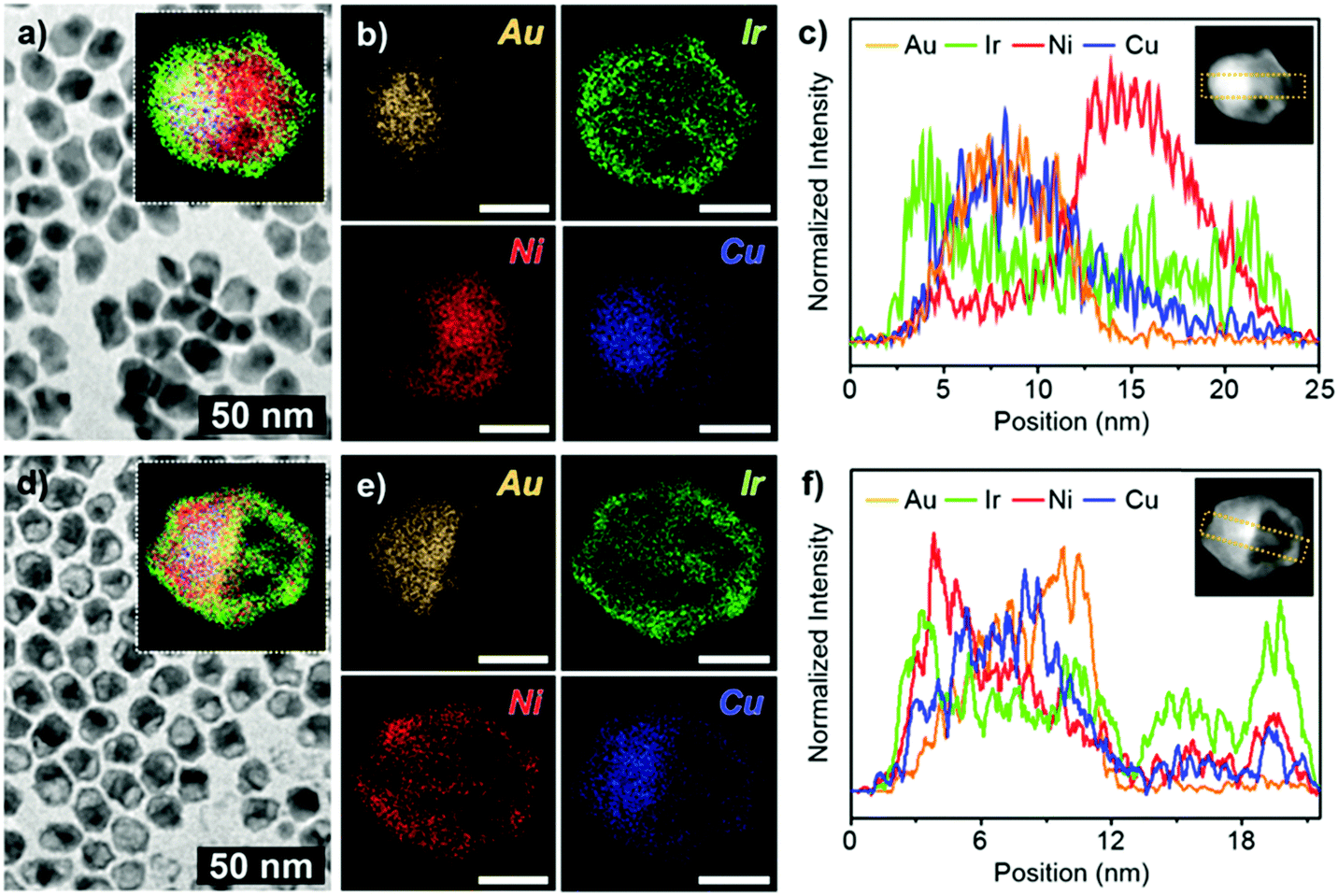 | ||
| Fig. 1 Characterization of PACIN-HF and ACIN-HF. (a) TEM, (b) corresponding elemental mapping analysis and (c) line profile analysis of PACIN-HF. (d) TEM, (e) corresponding elemental mapping analysis and (f) line profile analysis of ACIN-HF. Each color indicates Au (orange), Ir (green), Ni (red), and Cu (blue), respectively. The white scale bars in (b) and (e) indicate 10 nm. | ||
We found that the AuCu@IrNi core–shell nanoparticles (ACIN-CS) are obtained via co-decomposition of Cu, Ir, and Ni precursors in the presence of Au seeds, which is in stark contrast to the case of ACIN-HF formed by using the same precursors, but in the presence of AuCu alloy seeds. The representative TEM images and the corresponding elemental mapping analyses of ACIN-CS and ACIN-CS after etching are shown in Fig. 2. The line profile (Fig. S6, ESI†) and EDS analysis (Fig. S7, ESI†) indicate the core@shell structure of AuCu@IrNi for ACIN-CS. The core@shell AuCu@IrNi morphology remains largely intact after chemical etching; only a very small amount of Cu dissolution occurred. The Cu atoms, provided by decomposition of Cu precursors, were mixed with the Au seeds to form AuCu alloy seeds. The intermetallic AuCu alloy composition could be formed during the reaction by employing the diffusion of Cu atoms into presynthesized Au nanoparticles.40 After that, sequentially decomposed and reduced Ni and Ir precursors form an IrNi shell on the AuCu seed. The corresponding elemental mapping analysis of ACIN-CS and acid-etched ACIN-CS indicates that Cu atoms are found in the Au core. The HRTEM analysis of ACIN-CS (Fig. S8, ESI†) shows that it also has poly-crystalline nature because Au nanoparticles are twinned, similar to AuCu seeds. To elucidate the origin of polycrystallinity, we performed a contrast experiment without using Au or AuCu seeds. Similar to the results in the previous literature,14 we obtained Cu@IrNi single-frame nanostructures (CIN-SF); HRTEM analysis of CIN-SF indicates that CIN-SF is single-crystalline (Fig. S9, ESI†). Therefore, we concluded that the poly-crystalline structural feature of ACIN-HF and ACIN-CS was driven from the usage of twinned Au and AuCu seeds and heteroepitaxial growth on them.
 | ||
| Fig. 2 Characterization of AuCu@IrNi core@shell nanoparticles (ACIN-CS). TEM images and corresponding elemental mapping analysis of (a and b) ACIN-CS and (c and d) ACIN-CS after a chemical etching process. The white scale bars in (a)–(d) indicate 10 nm. | ||
The difference in morphologies of ACIN-HF and ACIN-CS might have originated from the different distributions of Cu on the AuCu seeds, depending on whether they are pre-formed or in situ generated (Fig. 3). In order to better understand the formation mechanism of ACIN-HF and ACIN-CS, we obtained the TEM images of intermediates and their corresponding EDS analyses (Fig. S10, ESI†). Interestingly, the preferred decomposition and reaction kinetics of Ni and Ir precursors is slightly different for ACIN-HF and ACIN-CS. At the initial stage of ACIN-HF, the reduction of the Ni precursor is preferred rather than the reduction of the Ir precursor because the existence of Au at the surface of seeds might act as a barrier to an alloying process. According to previous literature reports, Au–Ir alloying is an unfavourable process compared to Au–Ni.41,42 In addition, the outermost part of the AuCu core is mostly Au0.5Cu0.5 phase which is unfavourable to form Ni shell through epitaxial growth, due to the positive mixing enthalpies and segregation energies between Au/Ni.43,44 On the other hand, while the Ni/Cu system is known to form alloy phases of various Ni/Cu ratios,45,46 G. Guisbiers et al. showed that Cu–Ni nanoalloy can also take the hetero nanostructure depending on the reaction temperature and particle size.47 Therefore, this unprecedented formation of hetero structural feature of Ni–AuCu might be due to the synergy between the difficult Au/Ni mixing and the reaction conditions favouring the segregation of Cu/Ni phases. Consistent with the above finding, a hemi-core@frame structure could also be synthesized without using the Cu precursor as shown in Fig. S11 (ESI†). However, in this case, most of Ni atoms are dissolved during the chemical etching process by EDS (Fig. S12, ESI†) because a major fraction of Ni atoms has migrated to the Au–Ir interface. Therefore, the presence of Cu induced the thorough mixing between Ir and Ni and ultimately the stabilization of the IrNi alloy phase. From the electrocatalyst view, the IrNi alloy has shown enhanced electrocatalytic performance over the pure Ir phase.48 Therefore, the introduction of Cu appears to be beneficial for the OER activity because it leads to the mixing between Ir and Ni phases.
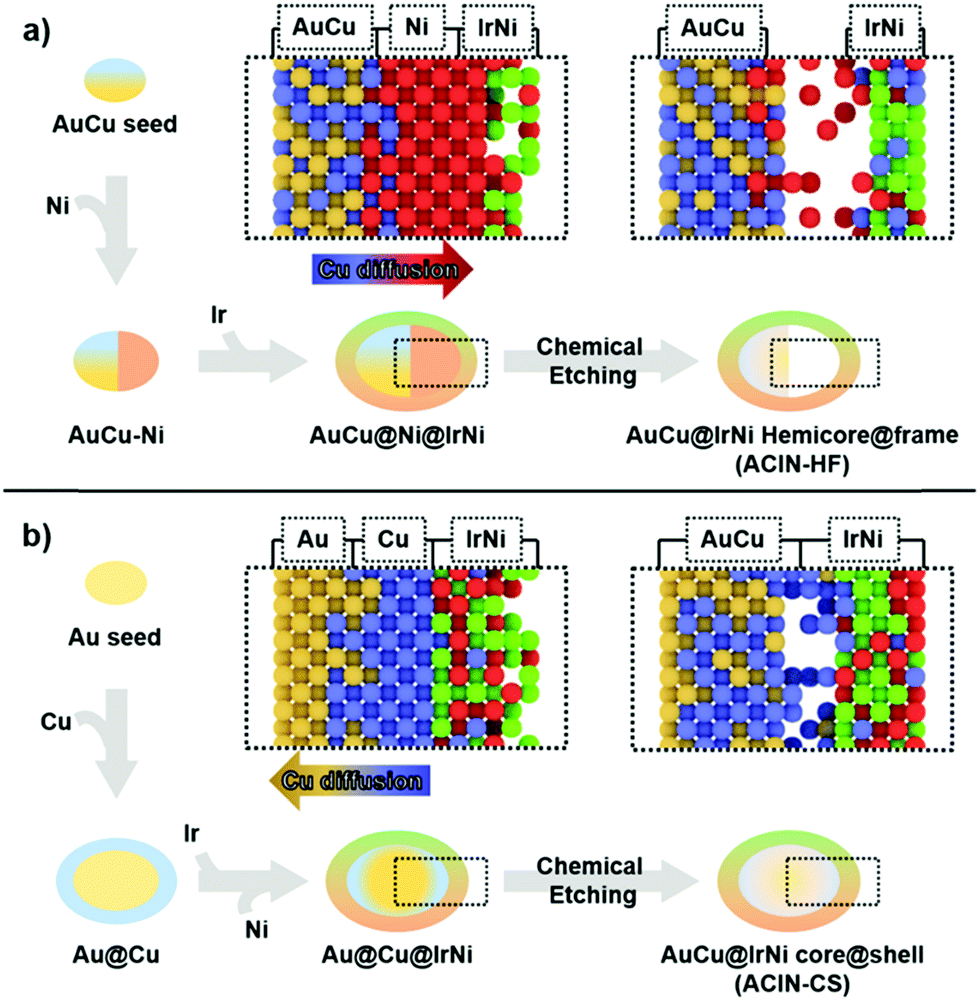 | ||
| Fig. 3 Schematic illustration of Cu diffusion pathway dependent on seed type. | ||
In the case of co-reduction of Ir, Ni, and Cu precursors with Au seeds to form ACIN-CS (Fig. 3b), the decomposition of Cu on Au seeds results in a Cu-rich shell at an initial stage. Due to the similar mixing enthalpy between Ir/Cu and Ir/Ni,44 the preferred reduction reaction is only determined by the discrepancy of reduction potential, so the reduction of Ir is facilitated compared to that of Ni. The outermost Cu-rich phase acts as an inter-diffusion layer for Ir and Ni phases so that Ir and Ni can mix thoroughly to result in the epitaxially grown IrNi shell. Consequently, the understanding of metal diffusion and surface segregation at the nanoscale is imperative to the preparation of rationally designed complex hetero-nanostructures.49–51
We carried out electrochemical measurements of OER and HER by Ir-based nanostructures to understand the nanostructural effects of catalysts on catalytic performance. The electrocatalytic performances of ACIN-CS/C, ACIN-HF/C, CIN-SF/C, commercial Ir/C and Pt/C toward the OER and HER in 0.1 M HClO4 (Merck, Suprapur grade) were evaluated as shown in Fig. 4. ACIN-CS, ACIN-HF, and CIN-SF nanoparticles were supported on carbon black (Vulcan XC 72) to afford ACIN-CS/C, ACIN-HF/C, and CIN-SF/C catalysts. The inductively coupled plasma atomic emission spectroscopy (ICP-AES) results of ACIN-HF/C, ACIN-CS/C, and CIN-SF/C were used to determine the precise metal contents in the catalysts (Table S1, ESI†). Several studies warned about the usage of Vulcan carbon as a support template,52,53 because the loss of carbon mass during the OER induced lower kinetics of electric pathways. The high corrosion rate of the carbon support, namely, the carbon oxidation reaction (COR), is accelerated when the material is used under conditions that require a large overpotential.54 Therefore, our prepared catalysts, which have low onset potential, would not be significantly affected by the COR. TEM images of the carbon supported catalysts are shown in Fig. S13 (ESI†). A three-electrode set-up was used with Ag/AgCl (filled with saturated KCl) and graphite electrodes as the reference and counter electrode, respectively. Thus far, an IrOx phase has shown the best electrocatalytic activity toward the OER and it is obtainable by the transformation from the pure Ir phase through the oxidation during the electrocycling test.55–57 To further understand the structure-dependent catalytic performance of catalysts of this study toward the OER and HER, we measured the electrochemically active surface area (ECSA) of the catalysts by a CO-stripping method (Fig. S14, ESI†). The ECSAs of ACIN-CS/C, ACIN-HF/C, CIN-SF/C, and Ir/C are 35.0, 36.8, 43.3, and 43.2 m2 g−1, respectively. Fig. 4a shows the OER polarization curves for ACIN-CS/C, ACIN-HF/C, CIN-SF/C, and commercial Ir/C in N2-saturated 0.1 M HClO4 at the scan rate of 5 mV s−1 normalized by the area of the rotating disk electrode (RDE). The ACIN-HF/C and CIN-SF/C exhibit better OER performance with the low overpotentials of 308 and 307 mV, respectively, than those of ACIN-CS (318 mV) and Ir/C (324 mV) (Fig. 4b). The Tafel slopes of ACIN-HF and CIN-SF/C were measured to be 58 and 56 mV dec−1, respectively, which are smaller than those of ACIN-CS (62 mV dec−1) and Ir/C (74 mV dec−1) (Fig. 4c). Therefore, the large surface of ACIN-HF was beneficial for driving efficient OER.
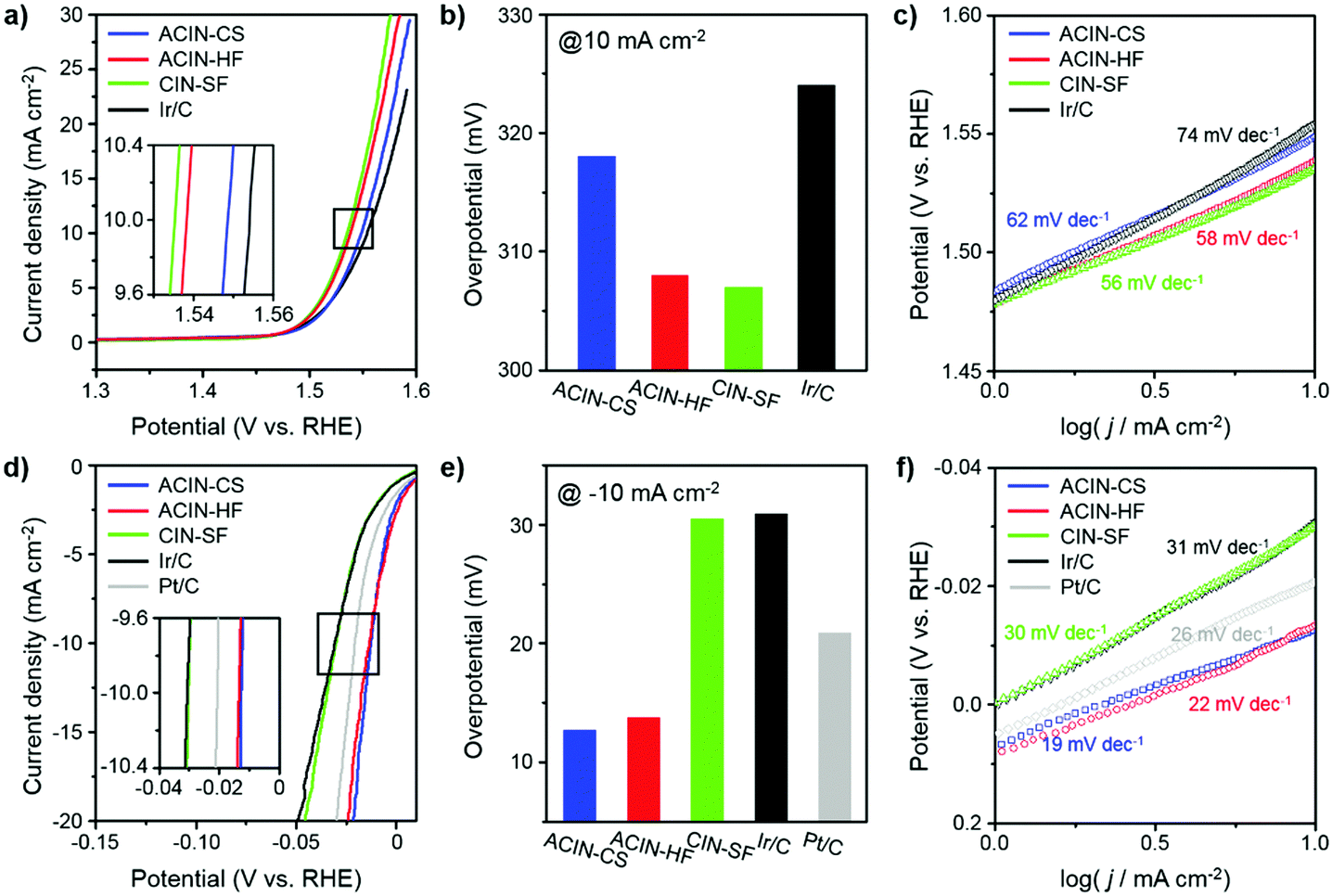 | ||
| Fig. 4 (a) The OER polarization curves, (b) overpotential bar columns graph measured at 10 mA cm−2 and (c) corresponding Tafel slopes of the ACIN-CS/C, ACIN-HF/C, CIN-SF/C, and commercial Ir/C catalysts in 0.1 M HClO4 solution. (d) The HER polarization curves, (e) overpotential bar columns graph measured at −10 mA cm−2 and (f) corresponding Tafel slopes of the ACIN-CS/C, ACIN-HF/C, CIN-SF/C, commercial Ir/C, and Pt/C catalysts in 0.1 M HClO4 solution. | ||
Thus far, Pt has been considered the ideal catalyst for the HER with near zero overpotential and low Tafel slope.13,58 Recently, Ir-based alloy nanocrystals also have shown comparable electrocatalytic activity with that of Pt for the HER.25–28 In contrast to the OER reaction, the ACIN-CS/C might serve as a promising HER catalyst due to the core–shell effect driven from the lattice mismatch between the AuCu core and IrNi shell. The polarization curves for the catalysts normalized by the area of the RDE are shown in Fig. 4d. As expected, the ACIN-CS/C exhibited the best performance for the HER, followed by ACIN-HF/C, commercial Pt/C, CIN-SF/C, and commercial Ir/C; the overpotentials of the current densities at −10 mA cm−2 were 12.7 mV, 13.7 mV, 30.5 mV, 30.9 mV, and 20.9 mV for ACIN-CS/C, ACIN-HF/C, CIN-SF/C, commercial Ir/C, and Pt/C, respectively (Fig. 4e). ACIN-CS/C and ACIN-HF/C exhibited very low Tafel slope values of 19 mV dec−1 and 22 mV dec−1, respectively, which are superior to that of commercial Pt/C (26 mV dec−1) (Fig. 4f). The Tafel slopes of CIN-SF/C were 30 mV dec−1, suggesting fast kinetics as compared to Ir/C (31 mV dec−1) toward the HER. However, the performance of CIN-SF/C significantly falls behind the performances of ACIN-CS/C, ACIN-HF/C, and Pt/C, thus making it unsuitable as a bifunctional water splitting catalyst. We also evaluated the HER activities of the prepared catalysts in a 0.1 M HClO4 electrolyte with H2 saturated conditions (purging pure H2 gas, 99.999%) in order to avoid Nernst-Shift from evolving H2 gas and compared it with the LSV curve obtained under N2 saturated conditions. As shown in Fig. S15 (ESI†), different shapes of plots were observed near 0 V, while similar overpotentials were required at the current densities over −10 mA cm−2. The positive current at a positive potential value under H2-saturated conditions seems to originate from a hydrogen oxidation reaction (HOR) on the catalyst surface and the negative current under N2-saturated conditions would be from double layer capacitance on the electrode surface.59 Since strong negative current was obtained at a low current density region of the sample under N2-saturated conditions, it is hard to evaluate a proper onset potential. However, we believe that comparable values can be obtained in a high current density region over −10 mA cm−2. In addition, we tested Au and AuCu cores for OER and HER (Fig. S16, ESI†). However, the electrocatalytic activities of Au and AuCu seeds were very poor toward the HER, indicating that the high HER activity of ACIN-HF or ACIN-CS has been driven from the Ir-based alloy compositions. The physical origins of the electrode kinetics were examined via electrochemical impedance spectroscopy (EIS) (Fig. S17, ESI†). Among our prepared catalysts, the ACIN-HF shows smaller semicircle radius measured for both the OER and HER, indicating the lowest electrical resistance and higher charge transfer rate compared with other catalysts,60–62 demonstrating that it is an ideal catalyst for improving the OER and HER kinetics.
We compared the overall overpotential of the catalysts measured at the 10 mA cm−2 current density in a water splitting reaction by adjoining the OER and HER results in Fig. 5a. The ACIN-HF/C has the lowest overall overpotential value among the tested catalyst samples. Remarkably, the measured overpotential of ACIN-HF/C (321.7 mV) is superior to the other recently reported state-of-the-art bifunctional electrocatalysts in acidic media (Tables S2–S4, ESI†).25–28 In order to gauge the electrocatalytic performance of catalysts measured under full cell conditions, we built a proton exchange membrane water electrolyzer (PEMWE) for overall water splitting. A two-electrode system for water splitting was built by applying each catalyst as both anode and cathode catalysts. All catalysts were supported on carbon fiber paper (CFP) and tested in N2-saturated 0.5 M H2SO4 solution. Fig. 5b shows the polarization curves for overall water-splitting performance. The ACIN-HF/CFP exhibited an excellent activity with a cell voltage of 1.585 V at the current density of 10 mA cm−2, which is lower than those of ACIN-CS/CFP (1.597 V), CIN-SF/CFP (1.603 V), and Ir/CFP (1.611 V). To probe the robustness of the catalysts during the catalytic cycling, we performed a durability test by using a chronopotentiometry technique (Fig. 5c). All of the synthesized nanocrystals maintained their activities for 24 h, while Ir/CFP could not. In order to determine which conditions are responsible for the slow deactivation, we studied the durability tests separately for the HER and OER half reactions (Fig. S18, ESI†). The durability test of HER in Fig. S18a (ESI†) indicates that a little deactivation occurred at the initial stage (<4 h) of the HER, however, the values of overpotential were still maintained after 4 h of HER. In contrast to the HER, the values of overpotential measured during the OER are gradually increased (Fig. S18b, ESI†). The TEM images and ICP-AES analyses obtained after a durability test (Fig. S19 and Table S5, ESI†) show that while the morphologies of the nanostructures are well-preserved, the Ni and Cu contents were leached from the nanocatalysts due to the formation of the IrOx phase, indicating the slow composition change of the nanocatalysts during the durability test.14 Specifically, the elemental composition analysis of ACIN-HF after the durability test in Fig. S20 (ESI†) also indicates that the Ni atoms are slightly dissolved during the electrocycling test. According to a previous study, the dissolution of a small amount of Ni might generate reactive surface hydroxyls on the IrNi based catalyst surface, leading to retention of the high OER activity.63 Overall, the full cell overpotential trend based on the structural morphologies is similar to the sum of half-cell reactions, suggesting that the hemi-core@frame nanostructure shows the best electrocatalytic performance regardless of electrolyzers.
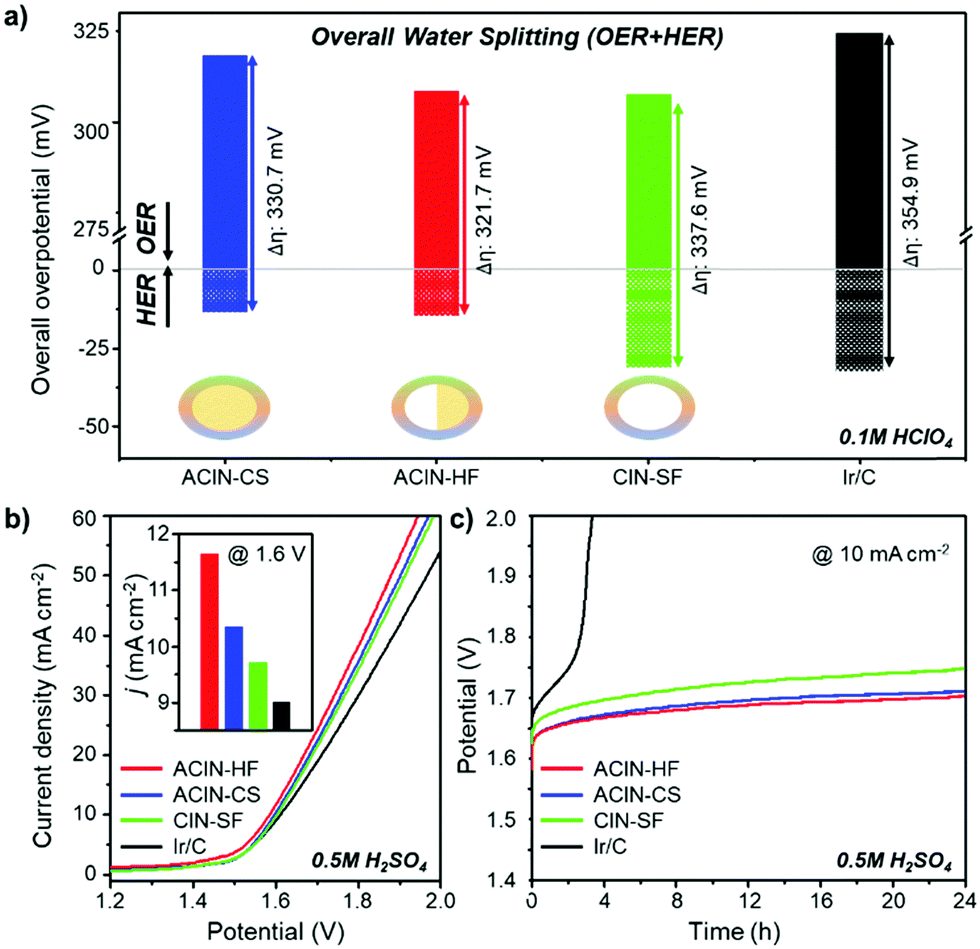 | ||
| Fig. 5 (a) Histogram of the overall overpotential diagram of the synthesized nanocrystals measured for the OER and HER in 0.1 M HClO4. Overall water splitting performance of synthesized nanocrystals. (b) Polarization curves of ACIN-HF/CFP, ACIN-CS/CFP, CIN-SF/CFP and Ir/CFP for overall water splitting reaction. (c) Chronopotentiometry test of the synthesized nanocrystals at 10 mA cm−2 in 0.5 M H2SO4 solution for overall water splitting reaction. | ||
In order to identify the surface composition and chemical state of the synthesized electrocatalysts, we performed an X-ray photoelectron spectroscopy (XPS) study (Fig. 6). The Ir 4f XPS peaks of ACIN-HF/C before the cycling test, after the HER durability test, and after the OER durability test are shown in Fig. 6a. The deconvoluted XPS analyses of these catalysts are described in Fig. S21 (ESI†) to quantify the oxidation states of Ir. The deconvolution peaks of Ir 4f XPS spectra of ACIN-HF/C indicate that the surface Ir atoms exist in a metallic state before and after the HER tests. On the other hand, a significant portion of Ir was transformed into higher valence Ir species after the OER test. Since this discrepancy of Ir oxidation state between after the OER and HER tests is caused by the conditions of the anode and cathode reactions; the catalytic performance of the OER has been driven from the IrO2 phase, which is known as the active phase for the OER, transformed from the Ir metal phase.55–57 The Ir 4f XPS peaks of the catalysts before electrocatalysis have similar oxidation states regardless of their different structural morphologies (Fig. 6b), and Ir exists mostly in a metallic Ir(0) state. This observation implies that the electronic states of Ir are only affected by chemical compositions of nanoparticles. We also carried out XPS analysis of ACIN-CS/C, ACIN-HF/C, CIN-SF/C, and Ir/C after durability tests. We found that all of the Ir 4f XPS peaks after the OER moved to higher binding energy compared to those of before the cycling tests, plausibly due to the oxidation during the electrocycling test (Fig. 6c). Furthermore, the overall oxidation states of Ir are dependent on the nanoparticle morphologies. Nanostructures with highly opened structural features such as frame and cage have a better chance to be oxidized than solid nanoparticles, which leads to more pronounced oxidation of Ir species and thus further enhanced OER activity. Therefore, CIN-SF/C and ACIN-HF/C catalysts exhibit enhanced electrocatalytic performance toward the OER compared to solid ACIN-CS/C because their highly opened inner surface area could act as active sites to be transformed to the IrOx phase. Although the ECSA of ACIN-HF/C is smaller than CIN-SF/C, the strain effect derived from the AuCu core might induce a lower value of overpotential toward the OER, resulting in similar electrocatalytic performance between ACIN-HF/C and CIN-SF/C. It was recently shown that the RuOx phase entrapping Pt nanoparticles and reduced Pt species exhibits excellent OER performance.6 Therefore, the IrOx surface with trapped AuCu nanoparticles of this study might be another case of a catalytically boosted oxide shell with a metallic hetero-core. Due to the extremely small size of Ir nanoparticles in commercial Ir/C, the overall Ir atoms in a commercial catalyst are converted from Ir to IrOx during the OER test, although they possess the solid nanoparticle feature.
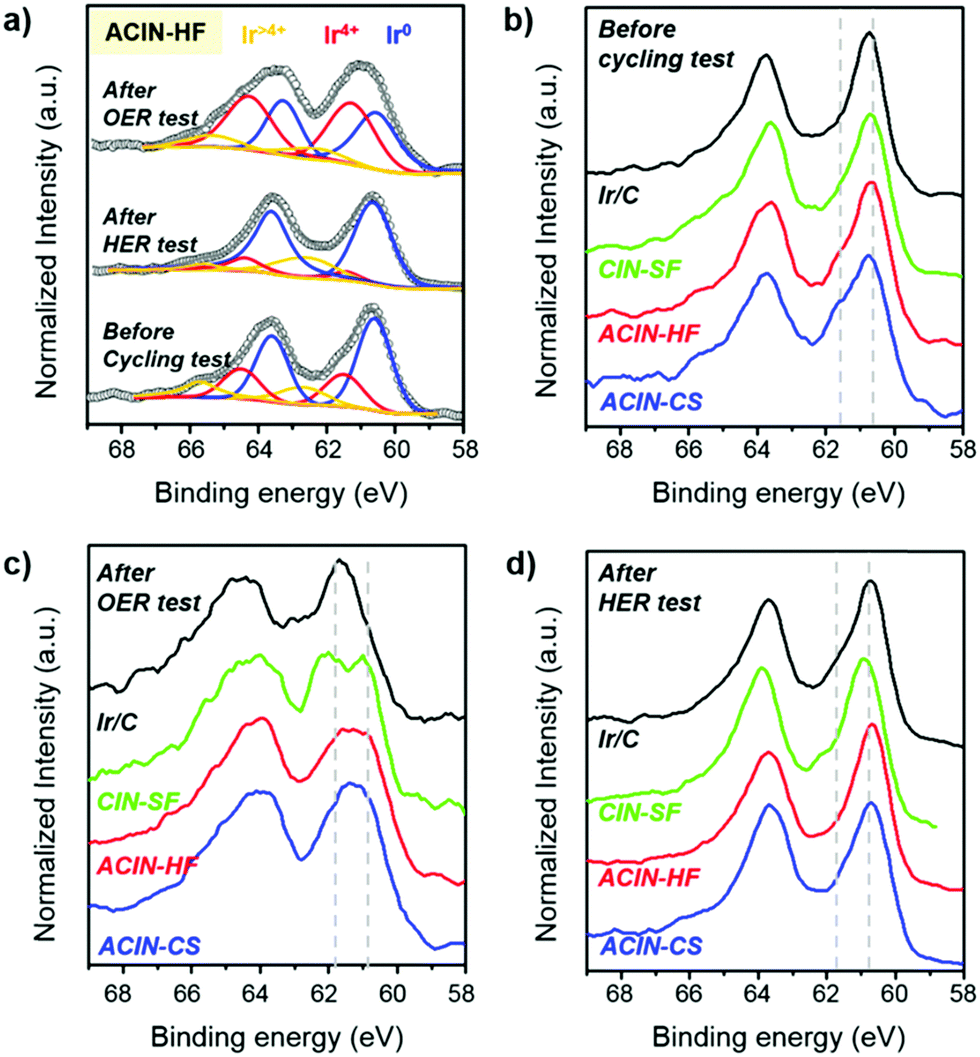 | ||
| Fig. 6 (a) Ir 4f XPS spectra of ACIN-HF/CFP catalysts before the durability test, and after the OER and HER durability tests. Ir 4f XPS spectra of ACIN-CS/CFP, ACIN-HF/CFP, CIN-SF/CFP, and Ir/CFP catalysts (b) before the durability test, (c) after the OER durability test, and (d) after the HER durability test. | ||
In contrast to the OER, the oxidation states of Ir are similar among all of the catalysts (Fig. 6d), having a zero valence state. Because of the reductive condition during the HER, the oxidation states of Ir are well maintained even after the durability test toward the HER. The XPS analysis indicates that the different electrocatalytic activities toward the HER might have originated from the structure-originated catalytic performance enhancement such as the core–shell effect. In order to clarify the role of AuCu (Au) seeds, we studied the Au 4f XPS analysis in Fig. S22 (ESI†). The shift of Au 4f XPS peaks of ACIN-CS/C and ACIN-HF/C is hardly discernible.64 In the case of Au-doped MoS2, the binding energy of Mo 3d peaks is negatively shifted and that of Au 4f is positively shifted compared to their pure chemical states, confirming the electron transfer from Au to MoS2. Therefore, the AuCu or Au core of our samples only decreased the surface areas of the catalysts without changing the electronic states of the catalyst surface. However, the AuCu or Au core can still generate the lattice mismatch between the IrNi shell and Au-based core, leading to the catalytic performance-boosting surface strain. The surface strain is expected to cause weakening of the surface-hydrogen binding strength, resulting in enhanced electrocatalytic performance toward the HER.65,66 Accordingly, ACIN-HF and ACIN-CS exhibit lower overpotential toward the HER although the oxidation states of Ir are similar to those of CIN-SF and Ir/C.
Conclusions
In summary, we have developed a synthetic route to an Ir-based hemi-core@frame nanostructure (ACIN-HF), which showed excellent electrocatalytic activity and stability toward the overall water splitting reaction in acidic media. ACIN-HF exhibits a very low cell voltage of 1.585 V of ACIN-HF for overall water splitting measured at the current density of 10 mA cm−2 in a 0.5 M H2SO4 electrolyte. The catalyst ACIN-HF possesses both structural features of a surface-area enhancing nanoframe and lattice-mismatch inducing core–shell, which contributed to the enhancement of catalytic activity toward the OER and HER, respectively. This study shows that combination of rather disparate nanostructural features can lead to unusual synergy in terms of both catalytic activity and stability. We expect that the synthetic strategy can be further extended to other important multicomponent alloy phases for the development of active and robust multifunctional electrocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by NRF-2017R1A2B3005682, NRF-2018R1A6A3A01013426, the Korea Basic Science Institute (KBSI) R&D program (Project No. C38530) supervised by the Ministry of Science, and Korea University Future Research Grant. The authors thank KBSI for the usage of their HRTEM instrument.References
- Y. Jin, H. Wang, J. Li, X. Yue, Y. Han, P. K. Shen and Y. Cui, Adv. Mater., 2016, 28, 3785 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- Y. Zhu, W. Zhou, Y. Zhong, Y. Bu, X. Chen, Q. Zhong, M. Liu and Z. Shao, Adv. Energy Mater., 2017, 7, 1602122 CrossRef.
- C. Ling, L. Shi, Y. Ouyang, X. C. Zeng and J. Wang, Nano Lett., 2017, 17, 5133–5139 CrossRef CAS PubMed.
- J. Joo, H. Jin, A. Oh, B. Kim, J. Lee, H. Baik, S. H. Joo and K. Lee, J. Mater. Chem. A, 2018, 6, 16130–16138 RSC.
- A. Oh, H. Y. Kim, H. Baik, B. Kim, N. K. Chaudhari, S. H. Joo and K. Lee, Adv. Mater., 2019, 31, 1805546 CrossRef PubMed.
- P. Kuang, B. Zhu, Y. Li, H. Liu, J. Yu and K. Fan, Nanoscale Horiz., 2018, 3, 317–326 RSC.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- A. Oh, H. Baik, D. S. Choi, J. Y. Cheon, B. Kim, H. Kim, S. J. Kwon, S. H. Joo, Y. Jung and K. Lee, ACS Nano, 2015, 9, 2856–2867 CrossRef CAS PubMed.
- Y. Wu, D. Wang, G. Zhou, R. Yu, C. Chen and Y. Li, J. Am. Chem. Soc., 2014, 136, 11594–11597 CrossRef CAS PubMed.
- Y. Wu, D. Wang, X. Chen, G. Zhou, R. Yu and Y. Li, J. Am. Chem. Soc., 2013, 135, 12220–12223 CrossRef CAS PubMed.
- J. Park, T. Kwon, J. Kim, H. Jin, H. Y. Kim, B. Kim, S. H. Joo and K. Lee, Chem. Soc. Rev., 2018, 47, 8173–8202 RSC.
- D. Yoon, B. Seo, J. Lee, K. S. Nam, B. Kim, S. Park, H. Baik, S. H. Joo and K. Lee, Energy Environ. Sci., 2016, 9, 850–856 RSC.
- J. Park, Y. J. Sa, H. Baik, T. Kwon, S. H. Joo and K. Lee, ACS Nano, 2017, 11, 5500–5509 CrossRef CAS PubMed.
- E. Antolini, ACS Catal., 2014, 4, 1426–1440 CrossRef CAS.
- J. Guan, D. Li, R. Si, S. Miao, F. Zhang and C. Li, ACS Catal., 2017, 7, 5983–5986 CrossRef CAS.
- H. G. S. Casalongue, M. L. Ng, S. Kaya, D. Friebel, H. Ogasawara and A. Nilsson, Angew. Chem., Int. Ed., 2014, 53, 7169–7172 CrossRef PubMed.
- D. Lebedev, M. Povia, K. Waltar, P. M. Abdala, I. E. Castelli, E. Fabbri, M. V. Blanco, A. Fedorov, C. Copéret, N. Marzari and T. J. Schmidt, Chem. Mater., 2017, 29, 5182–5191 CrossRef CAS.
- Y. Zhao, E. A. Hernandez-Pagan, N. M. Vargas-Barbosa, J. L. Dysart and T. E. Mallouk, J. Phys. Chem. Lett., 2011, 2, 402–406 CrossRef CAS.
- T. Kwon, H. Hwang, Y. J. Sa, J. Park, H. Baik, S. H. Joo and K. Lee, Adv. Funct. Mater., 2017, 27, 1604688 CrossRef.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- N. Cheng, S. Stambula, D. Wang, M. N. Banis, J. Liu, A. Riese, B. Xiao, R. Li, T.-K. Sham, L.-M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2016, 7, 13638 CrossRef CAS PubMed.
- Y. Shen, A. C. Lua, J. Xi and X. Qiu, ACS Appl. Mater. Interfaces, 2016, 8, 3464–3472 CrossRef CAS PubMed.
- Z.-J. Chen, G.-X. Cao, L.-Y. Gan, H. Dai, N. Xu, M.-J. Zang, H.-B. Dai, H. Wu and P. Wang, ACS Catal., 2018, 8, 8866–8872 CrossRef CAS.
- Y. Pi, Q. Shao, P. Wang, J. Guo and X. Huang, Adv. Funct. Mater., 2017, 27, 1700886 CrossRef.
- L. Fu, F. Yang, G. Cheng and W. Luo, Nanoscale, 2018, 10, 1892–1897 RSC.
- L. Fu, G. Cheng and W. Luo, J. Mater. Chem. A, 2017, 5, 24836–24841 RSC.
- J. Feng, F. Lv, W. Zhang, P. Li, K. Wang, C. Yang, B. Wang, Y. Yang, J. Zhou, F. Lin, G. C. Wang and S. Guo, Adv. Mater., 2017, 29, 1703798 CrossRef PubMed.
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J.-Y. Ma, D. Su and X. Huang, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed.
- Y.-C. Hsieh, Y. Zhang, D. Su, V. Volkov, R. Si, L. Wu, Y. Zhu, W. An, P. Liu, P. He, S. Ye, R. R. Adzic and J. X. Wang, Nat. Commun., 2013, 4, 2466 CrossRef PubMed.
- R. Chattot, T. Asset, P. Bordet, J. Drnec, L. Dubau and F. Maillard, ACS Catal., 2017, 7, 398–408 CrossRef CAS.
- C. Wang, X. Sang, J. T. L. Gamler, D. P. Chen, R. R. Unocic and S. E. Skrabalak, Nano Lett., 2017, 17, 5526–5532 CrossRef CAS PubMed.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- Y. Qin, M. Luo, Y. Sun, C. Li, B. Huang, Y. Yang, Y. Li, L. Wang and S. Guo, ACS Catal., 2018, 8, 5581–5590 CrossRef CAS.
- H. Kwon, M. K. Kabiraz, J. Park, A. Oh, H. Baik, S.-I. Choi and K. Lee, Nano Lett., 2018, 18, 2930–2936 CrossRef CAS PubMed.
- J. Park, M. K. Kabiraz, H. Kwon, S. Park, H. Baik, S.-I. Choi and K. Lee, ACS Nano, 2017, 11, 10844–10851 CrossRef CAS PubMed.
- J. L. Fenton, B. C. Steimle and R. E. Schaak, Science, 2018, 360, 513–517 CrossRef CAS PubMed.
- H. Hwang, T. Kwon, H. Y. Kim, J. Park, A. Oh, B. Kim, H. Baik, S. H. Joo and K. Lee, Small, 2018, 14, 1702353 CrossRef PubMed.
- J. M. Hodges and R. E. Schaak, Acc. Chem. Res., 2017, 50, 1433–1440 CrossRef CAS PubMed.
- W. Chen, R. Yu, L. Li, A. Wang, Q. Peng and Y. Li, Angew. Chem., Int. Ed., 2010, 49, 2917–2921 CrossRef CAS PubMed.
- H. Okamoto and T. B. Massalski, Bull. Alloy Phase Diagrams, 1984, 5, 381 Search PubMed.
- P. Nasa, Phase Diagrams of Binary Nickel Alloys, ASM International, 1991, p. 304 Search PubMed.
- A. V. Ruban, H. L. Skriver and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 15990 CrossRef.
- L.-L. Wang and D. D. Johnson, J. Am. Chem. Soc., 2009, 131, 14023–14029 CrossRef CAS PubMed.
- Z. Lin, J. Li, L. Li, L. Yu, W. Li and G. Yang, J. Mater. Chem. A, 2017, 5, 773–781 RSC.
- M. Wang, L. Wang, H. Li, W. Du, M. U. Khan, S. Zhao, C. Ma, Z. Li and J. Zeng, J. Am. Chem. Soc., 2015, 137, 14027–14030 CrossRef CAS PubMed.
- G. Guisbiers, S. Khanal, F. Ruiz-Zepeda, J. Roque de la Puente and M. Jose-Yacaman, Nanoscale, 2014, 6, 14630–14635 RSC.
- H. Jin, Y. Hong, J. Yoon, A. Oh, N. K. Chaudhari, H. Baik, S. H. Joo and K. Lee, Nano Energy, 2017, 42, 17–25 CrossRef CAS.
- Z. Gao, H. Ye, D. Tang, J. Tao, S. Habbi, A. Minerick, D. Tang and X. Xia, Nano Lett., 2017, 17, 5572–5579 CrossRef CAS PubMed.
- H. Liao, A. Fisher and Z. J. Xu, Small, 2015, 11, 3221 CrossRef CAS PubMed.
- N. M. Bedford, A. R. Showalter, T. J. Woehl, Z. E. Hughes, S. Lee, B. Reinhart, S. P. Ertem, E. B. Coughlin, Y. Ren, T. R. Walsh and B. A. Bunker, ACS Nano, 2016, 10, 8645–8659 CrossRef CAS PubMed.
- S. M. Alia, B. Rasimick, C. Ngo, K. C. Neyerlin, S. S. Kocha, S. Pylypenko, H. Xu and B. S. Pivovar, J. Electrochem. Soc., 2016, 163, F3105–F3112 CrossRef CAS.
- J. Wang, G. Yin, Y. Shao, S. Zhang, Z. Wang and Y. Gao, J. Power Sources, 2007, 171, 331–339 CrossRef CAS.
- J. Yang, S. Park, K. Y. Choi, H.-S. Park, Y.-G. Cho, H. Ko and H.-K. Song, ACS Sustainable Chem. Eng., 2018, 6, 9566–9571 CrossRef CAS.
- T. Reier, H. N. Nong, D. Teschner, R. Schlögl and P. Strasser, Adv. Energy Mater., 2017, 7, 1601275 CrossRef.
- E. Fabbri, A. Habereder, K. Waltar, R. Kotz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC.
- H. N. Nong, L. Gan, E. Willinger, D. Teschner and P. Strasser, Chem. Sci., 2014, 5, 2955–2963 RSC.
- M.-R. Gao, M. K. Y. Chan and Y. Sun, Nat. Commun., 2015, 6, 7493 CrossRef PubMed.
- J. Zhang, PEM Fuel Cell Electrocatalysts and Catalyst Layers: Fundamentals and Applications, Springer Science & Business Media, New York, 2008 Search PubMed.
- I. K. Mishra, H. Zhou, J. Sun, F. Qin, K. Dahal, J. Bao, S. Chen and Z. Ren, Energy Environ. Sci., 2018, 11, 2246–2252 RSC.
- J. Su, R. Ge, K. Jiang, Y. Dong, F. Hao, Z. Tian, G. Chen and L. Chen, Adv. Mater., 2018, 30, 1801351 CrossRef PubMed.
- H. Guo, N. Youliwasi, L. Zhao, Y. Chai and C. Liu, Appl. Surf. Sci., 2018, 435, 237–246 CrossRef CAS.
- T. Reier, Z. Pawolek, S. Cherevko, M. Bruns, T. Jones, D. Teschner, S. Selve, A. Bergmann, H. N. Nong, R. Schlögl, K. J. J. Mayrhofer and P. Strasser, J. Am. Chem. Soc., 2015, 137, 13031–13040 CrossRef CAS PubMed.
- Y. Shi, J. Wang, C. Wang, T.-T. Zhai, W.-J. Bao, J.-J. Xu, X.-H. Xia and H.-Y. Chen, J. Am. Chem. Soc., 2015, 137, 7365–7370 CrossRef CAS PubMed.
- K. Yan, S. K. Kim, A. Khorshidi, P. R. Guduru and A. A. Peterson, J. Phys. Chem. C, 2017, 121, 6177–6183 CrossRef CAS.
- J. Yang, X. Chen, X. Yang and J. Y. Ying, Energy Environ. Sci., 2012, 5, 8976–8981 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nh00520f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |