
Aromatic donor–acceptor interaction promoted catalyst assemblies for hydrolytic kinetic resolution of epichlorohydrin†
Daniel R.
Blechschmidt
,
Matthew D.
Woodhouse
,
Sebastien
Inagaki
,
Melita
Whitfield
,
Ayokunnumi
Ogunsanya
,
Aaron
Yoder
,
Daniel
Lilly
,
Eric W.
Heim
 ,
Luke N.
Soucie
,
Jian
Liang
* and
Yu
Liu
*
,
Luke N.
Soucie
,
Jian
Liang
* and
Yu
Liu
*
Department of Chemistry, Northern Michigan University, 1401 Presque Isle Ave., Marquette, MI 49855, USA. E-mail: jliang@nmu.edu; liuyu@nmu.edu
First published on 3rd December 2018
Abstract
Three generations of Co(III)–salen complexes containing electron-deficient aromatic moieties (acceptors) have been synthesized. When electron-rich aromatic compounds (donors) were introduced, these complexes were designed to form catalyst assemblies through aromatic donor–acceptor interaction. For all three generations of complexes, the addition of a proper donor led to higher catalytic efficiency in the hydrolytic kinetic resolution (HKR) of epichlorohydrin. The reaction rates are in the following order: Generation 3 > Generation 2 > Generation 1. The aromatic donor–acceptor interaction was verified by NMR spectroscopy and UV-vis absorption spectroscopy studies. These results demonstrated that aromatic donor–acceptor interaction can be a valuable driving force in the assembly of supramolecular catalysts.
Introduction
Supramolecular catalysts, inspired by enzymes, refer to multicomponent assemblies that are stabilized by noncovalent interactions to achieve catalytic systems with high selectivity and catalytic efficiency.1 Noncovalent interactions applied in the assembly of supramolecular catalysts2 include metal–ligand complexation,3 hydrogen bonding,4 cation–anion attraction,5 ion-dipole interaction6 and solvophobic effect.7 In contrast, the aromatic interaction,8 although ubiquitous in nature, plays a minor role in the construction of supramolecular catalysts.9 The main limitation of the aromatic interaction is the lack of the precise control of spatial arrangement and orientation. The aromatic interaction can have edge-to-face, offset-stacked or face-to-face geometry10 making it challenging to design structurally well-defined supramolecular catalysts. However, the aromatic donor–acceptor interaction (charge-transfer interaction),11 one special aromatic interaction, aroused our interest. It has an enhanced affinity resulting from the electrostatic attraction between electron-deficient and electron-rich aromatic molecules. The aromatic donor–acceptor interaction also provides only a face-centered geometry.10a,12 In addition, it tolerates a wide range of solvents including cyclohexane,13 CH2Cl2,14 CHCl3,15 CH3CN,16 CH3OH17 and H2O.17 This type of interaction has been utilized extensively in a variety of supramolecular assemblies11b,18 including molecular tweezers,19 catenanes/rotaxanes,20 organogels,21 foldamers,22 vesicle,23 liquid crystal24 and ion channels.25 The aromatic donor–acceptor interaction was also applied in the construction of supramolecular bidentate Rh complexes26 and Cu complexes,27 which catalyzed asymmetric hydrogenation and Diels–Alder reaction, respectively. Here, we report the assembly of Co(III)–salen dinuclear catalysts based on the aromatic donor–acceptor interaction and their application in the hydrolytic kinetic resolution (HKR) of epoxides (Fig. 1). Since H2O was one of the reagents, HKR served as an ideal reaction to examine the compatibility of the aromatic donor–acceptor interaction with H2O in the assembly of supramolecular catalysts.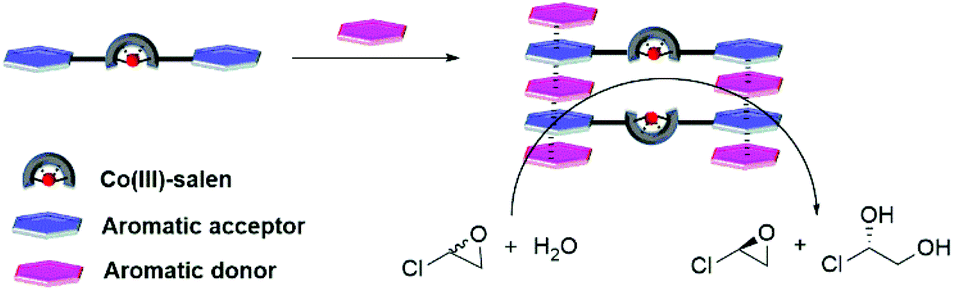 | ||
| Fig. 1 Bis-acceptor functionalized Co(III)–salen complexes assemble into bimetallic catalyst by introducing donor compounds and catalyse hydrolytic kinetic resolution of epichlorohydrin. | ||
Co(III)-Salen complex catalyzed HKR of epoxides follows a bimetallic mechanism, in which one Co(III)–salen activates the epoxide as a Lewis acid and another Co(III)–salen binds to hydroxide.28 To promote the cooperativity of Co(III)–salen complexes, many efforts were made to covalently attach Co(III)–salen onto a variety of supports including oligomers,29 polymers,30 dendrimers,31 silica,30a resins32 and zeolites.33 Hong and co-workers reported a noncovalent approach. The Co(III)–salen core was flanked by a urea pair. The dinuclear Co(III)–salen supramolecular catalysts, assembled through hydrogen-bonding of the urea pairs, achieved a rate acceleration in the HKR of epoxides.34 Our approach was to develop the bis-acceptor functionalized Co(III)–salen complex. The addition of a proper donor compound was expected to enhance cooperativity of the two Co(III)–salen complexes by bringing them in close proximity via the aromatic donor–acceptor interaction, and consequently accelerates the rate of the HKR of epoxides. X-ray crystal structures12a,b and computation study35 have showed that the total spacing between acceptor–donor–acceptor is about 7–8 Å, which falls in the operating distance of Co(III)–salen cores for the bimetallic pathway based HKR of epoxides,36 indicating two catalytic cores in our sandwich design should reach the desired distance for HKR. We chose aromatic diimides as the acceptor unit attached to the Co(III)–salen core, because the synthesis of the asymmetric diimide from the corresponding dianhydride has been well developed.37 In this work, three generations of bis-diimide functionalized Co(III)–salen complexes were developed (Chart 1). Commercially available polycyclic aromatic hydrocarbon based donor compounds were used as the additives.
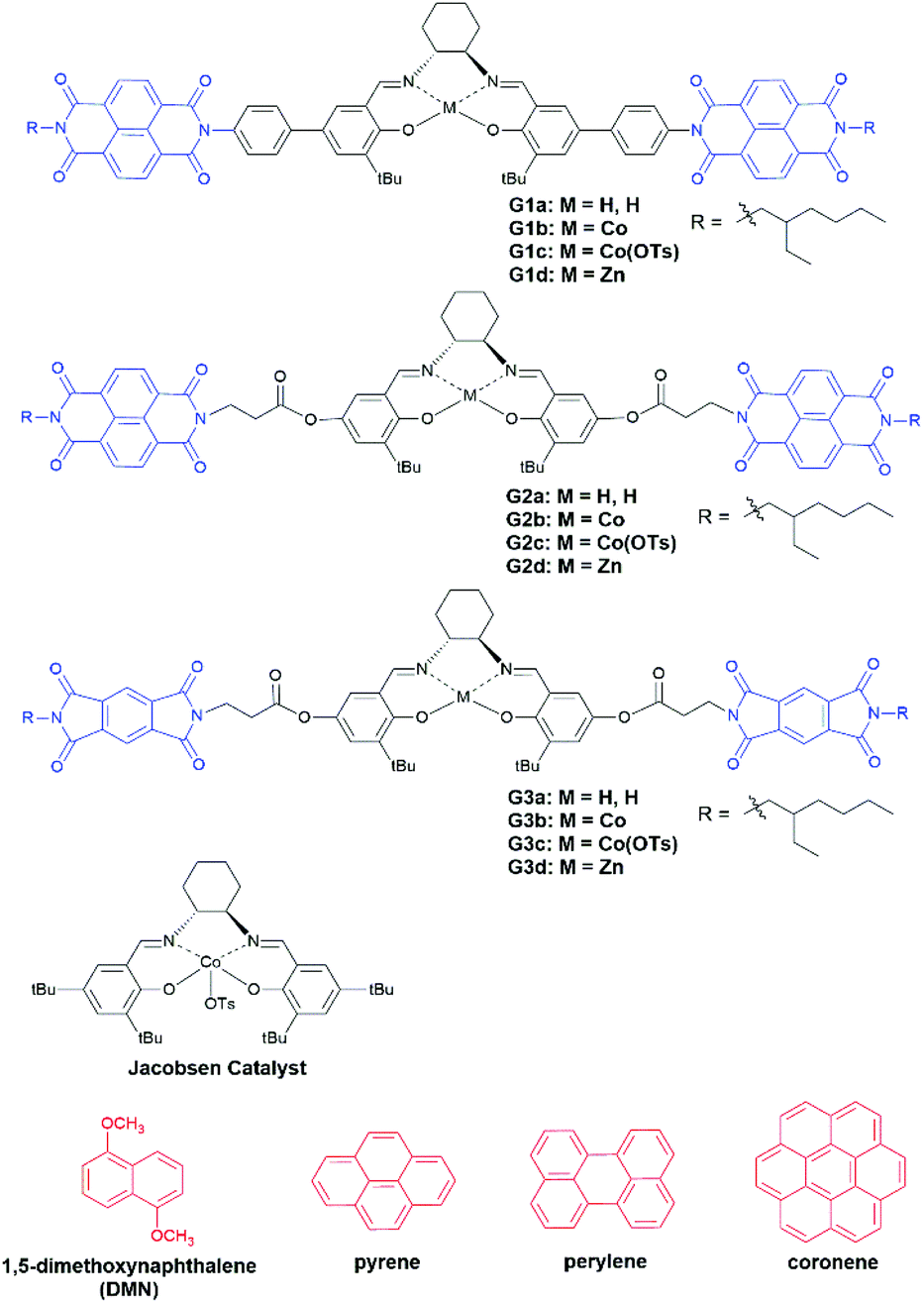 | ||
| Chart 1 Salen ligands, complexes and aromatic donor compounds used in this study. | ||
Results and discussion
Wilson and co-workers revealed electron-rich pyrene and electron-deficient 1,4,5,8-naphthalenediimide (NDI) had a highly congruent orbital interaction and were the best donor–acceptor pair based on systematic binding studies on nine combinations of donor and acceptor compounds.38 Hence, Generation 1 Co(III)–salen complex (G1c) was designed and synthesized with a pair of NDI as the aromatic acceptor units, which connect to the central Co(III)–salen core via a rigid phenyl linker. Pyrene was first tested as the donor compound. The major challenge in the complex preparation was the oxidation of Co(II)–salen precursor (G1b) to the active Co(III)–salen complex (G1c) in a p-toluenesulfonic acid solution. The Co(II)–salen precursor suffered from poor solubility. We tested different solvents and solvent combinations. A THF/acetonitrile (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture gave the best result, keeping the complex soluble during the oxidation. A CLEAR reddish green solution39 indicated the complete oxidation to the Co(III)–salen complex, which is crucial for a successful HKR of epoxides! After removal of the solvent by azeotropic drying with acetonitrile and high vacuum drying, the final catalyst was obtained as a dark green solid.
:1) mixture gave the best result, keeping the complex soluble during the oxidation. A CLEAR reddish green solution39 indicated the complete oxidation to the Co(III)–salen complex, which is crucial for a successful HKR of epoxides! After removal of the solvent by azeotropic drying with acetonitrile and high vacuum drying, the final catalyst was obtained as a dark green solid.
The catalyst assembly and HKR of G1c were evaluated with epichlorohydrin as the model epoxide. We screened an array of reaction conditions by comparison of conversions and enantiomeric excesses (ee) (Table 1). HKR of epichlorohydrin was first examined under solvent-free conditions28a,40 with 3.5 equivalents of pyrene to G1c. After 42.5 hours, the catalytic reaction with pyrene showed a higher ee value than the control one without pyrene (Table 1, entries 1 and 2). However, as the reaction proceeded, a cloudy suspension gradually appeared, resulting in a gelatinous precipitation in the solution. There was no further increase of ee values, even by extending the reaction time. We suspected that the formation of diol product had an impact on the solubility of the Co(III)–salen catalyst. In order to keep G1c soluble during the reaction process, two solvents were tested: THF, a typical solvent for HKR,34,41 and CHCl3, utilized in the study of the aromatic donor–acceptor interaction.15 THF led to a slight drop of the ee value (Table 1, entry 3), while CHCl3 afforded a better result: the HKR with pyrene was complete in 42.5 hours (Table 1, entry 6). In addition, G1c with pyrene outperformed the control without pyrene again in CHCl3 (Table 1, entry 4, Fig. 2) suggesting a positive effect of pyrene on the catalyst activity of G1c. To further investigate the effect of pyrene, we also tested different pyrene loading. One equivalent of pyrene (Table 1, entry 5) afforded a very similar result as the control without pyrene. The increase of pyrene amount led to the decline of G1c activity with 89% ee for 10 equivalents of pyrene and 78% ee for 30 equivalents of pyrene (Table 1, entries 7 and 8), suggesting an inhibition of the bimetallic catalyst formation.42 Other aromatic donor compounds were also examined including 1,5-dimethoxynaphthalene (DMN), perylene and coronene (Table 1, entries 9, 10 and 11). Perylene gave a slightly lower ee value and conversion than pyrene, followed by the further decreasing reactivity by coronene. DMN afforded the lowest activity for G1c. Jacobsen catalyst was also tested under the G1c optimized reaction condition as a comparison. The addition of pyrene had a negative impact on the catalyst activity of Jacobsen catalyst (Table 1, entries 12 and 13, Fig. 2). The results also showed that the G1c-pyrene catalytic system did not exhibit an advantage over Jacobsen catalyst in both the reaction rate and enantioselectivity. One possible reason was the rigid phenyl linker between NDI and the Co(III)–salen core in G1c. It has been reported that the supported metal–salen catalyst with a flexible linker had higher catalytic efficiency than the one with a rigid linker.32a,43 To improve the catalyst activity and enantioselectivity, we designed and synthesized the second generation of bis-NDI functionalized Co(III)–salen complex (G2c). It is featured with a flexible and non-conjugated alkyl ester link (Chart 1).
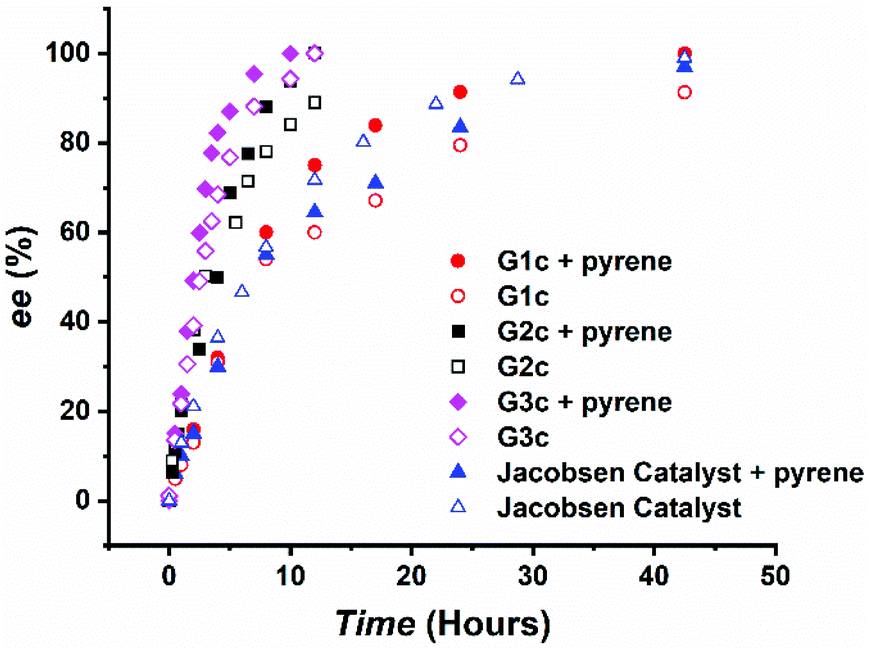 | ||
| Fig. 2 Kinetic plot of the HKR of epichlorohydrin by G1c, G2c, G3c and Jacobsen Catalyst (0.05 mol% catalyst loading; CHCl3 as the solvent; pyrene in 3.5 equivalents to catalysts if applied). | ||
a
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Aromatic donor compounds | Donor compounds: G1c (equiv.) | Solvents | Conversionsb (%) | eec (%) |
| a Reactions were carried at room temperature for 42.5 hours with 0.05% (mol) G1c loading. b Conversions were calculated based on chlorobenzene, the internal standard. The isolate yield of epichlorohydrin is listed in paranthesis. c ee were determined by chiral GC analysis (Chiraldex γ-TA). The 17-hour ee are listed in parentheses. | ||||||
| 1 | G1c | None | 0 | None | 45 | 75 |
| 2 | G1c | Pyrene | 3.5 | None | 50 | 91 |
| 3 | G1c | Pyrene | 3.5 | THF | 54 | 88 |
| 4 | G1c | None | 0 | CHCl3 | 49 | 91 |
| 5 | G1c | Pyrene | 1 | CHCl3 | 49 | 90 |
| 6 | G1c | Pyrene | 3.5 | CHCl3 | 54 (44) | >99 |
| 7 | G1c | Pyrene | 10 | CHCl3 | 49 | 89 |
| 8 | G1c | Pyrene | 30 | CHCl3 | 46 | 78 |
| 9 | G1c | DMN | 3.5 | CHCl3 | 48 | 89 (68) |
| 10 | G1c | Perylene | 3.5 | CHCl3 | 51 | 97 (79) |
| 11 | G1c | Coronene | 3.5 | CHCl3 | 50 | 94 (74) |
| 12 | Jacobsen catalyst | None | 0 | CHCl3 | 53 | 99 |
| 13 | Jacobsen catalyst | Pyrene | 3.5 | CHCl3 | 52 | 97 |
The optimized G1c reaction condition (3.5 equivalents of pyrene, CHCl3 as the solvent) was applied in the HKR of epichlorohydrin by G2c. G2c demonstrated a remarkable rate acceleration (Table 2, entry 2) compared to G1c. The reaction was complete in 12 hours (>99% ee). G2c-pyrene also surpasses the control, G2c without pyrene, in the reaction rate and ee values (Table 2, entry 1, Fig. 2). Hence, similar to G1c, the addition of pyrene promoted G2c activity and selectivity. We then tested stoichiometric influence of pyrene on G2c (Table 2, entries 3 and 4). Different from G1c, 10 equivalents of pyrene afforded a slightly higher ee value in 10 hours, but further increasing to 30 equivalents led to a lower ee value. Donor compound tests of G2c (Table 2, entries 5, 6 and 7) also showed different results from those of G1c. G2c-DMN could accomplish the HKR in 10 hours, faster than G2c-pyrene. In 10 hours, perylene and coronene gave 91% ee and 83% ee, respectively. It is interesting to note that for G2c the decrease of enantioselectivity (ee values) is in accordance with the increase of aromatic conjugation size of donor compounds.
a
|
|
||||
|---|---|---|---|---|
| Entry | Aromatic donor compounds | Donor: G2c (eq.) | Conv.b (%) | eec (%) |
| a Reactions were carried at in CHCl3 room temperature for 10 hours with 0.05% (mol) G2c loading. b Conversions were calculated based on chlorobenzene, the internal standard. The isolate yield of epichlorohydrin is listed in paranthesis. c ee were determined by chiral GC analysis (Chiraldex γ-TA). | ||||
| 1 | None | 0 | 46 | 84 |
| 2 | Pyrene | 3.5 | 52 (42) | 95 |
| 3 | Pyrene | 10 | 53 | 97 |
| 4 | Pyrene | 30 | 49 | 88 |
| 5 | DMN | 3.5 | 56 | >99 |
| 6 | Perylene | 3.5 | 48 | 91 |
| 7 | Coronene | 3.5 | 44 | 83 |
To further enhance the cooperativity between Co(III)–salen catalytic cores, we incorporated another common aromatic acceptor compound, pyromellitic diimide (PDI),12b,44 into the complex. We designed and synthesized the third generation of bis-imide functionalized Co(III)–salen complex (G3c), in which PDI replaced NDI as the acceptor units and the alkyl ester still acted as the linker (Chart 1). Under the same reaction condition as G1c and G2c (3.5 equiv. of pyrene, CHCl3), G3c exhibited a further reaction rate enhancement of the HKR of epichlorohydrin. The reaction was completed (>99% ee) in 10 hours (Table 3, entry 2, Fig. 2). In the same reaction time, the control without pyrene reached 94% ee (Table 3, entry 1). Once again, these results indicated the positive effect of pyrene on catalyst activity and selectivity. The increase of pyrene loading to 10 equivalents did not show much impact on the HKR outcome, but the further increase to 30 equivalents of pyrene led to a significant loss of the G3c activity (Table 3, entries 3 and 4). In the end, the tests with other donor compounds showed that perylene afforded a similar HKR result as pyrene (Table 3, entry 6). However, when DMN and coronene acted as the donor, there were marked drops of ee values and conversions (Table 3, entries 5 and 7), which were even lower than the control without any donor compound.
|
|
||||
|---|---|---|---|---|
| Entry | Aromatic donor compounds | Donor: G3c (eq.) | Conv.b (%) | eec (%) |
| a Reactions were carried at in CHCl3 room temperature for 10 hours with 0.05% (mol) G3c loading. b Conversions were calculated based on chlorobenzene, the internal standard. The isolate yield of epichlorohydrin is listed in paranthesis. c ee were determined by chiral GC analysis (Chiraldex γ-TA). | ||||
| 1 | None | 0 | 51 | 94 |
| 2 | Pyrene | 3.5 | 52 (43) | >99 |
| 3 | Pyrene | 10 | 52 | 98 |
| 4 | Pyrene | 30 | 46 | 78 |
| 5 | DMN | 3.5 | 48 | 80 |
| 6 | Perylene | 3.5 | 54 | >99 |
| 7 | Coronene | 3.5 | 49 | 87 |
The HKR results of G1c, G2c and G3c showed the influence of the aromatic donor–acceptor interaction on reaction rates and selectivity. When a proper donor compound, such as pyrene, was introduced in a proper amount, higher catalytic efficiency was obtained for all three complexes, suggesting that the aromatic donor–acceptor interaction could promote the cooperativity between Co(III)–salen cores. Under the same reaction conditions (3.5 equiv. of pyrene, CHCl3), the HKR rates were in the following order: G3c > G2c > G1c. To better understand the catalytic property differences of all three complexes and to further verify the role of aromatic donor–acceptor interactions in the supramolecular catalyst assembly, we did a series of UV-vis spectroscopy titration and NMR spectroscopy titration experiments. Because of its excellent solubility, pyrene was used in all spectroscopy studies.
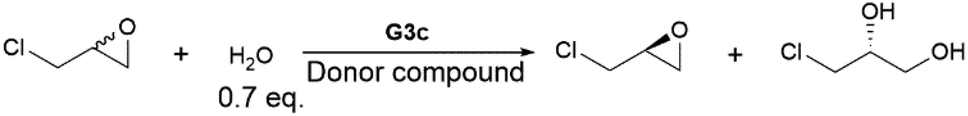
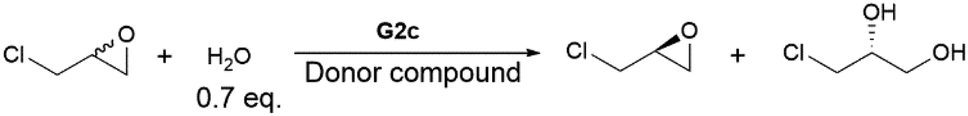
The aromatic donor–acceptor interaction can be readily detected by the appearance of a charge-transfer band.11b However, the Co(II)–salen absorption bands overlap with the charge-transfer band, so the corresponding Zn(II)–salen complexes (G1d, G2d and G3d) were used in all UV-vis absorption studies. The binding properties were investigated by titration of Zn(II)–salen complex solutions (10 mM) with varying amounts of pyrene in chloroform. Fig. 3a and b show the evolution of broad absorption bands with a maximum at 505 nm for G1d and 510 nm for G2d, upon addition of pyrene, characteristic of the charge-transfer transition. The appearance of a charge-transfer absorption band also renders solutions of G1d-pyrene and G2d-pyrene red in color, distinct from the light yellow of G1d and G2d initial solutions. However, G3d-pyrene displayed different UV-vis curves with the same titration process. No clear charge-transfer band was observed, which was also demonstrated by almost no color change after the addition of pyrene (Fig. 3c). These results suggested the aromatic donor–acceptor interaction between pyrene and the NDI units of G1d and G2d. However, the UV-vis titration study did not produce any evidence of the interaction between pyrene and G3d. It has been reported that the charge transfer band of PDI-pyrene was observed at 430 nm,45 so the CT band of G3d-pyrene could overlap with the absorption of other aromatic components. To investigate the binding stoichiometry between Co(III)–salen complexes and pyrene, Job plot experiments were performed for G1d and G2d. Interestingly, for both complexes the highest absorption was reached at approximately a 1:1 ratio (Fig. 3d and e). Since each salen complex has two acceptor units, it is less likely that 1 + 1 is the real stoichiometry between salen complex and pyrene. Instead, a sandwiched 2 + 2 assembly might be the possible binding form.46 Another possible explanation was that in the solution, salen complexes and pyrene may form multiple binding species, such as 1 + 1, 1 + 2, 2 + 1, 2 + 2 and even aggregations. These Job plots could illustrate a combination effect.47 We also attempted to extract association constants from UV-vis titration data by non-linear curve fitting.47b However, the derived association constants were either far away from the reasonable range of the association constants of the aromatic donor–acceptor interaction11b or had significant errors.
 | ||
| Fig. 3 UV-vis titration spectra of G1d, G2d and G3d. (a) The titration curves of G1d with pyrene (the starting curve and the end curve are highlighted in bold). Insert: The photographs of G1d solution (left) and the titration ending G1d-pyrene solution (right). (b) The titration curves of G2d with pyrene (the starting curve and the end curve are highlighted in bold). Insert: the photographs of G2d solution (left) and the titration ending G2d-pyrene solution (right). (c) The titration curves of G3d with pyrene (the starting curve and the end curve are highlighted in bold). Insert: The photographs of G3d solution (left) and the titration ending G3d-pyrene solution (right). (d) Job plot for stoichiometry study of G1d-pyrene binding. (e) Job plot for stoichiometry study of G2d-pyrene binding. | ||
1H NMR spectra can provide additional evidence of the aromatic donor–acceptor interaction.11b The salen ligands G1a, G2a and G3a were used in NMR titration experiments with pyrene as the donor compound and CDCl3 as the solvent. Acceptor protons of all three salen ligands showed upfield shifts upon the addition of pyrene. NDI protons in G1a and G2a shifted from 8.81 ppm to 8.20 ppm, and from 8.75 ppm to 8.21 ppm, respectively, when pyrene was gradually added up to 10 equivalents (Fig. 4, S5 and S6†). For PDI protons in G3a, a shift of 8.26 ppm to 7.89 ppm was detected with the same titration process (Fig. S7†). Pyrene protons also had smaller upfield shifts (<0.1 ppm) during the titration experiments with G1a, G2a and G3a. These chemical shift changes confirmed the face-to-face binding of aromatic donor and acceptor due to the shielding effect.48 On the other hand, NMR dilution experiments in CDCl3 from 10 mM to 0.1 mM revealed no significant chemical shift changes (≤0.02 ppm) for acceptor protons in G1a, G2a and G3a, indicating that no significant self-stacking took place between the acceptor units.49 Although the charge transfer band was not observed in the UV-vis absorption titration, the aromatic donor–acceptor interaction between G3a and pyrene was confirmed by the NMR titration experiment. The NMR titration data were also used to extract association constants by non-linear curve fitting. For G1a-pyrene, association constants of 16.7 M−1 (3.18% error) and <0.01 M−1 were derived for the 1 + 1 and 1 + 2 bindings, respectively. G2a-pyrene had much higher association constants, 242 M−1 (8.55% error) for the 1 + 1 binding and 7.63 M−1 (1.59% error) for the 1 + 2 binding. G3a-pyrene fell in the middle, 44.2 M−1 (3.65% error) for the 1 + 1 binding and 6.39 M−1 (1.78% error) for the 1 + 2 binding.50
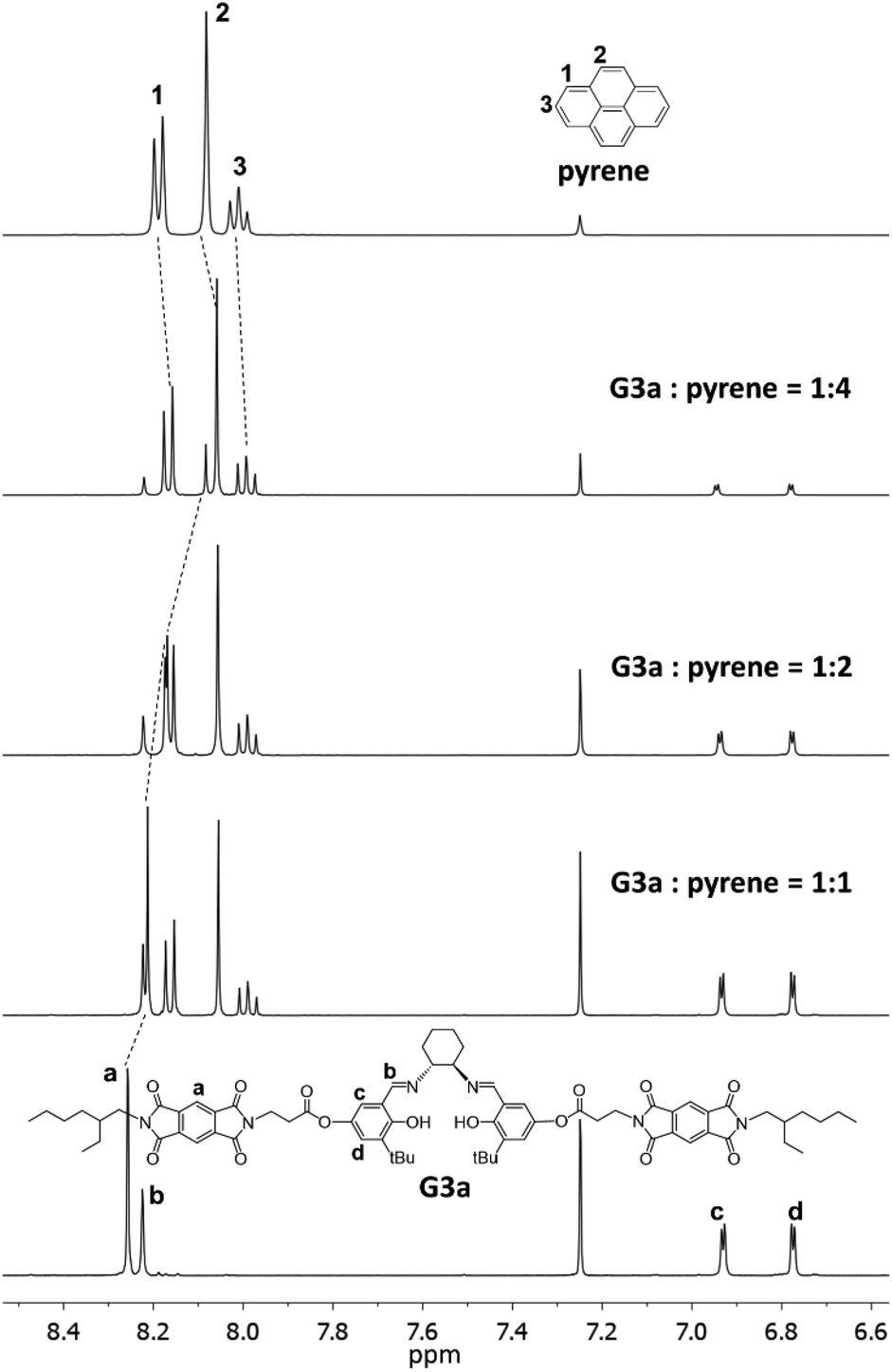 | ||
| Fig. 4 1H NMR spectra of the titration of G3a with pyrene in CDCl3 (See ESI† for all NMR titration spectra). | ||
The low association constants of G1a with pyrene could one reason for G1c low HKR activity. The relatively weak aromatic donor–acceptor interaction of G1c-pyrene did not promote the cooperativity of Co(III)–salen cores significantly. However, the different linkers of the three complexes may be a critical factor in their catalytic activities. The alkyl ester linker in G2c and G3c could contribute in two aspects, compared to the phenyl linker in G1c. First, the alkyl ester is flexible, and therefore allows the Co(III)–salen cores to reach the preferred conformation and distance for a cooperative pathway.43,51 Second, the phenyl linkage of G1c is a conjugated linker, so the electron-withdrawing effect of NDI can be extended to the Co(III)–salen catalytic core. This was verified by 1H NMR spectra of G1a, G2a and G3a ligands. The two phenyl protons in the salen core of G2a had chemical shifts of 6.96 ppm and 6.79 ppm, respectively (protons c and d in Fig. 4). Similar to G2a, the same protons in G3a displayed the shifts at 6.93 ppm and 6.78 ppm. However, the same protons in G1a showed downfield shifts at 7.58 ppm and 7.31 ppm, indicating a strong deshielding effect executed by NDI. It has been reported that electron-withdrawing groups/electron-donating groups have a significant effect on the catalytic properties of metal–salen complexes.52 With pyrene, G3c had a slightly higher HKR rate than G2c in spite of lower association constants. We were uncertain about the cause, but we noticed that, during the HKR of epichlorohydrin, G3c showed better solubility than G2c. For G2c and G3c, the controls without pyrene also showed high HKR rates. Since no regular aromatic interaction (π-stacking) was detected in the NMR dilution experiments, the possible explanation was solvophobic effect. The highly polar reagents, such as H2O, could drive complexes into close proximity without donor compounds.17
After the successful HKR test with epichlorohydrin, we also carried the HKR substrate scope tests for G2c and G3c with different epoxides including allyl glycidyl ether, styrene oxide and 1,2-epoxyhexane. However, the catalyst either could not dissolve in the epoxide solutions or precipitated during the reactions, resulting in poor resolutions of epoxides no matter pyrene was added or not. Currently, we are working on the development of a new generation of catalyst with higher solubility and a broader substrate scope.
This study mainly focused on the influence of the aromatic donor–acceptor interaction on catalyst assembly and efficiency. Our results suggested that care should be taken in choosing the aromatic donor compound. In the HKR by G1c, pyrene had the best rate enhancement effect. In contrast, DMN afforded the highest rate for G2c. As for G3c, pyrene and perylene showed similarly high rates. The addition of a proper donor compound can benefit the assembly of the cooperative Co(III)–salen catalyst in two aspects. First, the aromatic donor–acceptor interaction helped to bring complexes in close proximity. Second, the face-to-face stacking of acceptor/donor/acceptor could align Co(III)–salen cores in the optimal geometry for HKR. The salen complex catalyzed asymmetric reactions are sensitive to the orientation and geometry arrangement of the two catalytic cores.40,51,53 Our results also showed that the solubility of catalyst can have a significant impact to the catalyst activity, for instance, the higher reaction rate of G3c.
Conclusions
In summary, three generations of Co(III)–salen complexes containing aromatic acceptor units were designed and synthesized. The addition of a proper aromatic donor compound in a proper amount led to enhancement in the reaction rate and enantioselectivity in the HKR of epichlorohydrin for all three complexes. The structural difference of three generations played an important role in their catalytic activity. The second and third generations of complexes containing a flexible linker demonstrated a much higher reaction rate than the first generation containing a rigid and conjugated linker. The UV-vis absorption and 1H NMR spectroscopy studies verified the presence of the aromatic donor–acceptor interaction between Co(III)–salen complexes and pyrene. These results suggest that the aromatic donor–acceptor interaction can be a valuable noncovalent interaction in supramolecular catalyst development.Experimental section
Preoxidation of Co(II)–salen complex to Co(III)(OTs)–salen complex
(Note: For a successful HKR, it is important to fully oxidize Co(II)–salen complex to Co(III)(OTs)–salen.) For G1c, a full oxidation is indicated by a CLEAR reddish green solution (Fig. S4a†). For G2c a full oxidation is indicated by a CLEAR brownish green solution (Fig. S4b†). For G3c a full oxidation is indicated by a CLEAR greenish brown solution (Fig. S4c†).
G1c: G1b (4.9 mg, 0.0035 mmol) was weighed into a 20 mL vial. A solution of p-toluenesulfonic acid monohydrate in THF (0.01 M, 385 μL, 0.0039 mmol, 1.1 eq.) was added. The reaction mixture was stirred for 1 minute, and then anhydrous THF (4 mL) and anhydrous acetonitrile (1.5 mL) were added sequentially. The reaction mixture was further stirred at room temperature for 1 hour, during which time most solid was dissolved and the mixture color changed from brownish red to brownish green. The solvent was removed by rotary evaporation. The residue was redissolved in THF (3 mL) and acetonitrile (0.5 mL) and the solution was stirred at room temperature for 30 minutes. A CLEAR dark green solution was obtained. The solvent was removed by rotary evaporation. Residual solvent was further removed azeotropically with acetonitrile (2 × 1.0 mL) and the remaining dark green solid was dried under high vacuum. Due to broadening in the 1H NMR spectrum, accurate integrals could not be obtained, and therefore approximate proton counts are provided. 1H NMR (400 MHz, CDCl3) δ (ppm) 9.09 (br, 2H, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 8.80 (br, 8 H, Ar–H), 7.94–7.32 (m, br, 16H, Ar–H), 4.17 (br, 4H), 3.74 (br, 2H), 2.51 (br, 3H), 2.00–1.95 (m, br, 6 H), 1.63–1.40 (m, br, 38 H), 0.95 (br, 3 H, CH3CH2CH), 0.89 (br, 3H, CH3CH2CH2CH2); 13C NMR (100 MHz, CDCl3) δ (ppm) 163.3 (overlapping signals), 134.1 (overlapping signals), 131.6, 131.5, 131.2, 128.8, 128.7, 128.6, 127.5, 127.2, 127.0 (overlapping signals), 126.8, 119.2, 44.8, 38.0, 30.8, 29.8, 29.7, 28.7, 24.2, 24.1, 23.1, 22.9, 14.2, 10.7; IR (Zn/Se ART, cm−1) 2958, 2930, 2872, 1708 (imide asymmetrical CO), 1668 (imide symmetrical CO), 1343 (SO), 1192, 910, 769. HS-MS (ESI) calcd for C84H84CoN6O10 (no tosylate counter ion) [M]+: 1395.5576; found, 1395.5581.
CH), 8.80 (br, 8 H, Ar–H), 7.94–7.32 (m, br, 16H, Ar–H), 4.17 (br, 4H), 3.74 (br, 2H), 2.51 (br, 3H), 2.00–1.95 (m, br, 6 H), 1.63–1.40 (m, br, 38 H), 0.95 (br, 3 H, CH3CH2CH), 0.89 (br, 3H, CH3CH2CH2CH2); 13C NMR (100 MHz, CDCl3) δ (ppm) 163.3 (overlapping signals), 134.1 (overlapping signals), 131.6, 131.5, 131.2, 128.8, 128.7, 128.6, 127.5, 127.2, 127.0 (overlapping signals), 126.8, 119.2, 44.8, 38.0, 30.8, 29.8, 29.7, 28.7, 24.2, 24.1, 23.1, 22.9, 14.2, 10.7; IR (Zn/Se ART, cm−1) 2958, 2930, 2872, 1708 (imide asymmetrical CO), 1668 (imide symmetrical CO), 1343 (SO), 1192, 910, 769. HS-MS (ESI) calcd for C84H84CoN6O10 (no tosylate counter ion) [M]+: 1395.5576; found, 1395.5581.
G2c: G2b (4.9 mg, 0.0035 mmol) was weighed into a 20 mL vial. A solution of p-toluenesulfonic acid monohydrate in THF (0.01 M, 385 μL, 0.0039 mmol, 1.1 eq.) was added. The reaction mixture was stirred for 1 minute, and then anhydrous THF (3 mL) was added. The reaction mixture was further stirred at room temperature until all solid was dissolved (usually it takes 1.5–2 hours). Anhydrous acetonitrile (1.5 mL) was added and a CLEAR brownish green solution was obtained. The solvent was removed by rotary evaporation. Residual solvent was further removed azeotropically with acetonitrile (2 × 1.0 mL) and the remaining brownish green solid was dried under high vacuum. Due to broadening in the 1H NMR spectrum, accurate integrals could not be obtained, and therefore approximate proton counts are provided. 1H NMR (400 MHz, CDCl3) δ (ppm) 8.69 (br, 8 H, Ar–H), 8.17 (br, 2H, NCH), 7.19 (br, 4H), 6.92 (br, 2 H, Ar–H), 6.75 (br, 2 H, Ar–H), 4.55 (br, 4 H, NCH2CH2), 4.07 (br, 4H), 3.68 (br, 3H), 3.29 (br, 2H, NCHCH2), 2.91 (br, 4 H, NCH2CH2), 1.86 (br, 4 H, NCHCH2), 1.78 (br, 2 H, CH2CH (CH2)2), 1.23–1.46 (overlapping signals, br, 38 H, 5 × CH2 and 2 × C(CH3)3), 1.19 (br, 3 H, CH3CH2CH), 0.81 (br, 3H, CH3CH2CH2CH2); 13C NMR (100 MHz, CDCl3) δ (ppm) 170.2, 164.7, 163.2, 162.5, 158.3, 141.3, 138.5, 131.2 (overlapping signals), 127.1, 126.1, 123.1, 121.3, 72.4, 44.7, 37.8, 36.7, 34.8, 33.3, 32.7, 30.9, 29.2, 28.5, 25.6, 24.3, 22.8, 14.4, 10.7; IR (Zn/Se ART, cm−1) 2958, 2929, 2870, 1755 (ester CO), 1705 (imide asymmetrical CN), 1662 (imide symmetrical CN), 1337 (SO), 1182, 910, 733; HS-MS (ESI) calcd for C78H84CoN6O14 (no tosylate counter ion) [M]+: 1387.5378; found, 1387.5381.
G3c: G3b was oxidized to G3c in a similar way as the preparation of G2c. Due to broadening in the 1H NMR spectrum, accurate integrals could not be obtained, and therefore approximate proton counts are provided. 1H NMR (400 MHz, CDCl3) δ (ppm) 8.52 (br, 2H, 2 × NCH), 8.26 (br, 4H, Ar–H), 8.13 (br, Ar–H), 6.93 (br, 2H, Ar–H), 6.79 (br, 2H, Ar–H), 4.17 (br, 4H, 2 × NCH2CH2), 3.73 (br, 2H, 2 × NCHCH2), 3.63 (br, 4H, 2 × NCH2CH), 2.99 (br, 4H, 2 × NCH2CH2), 2.48 (br, 3H, CH3), 1.84–1.98 (overlapping signals, br, 10H, 2 × NCH2CH, 2 × NCHCH2CH2), 1.28 (br, 18H, 2 × C(CH3)3), 1.18–1.63 (br, 16H, 8 × CH2), 0.88 (br, 6H, 2 × CH3); 13C NMR (100 MHz, CDCl3) δ (ppm) 166.6, 166.5, 161.5, 160.1, 137.6, 137.4, 137.1, 127.1, 121.3 (overlapping signals), 118.7, 118.4, 68.0, 42.7, 38.4, 37.5, 35.1, 34.4, 30.6, 29.4 (overlapping signals), 29.2, 28.5, 23.9, 23.0, 14.1, 10.4; IR (Zn/Se ART, cm−1) 2961, 2939, 2869, 1723 (ester CO), 1700 (imine, CN), 1362 (SO), 1183, 913, 729; HS-MS (ESI) calcd for C70H80CoN6O14 (no tosylate counter ion) [M]+: 1287.5065; found, 1287.5078.
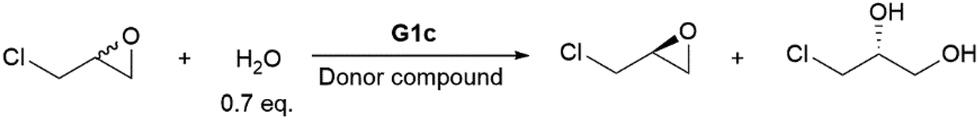
General procedure for hydrolytic kinetic resolution (HKR) of epichlorohydrin
Pyrene or other donor compounds was added to the vial holding Co(III)(OTs)–salen complex. Epichlorohydrin (548 μL, 7.0 mmol), CHCl3 (250 μL) and chlorobenzene (15 μL, as an internal standard) were added. The reaction mixture was stirred at room temperature for 5 minutes. All solid was dissolved and the solution is in a dark red color. The vial was immersed into a water bath at room temperature, and deionized water (88 μL, 4.9 mmol, 0.7 eq.) was added to start the reaction. The resolution reaction was monitored by chiral GC. After the reaction was complete, the remaining epoxide was isolated by vacuum transfer (rt, 0.5 Torr) into a receiving flask pre-cooled at −78 °C to obtain the yield. For kinetic studies, aliquots (6 μL) were taken at certain times and diluted with 1 mL diethyl ether/hexane (3/5) mixed solvent. The solution was then passed through a plug of silica gel packed in a Pasteur pipet. The silica plug was washed with 1 mL diethyl ether/hexane (3/5) twice. The combined filtrates were concentrated to about 500 μL and subject to chiral GC analyses. The absolute configuration of resolved epichlorohydrin was assigned by comparing it to authentic samples. Chiraldex γ-TA, 40 °C, isothermal, tR(R, minor) = 15.5 min, tR(S, major) = 17.0 min.For more experimental details, copies of NMR spectra, and nonlinear curve-fitting analyses see ESI.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Acknowledgment is made to the donors of The American Chemical Society Petroleum Research Fund for support of this research (55237-UNI1). This work was also supported partially by Faculty Research Grant of Northern Michigan University. We thank Prof. Eugene Wickenheiser for the help in the ICP-OES measurements.Notes and references
- (a) P. W. N. M. van Leeuwen, Supramolecular Catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed; (b) D. M. Vriezema, M. Comellas Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1490 CrossRef CAS PubMed; (c) J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed.
- (a) R. Bellini, J. I. van der Vlugt and J. N. H. Reek, Isr. J. Chem., 2012, 52, 613–629 CrossRef CAS; (b) B. Breit, Angew. Chem., Int. Ed., 2005, 44, 6816–6825 CrossRef CAS PubMed; (c) A. J. Sandee and J. N. H. Reek, Dalton Trans., 2006, 3385–3391 RSC.
- For example see: J. M. Takacs, D. S. Reddy, S. A. Moteki, D. Wu and H. Palencia, J. Am. Chem. Soc., 2004, 126, 4494–4495 CrossRef CAS PubMed.
- For example see: B. Breit and W. Seiche, J. Am. Chem. Soc., 2003, 125, 6608–6609 CrossRef CAS PubMed.
- For example see: T. S. Koblenz, H. L. Dekker, C. G. de Koster, P. W. N. M. van Leeuwen and J. N. H. Reek, Chem. Commun., 2006, 1700–1702 RSC.
- For example see: R. Cacciapaglia, S. Di Stefano, E. Kelderman and L. Mandolini, Angew. Chem., Int. Ed., 1999, 38, 348–351 CrossRef CAS PubMed.
- For example see: S. Liu, H. Gan, A. T. Hermann, S. W. Rick and B. C. Gibb, Nat. Chem., 2010, 2, 847–852 CrossRef CAS PubMed.
- In this report the aromatic interaction is defined as the intermolecular interaction between the neutral aromatic compounds. Cation–π, anion–π and radical–π interactions are not included. There are some meaningful discussions on the usage of the term of “π–π stacking” or “π–π interaction” ( C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191–2201 RSC ). A more general term “aromatic interaction” is used in this report.
- (a) R. F. D'Vries, N. Snejko, E. Gutiérrez-Puebla, M. Iglesias and M. Angeles Monge, Inorg. Chim. Acta, 2012, 382, 119–126 CrossRef; (b) S. E.-d. H. Etaiw and M. M. El-bendary, Inorg. Chim. Acta, 2015, 435, 167–173 CrossRef CAS; (c) S. E. H. Etaiw and S. N. Abdou, J. Inorg. Organomet. Polym., 2012, 22, 780–790 CrossRef CAS.
- (a) C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS; (b) M. L. Waters, Curr. Opin. Chem. Biol., 2002, 6, 736–741 CrossRef CAS PubMed.
- (a) R. Foster, Organic Charge-Transfer Complex, Academic Press, London, 1969 Search PubMed; (b) A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038–2054 CrossRef CAS PubMed.
- (a) R. Scott Lokey and B. L. Iverson, Nature, 1995, 375, 303–305 CrossRef; (b) H. M. Colquhoun, Z. Zhu, D. J. Williams, M. G. B. Drew, C. J. Cardin, Y. Gan, A. G. Crawford and T. B. Marder, Chem. – Eur. J., 2010, 16, 907–918 CrossRef CAS PubMed; (c) B. W. Greenland, S. Burattini, W. Hayes and H. M. Colquhoun, Tetrahedron, 2008, 64, 8346–8354 CrossRef CAS.
- R. Foster, J. Chem. Soc., 1960, 1075–1077 RSC.
- L. M. Klivansky, D. Hanifi, G. Koshkakaryan, D. R. Holycross, E. K. Gorski, Q. Wu, M. Chai and Y. Liu, Chem. Sci., 2012, 3, 2009–2014 RSC.
- J. S. Park, K. Y. Yoon, D. S. Kim, V. M. Lynch, C. W. Bielawski, K. P. Johnston and J. L. Sessler, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 20913–20917 CrossRef CAS PubMed.
- M. B. Nielsen, J. O. Jeppesen, J. Lau, C. Lomholt, D. Damgaard, J. P. Jacobsen, J. Becher and J. F. Stoddart, J. Org. Chem., 2001, 66, 3559–3563 CrossRef CAS PubMed.
- M. S. Cubberley and B. L. Iverson, J. Am. Chem. Soc., 2001, 123, 7560–7563 CrossRef CAS PubMed.
- Y. H. Ko, E. Kim, I. Hwang and K. Kim, Chem. Commun., 2007, 1305–1315 RSC.
- M. Hardouin-Lerouge, P. Hudhomme and M. Salle, Chem. Soc. Rev., 2011, 40, 30–43 RSC.
- A. C. Fahrenbach, C. J. Bruns, D. Cao and J. F. Stoddart, Acc. Chem. Res., 2012, 45, 1581–1592 CrossRef CAS PubMed.
- U. Maitra, P. Vijay Kumar, N. Chandra, L. J. D'Souza, M. D. Prasanna and A. R. Raju, Chem. Commun., 1999, 595–596 RSC.
- J. Q. Nguyen and B. L. Iverson, J. Am. Chem. Soc., 1999, 121, 2639–2640 CrossRef CAS.
- C. Wang, S. Yin, S. Chen, H. Xu, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2008, 47, 9049–9052 CrossRef CAS PubMed.
- H. Ringsdorf, R. Wüstefeld, E. Zerta, M. Ebert and J. H. Wendorff, Angew. Chem., Int. Ed. Engl., 1989, 28, 914–918 CrossRef.
- P. Talukdar, G. Bollot, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2005, 127, 6528–6529 CrossRef CAS PubMed.
- B. Lynikaite, J. Cvengroš, U. Piarulli and C. Gennari, Tetrahedron Lett., 2008, 49, 755–759 CrossRef CAS.
- O. Chuzel, C. Magnier-Bouvier and E. Schulz, Tetrahedron: Asymmetry, 2008, 19, 1010–1019 CrossRef CAS.
- (a) M. Tokunaga, J. F. Larrow, F. Kakiuchi and E. N. Jacobsen, Science, 1997, 277, 936–938 CrossRef CAS PubMed; (b) S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307–1315 CrossRef CAS PubMed; (c) L. P. C. Nielsen, C. P. Stevenson, D. G. Blackmond and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 1360–1362 CrossRef CAS PubMed.
- (a) J. M. Ready and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 2687–2688 CrossRef CAS PubMed; (b) X. L. Zheng, C. W. Jones and M. Weck, J. Am. Chem. Soc., 2007, 129, 1105–1112 CrossRef CAS PubMed; (c) Y. Liu, J. Rawlston, A. T. Swann, T. Takatani, C. D. Sherrill, P. J. Ludovice and M. Weck, Chem. Sci., 2011, 2, 429–438 RSC; (d) F. Wu, K. Wang, Z. Li and X. Zhu, Catal. Commun., 2015, 68, 101–104 CrossRef CAS.
- (a) D. A. Annis and E. N. Jacobsen, J. Am. Chem. Soc., 1999, 121, 4147–4154 CrossRef CAS; (b) Y. M. Song, X. Q. Yao, H. L. Chen, C. M. Bai, X. Q. Hu and Z. Zheng, Tetrahedron Lett., 2002, 43, 6625–6627 CrossRef CAS; (c) M. A. Kwon and G. J. Kim, Catal. Today, 2003, 87, 145–151 CrossRef CAS; (d) B. M. Rossbach, K. Leopold and R. Weberskirch, Angew. Chem., Int. Ed., 2006, 45, 1309–1312 CrossRef CAS PubMed; (e) M. Beigi, S. Roller, R. Haag and A. Liese, Eur. J. Org. Chem., 2008, 2135–2141, DOI:10.1002/ejoc.200701230; (f) X. L. Zheng, C. W. Jones and M. Weck, Adv. Synth. Catal., 2008, 350, 255–261 CrossRef CAS; (g) X. Hong, L. Billon, M. Mellah and E. Schulz, Catal. Sci. Technol., 2013, 3, 723–729 RSC.
- R. Breinbauer and E. N. Jacobsen, Angew. Chem., Int. Ed., 2000, 39, 3604–3607 CrossRef CAS PubMed.
- (a) P. Goyal, X. Zheng and M. Weck, Adv. Synth. Catal., 2008, 350, 1816–1822 CrossRef CAS; (b) M. G. C. Kahn, J. H. Stenlid and M. Weck, Adv. Synth. Catal., 2012, 354, 3016–3024 CrossRef CAS.
- X. F. Guo, Y. S. Kim and G. J. Kim, Top. Catal., 2009, 52, 153–160 CrossRef CAS.
- J. Park, K. Lang, K. A. Abboud and S. Hong, Chem. – Eur. J., 2011, 17, 2236–2245 CrossRef CAS PubMed.
- A. Das, M. R. Molla, B. Maity, D. Koley and S. Ghosh, Chem. – Eur. J., 2012, 18, 9849–9859 CrossRef CAS PubMed.
- D. D. Ford, L. P. C. Nielsen, S. J. Zuend, C. B. Musgrave and E. N. Jacobsen, J. Am. Chem. Soc., 2013, 135, 15595–15608 CrossRef CAS PubMed.
- E. Buncel, N. L. Mailloux, R. S. Brown, P. M. Kazmaier and J. Dust, Tetrahedron Lett., 2001, 42, 3559–3562 CrossRef CAS.
- N. S. S. Kumar, M. D. Gujrati and J. N. Wilson, Chem. Commun., 2010, 46, 5464–5466 RSC.
- See ESI.†.
- E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421–431 CrossRef CAS PubMed.
- M. E. Furrow, S. E. Schaus and E. N. Jacobsen, J. Org. Chem., 1998, 63, 6776–6777 CrossRef CAS PubMed.
- E. Peris, Chem. Commun., 2016, 52, 5777–5787 RSC.
- Y. Chen, R. Tan, Y. Zhang, G. Zhao and D. Yin, ChemCatChem, 2015, 7, 4066–4075 CrossRef CAS.
- (a) Q.-Z. Zhou, X.-K. Jiang, X.-B. Shao, G.-J. Chen, M.-X. Jia and Z.-T. Li, Org. Lett., 2003, 5, 1955–1958 CrossRef CAS PubMed; (b) T. Nakagaki, A. Harano, Y. Fuchigami, E. Tanaka, S. Kidoaki, T. Okuda, T. Iwanaga, K. Goto and T. Shinmyozu, Angew. Chem., Int. Ed., 2010, 49, 9676–9679 CrossRef CAS PubMed.
- S. Amemori, K. Kokado and K. Sada, Angew. Chem., Int. Ed., 2013, 52, 4174–4178 CrossRef CAS PubMed.
- H. L. Anderson, Inorg. Chem., 1994, 33, 972–981 CrossRef CAS.
- (a) F. Ulatowski, K. Dąbrowa, T. Bałakier and J. Jurczak, J. Org. Chem., 2016, 81, 1746–1756 CrossRef CAS PubMed; (b) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- A. J. Zych and B. L. Iverson, J. Am. Chem. Soc., 2000, 122, 8898–8909 CrossRef CAS.
- C. Tung and Y. Wang, J. Am. Chem. Soc., 1990, 112, 6322–6328 CrossRef CAS.
- There is a possibility of multiple equilibriums between binding species (salen + pyrene, salen + 2pyrene, 2 salen + pyrene and 2 salen + 2 pyrene, etc.). However, non-linear curve fitting of higher order complexation has considerable inherent error and uncertainty.
- K. Kinslow, A. M. Sevde, J. Liang and Y. Liu, Tetrahedron: Asymmetry, 2015, 26, 385–392 CrossRef CAS.
- (a) X. Yao, M. Qiu, W. Lü, H. Chen and Z. Zheng, Tetrahedron: Asymmetry, 2001, 12, 197–204 CrossRef CAS; (b) H. Guo, Y. Pan, D. Ma and P. Yan, Wuji Huaxue Xuebao, 2013, 29, 1447–1453 CAS.
- R. G. Konsler, J. Karl and E. N. Jacobsen, J. Am. Chem. Soc., 1998, 120, 10780–10781 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Syntheses of new compounds, 1H and 13C NMR spectra of selected compounds, and nonlinear curve-fitting analyses of NMR titration experiments. See DOI: 10.1039/c8ob02249f |
| This journal is © The Royal Society of Chemistry 2019 |