
Appearance of SERS activity in single silver nanoparticles by laser-induced reshaping†
Kamalesh
Chaudhari‡
a,
Tripti
Ahuja‡
a,
Vasanthanarayan
Murugesan
a,
Vidhya
Subramanian
a,
Mohd Azhardin
Ganayee
a,
Thomas
Thundat
 *b and
Thalappil
Pradeep
*a
*b and
Thalappil
Pradeep
*a
aDST Unit of NanoScience (DST UNS) and Thematic Unit of Excellence (TUE), Department of Chemistry, Indian Institute of Technology Madras, Chennai 600 036, India. E-mail: pradeep@iitm.ac.in; Tel: +91044 -2257-4208
bDepartment of Chemical and Materials Engineering, University of Alberta, Edmonton, Alberta T6G 2 V4, Canada. E-mail: thundat@ualberta.ca
First published on 16th November 2018
Abstract
We report simultaneous plasmonic scattering and Raman spectroscopic observations of single citrate capped silver nanoparticles (AgNPs) which exhibit surface enhanced Raman scattering (SERS) upon meeting specific conditions induced by laser (532 nm) exposure. We show that nanoparticles which are not initially SERS active become SERS active by laser-induced reshaping/reorientation. A set-up developed for these observations enabled in situ high speed time-lapse characterization using plasmonic and Raman spectroscopies in conjunction with dark-field microscopy (DFM). Changes in the AgNPs were confirmed by monitoring plasmonic scattering spectra and DFM images. Time-lapse observations have shown that laser-induced changes in the plasmonic properties of AgNPs resulted in the appearance of SERS. Spectral matching between plasmon resonance and downward molecular vibronic transitions for molecules adsorbed on the surface of plasmonic nanomaterials is attributed to the nanoparticle SERS. We have further shown that the release of silver ions by silver nanoparticles can be the probable reason for their plasmonic changes. Gold nanoparticles inert to such mild (850 μW, 532 nm) laser-induced changes do not exhibit the appearance of SERS.
Introduction
The development of new nanosensors with multiplexed properties requires spatial and temporal correlation.1–4 Such correlation is necessary for dual confirmation of sensing signals.5,6 Each signal provides specific information about the analyte and its interaction with the sensing element. Multiple signals from the same system can be used to assure the detection of the analyte with high specificity. For example, due to the recent advances in photothermal cantilever deflection spectroscopy, multiple properties of analytes (mass, electrical and optical properties) can be measured simultaneously.7,8 Other examples include electroluminescence or chemiresistance based sensors.9,10 Such sensors can provide electrical readout along with other physico-chemical parameters. At various stages in material characterization, it is necessary to find correlation between multiple properties of the material. For example, imaging of nanoparticles by diffraction-limited optical techniques cannot provide information about their actual size but on correlating the optical data with high resolution imaging techniques such as scanning electron microscopy,11 transmission electron microscopy12 or atomic force microscopy,13 structural information of the nanoparticles has been obtained. Such complementary techniques are helpful in correlating optical properties of nanomaterials with their size, shape and structural arrangements. Various integrated techniques and instruments have been developed for such applications in the past.1,2,14 In such applications, when it comes to visualizing plasmonic nanoparticles in native environments, spectroscopic observations are usually combined with DFM.1,2,6 It allows the observation of single nanoparticles with a high signal to noise ratio, in real-time, in a native environment where interactions between the particles and analyte molecules take place. Observations with scanning electron microscopy11 or transmission electron microscopy12 can provide high resolution images for correlation studies but these observations are possible only under high vacuum which is not suitable for many studies and particles do not remain in their native state. Considering the difficulties in the use of microcantilevers in liquid media, such experiments are tedious in terms of obtaining good resolution for every sample and it is difficult to couple the same with molecular identification techniques such as confocal Raman microspectroscopy (CRM). Hence a combination of DFM with CRM can be considered as an efficient combination for the observation of samples in real-time, and in native-environments with enhanced spatial and temporal resolution.1,2,4,6,15 Examples from the past include demonstrations by the El-Sayed group where they have shown applications of such combined Rayleigh and Raman scattering spectroscopy to monitor live cell division and concomitant molecular changes due to drug delivery to the cells.16–18 In the recent past, our group has demonstrated a combination of DFM and CRM for the localization of molecules in bacterial and mammalian cells with silver4 and gold6 nanoparticles. The set-ups reported in the aforementioned studies were able to do measurements only on fixed samples without single particle tracking and repositioning. For the kind of study reported in the present manuscript, we have modified the set-up such that it can be used for in situ time dependent plasmonic and Raman scattering spectroscopic measurements with particle tracking capabilities, on single nanoparticles.The effect of temperature and laser excitation on SERS has been studied in the past.19,20 In these studies, either laser heating or changes in the substrate temperature were used as control parameters which can affect SERS blinking in single nanoparticles. However, the real-time fate of nanoparticles and the corresponding nanoplasmonic property effect on SERS blinking were not studied. In our study, we have periodically exposed nanoparticles to a Raman excitation laser where plasmonic changes after each Raman spectrum collection were monitored in situ. In the present work, we focus on the appearance of laser-induced SERS activity in citrate capped AgNPs without the aid of aggregating agents. Briefly, we call this technique Particle Dynamics Induced-Plasmon Enhanced Raman Scattering (PDI-PERS).
Various routes have been explored to control the SERS activity of nanoparticles in the past.21–27 These routes are mainly based on controlling the plasmonic nanocavity14 by changing the gap between nanoparticles such as aggregation, using linker molecules of different lengths23etc. We report our observations on the possibility of appearance of SERS activity in single AgNPs by laser-induced changes that can help particles to reshape/reorient resulting in SERS favorable on-resonance situations. This supports previously reported steady state observations by Zhang et al. which have shown that whenever there is spectral matching between plasmon resonance and downward molecular vibronic transitions, such situations are favorable for SERS and are called on-resonance situations.14 To observe this effect, we have performed PDI-PERS measurements on immobilized single AgNPs. With the help of immobilization, the translational motion of particles was ceased but it allowed monitoring in situ shape and orientation related changes in the particles with the help of DF imaging and spectroscopic measurements. We have also demonstrated the release of silver ions28 as a probable reason for the appearance of SERS. Details of the data obtained are discussed in the next section.
Results and discussion
Set-up for PDI-PERS
A preliminary form of the set-up for combined plasmonic and Raman scattering microspectroscopy was reported previously by our group.6 In the previous set-up, plasmonic and Raman spectroscopic measurements were possible with a conventional optical microscopy camera and fixed samples, in the absence of particle tracking, which required manual positioning of the samples during measurements. Also, the absence of automated shutters made it difficult to reposition samples for repetitive measurements. In the set-up developed for the present study (schematic shown in Fig. 1A), we have incorporated a high resolution camera coupler with two automated shutters (SH1 & SH2). These shutters control the signal passing towards the camera and the CCD spectrometer. The intensity of light used to expose the sample is controlled using automated ND filter wheels (FW1 & FW2). Fig. 1B shows the UV-Vis spectrum, TEM image and DFM image of AgNPs used in this study. The AgNPs are polydisperse in size and shape which is also reflected in the DFM image. The existence of single nanoparticles immobilized on an ITO coated glass slide was verified by field emission scanning electron microscopy (FESEM) (Fig. S1†). Single particles as well as small aggregates composed of 2–3 nanoparticles were observed in such images. DFM images on a similar length scale show mostly well separated single nanoparticles, supporting their immobilization (Fig. S1†). Some of the high magnification FESEM images at different locations of the sample are also shown in Fig. S2† which clearly depicts different morphologies of immobilized nanoparticles such as spheres, rods and faceted spheres. The table in Fig. 1C shows the sequence of shutters and ND filter wheels along with their outcomes that were followed during the time-lapse measurements of the immobilized AgNPs. Briefly, the sample was first exposed to white light and a DF image was collected (Sr no. 1, Fig. 1C). This step was followed by position correction using a particle tracking software (if necessary) and then the scattering spectrum was collected (Sr no. 2, Fig. 1C). Then the camera shutter was closed to prevent exposure of CCD to high intensity laser light and the sample was exposed to the Raman excitation laser to collect the spectrum (Sr no. 3, Fig. 1C). After that, all the shutters and ND filter wheels were brought to a close position before opening the camera shutter for the next cycle of measurements (Sr no. 4, Fig. 1C). Repeating this sequence periodically exposes AgNPs under observation to the laser of ∼850 μW intensity. The particle of interest was brought to the confocal volume by manual positioning before starting the measurement sequence and particle tracking was turned on. We observed from initial measurements that, upon exposure to periodic laser pulses, the plasmonic scattering spectrum and the color of the particle in the DFM image exhibited changes. These changes were attributed to the laser-induced reshaping/reorientation of AgNPs. In the confocal set-up, the particle under observation alone was exposed to the laser. Hence, photoreduction and heating induced Ostwald ripening was seen only in the particle under observation. As the nanoparticles were linked to the substrate through mercaptopropyltrimethoxy silane, the linker allowed limited movement. Due to the lower power of the white light source in contrast to the laser, even if particles were exposed for long, they did not exhibit any change in color. These data along with the corresponding changes in the SERS activity of AgNPs are discussed in the next section.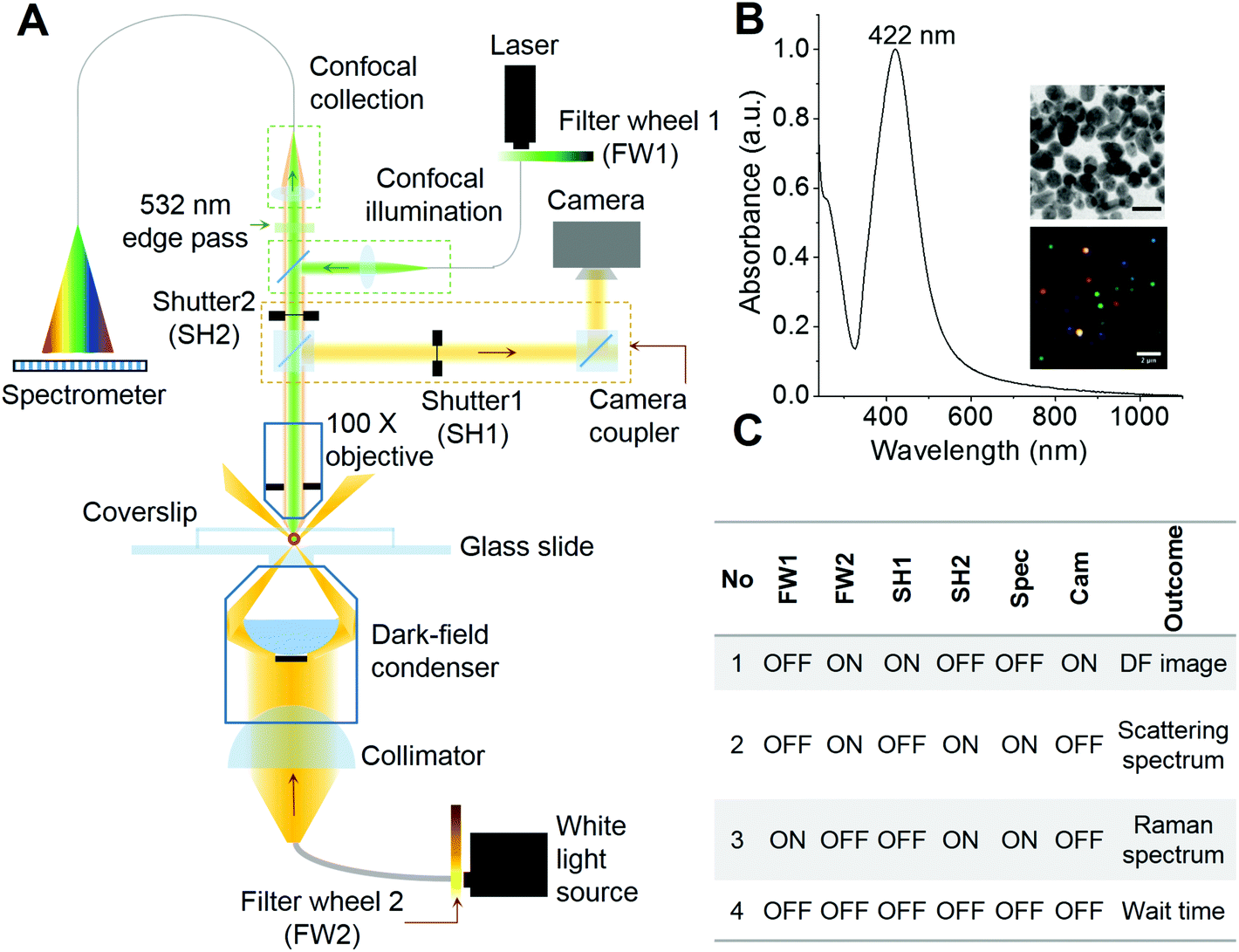 | ||
| Fig. 1 (A) Schematic of the set-up for PDI-PERS. Set-up also facilitates high resolution dark field imaging and real-time tracking of single nanoparticles for continuous monitoring of the same nanoparticle. Filter wheels (FW1 and FW2) and shutters (SH1 and SH2) control the intensity of light sources and exposure of spectrometer/camera, respectively. (B) UV-Vis spectrum of the AgNPs used in this study. Insets show TEM (scale bar – 20 nm, top) and hyperspectral dark field image (scale bar – 2 μm, bottom) of AgNPs. (C) Time-lapse sequence of neutral density (ND) filter wheels, shutters and the outcomes of such combinations. | ||
Observing changes in plasmonic scattering and SERS activity of AgNPs using PDI-PERS
In this section, we demonstrate the PDI-PERS set-up to account for the observed dynamic changes in the shape and size of single nanoparticles and correlate the same with SERS activity. We also discuss the importance and necessity of DFM and plasmonic scattering spectroscopic measurements over the other. We have used water as the medium in these experiments as the refractive index of water (1.33) is close to that of glass (1.5) and this helps the use of a high magnification oil immersion 100× objective for the same. But the use of water as a surrounding medium and an oil immersion objective for real-time observations limits the verification of changes in size and shape of AgNPs by other techniques such as AFM or SEM. Hence, we have discussed our observations of shape and size changes in AgNPs with DFM and plasmonic scattering spectra alone which have been performed in situ.Fig. 2 shows the temporal changes in the plasmonic scattering intensity (gray curve, integrated over DFM images) of a silver nanoparticle monitored with the aforementioned sequence of measurements (Fig. 1C). The scattering intensity of the particle decreases initially and then it increases gradually in two exponential steps. Previously reported simulations29 of plasmonic nanoparticles have shown that the red shift in the plasmon is mainly a function of the aspect ratio or plasmon length of a nanoparticle, whereas particles with a small aspect ratio exhibit plasmonic scattering in the green region. Along these lines, it has been also shown that particles with a larger volume exhibit a larger scattering intensity as compared to smaller particles. This suggests that in our observations, while there was red shift in the plasmon (time unit 100–200), the particle volume did not change as corroborated by a decrease in the plasmonic scattering but later when its volume started increasing, growth was mostly lateral which resulted in the blue shift and broadening of the plasmonic scattering spectrum along with an increase in the scattering intensity. Fig. S3† shows a montage of all the particle images captured during the observation. Changes in the scattering intensity and plasmon resonance can be seen clearly in this montage. It has been reported previously that changes in the plasmon resonance wavelength are due to changes in the plasmonic length (the length over which oscillations take place),30 and the aspect ratio of the nanostructure, whereas changes in the scattering intensity are mainly related to the volume of the nanoparticle.29 Such changes are highly dependent and directed by the nature of the ligand but in the context of this paper, we have not studied such details. The possible reason behind the exponential nature of changes is that the increase in the length and volume (lateral growth) of the particle can be complementary and once the changes in length saturate, then only changes in volume begin as indicated in the reported simulations.29,30 This suggests step by step changes in the shape of the particle upon laser exposure. Similar kinds of changes were observed when the plasmonic scattering intensity was integrated over a complete spectrum of particles (cyan curve, Fig. 2). A better correlation between DFM images and scattering spectral intensities can also be visualized in ESI Video S1.† This video shows that the blue shift observed in DFM images (in the time window of 120–200) is actually due to the NIR shift in the plasmonic scattering spectrum which was not observable in DFM due to lower quantum efficiency of the CCD camera in the NIR region as it responded well only to the blue part of the scattering spectrum. However, in prolonged PDI-PERS measurements, there can be minor changes in the position due to heating of the substrate and the particle may move away from the confocal volume. Hence, DFM of the particle is necessary to monitor and track the position of the particle to facilitate undisturbed and prolonged measurements. Other benefit of DFM based intensity monitoring is that the CCD device can capture images over large areas of the sample and other nanoparticles can be monitored as a control. The minor intensity variations due to occasional auto positioning create similar intensity variations in other particles which can be used for noise correction. Details on noise correction are explained in Fig. S4.† To conclude, DFM in conjunction with the information provided by scattering spectroscopy can be used to interpret smooth RGB channel-based changes resulting from the reshaping of the particle. Inset (i) of Fig. 2 shows a resolved SERS spectrum of the citrate ligand anchored on a AgNP. Assignments for the observed Raman bands are provided in Fig. S5† and corresponding Table S1.†
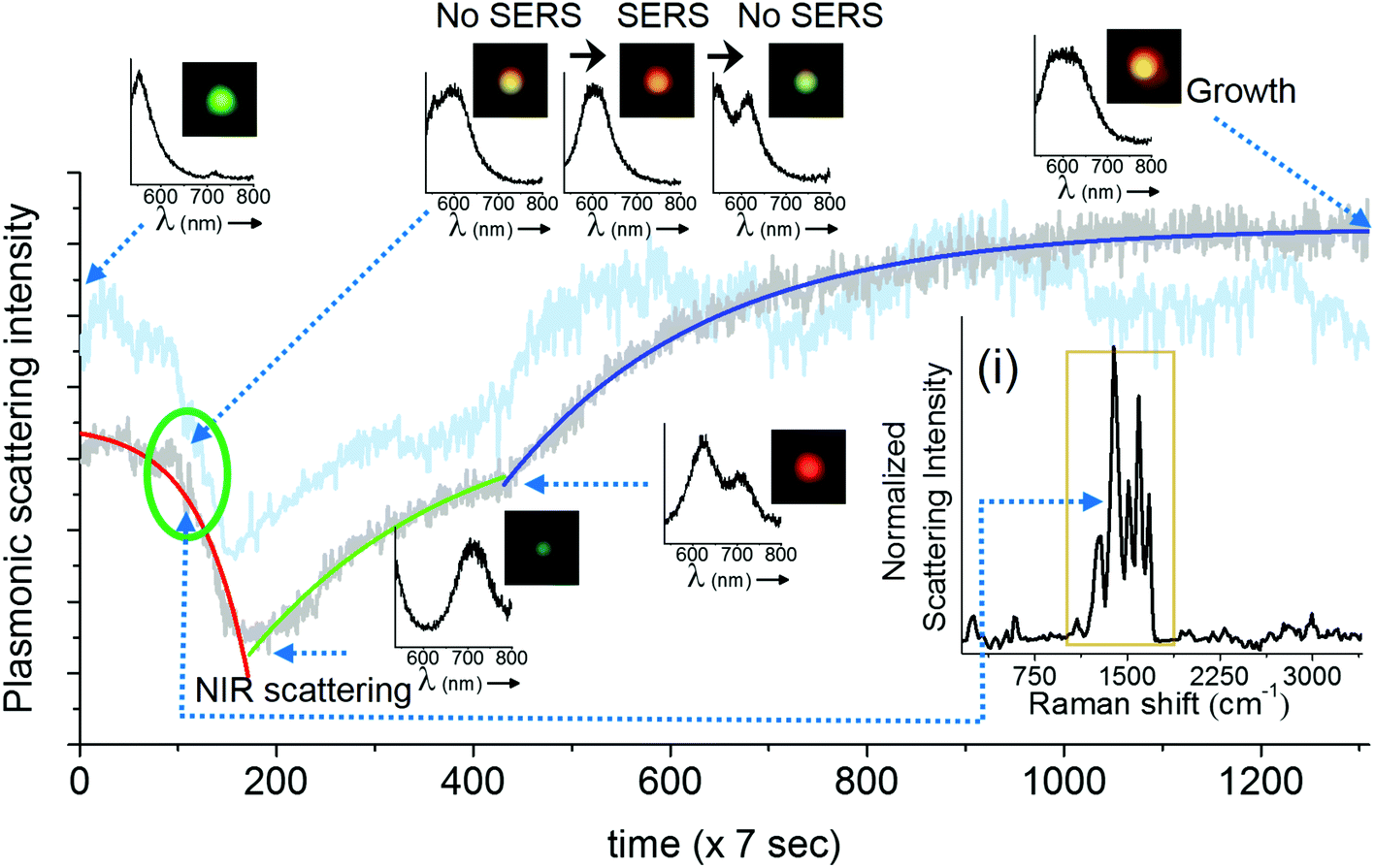 | ||
| Fig. 2 Variations in the plasmonic scattering intensity of a single isolated AgNP are plotted as a function of time (time-lapse interval – ×7 s). The gray curve shows intensity determined from DF images and the cyan curve shows the intensity integrated over temporal plasmonic spectra. The corresponding DF images and plasmonic scattering spectra of the particles are shown at the time points of interest. The green circle highlights the region where SERS activity of the particle was observed. Inset (i) shows the SERS spectrum of the citrate ligand on AgNPs, measured at the 108th time step. Beyond 900 seconds in the time axis, growth was saturated, thereafter no SERS was observed. | ||
Correlation between plasmonic scattering and SERS activity of AgNPs in time-lapse measurements
When the temporal intensity map of Raman spectra was correlated with the color map of plasmonic spectra (Fig. 3A), it was clear that the particle exhibited surface enhanced Raman activity whenever there was a red shift in the plasmon of AgNPs. The region of interest was expanded from the temporal plasmonic colormap shown in Fig. 3A.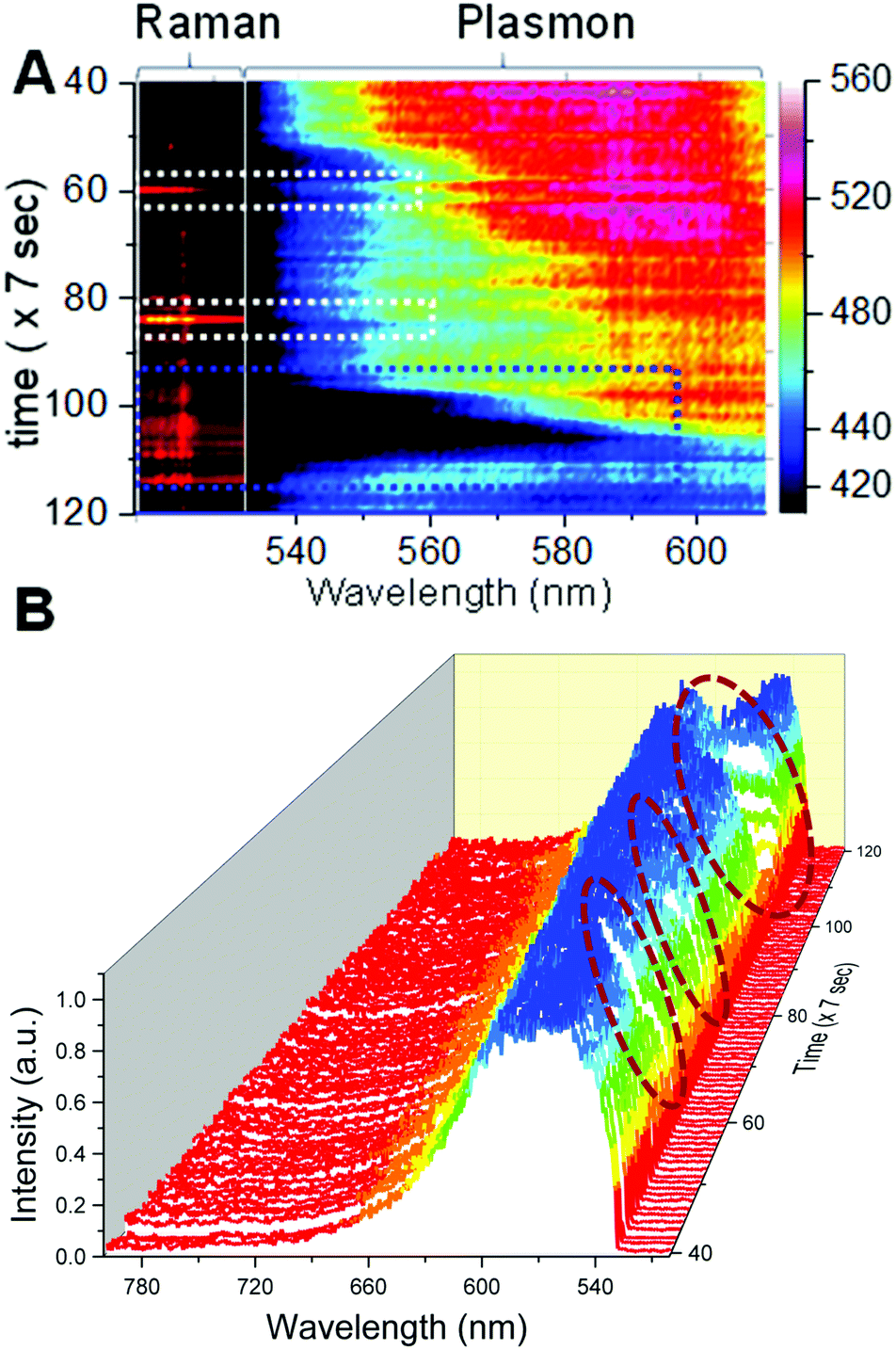 | ||
| Fig. 3 (A) A selected portion of intensity and color map showing correlated changes in the Raman and plasmonic scattering of AgNPs, respectively made with the help of Matlab algorithm. Dotted squares show that SERS activity was observed upon red shift in the plasmonic scattering of the particle. (B) A temporal plasmonic scattering waterfall plot of AgNPs shows the region (navy blue colored dotted box in (A) and wine colored circles in (B)) when SERS activity of the nanoparticle was observed. | ||
Dotted white and blue squares in the spectral maps show the temporal region when SERS was observed. Complete temporal maps of Raman and plasmonic spectra are shown in Fig. S6.† Correlated plasmonic and Raman scattering spectra corresponding to the time points of interest are provided in Fig. S8.† SERS activity is highly specific to the structure of the plasmonic particle and the position of the vibrating molecule with respect to the particle, hence all the red shifts in the plasmon do not give rise to the SERS spectrum but SERS occurs only when all the favorable conditions are met. Although in this case, only a weak correlation was observed, we conclude that laser-induced changes in plasmons resulted in spectral matching between plasmon resonance and molecular vibronic transitions14 responsible for maximum SERS activity observed at specific times. This phenomenon has been reported previously for plasmon enhanced Raman spectroscopy measurements performed on single molecules adsorbed on a silver substrate with spectrally matching and unmatching plasmons.14,31 Changes in the scattering spectra can be seen clearly from the temporal 3D waterfall plot (Fig. 3B) for the nanoparticle under observation, and a more prominent correlation between the red shift of the plasmon and SERS can be seen. This is shown by the wine colored circles in Fig. 3B. The RGB profile for the particle is shown in Fig. S7† which was calculated from the time dependent DFM images of the nanoparticle. However, this was not the case when the plasmon was shifted to the NIR region and it finally became broad with a span over both green and red regions, supporting the high specificity of SERS to the particle structure and the position of the analyte with respect to the hot spots in addition to the plasmonic scattering in a specific region. Single nanoparticles exhibited SERS only when an on-resonance condition is fulfilled in which (a) laser excitation and a high energy shoulder of sharp plasmon resonance match with the upward molecular vibronic transitions and (b) the plasmon resonance matches with the downward molecular vibronic transitions.14 The aforementioned condition was not fulfilled when there was broad plasmonic scattering and hence it led to an off-resonance situation. A major conclusion from these overall data is that although the nanoparticle did not exhibit SERS activity initially, upon laser-induced changes, reshaping/reorientation made it into a SERS-active state. Conclusions made from these data were verified by repeating similar measurements on multiple single silver nanoparticles. Results of the same are discussed in the next section.
Laser-induced appearance of the SERS activity in single AgNPs
Fig. 4 shows further measurements performed on single AgNPs. Fig. 4A, B, and C show a Raman intensity map, correlation between the integrated intensity profiles of the Raman intensity and plasmon colormap and a temporal plasmonic colormap (Raman – 1000–2000 cm−1, plasmon – 562–595 nm) for single AgNPs. Time-lapse Raman and plasmonic spectra corresponding to the Raman and plasmonic colormaps in Fig. 4A and C are shown in Fig. 4D and E respectively. The region of 1000–2000 cm−1 was selected because the most prominent vibrational features of citrate are in this region (refer to the inset of Fig. 2 and Table S1†). The corresponding region of plasmonic scattering is 562–595 nm. Data in Fig. 4B support the fact that SERS activity appeared whenever spectral matching between plasmon resonance and molecular vibronic transitions took place.14,31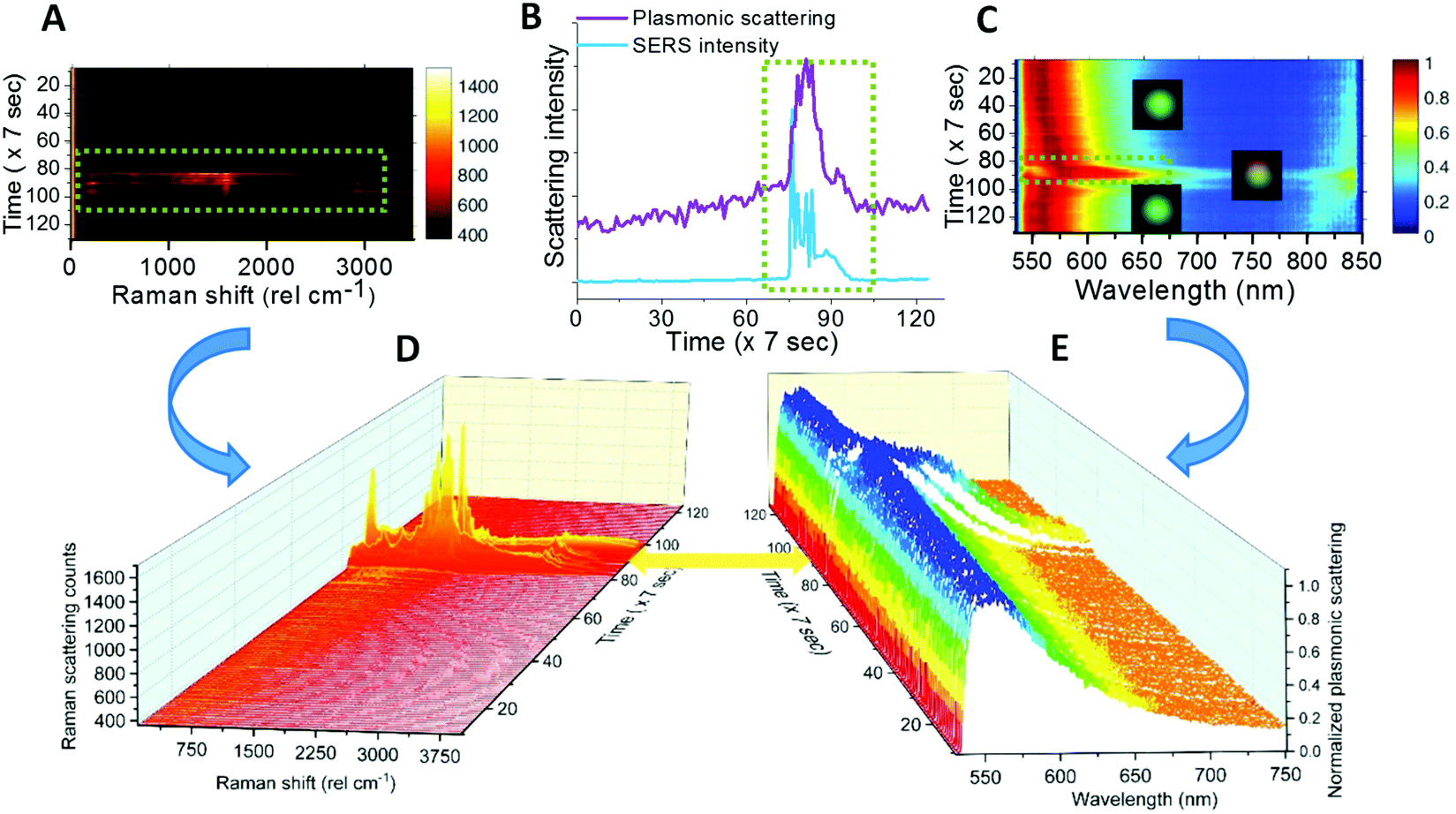 | ||
| Fig. 4 (A) Raman intensity map, (B) correlating integrated intensity profiles for Raman and plasmonic spectral match and (C) plasmonic scattering colormap and their corresponding time-lapse Raman and plasmonic scattering spectra (D&E) for single AgNPs. All the figures show that abrupt changes occurred during time 70 to 100 (×7 s), the whole spectra are presented in D & E. | ||
Reproducibility was checked on various single AgNPs and similar correlation between colormaps and spectra is shown for other particles in Fig. S9.†
The SERS favorable situations of spectral matching can be called on-resonance situations and others correspond to off-resonance.14 Plasmonic colormaps (Fig. 4C) justify that particles in the off-resonance situation can undergo laser-induced changes in structure and shape leading to on-resonance.
Temporal intensity profiles suggest that the on-resonance situation implies the possibility of SERS activity. As mentioned previously, fluctuations in the Raman intensity can be the outcome of high specificity of SERS to the position and orientations of molecules with respect to the hot spots on AgNPs. Despite the aforementioned fact, the appearance of SERS by laser-induced changes in single AgNPs is evident. It can be observed that due to intense periodic exposure to the laser, the particles keep reorienting and reshaping and because of this, the on-resonance situation cannot last for a long time. Hence, we have tried to control the intensity of the Raman excitation laser such that the SERS favorable on-resonance situation can be maintained for a longer period. These results are discussed in the next section.
Maintaining SERS activity for a longer time by controlling the laser intensity
Temporal maps of Raman intensity (Fig. 5A and C) and plasmonic scattering (Fig. 5B and D) for two single AgNPs are shown in Fig. 5. During these measurements, the particle was excited with a laser at 850 μW intensity initially. As soon as SERS activity was observed, the excitation laser intensity was reduced to 85 μW. White arrows (marked as SERS appeared) in Fig. 5A and C show the point in time when the laser intensity was reduced, and this can be seen by reduction in the intensity and width of the Rayleigh peak in the Raman spectrum. The corresponding changes in the plasmonic spectrum (black arrows, marked as Plasmon change) can be seen in the plasmonic color maps (Fig. 5B and D). In this way, we were able to slow down the changes in the plasmon position of AgNPs as well as orientation, so as to maintain the SERS activity for a longer time. However, this only slows down changes but does not stop them completely. Hence after sometime, upon changes in the plasmon (green arrows, marked as the plasmon change) back to the off-resonance situation, SERS activity of the AgNPs diminishes (yellow arrows, marked as SERS disappeared).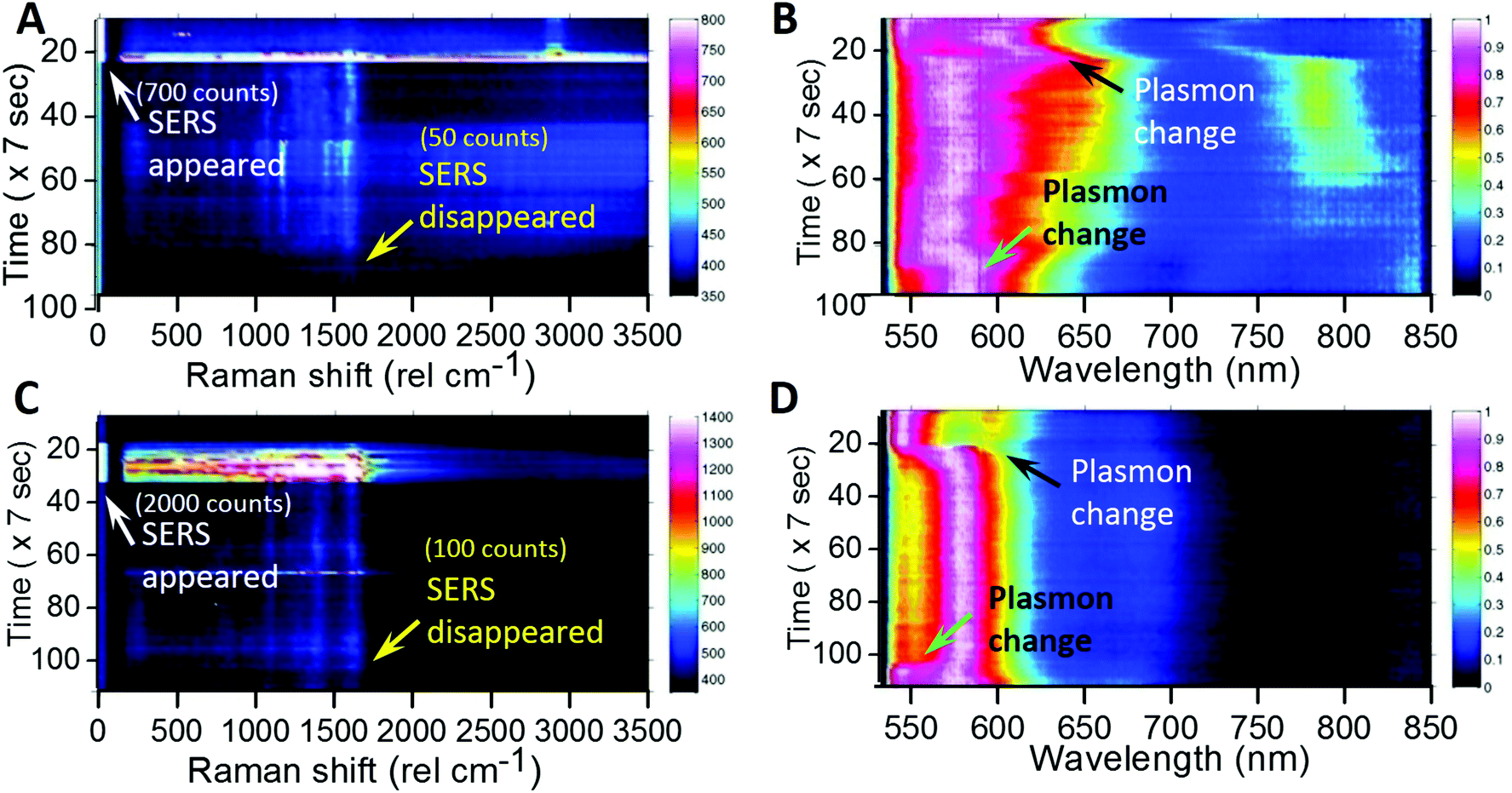 | ||
| Fig. 5 Raman intensity map (left) and plasmonic scattering colormap (right) for AgNP-3 (top, A, B) and AgNP-4 (bottom, C, D). White arrows show the point in time when laser intensity was decreased from 850 μW to 85 μW after the appearance of SERS. Yellow arrows show the point in time when SERS disappeared. The corresponding Raman intensity counts for the 1590 cm−1 peak are also shown within brackets. Black and green arrows show the corresponding points in time of plasmonic colormaps when SERS appeared and disappeared respectively. | ||
This proves the possibility of controlling SERS activity by slowing down the laser-induced changes in the SERS favorable on-resonance plasmon of AgNPs.
We have also performed such observations on single gold nanoparticles (Fig. S10†). For them, even after performing measurements for a long time, no laser-induced changes were observed due to better stability of AuNPs over AgNPs (Fig. S10†). Only constant luminescence instead of SERS was detected. In order to understand laser-induced changes, we have performed various control experiments by performing prolonged observations in the absence of laser exposure. In those cases, no such appearance of SERS was seen in AgNPs.
Role of silver ions released from AgNPs in the appearance of laser-induced SERS
In control observations, it was found that the growth and reshaping of AgNPs is assisted by the release of silver ions from nanoparticles in the surrounding region. To study this, time-lapse experiments were performed on the AgNPs washed extensively with DI water (four times at an interval of six hours (4 × 6 hours – 24 hours) after sample immobilization). These particles were referred to as washed particles. To prove the decrease in the release of silver ions with time and repeated washing steps, ICP-MS (inductively coupled plasma mass spectrometry) of the collected water was performed. These data are provided in Table S2.† The content of silver ions decreased after successive washing steps. Based on the ICP-MS data of release of silver ions, detailed calculations were performed corresponding to the number of atoms and nanoparticles formed and their effect on the increment of the volume of the nanoparticles. From calculations, it was found that the effective concentration of silver ions released (3.47 ppb) after 6 h is enough to form 2.5 × 1011 new nanoparticles of 50 nm diameter by photoreduction. The same quantity of ions is also enough to increase the diameter of 1.4 × 1011 nanoparticles to 70 nm from 50 nm. Therefore, an increase in the number and growth of nanoparticles is possible from the ions released. As photoreduction is limited to the confocal volume, we haven't observed an increase in the plasmonic particles elsewhere on the substrate.Washed AgNPs were almost inert to the laser-induced changes and no SERS was observed (Fig. 6A). A total of seventeen (twelve are shown here) particles were monitored and only one exhibited laser-induced SERS. The probability of appearance of SERS in washed AgNPs was as low as ∼6%. AgNPs which were washed once with DI water after immobilization exhibited good probability of the laser-induced SERS (∼44%). To differentiate from the washed AgNPs, these were referred to as unwashed AgNPs. The images of unwashed particles before and after laser exposure are shown in Fig. 6B which did not exhibit SERS. Fig. 6C shows images of unwashed particles before and after laser exposure which exhibited SERS. A total of thirty-six particles (twenty-four shown here) were monitored and fourteen exhibited laser-induced SERS. It can be seen from the images of washed and unwashed AgNPs before and after laser exposure that unwashed particles are more prone to changes in scattering as compared to washed particles. For unwashed particles, in some cases, drastic changes in the scattering images can be seen. Time dependent changes in the size and shape of nanoparticles can occur due to the Ostwald ripening process. Ostwald ripening of silver nanoparticles in a synthetic mixture has been reported in the past where excess silver ions assist a controlled growth of specific morphologies.32 In our observations, we have immobilized silver nanoparticles and no excess silver ions were added. Hence, Ostwald ripening is limited to the presence of silver ions released from the surrounding silver nanoparticles. To simplify these observations, changes in the red (R) and blue (B) channel scattering intensities relative to the green channel (R/G and B/G) were calculated and distributions of the percentage change in these quantities before and after laser exposure were plotted.
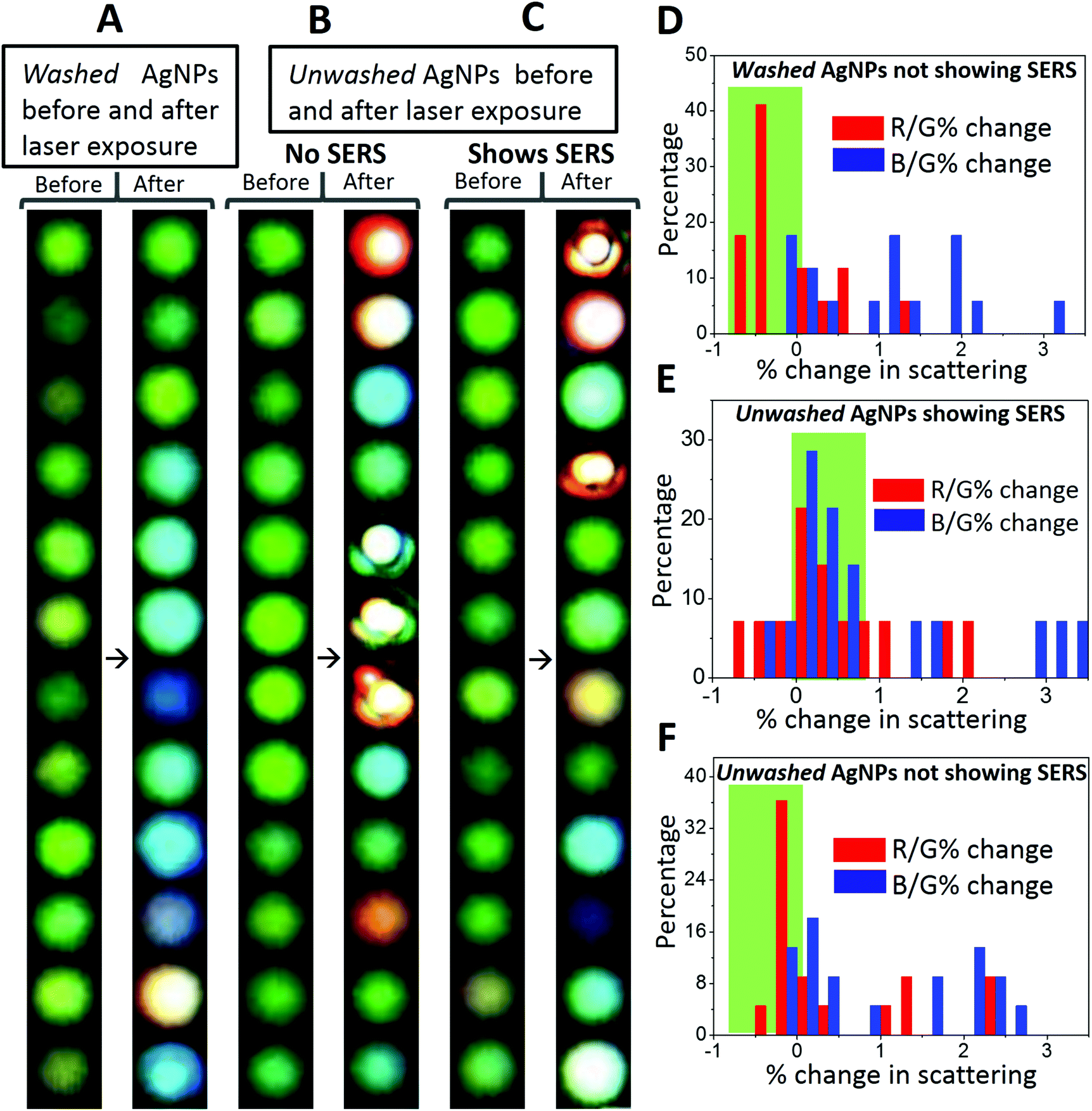 | ||
| Fig. 6 Stacks of single particle images for (A) washed AgNPs before and after laser exposure which did not exhibit SERS, (B) unwashed AgNPs before and after laser exposure which did not exhibit SERS and (C) unwashed AgNPs before and after laser exposure which exhibited SERS. Changes in the red (R) and blue (B) channel scattering intensities relative to the green channel (R/G and B/G) were calculated. Distributions of the percentage change in the aforementioned relative scattering intensities before and after laser exposure are plotted for AgNPs, (D) washed AgNPs, (E) unwashed AgNPs showing SERS, and (F) unwashed AgNPs not showing SERS. | ||
Details of the calculations are provided in the end of the ESI.† Direct changes in the RGB intensity were not a good parameter to group particles because RGB intensities vary for individual particles. These percentage changes are shown in Fig. 6D, E and F. For the AgNPs which do not exhibit SERS, %R/G change is mostly negative, whereas for the AgNPs which exhibit SERS, %R/G change is mostly positive. Negative and positive % changes are shaded in green color for proper identification. This suggests that laser-induced appearance of SERS is assisted by silver release. We conclude our study with certain observations as below.
Conclusions
We have developed a set-up which enables multispectral, high speed (8–10 s) time-lapse characterization of single nanoparticles by plasmonic and Raman scattering spectroscopy. Observations using this set-up have shown that AgNPs which are initially in the off-resonance situation can change into the on-resonance situation upon exposure to a Raman excitation laser. We note that effects of temperature and laser excitation on SERS blinking have been studied in the past.19,20 The real-time fate of nanoparticles and consequence of nanoplasmonic properties on SERS blinking are our contributions to this literature. We have also shown that once favorable conditions (like reshaping/reorientation) are obtained, the on-resonance situation can be maintained for longer times by lowering the intensity of the excitation laser from 850 μW to 85 μW. Supporting experiments28 have shown that such changes in AgNPs are assisted by the release of silver ions from the surrounding nanoparticles whereas gold nanoparticles are completely inert to such changes due to their relatively less reactivity. We have also performed experiments to correlate the release of silver ions from AgNPs with the probability of the appearance of SERS. It was found that washed AgNPs exhibited ∼6% SERS probability while unwashed AgNPs exhibited ∼44%. We have simplified these observations with the help of a schematic shown in Fig. 7.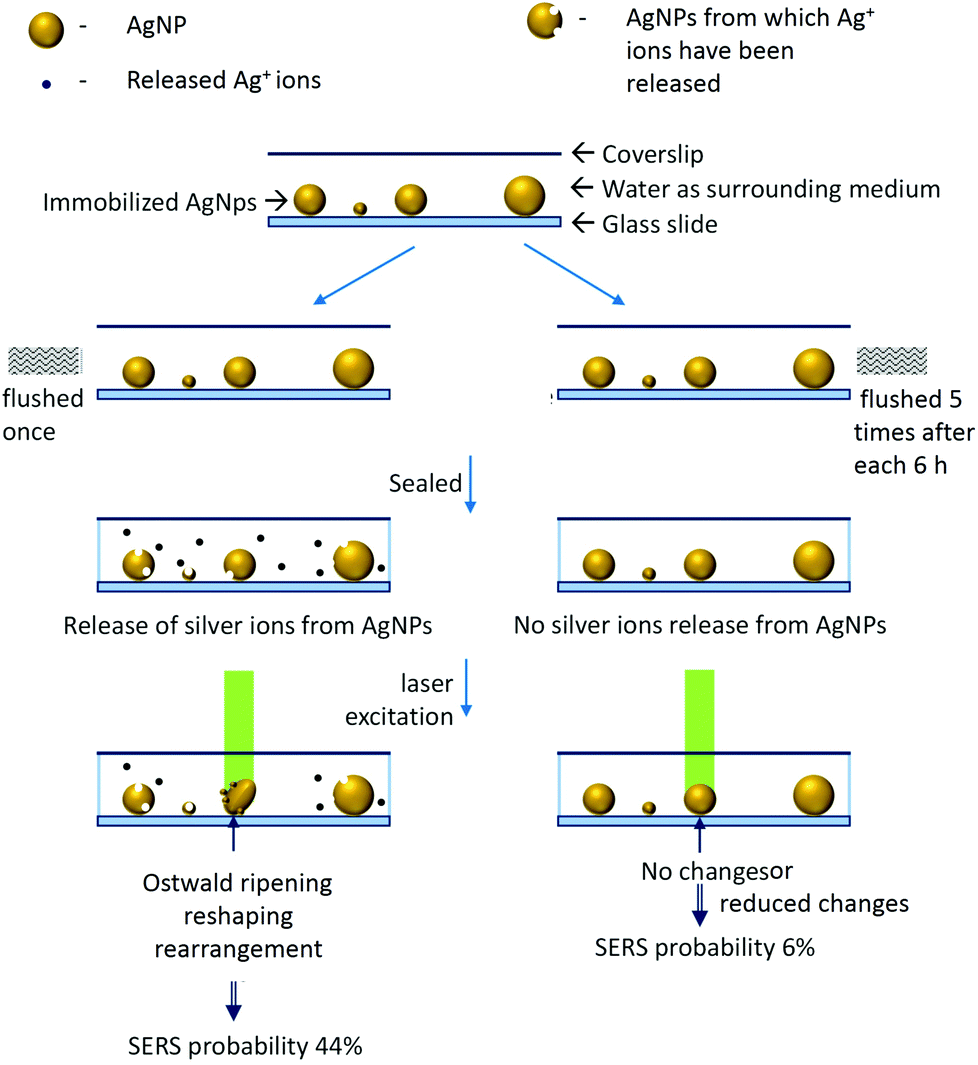 | ||
| Fig. 7 Schematic to explain the effect of laser irradiation and silver ion release from AgNPs on the appearance of SERS. | ||
This study opens up the possibility of multispectral observations of intermolecular, interparticle reactions in real-time at the single-particle/single-molecule level. In the presented study, although measurements with water as a surrounding medium open up various possibilities for investigating biological samples, the limitation was that one needs to rely on dark-field spectroscopy for inference on the morphology of nanoparticles and direct correlation with FESEM was not possible. Such observations will be able to provide new insights into the behavior of molecules on the surface of SERS active nanoparticles. We speculate that many temporal changes with SERS active nanoparticles may involve intermittent states of molecules/particles which may be revealed by studies using the presented strategy.
Experimental
Materials
Tetrachloroauric acid trihydrate (HAuCl4·3H2O) (99.9%) was made and purified in lab using pure gold purchased from the local market. Trisodium citrate (>99%) was purchased from Qualigens. 3-(Mercaptopropyl)trimethoxysilane was purchased from Sigma Aldrich. Silver nitrate (99.9%) was purchased from RANKEM, India. Deionized water (DI) (∼18.2 MΩ) obtained from Millipore was used throughout the experiments.Synthesis of citrate capped silver nanoparticles
The synthesis of AgNPs was done by the Turkevich method.33 Briefly, AgNO3 (17 mg in 100 mL DI water) solution was heated to boil and then trisodium citrate (40 mg) was added. Once the color of the solution changed to pale yellow, it was quenched under tap water. The synthesis of AgNPs was confirmed by UV-Vis spectroscopy and transmission electron microscopy (TEM) imaging. A strong localized surface plasmon resonance (LSPR) peak was observed at ∼422 nm in the UV-Vis spectrum (Fig. 1B). Synthesized AgNPs were polydisperse in size and their shape was studied by TEM.Synthesis of citrate capped gold nanoparticles
The synthesis of AuNPs was done by the Turkevich method.33 Briefly, 100 mL of HAuCl4 (1.25 mM in DI water) solution was heated to boil in a round bottom flask and stirred at 400 rpm. Upon boiling, 2 mL trisodium citrate (1%) solution was added. Gradually, the color of the solution changed to wine red. Then the solution was cooled at room temperature. The synthesis of AuNPs was confirmed by UV-Vis spectroscopy and TEM imaging (Fig. S11†). A strong LSPR peak was observed at ∼535 nm in the UV-Vis spectrum. Synthesized AuNPs were almost uniform with a diameter of ∼25–35 nm as observed under TEM.UV-Visible extinction spectroscopy
UV-Visible spectroscopic measurements were performed using a PerkinElmer Lambda 25 spectrophotometer in the range of 200–1100 nm.Transmission electron microscopy
Transmission electron microscopic (TEM) measurements were performed using a JEOL 3010, 300 kV instrument. Samples were spotted on carbon coated copper grids by drop casting followed by drying in ambient air.Field emission scanning electron microscopy (FESEM)
FESEM measurements were performed using an Inspect F50 from FEI, with 1.2 nm resolution at 30 kV in SE (Fig S1A†). Later, FESEM experiments were done using a Tescan-Mira 3 LMH instrument (Fig S2†). Samples were prepared by immobilizing AgNPs on ITO coated glass slides.Particle dynamics induced-plasmon enhanced Raman scattering (PDI-PERS)
The PDI-PERS set-up was built by integrating three separate instruments namely a Cytoviva™ high resolution dark field condenser, WiTec GmbH confocal Raman microscope and Dage Excel M cooled CCD camera. The core part of the integration was a coupler comprised of two automatic shutters and two automated ND filter wheels (Thorlabs) as shown in the schematic (Fig. 1A). These automated shutters and ND filter wheels were controlled using a home built controller and LABVIEW software. Particle tracking abilities were included in the LABVIEW software to take care of changes in the position of the sample, if any during measurements. Samples were mounted on a piezo stage for real-time positioning using a tracking software. The piezo stage can correct the position of the particle with ∼27 nm resolution. A frequency doubled Nd:YAG diode laser (532 nm) was used for Raman excitation of the sample. An L1090 halogen lamp from International Light Technologies Inc., USA was used for white light illumination (400–1000 nm) required for DFM. Samples were focused using a 100× oil immersion objective (UPLFLN, Olympus). The scattering signal after passing through a 535 nm edge pass filter was dispersed using a grating (150 grooves per mm) onto a 1024 pixel charge coupled device (CCD) spectrometer.Immobilization of nanoparticles
Immobilization of AgNPs and AuNPs on a glass slide was done by the pinpoint immobilization method.3 Briefly, an ultrasonically cleaned 1 mm thick glass slide (SCHOTT) was flushed with a 0.2 μM, 5 mL solution of (3-mercaptopropyl)trimethoxysilane (in ethanol). Then it was washed extensively with DI water followed by dropping of 10 μL (10 fold diluted) solution of nanoparticles over it and covering it with a clean glass coverslip. After incubation for 30 minutes, the coverslip was removed and the glass slide was washed extensively with DI water to remove unbound nanoparticles. Then 10 μL DI water was dropped on the immobilized region and covered with a 0.145 mm thick Nexterion® clean room cleaned coverslip. It was sealed with nail polish on its sides to avoid drying of the samples. In the overall procedure, care was taken to expose only one side of the slide to chemicals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Department of Science and Technology, Government of India for constantly supporting our research program on nanomaterials. We thank the University of Alberta and the Canada Excellence Research Chairs (CERC) for supporting our research program.Notes and references
- X. Shi, H. W. Li, Y. L. Ying, C. Liu, L. Zhang and Y. T. Long, Chem. Commun., 2016, 52, 1044–1047 RSC.
- M. Aioub and M. A. El-Sayed, J. Am. Chem. Soc., 2016, 138, 1258–1264 CrossRef CAS PubMed.
- K. Chaudhari and T. Pradeep, Sci. Rep., 2014, 4, 27–29 Search PubMed.
- S. Vishnupriya, K. Chaudhari, R. Jagannathan and T. Pradeep, Part. Part. Syst. Charact., 2013, 30, 1056–1062 CrossRef CAS.
- V. Subramanian, S. Jena, D. Ghosh, M. Jash, A. Baksi, D. Ray and T. Pradeep, ACS Omega, 2017, 2, 7576–7583 CrossRef CAS PubMed.
- K. Chaudhari and T. Pradeep, J. Biomed. Opt., 2015, 20, 046011 CrossRef PubMed.
- K. Prashanthi, A. Phani and T. Thundat, Nano Lett., 2015, 15, 5658–5663 CrossRef CAS PubMed.
- D. Lee, S. Kim and T. Thundat, Nano-Bio Sensing, Imaging, Spectrosc., 2013, 8879, 88790Q Search PubMed.
- C. Larson, B. Peele, S. Li, S. Robinson, M. Totaro, L. Beccai, B. Mazzolai and R. Shepherd, Science, 2016, 351, 1071–1074 CrossRef CAS PubMed.
- F. J. Ibañez and F. P. Zamborini, Small, 2012, 8, 174–202 CrossRef PubMed.
- M. Hu, C. Novo, A. Funston, H. Wang, H. Staleva, S. Zou, P. Mulvaney, Y. Xia and G. V. Hartland, J. Mater. Chem., 2008, 18, 1949 RSC.
- C. Rosman, S. Pierrat, A. Henkel, M. Tarantola, D. Schneider, E. Sunnick, A. Janshoff and C. Sönnichsen, Small, 2012, 8, 3683–3690 CrossRef CAS PubMed.
- Y. Song, P. D. Nallathamby, T. Huang, H. E. Elsayed-Ali and X.-H. N. Xu, J. Phys. Chem. C, 2010, 114, 74–81 CrossRef CAS PubMed.
- R. Zhang, Y. Zhang, Z. C. Dong, S. Jiang, C. Zhang, L. G. Chen, L. Zhang, Y. Liao, J. Aizpurua, Y. Luo, J. L. Yang and J. G. Hou, Nature, 2013, 498, 82–86 CrossRef CAS PubMed.
- K. Ock, W. Il Jeon, E. O. Ganbold, M. Kim, J. Park, J. H. Seo, K. Cho, S. W. Joo and S. Y. Lee, Anal. Chem., 2012, 84, 2172–2178 CrossRef CAS PubMed.
- M. R. K. Ali, Y. Wu, T. Han, X. Zang, H. Xiao, Y. Tang, R. Wu, F. M. Fernández and M. A. El-Sayed, J. Am. Chem. Soc., 2016, 138, 15434–15442 CrossRef CAS PubMed.
- B. Kang, L. A. Austin and M. A. El-Sayed, ACS Nano, 2014, 8, 4883–4892 CrossRef CAS PubMed.
- B. Kang, L. A. Austin and M. A. El-Sayed, Nano Lett., 2012, 12, 5369–5375 CrossRef CAS PubMed.
- S. R. Emory, R. A. Jensen, T. Wenda, M. Han and S. Nie, Faraday Discuss., 2006, 132, 249–259 RSC.
- Y. Maruyama, M. Ishikawa and M. Futamata, J. Phys. Chem. B, 2004, 108, 673–678 CrossRef CAS.
- M. F. Cardinal, E. Vander Ende, R. A. Hackler, M. O. McAnally, P. C. Stair, G. C. Schatz and R. P. Van Duyne, Chem. Soc. Rev., 2017, 46, 3886–3903 RSC.
- H. Ko, S. Singamaneni and V. V. Tsukruk, Small, 2008, 4, 1576–1599 CrossRef CAS PubMed.
- T. S. Sreeprasad and T. Pradeep, Langmuir, 2011, 27, 3381–3390 CrossRef CAS PubMed.
- N. H. Kim, S. J. Lee and M. Moskovits, Adv. Mater., 2011, 23, 4152–4156 CrossRef CAS PubMed.
- X. Yu, H. Cai, W. Zhang, X. Li, N. Pan, Y. Luo, X. Wang and J. G. Hou, ACS Nano, 2011, 5, 952–958 CrossRef CAS PubMed.
- N. M. B. Perney, F. J. García De Abajo, J. J. Baumberg, A. Tang, M. C. Netti, M. D. B. Charlton and M. E. Zoorob, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 1–5 CrossRef.
- V. K. Rao and T. P. Radhakrishnan, ACS Appl. Mater. Interfaces, 2015, 7, 12767–12773 CrossRef CAS PubMed.
- G. Lei, P. F. Gao, T. Yang, J. Zhou, H. Z. Zhang, S. S. Sun, M. X. Gao and C. Z. Huang, ACS Nano, 2017, 11, 2085–2093 CrossRef CAS PubMed.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–7248 CrossRef CAS PubMed.
- E. Ringe, M. R. Langille, K. Sohn, J. Zhang, J. Huang, C. A. Mirkin, R. P. Van Duyne and L. D. Marks, J. Phys. Chem. Lett., 2012, 3, 1479–1483 CrossRef CAS PubMed.
- W. Zhu and K. B. Crozier, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- X. Wu, P. L. Redmond, H. Liu, Y. Chen, M. Steigerwald and L. Brus, J. Am. Chem. Soc., 2008, 130, 9500–9506 CrossRef CAS PubMed.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: FESEM and HSI images of AgNPs for spacing correlation, DFM montage of time-lapse images, intensity profiles for noise correction, citrate SERS band assignment, temporal and plasmonic colormaps for AuNPs with their characterization by UV-Vis and HRTEM, ICP-MS data for Ag ion quantification and time-lapse video showing plasmonic and Raman variations at the single particle level. See DOI: 10.1039/c8nr06497k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |