
Alkoxyallene-based syntheses of preussin and its analogs and their cytotoxicity†
Arndt
Hausherr
a,
Gerhard
Siemeister
b and
Hans-Ulrich
Reissig
 *a
*a
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Takustr. 3, 14195 Berlin, Germany. E-mail: hans.reissig@chemie.fu-berlin.de
bBayer AG, Research & Development, Pharmaceuticals, Müllerstraße 178, 13353 Berlin, Germany
First published on 21st November 2018
Abstract
Short syntheses of oxa-preussin, racemic preussin and (−)-preussin are reported. Starting from a racemic 3-nonyl-substituted methoxyallene derivative, its lithiation and addition to phenylethanal provided the corresponding allenyl alcohol that was converted into two diastereomeric dihydrofuran derivatives by silver nitrate-catalyzed 5-endo-trig cyclization. The acid hydrolysis of the enol ether moiety gave heterocyclic ketones and subsequent highly stereoselective reductions with L-selectride furnished 2-benzyl-5-nonylfuran-3-ol derivatives in good overall yield. The major all-cis-diastereomer has the skeleton and relative configuration of preussin and is hence called oxa-preussin. An analogous sequence with the same allene, but an N-sulfonyl imine as the electrophile, finally led to racemic preussin. The stereoselectivities of the individual steps are discussed in detail. With an enantiopure 2-benzyl-5-nonylpyrrolidin-3-one intermediate the preparation of (−)-preussin with an enantiomeric ratio of >95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 could be accomplished in a few steps. The sign of the optical rotation of this product finally proved the absolute configurations of its precursors and demonstrated that our chiral auxiliary-based route led to the antipode of the natural product. The cytotoxicity of several of the prepared heterocycles against MCF-7 tumor cells was investigated and five compounds, including racemic and enantiopure (−)-preussin, were identified as highly cytotoxic with IC50 values in the range of 3–6 μM.
:5 could be accomplished in a few steps. The sign of the optical rotation of this product finally proved the absolute configurations of its precursors and demonstrated that our chiral auxiliary-based route led to the antipode of the natural product. The cytotoxicity of several of the prepared heterocycles against MCF-7 tumor cells was investigated and five compounds, including racemic and enantiopure (−)-preussin, were identified as highly cytotoxic with IC50 values in the range of 3–6 μM.
Introduction
Compounds with hydroxylated pyrrolidine, pyrrolizidine or indolizidine skeletons are frequently occurring natural products with many interesting biological activities.1 Applying suitably substituted alkoxyallenes2 and imines we developed short and stereoselective syntheses of the amino acid (−)-detoxinine3 and the biologically active alkaloids (−)-anisomycin4 and (+)-codonopsinine.5 Employing an arabinose-derived nitrone and the requisite alkoxyallenes the preparation of (+)-australine and (+)-casuarine6 or (−)-hyacinthacine B4 and hyacinthacine C5 epimers7 could be achieved in close collaboration with the Goti research group (Fig. 1). As a related interesting target compound we planned to prepare the pyrrolidin-3-ol natural product preussin and analogs such as its oxygen congener oxa-preussin.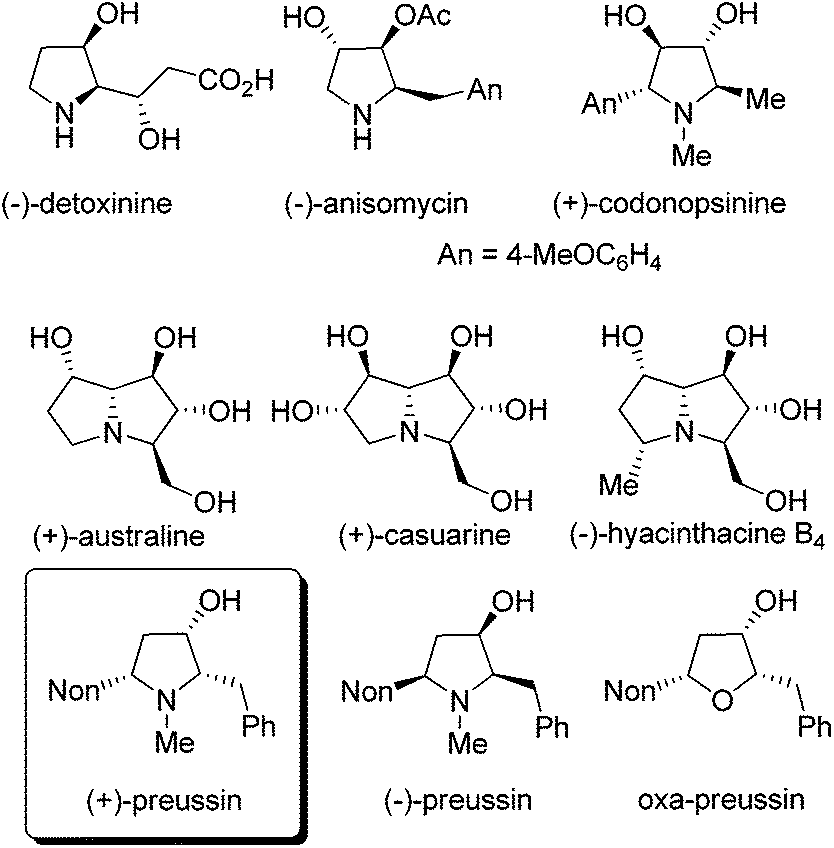 | ||
| Fig. 1 Hydroxylated pyrrolidine and pyrrolizidine natural products. | ||
The naturally occurring (+)-preussin was isolated independently by two research groups either from the fermentation broth of Aspergillus ochraceus (ATCC 22947)8 or that of a Preussia species.9 This pyrrolidin-3-ol alkaloid has a broad activity spectrum against bacteria and filamentous fungi8,9 and in 1997, Yoshida et al. also described its cytotoxic activity against rat fibroblast cells 3Y1.10 Later, Müller et al. reported that the compound directly interferes with the cell cycle and induces apoptosis in human tumor cells.11 It could be shown with different human cell lines that (+)-preussin inhibits in vitro the cyclin E kinase (CDK2-cyclin E)12 with an IC50 value of ca. 0.5 μM by blocking the cell cycle progression into S phase. It was also found that (+)-preussin inhibits the −1 programmed ribosomal frameshifting and virus propagation.13 More recently, the closely related (+)-preussin B (a compound with a 5-heptyl instead of a 5-nonyl substituent) was isolated from Simplicillium ianosoniveum and its biosynthesis was elucidated.14 In 2018, Kijjoa et al. isolated together with 14 other compounds a second close relative of (+)-preussin from the marine sponge-associated fungus Aspergillus candidus KUFA0062: preussin C is the N-demethylated version of (+)-preussin.15 These authors found that preussin C has cytotoxic effects against different tumor cell lines, but that the N-methyl group of (+)-preussin seems to be crucial for antibiotic and highly cytotoxic activities.
As a consequence of these interesting biological activities and the moderate structural complexity of this natural product, it became a popular target for the proof of new synthetic concepts or methods. A large number of syntheses of (+)-preussin, a few of its antipode (−)-preussin and of the racemic compound have been reported.16 Since most of these syntheses are based on chiral pool compounds, there is still room for improved selectivity and flexibility with respect to access to analogs. For the envisioned preparation of compounds with the preussin skeleton we planned the stereoselective reduction of ketones of type A that should be available from the corresponding heterocyclic compounds B bearing an enol ether moiety (Scheme 1). These 2,5-dihydrofuran or 2,5-dihydropyrrole derivatives B should be prepared from the corresponding allenyl alcohols17 or amines C,18 respectively, by suitable 5-endo-trig cyclizations.19 Hence this analysis leads to starting materials such as lithiated 3-nonyl-substituted alkoxyallenes E as crucial nucleophilic components and phenylethanal or its imine congener D as electrophiles.
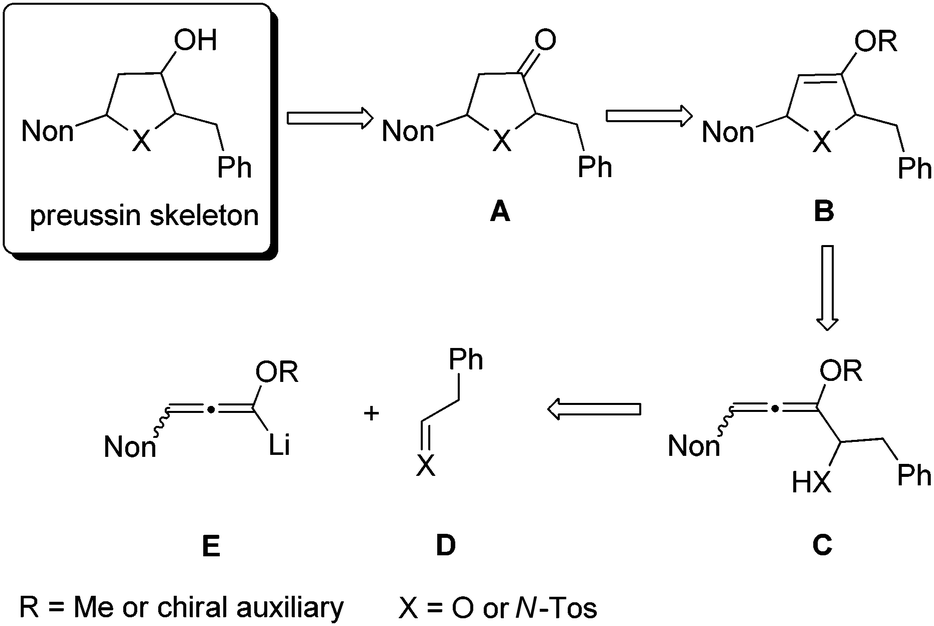 | ||
| Scheme 1 Retrosynthetic analysis of compounds with the preussin skeleton leading, via heterocyclic ketones A, enol ethers B, and allenyl alcohols or allenyl amines C, to nucleophilic building blocks E and electrophiles D. | ||
The synthesis and lithiation of 3-alkyl-substituted alkoxyallenes and the addition to electrophiles have been reported by our group in preceding publications.20 These model reactions revealed that only low diastereoselectivities in the additions of the axially chiral allenes to prochiral electrophiles are to be expected.21 In addition, we also investigated the feasibility of this [3 + 2] route to five-membered heterocycles using allenes with carbohydrate-derived auxiliaries at the oxygen, which led to highly enantio-enriched pyrrolidine building blocks.22
Results and discussion
First, the short synthesis of racemic oxa-preussin (X = O) is described. The axially chiral 3-nonyl-substituted methoxyallene 120 was used in the racemic form, lithiated with n-butyllithium under standard conditions and treated with phenylethanal (2) at −80 °C. After warm-up of the mixture and quench with water crude allenyl alcohol 3 was quantitatively obtained (Scheme 2). The low diastereoselectivity in the addition step was expected since in a model reaction of lithiated 1 or related allenes with various electrophiles similarly unselective additions were observed.21 The slim nonyl group of 1 seems to be too far away from the C–C bond forming event to have a strong influence on this step. Since allenyl alcohols such as 3 rapidly undergo decomposition, crude 3 was treated with 0.2 equivalents of silver nitrate in the presence of potassium carbonate17d to furnish the expected 2,5-dihydrofuran derivative 4. The diastereomeric ratio was slightly shifted in favor of the cis-compound (assignments after the next step). This is likely due to a partial equilibration at the allenyl alcohol stage by silver(I) catalysis and a faster cyclization of the pro-cis-allenyl alcohol 3 to furnish cis-4 in slight excess. This behavior has also been found and explained in allenyl amine cyclizations.22 Intermediate 4 was not purified but directly hydrolyzed to furan-3-one derivatives 5 obtained in 66% overall yield after column chromatography. This good yield demonstrates the efficacy of each step of this approach to specifically substituted furan-3-ones. The separation of the two isomers by HPLC provided cis-5 and trans-5 in 42% and 17% overall yields, respectively.
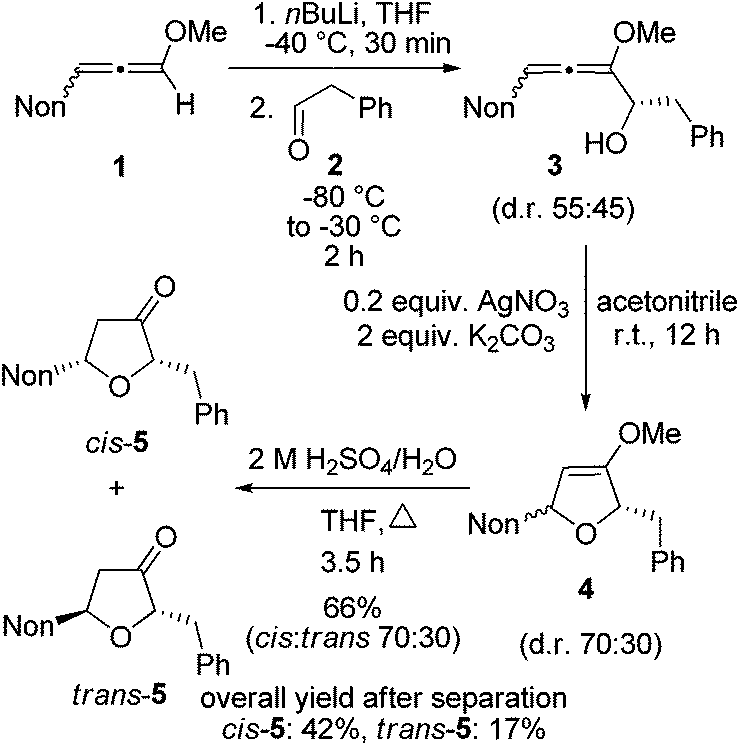 | ||
| Scheme 2 Synthesis of cis- and trans-furan-3-one 5 by lithiation of alkoxyallene 1 and addition to phenylethanal (2) followed by silver nitrate-promoted cyclization and acid hydrolysis of 4. (The shown formulas refer to the relative configurations; the compounds are racemic mixtures.) | ||
In order to obtain oxa-preussin, the stereoselective reductions of ketones cis-5 and trans-5 were studied (Scheme 3). Treatment of cis-5 with sodium borohydride in ethanol afforded a 3:1 mixture of the desired all-cis-6 and r-2,t-3,c-5-6, which were isolated after purification in 77% and 18% yields, respectively. Gratifyingly, the use of L-selectride in tetrahydrofuran at −78 °C exclusively gave all-cis-6 in essentially quantitative yield. A NOESY experiment confirmed the configurational assignment of all-cis-6. Starting from alkoxyallene 1, the synthesis of oxa-preussin (all-cis-6) was accomplished in four steps with an overall yield of 42%.
 | ||
| Scheme 3 Synthesis of oxa-preussin (all-cis-6) by stereoselective reduction of cis-5 and reduction of trans-5. (The shown formulas refer to the relative configurations; the compounds are racemic mixtures.) | ||
The reduction of trans-5 with L-selectride was less selective in furnishing a mixture of the two possible diastereomers in 62% and 8% yields (Scheme 3). The depicted configurations are based on our observations with related pyrrolidin-3-ones showing that in trans-compounds the substituent at C-5 has a stronger influence on the reduction than the C-3 substituent.21 The r-2,t-3,t-5 configuration of the major diastereomer of 6 is thus very likely.
For the synthesis of preussin we planned the use of N-tosyl imine 8 since this electrophile should be highly reactive. The N-tosyl group also leads to fairly stable products and on the other hand, it can easily be removed under mild conditions at later stages. However, compound 8 is not particularly stable due to its fast isomerization into the corresponding enamine tautomer.23 Therefore we examined the C,N-ditosyl amine 9 as a precursor, since under basic conditions this compound undergoes smooth elimination of sulfinate to provide in situ imine 8 that can be trapped by nucleophiles.24 In most cases the employed base and nucleophile are identical. In a first model reaction, two equivalents of methoxyallene (7) were lithiated and imine precursor 9 was added at −80 °C, giving the expected allenyl amine 10 quantitatively (Scheme 4). This intermediate was cyclized with silver nitrate to furnish the expected 2,5-dihydropyrrole derivative 11 in 60% overall yield. This experiment taught us that 9 should be a suitable building block for the preparation of the target compound preussin.
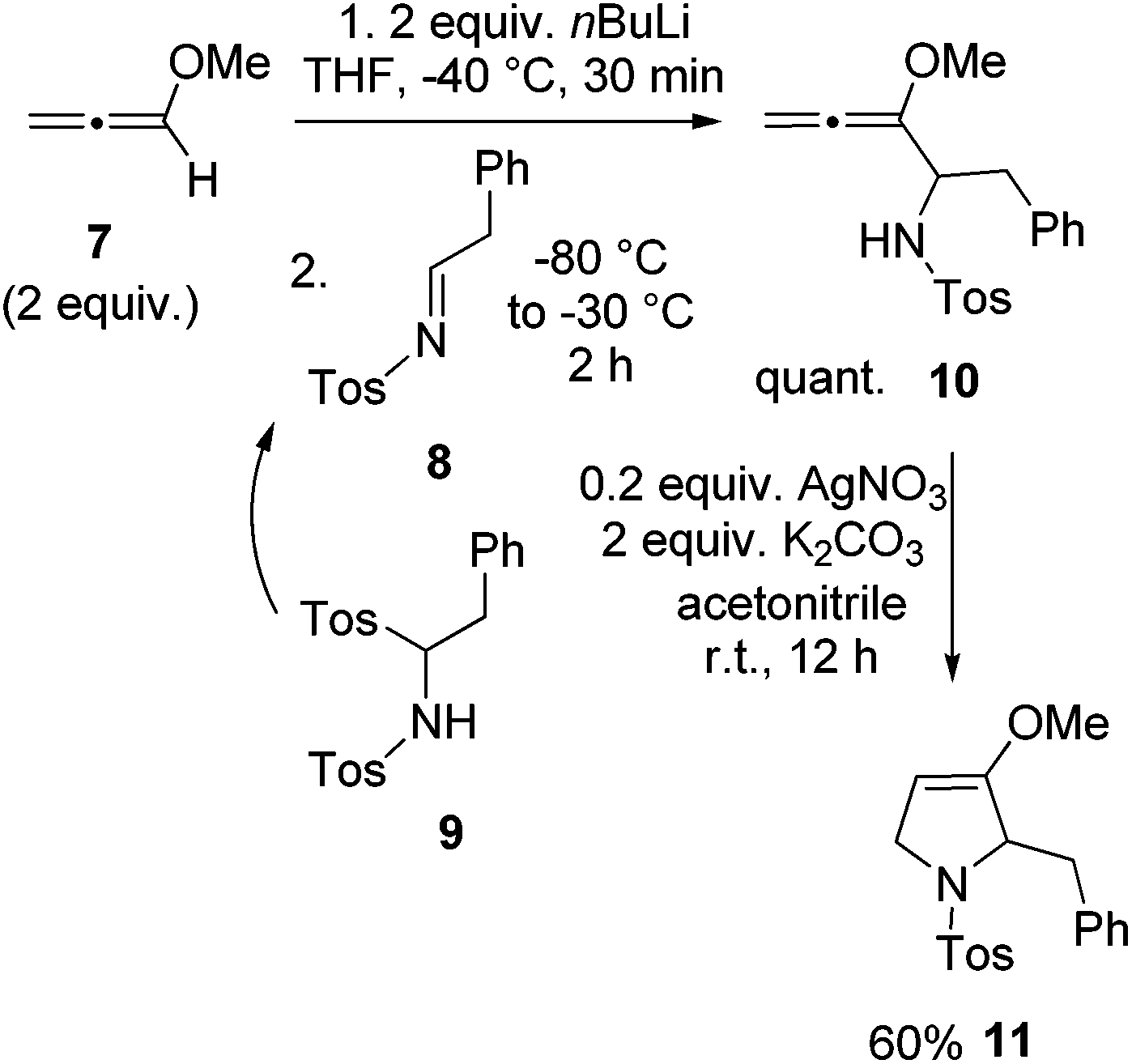 | ||
| Scheme 4 Synthesis of model compound 11 by lithiation and addition of methoxyallene (7) to in situ generated imine 8 and subsequent cyclization of allenyl amine 10. | ||
With the knowledge gained during the preparation of oxa-preussin and the model reaction above, we combined racemic 3-nonyl-substituted methoxyallene 1 with imine precursor 9 (Scheme 5). Two equivalents of lithiated 1 and 9 afforded the expected diastereomers pro-cis-12 and pro-trans-12 in good yields, but with no selectivity. The two isomers were separated by conventional column chromatography and both were isolated in good quantities in 36% yield. The excess of 1 required in this experiment could be re-isolated and used again.
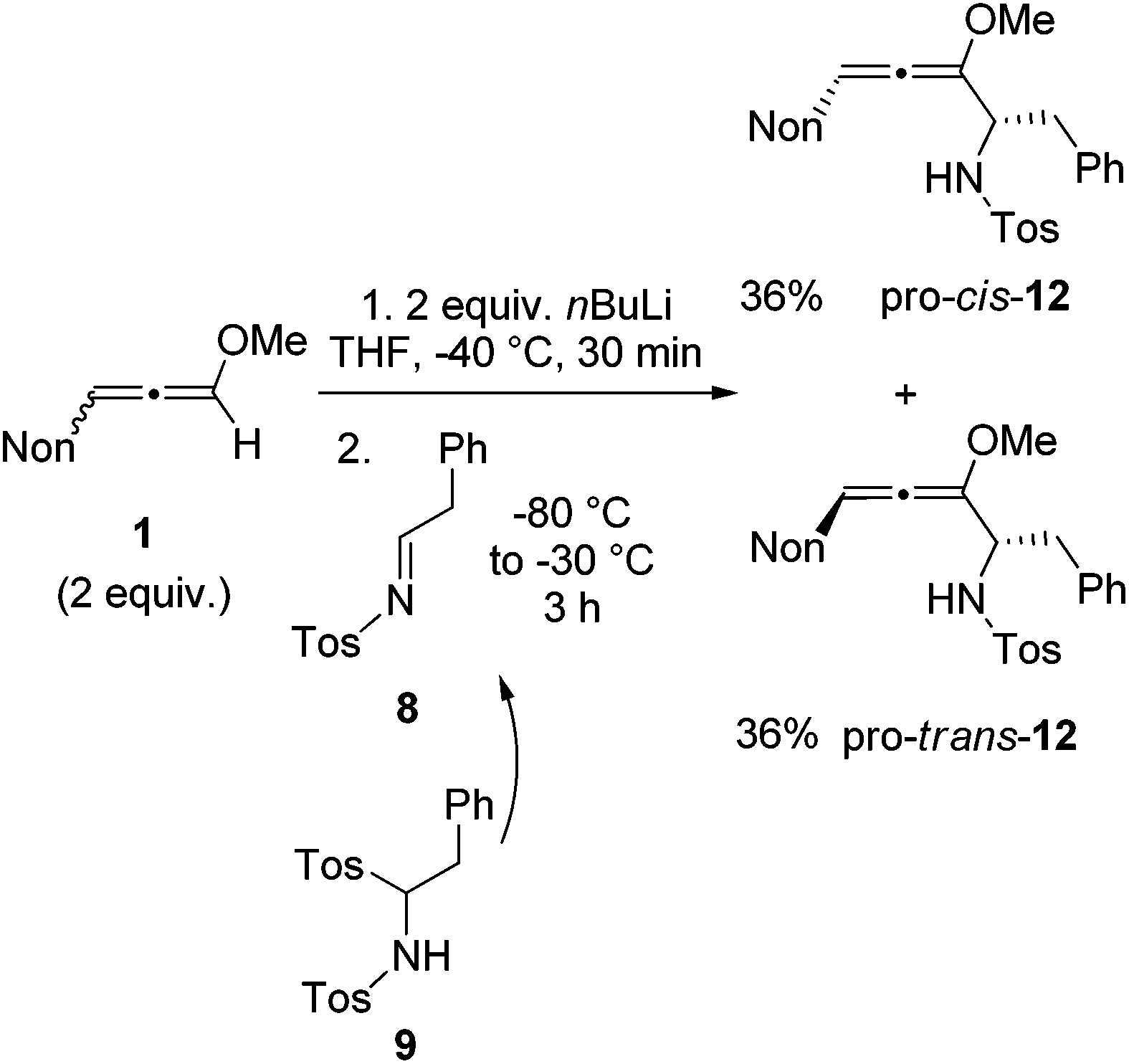 | ||
| Scheme 5 Synthesis of diastereomeric allenyl amines pro-cis-12 and pro-trans-12 by lithiation of 3-nonyl-substituted methoxyallene 1 and addition to in situ generated imine 8. (The shown formulas refer to the relative configurations; the compounds are racemic mixtures.) | ||
Next, two methods were used for the cyclization step in order to receive the required cis-13 from both diastereomeric precursors.21 The cyclization of pro-trans-12 with silver nitrate under buffered conditions led to a mixture of cis-13 and trans-13 in 87% yield (Scheme 6). The two isomers were separated by HPLC to furnish the two crystalline isomers in 42% and 30% yields, respectively. Whereas this method is not stereospecific due to the already mentioned equilibration at the allenyl amine stage, alternative cyclization under strongly basic conditions with potassium tert-butoxide in dimethyl sulfoxide proceeds stereospecifically.21 Starting with pro-cis-12 after 12 h the desired 2,5-disubstituted dihydropyrrole derivative cis-13 was isolated exclusively in good yield. The 1H NMR data allow for unambiguous assignment of the relative configurations of the compounds. Altogether, preussin precursor cis-13 was isolated in 43% overall yield when the results of Schemes 5 and 6 are combined.
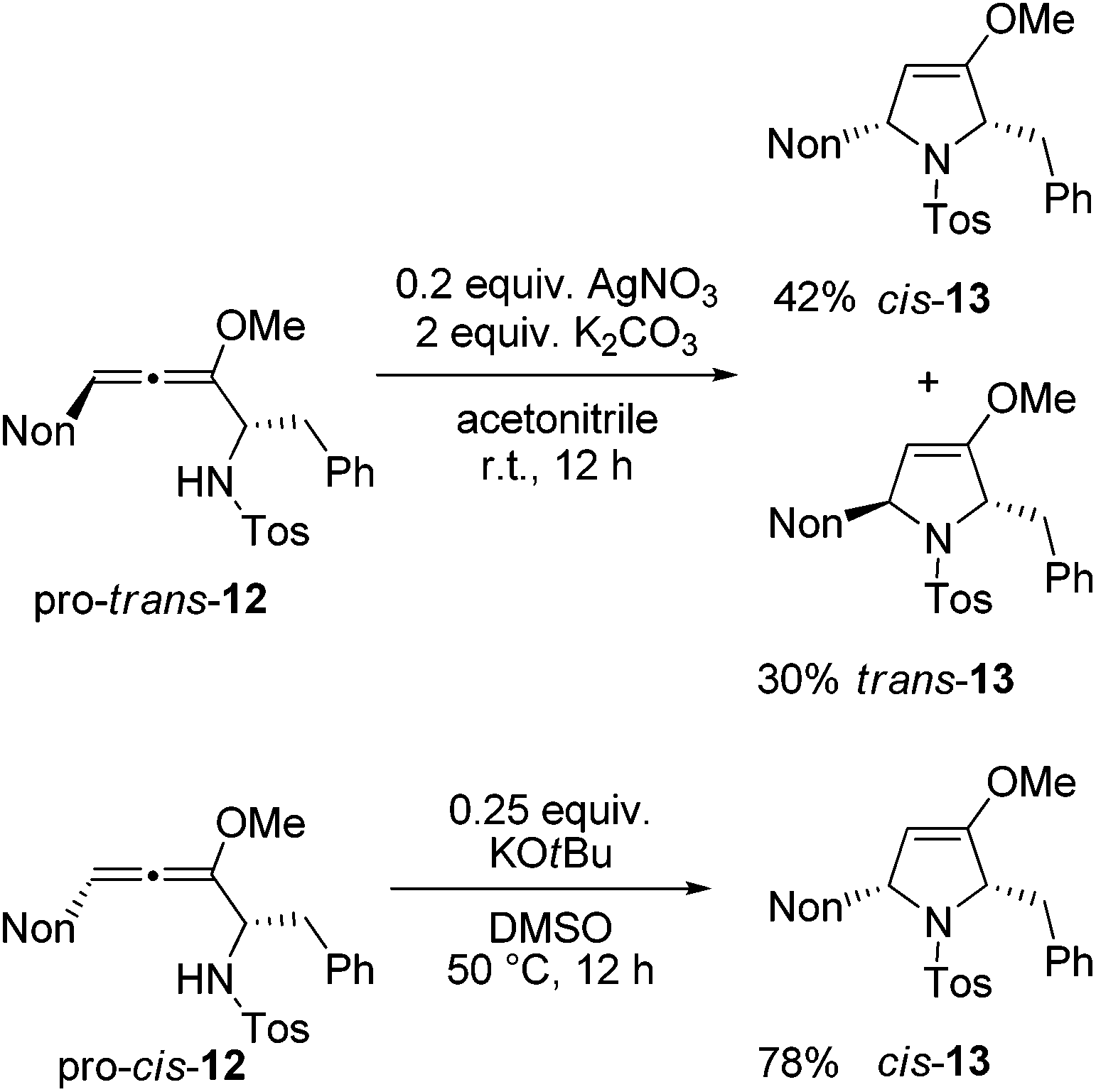 | ||
| Scheme 6 Cyclizations of diastereomeric allenyl amines 12 to cis- and trans-configured dihydropyrrole derivatives 13 under silver nitrate or potassium tert-butoxide promotion. (The shown formulas refer to the relative configurations; the compounds are racemic mixtures.) | ||
The hydrolysis of cis-13 and trans-13 could be routinely achieved with 20% aqueous sulfuric acid, furnishing the required 2,5-disubstituted pyrrolidin-3-ones cis-14 and trans-14 quantitatively (Scheme 7, eqn (1) and (2)). A cis/trans-equilibration via the corresponding enol form might be possible due to the CH acidic position at C-2; however, even under the relatively harsh hydrolysis conditions no cross-over between cis-14 and trans-14 was found. Since only cis-14 has the required relative configuration to approach preussin, we also examined the equilibration under basic conditions. Whereas no reaction was observed with weak bases (K2CO3 or NEt3), 1,8-diazabicyclo-[5.4.0]undec-7-ene (DBU) converted trans-14 within 2 h into the thermodynamically more stable cis-14. Unfortunately, a second compound was formed in similar quantities (Scheme 7, eqn (3)) whose NMR data reveal the constitution of product 15. Apparently, a base-promoted sulfinate elimination competes with the desired trans/cis-equilibration. The conversion into 15 could be completed by applying DBU for longer reaction times (Scheme 7, eqn (4)). Without the disturbing NMR signals of the compound cis-14 we could detect that compound 15 is in equilibrium (ratio of ca. 3:1) with its tautomer 16 (E/Z ca. 1:1).
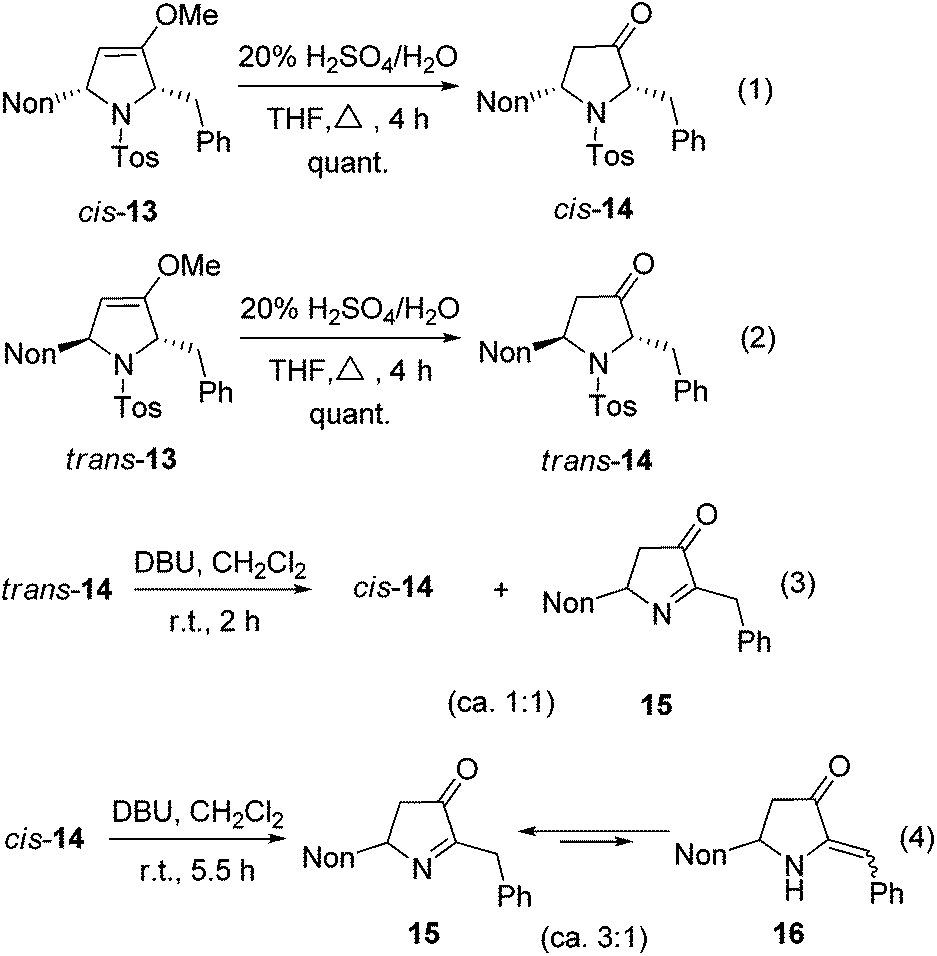 | ||
| Scheme 7 Hydrolysis of the enol ether moiety of 2,5-dihydropyrroles cis-13 and trans-13 to pyrrolidin-3-ones cis-14 and trans-14 and equilibration experiments finally leading to 15 and 16. (The shown formulas refer to the relative configurations; the compounds are racemic mixtures.) | ||
For the completion of the synthesis of rac-preussin the N-tosyl group of cis-13 has to be replaced by an N-methyl group. Among the several possibilities to reductively remove N-sulfonyl substituents we examined sodium naphthalenide as a reagent.25 At −78 °C compound cis-13 was smoothly converted into cis-17 when the mixture was quenched with water (Scheme 8). An analogous reaction, similarly executed but quenched with an excess of methyl iodide, furnished cis-18 in 53% yield. The moderate efficacy of this step is probably due to the formation of the quaternary ammonium salt. The compound cis-18 was transformed by acid hydrolysis into 2,5-disubstituted pyrrolidin-3-one derivative cis-19 in 60% yield. The final reduction with sodium borohydride provided an 80:20 mixture of the two possible diastereomers, but with L-selectride it proceeded with high diastereoselectivity and exclusively gave all-cis-20 in good yield. The NMR data of the sample were in full agreement with those published for preussin in the literature.26 In conclusion, we could accomplish a synthesis of rac-preussin in five steps (from alkoxyallene 1 and imine precursor 9). The moderate overall yield of 11% is mainly due to the unselective formation of pro-cis-12 from alkoxyallene 1 and imine 8. On the other hand, the isolation of pro-trans-12 in similar quantities should allow the preparation of diastereomeric analogs of rac-preussin with trans orientation of the benzyl and nonyl substituents at C-2 and C-5. Starting with differently 3-alkyl-substituted alkoxyallenes, the preparation of other preussin analogues, e.g. the new relative preussin B, should easily be possible.
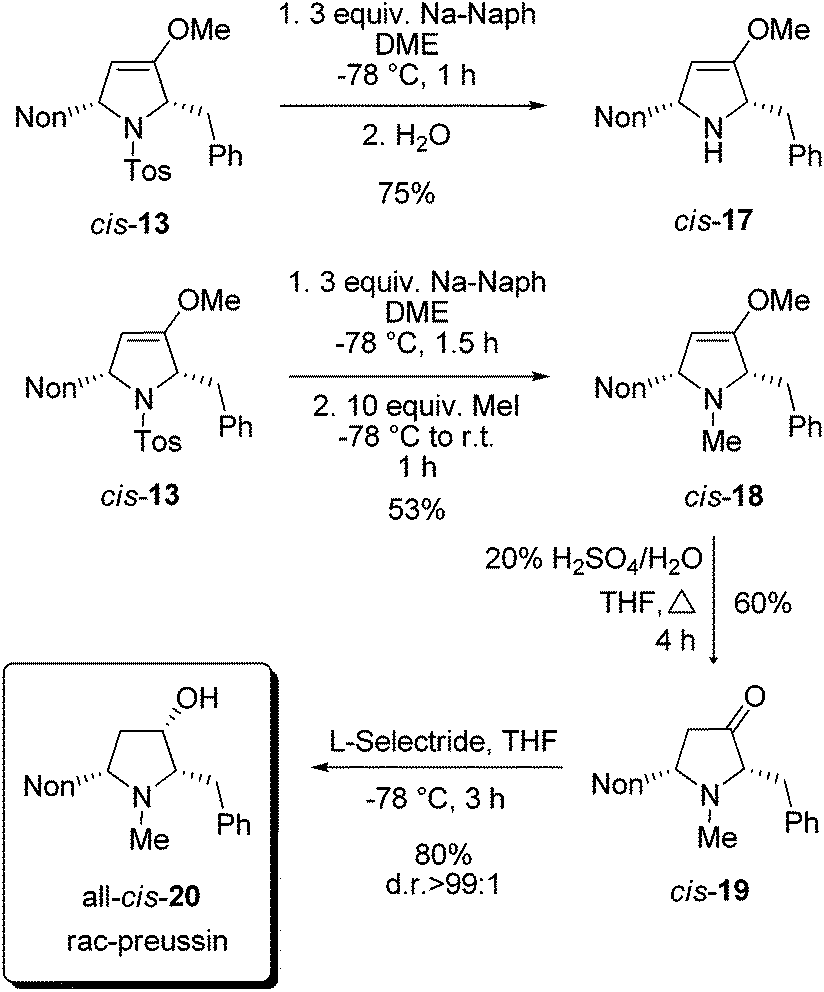 | ||
| Scheme 8 Synthesis of rac-preussin (all-cis-20) by reductive removal of the N-tosyl group of cis-13, N-methylation to cis-18, conversion into pyrrolidin-3-one cis-19 followed by stereoselective reduction. (The shown formulas refer to the relative configurations; the compounds are racemic mixtures.) | ||
As a result of our systematic studies of 3-alkyl-substituted alkoxyallenes bearing carbohydrate-derived auxiliaries, the pyrrolidin-3-one cis-14 was also available in an enantiopure form.22 Enantiomers 2R,5S-cis-14 and 2S,5R-cis-14 have been prepared in a three-step sequence employing the diacetone fructose-derived alkoxyallene congeners of 1 and imine 8 as crucial precursors. Without knowing the absolute configuration with certainty,22 we converted the obtained major isomer cis-14 into enantiopure preussin whose optical rotation showed that we have prepared the unnatural (−)-enantiomer (Scheme 9). Hence the configuration of the used cis-14 could finally be confirmed to be 2R,5S. For the preparation of (−)-preussin we slightly modified the sequence of steps, starting with the reduction of the ketone moiety and finalizing it by a reductive amination to introduce the N-methyl group. Whereas the reduction of 2R,5S-cis-14 was unselective with sodium borohydride giving an 80:20 mixture of the two diastereomers, the use of L-selectride exclusively afforded 2R,3R,5S-all-cis-21 in good yield. The subsequent reductive removal of the N-tosyl group furnished 2R,3R,5S-all-cis-22, which is the optical antipode of the recently isolated preussin C. Since we had learned during the synthesis of racemic preussin that the direct N-methylation of the intermediate amide anion with methyl iodide proceeded only with moderate efficacy (Scheme 8), we employed a literature known method27 for a reductive methylation with aqueous formaldehyde and sodium cyanoborohydride under acidic conditions. The obtained 2R,3R,5S-all-cis-20 was isolated in 80% yield and its NMR data agree very well with those of the literature.26
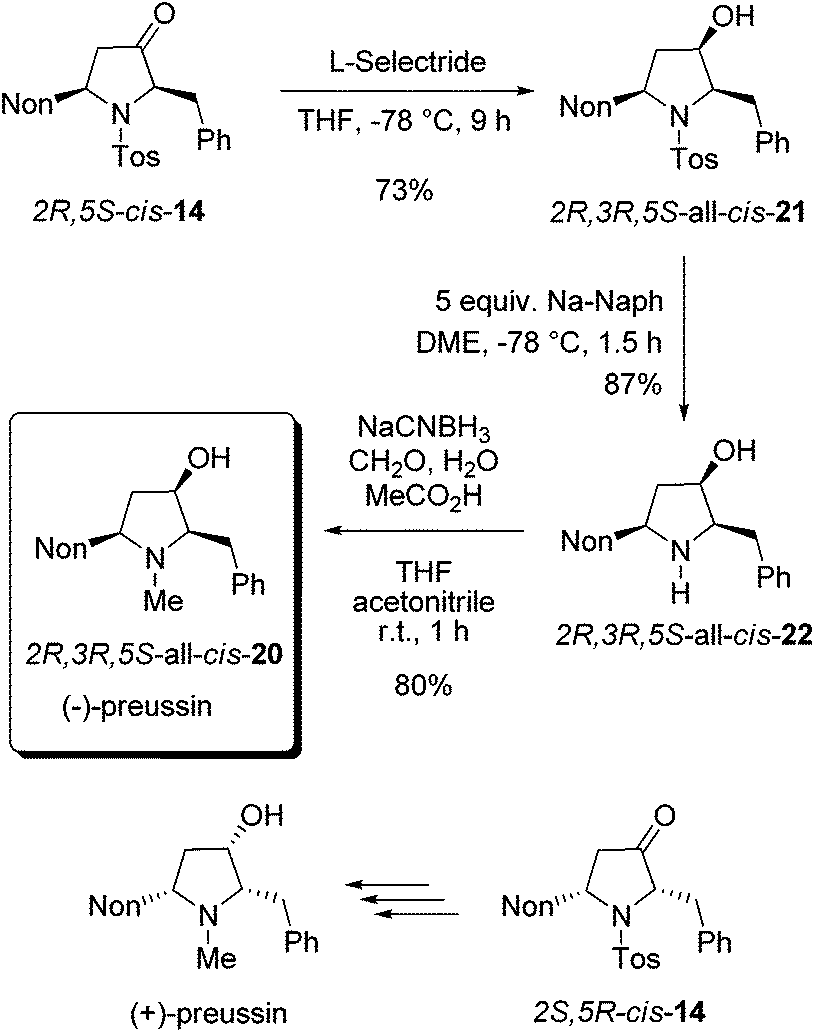 | ||
| Scheme 9 Synthesis of enantiopure (−)-preussin by stereoselective reduction of 2R,5S-cis-14 followed by transformations into all-cis-22 and all-cis-20. | ||
The negative sign of the optical rotation of the final product revealed that we have obtained the unnatural enantiomer and the absolute value of −25.8 (in chloroform) indicated high enantiomeric purity.28 The optical purity was further evidenced by converting the sample of all-cis-20 into its Mosher ester. NMR and HPLC analyses showed that the obtained product has a ratio of diastereomers of >95:5, indicating an ee of at least 90%.
Summing up, our route to (−)-preussin involved six steps (starting from the diacetone fructose-derived allene and imine precursor 9), but the overall yield of only 16% is due to the formation of diastereomers during the route to the required allenyl amine. It should also be mentioned that the 2S,5R-cis-14 isomer, also available in lower quantities by this route,22 will allow the preparation of the natural (+)-preussin in an analogous fashion.
Twenty-one racemic or enantiopure precursors and analogs of preussin (including oxa-preussin) obtained in this study and the preceding report were investigated in vitro for their cytotoxicity against MCF-7 tumor cells during 96 hours of incubation. Only five compounds were found to be cytotoxic with IC50 values in the range of 3 to 6 μM (Fig. 2). Racemic preussin showed an IC50 value of 6 μM whereas for (−)-preussin an IC50 value of 3.5 μM was determined. In the literature an IC50 value of 4.1 μM is reported for the naturally occurring (+)-preussin.11 These results indicate that the absolute configurations of the preussin enantiomers are not decisive for its cytotoxicity. This is also in accordance with an investigation of the eight stereoisomers with preussin constitution that revealed that the relative and absolute configurations of the compounds have no crucial effect on their inhibitory activity against the cell growth of fission yeast.28b
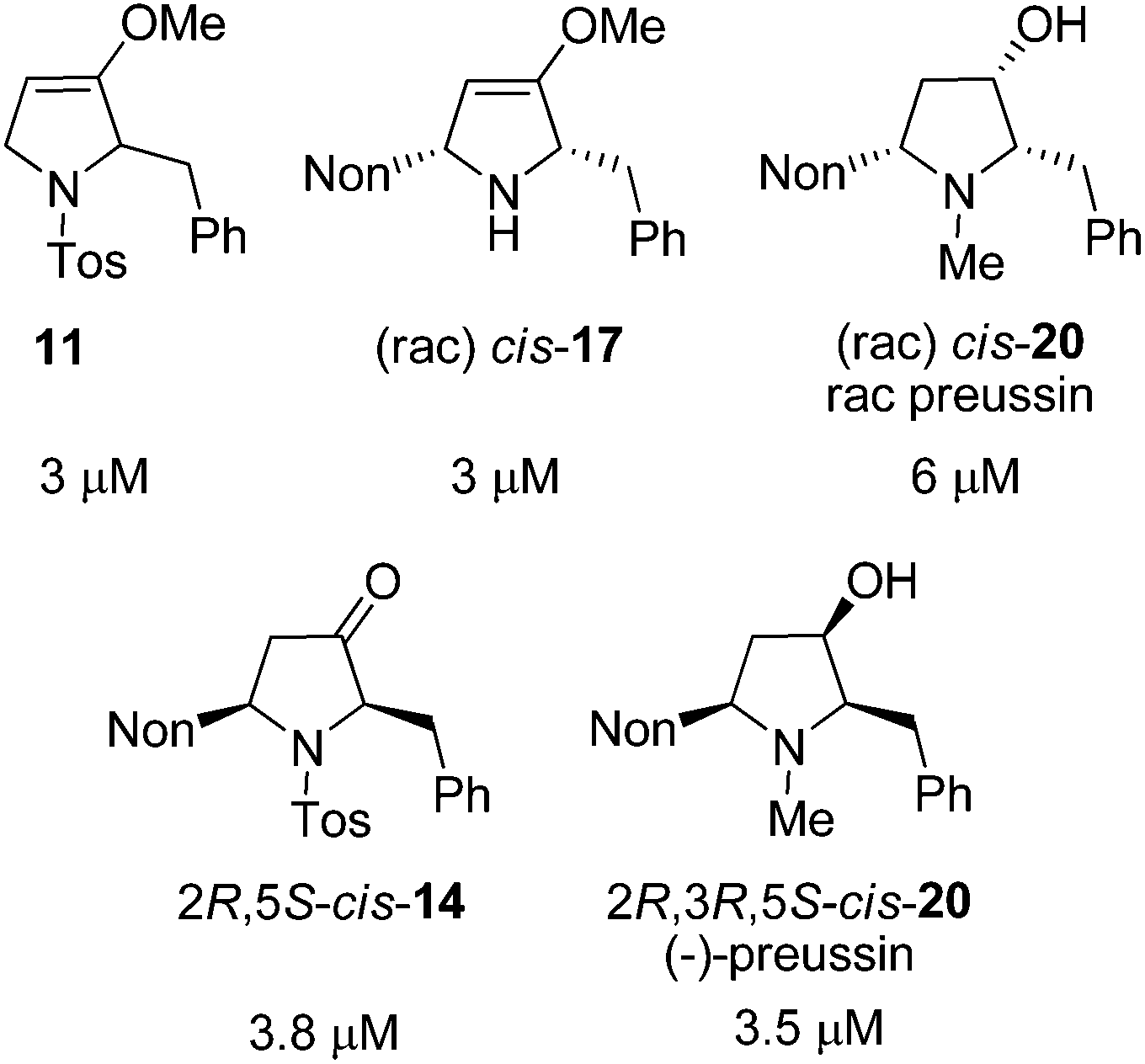 | ||
| Fig. 2 IC50 values of the cytotoxic compounds of this study against the human tumor cell line MCF-7. | ||
As mentioned in the introduction, it was reported that (+)-preussin directly intervenes in the cell cycle and inhibits the entry to the S phase.11 In order to confirm this finding, the five cytotoxic compounds depicted in Fig. 2 were additionally investigated in a cyclinE/CDK2-assay. However, none of the compounds inhibited this kinase. The cellular active compounds 11, cis-17 and cis-20 [(−)-preussin] were tested as inhibitors of other kinases, but none showed activity. Drawing a final conclusion about the mode of action of this type of compound is therefore not possible.
Conclusions
The routes to racemic preussin and its oxa-analog oxa-preussin based on 3-nonyl-substituted methoxyallene 1 as a key precursor are short and flexible. The diastereoselectivity of the addition of the lithiated allene to the electrophiles is low, but this stereodivergent step opens the way to the synthesis of diastereomeric target compounds. A synthesis of the antipode of the natural product – (−)-preussin – proceeds efficiently by starting from highly enantio-enriched 2R,5S-cis-14 and proves the absolute configuration of this compound and its precursors. On the way to synthesizing (−)-preussin, its demethylated precursor (−)-preussin C was prepared which is the optical antipode of a recently isolated natural product, (+)-preussin C. Our approach to compounds with the preussin skeleton is highly flexible since we do not follow a chiral pool but an auxiliary-based strategy. As a consequence, the 3-alkyl group of the starting allene and the C-substituent of the imine can easily be varied and a broad range of target compounds with differing substituents at C-2 and C-5 should easily be accessible, e.g. preussin B or related compounds. Racemic preussin and (−)-preussin showed IC50 values against MCF-7 tumor cells in the range of 3.5–6 μM, similar to the literature reported value for (+)-preussin. Unexpectedly, the absolute configuration of the compounds does not play an essential role in its cytotoxicity.Experimental
For general information and details of the remaining experiments, see the ESI.†Starting materials: 1-methoxydodeca-1,2-diene (1),20b imine precursor 9.24
General procedure for the addition of lithiated alkoxyallenes to aldehydes or imines (GP1)
For the generation of the lithiated alkoxyallene, the corresponding alkoxyallene was dissolved in THF and n-butyllithium was added at −40 °C. After 30 min, the solution was cooled to −80 °C and the corresponding electrophile was added. The mixture was allowed to warm up to −30 °C and stirred for the time given in the individual experiments. After quenching with saturated aqueous NaHCO3 solution (10 mL), the organic phase was separated and the aqueous phase was extracted with diethyl ether (3 × 15 mL per mmol of alkoxyallene). The combined organic phases were dried (Na2SO4), filtered and evaporated in vacuo to provide a crude product that was used directly or purified as given in the individual experiments. The yields refer to the amount of electrophile used.3-Methoxy-1-phenyltetradeca-3,4-diene-2-ol (3)
According to GP1, 1-methoxydodeca-1,2-diene (1) (0.620 g, 3.16 mmol), n-butyllithium (1.30 mL, 3.16 mmol of 2.43 M solution in hexanes) and phenylethanal (2) (0.380 g, 3.16 mmol) in THF (40 mL) provided after 2 h crude 3 (1.13 g, dr 55:45) as a light yellow oil that was cyclized without purification.
1H NMR (CDCl3, 270 MHz), major diastereomer: δ = 0.89 (t, J = 6.6 Hz, 3 H, Me), 1.15–1.40, 1.85–2.00 (2 m, 14 H, 2 H, CH2), 2.01 (d, J = 5.9 Hz, 1 H, OH), 2.88, 2.98 (AB part of the ABX system, JAB = 13.6 Hz, JAX = 7.4 Hz, JBX = 2.6 Hz, 1 H each, 1-H), 3.41 (s, 3 H, OMe), 4.28–4.44 (m, 1 H, 2-H), 5.84 (d, J = 7.0 Hz, 1 H, 5-H), 7.10–7.40 ppm (m, 5 H, Ph); the following signals of the minor diastereomer are distinguishable from those of the major isomer: δ = 2.15 (d, J = 5.9 Hz, 1 H, OH), 2.88, 3.00 (AB part of the ABX system, JAB = 13.6 Hz, JAX = 7.4 Hz, JBX = 3.3 Hz, 1 H each, 1-H), 5.89 ppm (td, J = 6.8 Hz, J = 1.7 Hz, 1 H, 5-H). 13C NMR (CDCl3, 67.9 MHz), major diastereomer: δ = 14.0 (q, Me), 22.6, 28.5, 29.1, 29.2, 29.4, 29.5, 31.4, 31.8 (8 t, CH2), 40.9 (t, C-1), 55.9 (q, OMe), 71.2 (d, C-2), 109.8 (d, C-5), 126.2, 128.1, 129.4 (3 d, Ph), 134.1 (s, C-3), 138.0 (s, Ph), 188.8 ppm (s, C-4); the following signals of the minor diastereomer are distinguishable from those of the major isomer: δ = 31.5 (t, CH2), 41.1 (t, C-1), 72.5 (d, C-2), 108.8 (d, C-5), 129.5 (d, Ph), 135.0 (s, C-3), 137.8 (s, Ph), 188.5 ppm (s, C-4). IR (film): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3460 (OH), 3030–2855 (C–H), 1950 cm−1 (C
= 3460 (OH), 3030–2855 (C–H), 1950 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CC). MS (EI, 80 eV): m/z (%) = 316 (1) [M+], 301 (1) [M+ − Me], 239 (21) [M+ − Ph], 227 (65), 91 (100) [Bn+].
CC). MS (EI, 80 eV): m/z (%) = 316 (1) [M+], 301 (1) [M+ − Me], 239 (21) [M+ − Ph], 227 (65), 91 (100) [Bn+].
General procedure for silver nitrate-promoted cyclization (GP2)
To a solution of the corresponding allenyl alcohol or amine in acetonitrile (5 mL per mmol) were added, under a stream of argon via a funnel, potassium carbonate and silver nitrate. The resulting mixture was stirred at room temperature under light exclusion for 12 h and then filtered and evaporated. The residue was dissolved in a small amount of ethyl acetate and filtered through a pad of Celite (elution with ethyl acetate). After removal of the solvents in vacuo, the crude product was purified as indicated in the individual experiments.2-Benzyl-3-methoxy-5-nonyl-2,5-dihydrofuran (4)
According to GP2, crude allenyl alcohol 3 (1.13 g, dr 55:45) in acetonitrile (40 mL) was treated with AgNO3 (0.107 g, 0.63 mmol) and K2CO3 (0.873 g, 6.30 mmol) for 12 h to provide crude 4 (1.11 g, dr 70:30) as a brown oil, which was directly hydrolyzed in the next step. The cis/trans assignments are based on the assignment of cis-5 and trans-5.
General procedure for acid-promoted hydrolysis of 2,5-dihydrofurans and 2,5-dihydropyrroles (GP3)
A solution of the corresponding substrate in THF was heated under reflux with aqueous sulfuric acid (2 M or 20%) for the time given in the individual experiments (the progress of conversion was followed by TLC). After cooling to room temperature the mixture was cautiously neutralized with a saturated aqueous NaHCO3 solution and the organic phase was separated. The aqueous phase was extracted with diethyl ether (2 × 15 mL). The combined organic phases were dried (Na2SO4), filtered and evaporated in vacuo. The purification is indicated in the individual experiments.2-Benzyl-5-nonyltetrahydrofuran-3-one (5)
According to GP3, crude 4 (1.11 g, dr 70:30) in THF (20 mL) and H2SO4 (14 mL of 2 M aqueous solution) provided after 3.5 h crude 5 (1.06 g, dr 70:30) as a yellow oil. Purification by column chromatography (silica gel, hexanes/ethyl acetate 10:1) furnished 5 (0.630 g, 66%, dr 70:30) as a colorless oil and subsequent separation of the diastereomers by HPLC (hexanes/ethyl acetate 97:3) afforded trans-5 (0.165 g, 17%, containing minor impurities) and cis-5 (0.402 g, 42%) as colorless oils. The overall yield for three steps: 59%.
Data of cis-5: 1H NMR (CDCl3, 270 MHz): δ = 0.88 (t, J = 6.9 Hz, 3 H, Me), 1.20–1.40, 1.47–1.60 1.62–1.70, (3 m, 14 H, 1 H each, CH2), 1.83 (dd, J = 17.9 Hz, J = 10.6 Hz, 1 H, 4-H), 2.45 (dd, J = 17.9 Hz, J = 5.6 Hz, 1 H, 4-H), 2.87, 3.08, 4.00 (ABX system, JAB = 14.4 Hz, JAX = 6.9 Hz, JBX = 3.9 Hz, 1 H each, PhCH2, 2-H), 4.06 (ddt, J = 10.6 Hz, J = 5.9 Hz, J = 5.6 Hz, 1 H, 5-H), 7.20–7.32 ppm (m, 5 H, Ph). 13C NMR (CDCl3, 67.9 MHz): δ = 14.1 (q, Me), 22.7, 25.2, 29.3, 29.4, 29.5*, 31.9, 35.5, 37.2, 41.2 (9 t, CH2, C-4, PhCH2,), 75.9, 81.9 (2 d, C-2, C-5), 126.5, 128.1, 129.7, 137.2 (3 d, s, Ph), 216.0 ppm (s, C-3); * signal with higher intensity. IR (film): = 3090–2855 (C–H), 1740 cm−1 (CO). MS (EI, 80 eV): m/z (%) = 302 (20) [M+], 181 (16), 91 (100) [Bn+]. C20H30O2 (302.5): calcd C 79.42, H 10.00; found C 79.34, H 9.75.
Data of trans-5: 1H NMR (CDCl3, 270 MHz): δ = 0.88 (t, J = 6.7 Hz, 3 H, Me), 1.20–1.40, 1.45–1.60, 1.60–1.75 (3 m, 14 H, 1 H each, CH2), 2.15, 2.34 (AB part of the ABX system, JAB = 18.0 Hz, JAX = 7.3 Hz, JBX = 6.8 Hz, 1 H each, 4-H), 2.90, 2.98, 4.23 (ABX system, JAB = 14.2 Hz, JAX = 6.8 Hz, JBX = 4.6 Hz, 1 H each, PhCH2, 2-H), 4.03–4.16 (m, 1 H, 5-H), 7.16–7.31 ppm (m, 5 H, Ph). 13C NMR (CDCl3, 67.9 MHz): δ = 14.1 (q, Me), 22.6, 25.4, 29.2, 29.4, 29.5*, 31.8, 35.6, 37.0, 42.6 (9 t, CH2, C-4, PhCH2), 75.5, 80.0 (2 d, C-2, C-5), 126.5, 128.3, 129.5, 137.1 (3 d, s, Ph), 216.4 ppm (s, C-3); * signal with higher intensity. IR (film): = 3065–2855 (C–H), 1755 cm−1 (CO). MS (EI, 80 eV): m/z (%) = 302 (6) [M+], 136 (16), 120 (18), 91 (100) [Bn+].
General procedure for sodium borohydride-promoted reductions of cyclic ketones (GP4)
The corresponding substrate was dissolved in dry ethanol and at 0 °C sodium borohydride was added under a stream of argon via a funnel. After stirring at room temperature for the time indicated, the mixture was treated with 2 N HCl until the precipitate dissolved and was extracted with diethyl ether (3 × 10 mL). The combined organic phases were washed with saturated aqueous NaHCO3 solution (until the extract is neutral), dried (Na2SO4), filtered and concentrated in vacuo. The crude product was purified by chromatography.(all-cis)- and (r-2,t-3,c-5)-2-Benzyl-5-nonyltetrahydrofuran-3-ol (6)
According to GP4, a solution of cis-5 (0.027 g, 0.09 mmol) and NaBH4 (0.007 g, 0.17 mmol) in EtOH (3 mL) provided, after 14 h, crude 6 (0.032 g, dr 75:25) as a colorless oil. Column chromatography (silica gel, hexanes/ethyl acetate 5:1) furnished all-cis-6 (0.021 g, 77%) as colorless crystals (m.p. 49–50 °C) and r-2,t-3,c-5-6 (0.005 g, 18%) as colorless crystals (m.p. 42–43 °C).
Data of all-cis-6: 1H NMR (CDCl3, 500 MHz): δ = 0.88 (t, J = 6.5 Hz, 3 H, Me), 1.20–1.40 (m, 14 H, CH2), 1.54 (ddd, J = 13.9 Hz, J = 6.5 Hz, J = 1.8 Hz, 1 H, 4-H), 1.51–1.58 (m, 1 H, CH2), 1.68 (d, J = 7.5 Hz, 1 H, OH), 1.66–1.78 (m, 1 H, CH2), 2.34 (ddd, J = 13.9 Hz, J = 8.2 Hz, J = 6.3 Hz, 1 H, 4-H), 3.01 (d, J = 7.1 Hz, 2 H, PhCH2), 3.75 (td, J = 7.1 Hz, J = 3.3 Hz, 1 H, 2-H), 3.79 (dq, J = 8.2 Hz, J = 6.5 Hz, 1 H, 5-H), 4.05–4.13 (m, 1 H, 3-H), 7.15–7.33 ppm (m, 5 H, Ph). 13C NMR (CDCl3, 125.8 MHz): δ = 14.1 (q, Me), 22.7, 26.2, 29.3, 29.5, 29.6*, 31.9, 36.8, (7 t, CH2), 35.1 (t, PhCH2), 41.6 (t, C-4), 72.3 (d, C-3), 77.8 (d, C-5), 83.8 (d, C-2), 126.2, 128.4, 129.2, 138.6 ppm (3 d, s, Ph); * signal with higher intensity. IR (KBr): = 3410 (O–H), 3030–2850 cm−1 (C–H). MS (EI, 80 eV): m/z (%) = 304 (10) [M+], 213 (100) [M+ − Bn], 177 (34) [M+ − C9H19]. C20H30O2 (304.5): calcd C 78.90, H 10.59; found C 78.73, H 10.57.
Data of r-2,t-3,c-5-6: 1H NMR (CDCl3, 270 MHz): δ = 0.88 (t, J = 6.6 Hz, 3 H, Me), 1.20–1.50 (m, 16 H, CH2), 1.62 (ddd, J = 13.1 Hz, J = 9.6 Hz, J = 6.6 Hz, 1 H, 4-H), 1.55–1.70 (m, 1 H, OH), 1.87 (ddd, J = 13.1 Hz, J = 5.7 Hz, J = 2.4 Hz, 1 H, 4-H), 2.71, 2.96, 3.94 (ABX system, JAB = 13.7 Hz, JAX = 7.4 Hz, JBX = 6.0 Hz, 1 H each, PhCH2, 2-H), 4.07 (dtd, J = 9.6 Hz, J = 6.0 Hz, J = 5.7 Hz, 1 H, 5-H), 4.08–4.16 (m, 1 H, 3-H), 7.20–7.35 ppm (m, 5 H, Ph). 13C NMR (CDCl3, 67.9 MHz): δ = 14.1 (q, Me), 22.7, 26.0, 29.3, 29.6*, 29.7, 31.9, 35.1, 35.8, 40.6 (9 t, CH2, C-4, PhCH2), 75.7 (d, C-3), 78.2 (d, C-5), 87.1 (d, C-2), 126.4, 128.4, 129.4, 137.9 ppm (3 d, s, Ph); * signal with higher intensity. IR (KBr): = 3410 (O–H), 3085, 3020, 2955, 2920, 2820 cm−1 (C–H). MS (EI, 80 eV): m/z (%) = 304 (13) [M+], 213 (100) [M+ − Bn], 177 (37) [M+ − C9H19]. HRMS (EI, 80 eV): calcd for C20H32O2: 304.2402; found 304.2444.
General procedure for the reduction of cyclic ketones with L-selectride (GP5)
The corresponding substrate was dissolved in THF and at −78 °C an excess of L-selectride (1 M in THF) was added dropwise. The mixture was stirred at this temperature for the time indicated and then quenched by the addition of water (10 mL). The aqueous phase was separated and the organic phase was extracted with ethyl acetate (3 × 10 mL). The combined organic phases were dried (Na2SO4) and filtered and the solvents were removed under vacuum. The crude product was purified by chromatography.(all-cis)-2-Benzyl-5-nonyltetrahydrofuran-3-ol (6)
According to GP5, a solution of cis-5 (0.036 g, 0.12 mmol) and L-selectride (0.18 mL of 1 M solution in THF, 0.18 mmol) in THF (3 mL) provided, after 3 h, crude 6 (0.061 g). Purification by column chromatography (silica gel, hexanes/ethyl acetate 5:1) afforded all-cis-6 (0.036 g, 99%) as colorless crystals (m.p. 49–50 °C). The spectroscopic data agree with those of the sample obtained before.
(r-2,t-3,t-5)- and (r-2,c-3,t-5)-2-Benzyl-5-nonyltetrahydrofuran-3-ol (6)
According to GP5, a solution of trans-5 (0.147 g, 0.49 mmol) and L-selectride (0.73 mL of 1 M solution in THF, 0.73 mmol) in THF (20 mL) provided, after 3 h, the crude product (0.250 g, dr 90:10). The ratio of diastereomers was determined by HPLC. Purification by column chromatography (silica gel, hexanes/ethyl acetate 5:1) and subsequent separation by HPLC (hexanes/ethyl acetate 5:1) afforded r-2,t-3,t-5-6 (0.091 g, 62%) as colorless crystals (m.p. 54–55 °C) and r-2,c-3,t-5-6 (0.010 g, 8%) as a colorless oil.
Data of r-2,t-3,t-5-6: H NMR (CDCl3, 270 MHz): δ = 0.88 (t, J = 6.5 Hz, 3 H, Me), 1.20–1.45 (m, 15 H, CH2), 1.55–1.73 (m, 2 H, OH, CH2), 1.70 (ddd, J = 13.4 Hz, J = 9.5 Hz, J = 4.4 Hz, 1 H, 4-H), 2.08 (dd, J = 13.4 Hz, J = 6.1 Hz, 1 H, 4-H), 2.96, 3.01, 4.05 (ABX system, JAB = 13.5 Hz, JAX = 7.7 Hz, JBX = 6.7 Hz, 1 H each, PhCH2, 2-H), 4.09–4.17 (m, 1 H, 3-H), 4.28 (dq, J = 9.5 Hz, J = 6.1 Hz, 1 H, 5-H), 7.17–7.33 ppm (m, 5 H, Ph). 13C NMR (CDCl3, 67.9 MHz): δ = 14.1 (q, Me), 22.7, 26.0, 29.3, 29.5, 29.6*, 31.9, 35.3, 36.3, 41.6 (9 t, CH2, C-4, PhCH2), 72.7 (d, C-3), 77.5 (d, C-5), 83.6 (d, C-2), 126.2, 128.4, 129.2, 138.5 ppm (3 d, s, Ph); * signal with higher intensity. IR (KBr): = 3420 (O–H), 3035–2850 cm−1 (C–H). MS (EI, 80 eV): m/z (%) = 304 (18) [M+], 213 (100) [M+ − Bn], 91 (81) [Bn+]. HRMS (EI, 80 eV): calcd for C20H32O2: 304.2402; found 304.2434.
Data of r-2,c-3,t-5-6: 1H NMR (CDCl3, 270 MHz): δ = 0.87 (t, J = 6.9 Hz, 3 H, Me), 1.15–1.73 (m, 17 H, OH, CH2), 1.57 (ddd, J = 13.2 Hz, J = 7.3 Hz, J = 5.6 Hz, 1 H, 4-H), 2.33 (dd, J = 13.2 Hz, J = 6.3 Hz, 1 H, 4-H), 2.71, 2.92 (AB part of the ABX system, JAB = 13.6 Hz, JAX = 7.2 Hz, JBX = 6.3 Hz, 1 H each, PhCH2), 3.95–4.05, 4.08–4.16 (2 m, 3 H, 2-H, 3-H, 5-H), 7.17–7.35 ppm (m, 5 H, Ph). 13C NMR (CDCl3, 67.9 MHz): δ = 14.1 (q, Me), 22.7, 26.0, 29.3, 29.5, 29.6, 29.7, 31.9, 36.6, 39.4, 40.3 (10 t, CH2, C-4, PhCH2), 75.9, 77.4, 85.0 (3 d, C-2, C-3, C-5), 126.4, 128.5, 129.3, 137.8 ppm (3 d, s, Ph). IR (film): = 3410 (O–H), 3085–2855 cm−1 (C–H). MS (EI, 80 eV): m/z (%) = 304 (11) [M+], 213 (100) [M+ − Bn], 91 (96) [Bn+]. HRMS (EI, 80 eV): calcd for C20H32O2: 304.2402; found 304.2439.
N-(3-Methoxy-1-phenyltetradeca-3,4-dien-2-yl)-p-toluenesulfonamide (12)
According to GP1, 1-methoxydodeca-1,2-diene (1) (1.32 g, 6.71 mmol), n-butyllithium (2.80 mL, 6.71 mmol of 2.40 M solution in hexanes) and 9 (1.44 g, 3.35 mmol) in THF (50 mL) provided, after 3 h, crude 12 (2.34 g, dr 50:50) as a light yellow oil. The crude product was purified by column chromatography (alumina III, hexanes/ethyl acetate 5:1) to give 12 (0.778 g), pro-cis-12 (0.569 g, 36%) as a light yellow solid (melting range 36–40 °C) and pro-trans-12 (0.566 g, 36%) as light yellow crystals (m.p. 48 °C).
Data of pro-cis-12: 1H NMR (CDCl3, 270 MHz): δ = 0.89 (t, J = 6.3 Hz, 3 H, Me), 1.00–1.40, 1.60–1.70 (2 m, 14 H, 2 H, CH2), 2.40 (s, 3 H, Tos-Me), 2.92, 2.99 (AB part of the ABX system, JAB = 13.8 Hz, JAX = 7.0 Hz, JBX = 5.9 Hz, 1 H each, 1-H), 3.20 (s, 3 H, OMe), 4.05–4.18 (m, 1 H, 2-H), 4.56 (d, J = 9.6 Hz, 1 H, NH), 5.45 (t, J = 5.9 Hz, 1 H, 5-H), 7.09 (d, J = 7.4 Hz, 2 H, Ph), 7.15–7.30 (m, 5 H, Ph, Tos), 7.66 ppm (d, J = 8.1 Hz, 2 H, Tos). 13C NMR (CDCl3, 67.9 MHz): δ = 14.1 (q, Me), 21.5 (q, Tos-Me), 22.7, 28.7, 29.2, 29.3, 29.4, 29.6, 31.2, 31.9 (8 t, CH2), 40.1 (t, C-1), 58.8, 55.4 (q, d, OMe, C-2), 109.7 (d, C-5), 126.5, 127.1, 128.1, 129.3, 129.8 (5 d, Ph, Ts), 131.2 (s, C-3), 136.5*, 142.9 (2 s, Ph, Ts), 189.0 ppm (s, C-4); * signal with higher intensity. IR (KBr): = 3255 (N–H), 3065, 2925, 2850 (C–H), 1965 (CC), 1325, 1165 cm−1 (Tos-N). MS (pos. FAB): m/z (%) = 470 (9) [M+ + H], 456 (11) [M+ + H − Me], 378 (5) [M+ − Bn], 274 (34), 154 (73), 136 (66), 91 (100) [Bn+].
Data of pro-trans-12: 1H NMR (CDCl3, 500 MHz): δ = 0.89 (t, J = 7.0 Hz, 3 H, Me), 1.00–1.45, 1.50–1.60 (2 m, 14 H, 2 H, CH2), 2.40 (s, 3 H, Tos-Me), 2.89, 2.97 (AB part of the ABX system, JAB = 13.2 Hz, JAX = 8.8 Hz, JBX = 5.7 Hz, 1 H each, 1-H), 3.06 (s, 3 H, OMe), 4.05–4.13 (m, 1 H, 2-H), 4.86 (d, J = 9.4 Hz, 1 H, NH), 5.42 (t, J = 6.7 Hz, 1 H, 5-H), 7.08 (d, J = 7.1 Hz, 2 H, Ph), 7.10–7.30 (m, 5 H, Ph, Ts), 7.67 ppm (d, J = 8.4 Hz, 2 H, Ts). 13C NMR (CDCl3, 125.8 MHz): δ = 14.2 (q, Me), 21.5 (q, Tos-Me), 22.7, 28.4, 29.1, 29.3, 29.4, 29.5, 30.9, 31.9 (8 t, CH2), 40.1 (t, C-1), 55.5, 57.4 (q, d, OMe, C-2), 108.0 (d, C-5), 126.4, 128.6, 128.9, 129.1, 130.0 (5 d, Ph, Ts), 136.9, 137.8, 142.9 (3 s, Ph, Ts)*, 189.3 ppm (s, C-4) ppm; * signal of C-3 is hidden by the aryl signals. IR (KBr): = 3270 (N–H), 3065, 2925, 2855 (C–H), 1965 (CC), 1335, 1160 cm−1 (Tos-N). MS (pos. FAB): m/z (%) = 470 (21) [M+ + H], 438 (7) [M+ − OMe], 378 (12) [M+ − Bn], 314 (22), 299 (44), 91 (100) [Bn+].
cis- and trans-2-Benzyl-3-methoxy-5-nonyl-1-tosyl-2,5-dihydro-pyrrole (13)
According to GP2, allenyl amine pro-trans-12 (0.200 g, 0.43 mmol) in acetonitrile (8 mL) was treated with AgNO3 (0.015 g, 0.09 mmol) and K2CO3 (0.118 g, 0.86 mmol) for 12 h to provide crude 13 (0.220 g, dr 55:45) as a brown oil that was purified by column chromatography (silica gel, hexanes/ethyl acetate 9:1) to give the pure diastereomers of 13 (0.174 g, 87%). Separation by HPLC (hexanes/ethyl acetate 93:7) furnished trans-13 (0.060 g, 30%) as colorless crystals (m.p. 66 °C) and cis-13 (0.083 g, 42%) as colorless crystals (m.p. 57–58 °C).
Data of cis-13: 1H NMR (CDCl3, 500 MHz): δ = 0.89 (t, J = 6.6 Hz, 3 H, Me), 0.95–1.41 (m, 16 H, CH2), 2.42 (s, 3 H, Tos-Me), 3.06, 3.17 (AB part of the ABX system, JAB = 13.6 Hz, JAX = 2.6 Hz, JBX = 5.5 Hz, 1 H each, PhCH2), 3.54 (s, 3 H, OMe), 3.94–4.02 (m, 1 H, 5-H), 4.27 (s, 1 H, 4-H), 4.37–4.43 (m, 1 H, 2-H), 7.15–7.27 (m, 5 H, Ph), 7.30, 7.71 ppm (2 d, J = 8.1 Hz, 2 H each, Tos). 13C NMR (CDCl3, 125.8 MHz): δ = 14.0 (q, Me), 21.5 (q, Tos-Me), 22.7, 28.7, 25.6, 28.7, 29.3, 29.4, 29.5, 31.9 (8 t, CH2), 38.8 (t, PhCH2), 56.6 (q, OMe), 65.0, 65.2 (2 d, C-2, C-5), 94.4 (d, C-4), 126.3, 127.5, 127.7, 129.8, 131.0 (5 d, Ph, Tos), 134.3, 136.4, 143.3 (3 s, Ph, Tos), 154.0 ppm (s, C-3). IR (KBr): = 3060, 3030, 2925, 2855 (C–H), 1670 (CC), 1345, 1165 cm−1 (Tos-N). MS (EI, 80 eV): m/z (%) = 469 (0.3) [M+], 378 (100) [M+ − Bn], 342 (20) [M+ − C9H19], 91 (100) [Bn+]. C28H39NO3S (469.3): calcd C 71.61, H 8.37, N 2.98; found C 71.62, H 8.22, N 2.91.
Data of trans-13: 1H NMR (CDCl3, 500 MHz): δ = 0.88 (t, J = 7.1 Hz, 3 H, Me), 0.90–1.31, 1.80–1.88 (2 m, 15 H, 1 H, CH2), 2.42 (s, 3 H, Tos-Me), 3.01 (dd, J = 13.7 Hz, J = 2.2 Hz, 1 H, PhCH2), 3.56 (s, 3 H, OMe), 3.74 (dd, J = 13.7 Hz, J = 4.5 Hz, 1 H, PhCH2), 3.97–4.02 (m, 1 H, 5-H), 4.23 (s, 1 H, 4-H), 4.71 (sbr, 1 H, 2-H), 7.19–7.28 (m, 3 H, Ph), 7.27 (d, J = 8.1 Hz, 2 H, Tos), 7.35 (d, J = 6.9 Hz, 2 H, Ph), 7.77 ppm (d, J = 8.1 Hz, 2 H, Tos). 13C NMR (CDCl3, 125.8 MHz): δ = 14.1 (q, Me), 21.4 (q, Tos-Me), 22.6, 25.1, 29.2, 29.4*, 29.5, 31.8, 33.9, (8 t, CH2), 38.4 (t, PhCH2), 56.3 (q, OMe), 65.7 (2 d, C-2, C-5), 94.6 (d, C-4), 126.1, 126.6, 127.6, 129.3, 130.5 (5 d, Ph, Tos), 136.5, 140.1, 142.6 (3 s, Ph, Tos), 153.8 ppm (s, C-3); signal with higher intensity. IR (KBr): = 3030, 2925, 2855 (C–H), 1675 (CC), 1335, 1160 cm−1 (Tos-N). MS (EI, 80 eV): m/z (%) = 469 (0.5) [M+], 378 (100) [M+ − Bn], 342 (12) [M+ − C9H19], 91 (50) [Bn+]. HRMS (EI, 80 eV): calcd for C28H39NO2S: 469.2651; found 469.2636. C28H39NO3S (469.3): calcd C 71.61, H 8.37, N 2.98; found C 71.65, H 8.33, N 2.95.
2-Benzyl-3-methoxy-5-nonyl-1-tosyl-2,5-dihydropyrrole (cis-13)
A solution of pro-cis-12 (0.520 g, 1.11 mmol) in 15 mL of dry DMSO was heated to 50 °C. In a stream of argon, freshly sublimed KOtBu (0.031 g, 0.28 mmol) was added via a funnel and the mixture was stirred at this temperature for 12 h. After cooling to room temperature, a saturated aqueous NaHCO3 solution (10 mL) was added. The organic phase was separated and the aqueous phase was extracted with ethyl acetate (2 × 20 mL). The combined organic phases were washed with a saturated aqueous NaHCO3 solution (2 × 20 mL), dried (Na2SO4), filtered and evaporated in vacuo to provide crude cis-13 (0.534 g) as a yellow oil. The crude product was purified by column chromatography (silica gel, hexanes/ethyl acetate) to furnish cis-13 (0.405 g, 78%) as colorless crystals (m.p. 57–58 °C). For the analytical data see above.2-Benzyl-5-nonyl-1-tosylpyrrolidin-3-one (cis-14)
According to GP3, cis-13 (0.120 g, 0.26 mmol) in THF (6 mL) and 20% aqueous H2SO4 (4 mL) provided, after 4 h, crude cis-14 (0.189 g) as a light yellow oil. Purification by column chromatography (silica gel, hexanes/ethyl acetate 7:1) furnished cis-14 (0.116 g, quant.) as colorless crystals (m.p. 63–65 °C).
1H NMR (CDCl3, 270 MHz): δ = 0.90 (t, J = 6.7 Hz, 3 H, Me), 0.90–1.40 (m, 16 H, CH2), 1.76, 2.14 (AB part of the ABX system, JAB = 18.2 Hz, JAX = 3.0 Hz, JBX = 9.3 Hz, JB,CH = 1.3 Hz, 1 H each, 4-H), 2.43 (s, 3 H, Tos-Me), 3.22, 3.27, 3.93 (ABX system, JAB = 13.5 Hz, JAX = 5.7 Hz, JBX = 4.0 Hz, 1 H each, PhCH2, 2-H), 3.77–3.88 (m, 1 H, 5-H), 7.20–7.30 (m, 5 H, Ph), 7.33, 7.73 ppm (2 d, J = 8.3 Hz, 2 H each, Tos). 13C NMR (CDCl3, 67.9 MHz): δ = 14.1 (q, Me), 21.5 (q, Tos-Me), 22.6, 25.8, 29.0, 29.2, 29.3, 29.4, 31.8, 37.0, 37.9, 42.2 (10 t, CH2, PhCH2, C-4,), 56.8, 65.4 (2 d, C-2, C-5), 126.9, 127.5, 128.2, 130.0, 130.9 (5 d, Ph, Tos), 134.1, 136.2, 144.1 (3 s, Ph, Tos), 211.3 ppm (s, C-3). IR (KBr): = 3060–2855 (CH), 1760 (CO), 1355, 1155 cm−1 (Tos-N). MS (EI, 80 eV): m/z (%) = 455 (7) [M+], 364 (100) [M+ − Bn], 155 (88) [Tos+], 91 (82) [Bn+].
2-Benzyl-5-nonyl-1-tosylpyrrolidin-3-one (trans-14)
According to GP3, trans-13 (0.060 g, 0.13 mmol) in THF (4 mL) and 20% aqueous H2SO4 (2 mL) provided, after 4 h, crude trans-14 (0.058 g, quant.) as a light yellow solid that was not purified but directly used in the epimerization experiments.1H NMR (CDCl3, 270 MHz): δ = 0.87 (t, J = 6.8 Hz, 3 H, Me), 0.90–1.35, 1.60–1.70 (2 m, 15 H, 1 H, CH2), 1.65, 1.99 (AB part of the ABX system, JAB = 17.2 Hz, JAX = 1.0 Hz, JBX = 9.0 Hz, 1 H each, 4-H), 2.44 (s, 3 H, Tos-Me), 3.12 (dd, J = 13.7 Hz, J = 3.2 Hz, 1 H, PhCH2), 3.64 (dd, J = 13.7 Hz, J = 5.1 Hz, 1 H, PhCH2), 3.94 (dd, J = 5.0 Hz, J = 3.2 Hz, 1 H, 2-H), 4.00–4.10 (m, 1 H, 5-H), 7.20–7.36 (m, 7 H, Ph, Tos), 7.77 ppm (d, J = 8.3 Hz, 2 H, Tos). 13C NMR (CDCl3, 67.9 MHz): δ = 14.1 (q, Me), 21.5 (q, Tos-Me), 22.6, 24.6, 27.4, 29.3,* 29.4, 31.8, 33.2, 37.9, 42.8 (9 t, CH2, PhCH2, C-4), 57.6, 64.8 (2 d, C-2, C-5), 127.0, 127.2, 128.3, 129.7, 130.6 (5 d, Ph, Tos), 135.3, 137.8, 143.6 (3 s, Ph, Tos), 210.9 ppm (s, C-3); * signal with higher intensity.
2-Benzyl-3-methoxy-1-methyl-5-nonyl-2,5-dihydropyrrole (cis-18)
A solution of sodium naphthalenide (0.98 mL of a 1 M solution in DME, prepared according to ref. 25) was added to a solution of cis-13 (0.153 g, 0.33 mmol) in DME (17 mL) at −78 °C. After 1.5 h the mixture was quenched at this temperature with methyl iodide (0.468 g, 3.30 mmol), and then stirred at room temperature for 1 h and concentrated in vacuo. The residue was taken up in a saturated aqueous NaHCO3 solution (20 mL) and extracted with ethyl acetate (3 × 10 mL). The combined organic phases were dried (Na2SO4), filtered and evaporated in vacuo to obtain the crude product (0.250 g) as a yellow solid. Purification by column chromatography (alumina III, hexanes/ethyl acetate 25:1 + 1% of NEt3) gave cis-18 (0.057 g, 53%) as a yellow oil.
1H NMR (CDCl3, 270 MHz): δ = 0.88 (t, J = 6.6 Hz, 3 H, Me), 1.05–1.40 (m, 16 H, CH2), 2.19 (s, 3 H, Me), 2.67, 2.95 (AB part of the ABX system, JAB = 13.2 Hz, JAX = 5.9 Hz, JBX = 4.4 Hz, 1 H each, PhCH2), 3.08–3.17, 3.46–3.53 (2 m, 1 H each, 2-H, 5-H), 3.57 (s, 3 H, OMe), 4.39 (s, 1 H, 4-H), 7.10–7.30 ppm (m, 5 H, Ph). 13C NMR (CDCl3, 67.9 MHz): δ = 14.1 (q, Me), 22.7, 25.7, 29.4, 29.6, 29.7, 30.0, 31.9, 36.8 (8 t, CH2), 40.1, 40.9 (t, q, PhCH2, NMe), 56.5 (q, OMe), 69.5, 71.7 (2 d, C-2, C-5), 95.0 (d, C-4), 125.5, 127.6, 130.1, 139.5 (3 d, s, Ph), 157.7 ppm (s, C-3). IR (film): = 3085–2775 (C–H), 1660 cm−1 (CC). MS (EI, 80 eV): m/z (%) = 329 (0.7) [M+], 328 (3) [M+ − H], 238 (100) [M+ − Bn], 202 (48) [M+ − C9H19], 91 (25) [Bn+].
2-Benzyl-1-methyl-5-nonylpyrrolidin-3-one (cis-19)
According to GP3, cis-18 (0.054 g, 0.16 mmol) in THF (4 mL) and 20% aqueous H2SO4 (4 mL) provided after 4 h crude cis-19 (0.053 g) as a yellow oil. Purification by column chromatography (alumina III, hexanes/ethyl acetate 20:1 + 1% of NEt3) furnished cis-19 (0.031 g, 60%) as a light yellow oil.
1H NMR (CDCl3, 270 MHz): δ = 0.88 (t, J = 6.7 Hz, 3 H, Me), 1.10–1.38, 1.68–1.84 (2 m, 15 H, 1 H, CH2), 1.76 (dd, J = 17.4 Hz, J = 10.3 Hz, 1 H, 4-H), 2.31 (s, 3 H, NMe), 2.38 (dd, J = 17.4 Hz, J = 5.9 Hz, 1 H, 4-H), 2.37–2.53 (m, 1 H, 5-H), 2.75 (X part of the ABX system, JAX = 5.1 Hz, JBX = 4.7 Hz, 1 H, 2-H), 2.84, 3.05 (AB part of the ABX system, JAB = 14.2 Hz, JAX = 5.1 Hz, JBX = 4.7 Hz, 1 H each, PhCH2), 7.14–7.29 ppm (m, 5 H, Ph). 13C NMR (CDCl3, 67.9 MHz): δ = 14.1 (q, Me), 22.7, 25.6, 29.3, 29.5, 29.6, 29.8, 31.9, 32.9, 35.9, 42.8 (10 t, CH2, PhCH2, C-4), 39.2 (q, NMe), 62.5, 74.3 (2 d, C-2, C-5), 126.1, 128.0, 129.7, 138.5 (3 d, s, Ph), 214.8 ppm (s, C-3). IR (film): = 3060–2855 (C–H), 1755 cm−1 (CO). MS (EI, 80 eV): m/z (%) = 315 (1.5) [M+], 224 (100) [M+ − Bn], 91 (21) [Bn+]. HRMS (EI, 80 eV): calcd for C21H33NO [M+]: 315.2562; found 315.2539; calcd for C14H26NO [M+ − Bn] 224.2014; found 224.2042.
2-Benzyl-1-methyl-5-nonylpyrrolidin-3-ol (all-cis-20), rac-preussin
According to GP5, a solution of cis-19 (0.010 g, 0.04 mmol) and L-selectride (0.06 mL of 1 M solution in THF, 0.73 mmol) in THF (4 mL) provided after 3 h the crude product (0.038 g) as a yellow oil. Purification by column chromatography (silica gel, dichloromethane/methanol 9:1) provided pure all-cis-20 (0.008 g, 80%, dr > 99:1 according to HPLC) as a light yellow oil.
1H NMR (CDCl3, 270 MHz): δ = 0.88 (t, J = 6.6 Hz, 3 H, Me), 1.15–1.36 (m, 16 H, CH2), 1.42 (ddd, J = 13.0 Hz, J = 5.5 Hz, J = 1.4 Hz, 1 H, 4-H), 1.65–1.78 (m, 1 H, OH), 2.05–2.15 (m, 1 H, 5-H), 2.18 (dd, J = 13.0 Hz, J = 6.4 Hz, 1 H, 4-H), 2.27, 2.84, 2.89 (X part and AB part of the ABX system, JAB = 13.4 Hz, JAX = 5.3 Hz, JBX = 9.1 Hz, 1 H each, 2-H, PhCH2), 2.34 (s, 3 H, NMe), 3.76–3.84 (m, 1 H, 3-H), 7.16–7.32 ppm (m, 5 H, Ph). 13C NMR (CDCl3, 67.9 MHz): δ = 22.7, 26.3, 29.3, 29.5, 29.6, 29.9, 31.9, 33.7, 34.9, 39.2 (10 t, CH2, PhCH2, C-4), 38.6 (q, NMe), 65.8, 70.4, 73.6 (3 d, C-2, C-3, C-5), 126.0, 128.4, 129.3, 139.4 ppm (3 d, s, Ph). The spectroscopic data of the sample agree with those reported in the literature.26
(2R,3R,5S)-2-Benzyl-5-nonyl-1-tosylpyrrolidin-3-ol (all-cis-21)
According to GP5, a solution of 2R,5S-cis-14 (0.055 g, 0.12 mmol) and L-selectride (0.24 mL of 1 M solution in THF, 0.24 mmol) in THF (5 mL) provided after 6 h the crude product that still contained the starting material. It was treated again with L-selectride (0.96 mL of 1 M solution in THF, 0.96 mmol) in THF (5 mL) for 3 h and finally furnished crude 2R,3R,5S-all-cis-21 (0.080 g) as a yellow oil (dr 90:10). Pre-purification by column chromatography (silica gel, hexanes/ethyl acetate 4:1) and purification by HPLC (hexanes/ethyl acetate 4:1) afforded diastereomerically pure 2R,3R,5R-all-cis-21 (0.040 g, 73%) as a colorless oil.
Optical rotation: [α]20D = −20.4 (c = 0.8, CHCl3). 1H NMR (CDCl3, 270 MHz): δ = 0.89 (t, J = 6.4 Hz, 3 H, Me), 1.20–1.40, 1.50–1.80, 1.90–2.10 (3 m, 14 H, 4 H, 1 H, CH2, 4-H, OH), 2.42 (s, 3 H, Tos-Me), 3.08, 3.24 (AB part of the ABX system, JAB = 13.9 Hz, JAX = 9.0 Hz, JBX = 3.9 Hz, 1 H each, PhCH2), 3.55–3.70 (m, 1 H, 5-H), 3.80–3.90 (m, 2 H, 2-H, 3-H), 7.17–7.30 (m, 7 H, Ph, Tos), 7.71 ppm (d, J = 8.0 Hz, 2 H, Tos). 13C NMR (CDCl3, 67.9 MHz): δ = 14.1 (q, Me), 21.5 (q, Tos-Me), 22.7, 26.5, 29.3, 29.4, 29.5, 29.6, 31.9, 37.1, 37.3, 37.8 (10 t, CH2, C-4, PhCH2), 59.9, 65.6, 71.6 (3 d, C-2, C-3, C-5), 126.3, 127.6, 128.5, 129.4, 129.7 (5 d, Ph, Tos), 135.0, 128.9, 143.4 ppm (3 s, Ph, Tos). IR (film): = 3520 (OH), 3085–2855 (C–H), 1600 (Ph), 1340, 1160 cm−1 (Tos-N). MS (EI, 80 eV): m/z (%) = 457 (0.1) [M+], 456 (0.2) [M+ − H], 366 (100) [M+ − Bn], 155 (20) [Tos+], 91 (52) [Bn+]. HRMS (EI, 80 eV): calcd for C27H39NO3S [M+]: 457.2651; found 457.2677; calcd for C27H38NO3S [M+ − H]: 456.2572; found 456.2547.
(2R,3R,5S)-2-Benzyl-5-nonylpyrrolidin-3-ol (all-cis-22)
A solution of sodium naphthalenide (0.44 mL of a 1 M solution in DME, 0.44 mmol, prepared according to ref. 25) was added to a solution of all-cis-21 (0.040 g, 0.09 mmol) in DME (7 mL) at −78 °C. After 1.5 h the mixture was quenched with water (5 mL) and concentrated in vacuo. The residue was taken up in saturated aqueous NaHCO3 solution (20 mL) and extracted with ethyl acetate (3 × 10 mL). The combined organic phases were dried (Na2SO4), filtered and evaporated in vacuo to obtain the crude product (0.090 g) as a yellow solid. Purification by column chromatography (silica gel, hexanes/ethyl acetate 1:3, +1% of NEt3) gave all-cis-22 (0.021 g, 87%) as light yellow crystals (melting range 96–100 °C; ref. 29: m.p. of the enantiomer: 101–102 °C). The spectroscopic data agree with those of the enantiomer.29
Optical rotation: [α]20D = +14.1 (c = 0.4, CHCl3); for the enantiomer [α]20D = −15.6 (c = 1.0, MeOH).291H NMR (CDCl3, 270 MHz): δ = 0.86 (t, J = 6.6 Hz, 3 H, Me), 1.20–1.38 (m, 14 H, CH2), 1.35 (ddd, J = 14.2 Hz, J = 6.7 Hz, J = 1.5 Hz, 1 H, 4-H), 1.45–1.60 (m, 2 H, CH2), 2.08–2.25 (m, 2 H, OH, NH), 2.26 (ddd, J = 14.2 Hz, J = 8.6 Hz, J = 6.2 Hz, 1 H, 4-H), 2.85, 2.94 (AB part of the ABX system, JAB = 13.0 Hz, JAX = 7.4 Hz, JBX = 6.6 Hz, 2 H, PhCH2), 2.89–3.07 (m, 2 H, 2-H, 5-H), 3.99 (ddd, J = 6.2 Hz, J = 3.4 Hz, J = 1.5 Hz, 1 H, 3-H), 7.16–7.36 ppm (m, 5 H, Ph). 13C NMR (CDCl3, 67.9 MHz): δ = 14.0 (q, Me), 22.7, 27.2, 29.3, 29.5, 29.6, 29.7, 31.9, 35.6, 37.5, 42.0 (10 t, CH2, PhCH2, C-4), 57.0, 65.7, 72.2 (3 d, C-2, C-3, C-5), 126.1, 128.5, 128.9, 139.9 ppm (3 d, s, Ph). IR (KBr): = 3420 (N–H), 3065, 3030, 2925, 2855 cm−1 (C–H). MS (EI, 80 eV): m/z (%) = 303 (0.5) [M+], 302 (2) [M+ − H], 212 (100) [M+ − Bn], 176 (22) [C9H19+], 91 (16) [Bn+]. HRMS (EI, 80 eV): calcd for C18H20NO [M+]: 303.2562; found 303.2575; calcd for C18H19NO [M+ − H]: 302.2484; found 302.2442.
(2R,3R,5S)-2-Benzyl-1-methyl-5-nonylpyrrolidin-3-ol (all-cis-20), (−)-preussin
2R,3R,5S-all-cis-22 (0.019 g, 0.06 mmol) was dissolved in a 1:1 mixture of THF/acetonitrile (4 mL) and aqueous formaldehyde (0.03 mL, 35%, 0.38 mmol) was added dropwise. After 5 min sodium cyanoborohydride (0.008 g, 0.13 mmol) and after 1 h stirring at room temperature acetic acid (0.5 mL) were added. After 30 min, solid NaHCO3 was added, using aqueous 2 N NaOH the solution pH was adjusted to pH 10 and the organic phase was separated. The aqueous phase was extracted with dichloromethane (3 × 10 mL) and the combined organic phases were dried (Na2SO4). After filtration and evaporation of solvents in vacuo the residue was purified by column chromatography (silica gel, dichloromethane/methanol 9:1) to afford pure 2R,3R,5S-all-cis-20 (0.016 g, 80%) as a yellow oil.
Optical rotation: [α]20D = −25.8 (c = 0.8, CHCl3); for the literature values see ref. 28. The NMR data agree with those of rac-preussin (see above).
MCF-7 cell proliferation assay
MCF-7 cells (ATCC HTB-22, USA) were seeded in RPMI 1640 medium (Biochrome, Germany) supplemented with fetal calf serum (10%; PAA, Austria), L-glutamine (2 mM), estradiol (0.1 nM), and insulin (1 U mL−1) at 5000 cells per well in 96-well plates. Cells were allowed to adhere for 24 h and then a fresh growth medium plus compound was added. The final concentration of DMSO was 0.5%. After four days of continuous incubation, the cells were fixed with glutaraldehyde and stained with crystal violet, and the absorbance was recorded at 595 nm using Tecan Sunrise equipment. All measurements were performed in quadruplicate. The values were normalized to the absorbance of solvent-treated cells (100%) and the absorbance of a reference plate, which was fixed at the time point of compound application (0%). Half-maximal growth inhibition (IC50) was determined as the compound concentration required to achieve 50% inhibition of cell growth.Conflicts of interest
Gerhard Siemeister is employee of Bayer AG.Acknowledgements
The generous support for this work from the Fonds der Chemischen Industrie (Kekulé fellowship for Arndt Hausherr), the Deutsche Forschungsgemeinschaft and Schering AG is most gratefully acknowledged. We thank Winfried Münch and Christiane Groneberg for their assistance in analytical work and Dr Reinhold Zimmer for his great help during the preparation of the manuscript.Notes and references
- (a) D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435–446 RSC; (b) N. Asano, R. J. Nash, R. J. Molyneux and G. W. Fleet, Tetrahedron: Asymmetry, 2000, 11, 1645–1680 CrossRef CAS; (c) J. R. Liddell, Nat. Prod. Rep., 2002, 19, 773–781 RSC; (d) H. Yoda, Curr. Org. Chem., 2002, 6, 223–243 CrossRef CAS; (e) P. Compain and O. R. Martin, Iminosugars: from Synthesis to Therapeutic Applications, Wiley-VCH, Weinheim, New York, 2007 CrossRef; (f) J. P. Michael, Nat. Prod. Rep., 2007, 24, 191–222 RSC; (g) V. Desvergnes and Y. Landais, Stud. Nat. Prod. Chem., 2014, 42, 373–419 CAS.
- Reviews on the use of alkoxyallenes in organic synthesis: (a) R. Zimmer, Synthesis, 1993, 165–178 CrossRef CAS; (b) M. Brasholz, H.-U. Reissig and R. Zimmer, Acc. Chem. Res., 2009, 42, 45–56 CrossRef CAS PubMed; (c) F. Pfrengle and H.-U. Reissig, Chem. Soc. Rev., 2010, 39, 549–557 RSC; (d) A. Nedolya, O. Tarasova, O. G. Volostnykh, A. L. Albanov, L. V. Klyba and B. A. Trofimov, Synthesis, 2011, 2192–2204 CrossRef; (e) R. Zimmer and H.-U. Reissig, Chem. Soc. Rev., 2014, 43, 2888–2903 RSC; (f) M. A. Tius, Chem. Soc. Rev., 2014, 43, 2979–3002 RSC; (g) H.-U. Reissig and R. Zimmer, Synthesis, 2017, 49, 3291–3302 CrossRef CAS.
- O. Flögel, M. G. Okala Amombo, H.-U. Reissig, G. Zahn, I. Brüdgam and H. Hartl, Chem. – Eur. J., 2003, 9, 1405–1415 CrossRef PubMed.
- S. Kaden, M. Brockmann and H.-U. Reissig, Helv. Chim. Acta, 2005, 88, 1826–1838 CrossRef CAS.
- M. A. Chowdhury and H.-U. Reissig CAS, to be published. For a synthesis of the racemic compound, see: M. A. Chowdhury and H.-U. Reissig, Synlett, 2006, 2383–2386 CAS.
- C. Parmeggiani, F. Cardona, L. Giusti, H.-U. Reissig and A. Goti, Chem. – Eur. J., 2013, 19, 10595–10604 CrossRef CAS PubMed.
- T. Pecchioli, F. Cardona, H.-U. Reissig, R. Zimmer and A. Goti, J. Org. Chem., 2017, 83, 5835–5844 CrossRef PubMed.
- R. E. Schwartz, J. Liesch, O. Hensens, L. Zitano, S. Honeycutt, G. Garrity, R. A. Fromtling, J. Onishi and R. Monagahn, J. Antibiot., 1988, 41, 1774–1779 CrossRef CAS.
- J. H. Johnson, D. W. Phillipson and A. D. Kahle, J. Antibiot., 1989, 42, 1184–1185 CrossRef CAS.
- K. Kasahara, M. Yoshida, J. Eishima, K. Takesako, T. Beppu and S. Horinouchi, J. Antibiot., 1997, 50, 267–269 CrossRef CAS.
- T. V. Achenbach, E. P. Slater, H. Brummerhop, T. Bach and R. Müller, Antimicrob. Agents Chemother., 2000, 44, 2794–2801 CrossRef CAS.
- For selected reviews on cyclin-dependent kinase inhibitors, see: (a) T. M. Sielecki, J. F. Boylan, P. A. Benfield and G. L. Trianor, J. Med. Chem., 2000, 43, 1–18 CrossRef CAS; (b) P. L. Toogood, Med. Res. Rev., 2001, 21, 487–498 CrossRef CAS PubMed; (c) A. Huwe, R. Mazitschek and A. Giannis, Angew. Chem., 2003, 115, 2170–2187 ( Angew. Chem., Int. Ed. , 2003 , 42 , 2122–2138 ) CrossRef; (d) Q. Wang, L. Su, N. Liu, L. Zhang, W. Xu and H. Fang, Curr. Med. Chem., 2011, 18, 2025–2043 CrossRef CAS; (e) M. Tutone and A. M. Almerico, Eur. J. Med. Chem., 2017, 142, 300–315 CrossRef CAS.
- T. G. Kinzy, J. W. Harper, A. Carr-Schmid, J. Kwon, M. Shastry, M. Justice and J. D. Dinman, Virology, 2002, 300, 60–70 CrossRef.
- T. Fukuda, Y. Sudoh, Y. Tsuchiya, T. Okuda and Y. Igarashi, J. Nat. Prod., 2014, 77, 813–817 CrossRef CAS PubMed.
- S. Buttachon, A. A. Ramos, A. Inácio, T. Dethoup, L. Gales, M. Lee, P. M. Costa, A. M. S. Silva, N. Sekeroglu, E. Rochan, M. M. M. Pinto, J. A. Pereira and A. Kijjoa, Mar. Drugs, 2018, 16, 119 CrossRef.
- For an excellent compilation and analysis of preussin syntheses including the year 2002, see (a) B. Basler, A. Spiegel and T. Bach, Top. Curr. Chem., 2005, 243, 1–42 CrossRef CAS (for a list of these references see the ESI†). For new syntheses including formal total syntheses since 2003 (abbreviations for the type of synthesis: CP = chiral pool; AUX = auxiliary-based; AC = asymmetric catalysis; KR = kinetic resolution; rac = synthesis of the racemic compound), see (b) S. Raghavan and M. A. Rasheed, Tetrahedron, 2003, 59, 10307–10312 CrossRef CAS (CP); (c) D. K. Dikshit, L. N. Goswami and V. S. Singh, Synlett, 2003, 1737–1739 CrossRef CAS (CP); (d) P.-Q. Huang, T.-J. Wu and Y.-P. Ruan, Org. Lett., 2003, 5, 4341–4344 CrossRef CAS (AUX); (e) F. A. Davis and J. Deng, Tetrahedron, 2004, 60, 5111–5115 CrossRef CAS (AUX); (f) S. Canova, V. Bellosta and J. Cossy, Synlett, 2004, 1811–1813 CAS (AUX); (g) M. Bertrand and J. P. Wolfe, Org. Lett., 2006, 8, 2353–23256 CrossRef PubMed (AUX); (h) N. Gogoi, J. Boruwa and N. C. Barua, Eur. J. Org. Chem., 2006, 1722–1725 CrossRef CAS (AC); (i) J. J. Caldwell, D. Craig and S. P. East, ARKIVOC, 2007, 12, 67–90 Search PubMed (CP); (j) M. Bertrand, M. L. Leathen and J. P. Wolfe, Org. Lett., 2007, 9, 457–460 CrossRef CAS (rac); (k) F. A. Davis, J. Zhang, H. Qiu and Y. Wu, Org. Lett., 2008, 10, 1433–1436 CrossRef CAS PubMed (AUX); (l) R. Chowdhury and S. K. Ghosh, Org. Lett., 2009, 11, 3270–3273 CrossRef CAS PubMed (AC); (m) J. A. Draper and R. Britton, Org. Lett., 2010, 12, 4034–4037 CrossRef CAS PubMed (AC); (n) K.-J. Xiao, Y. Wang, K.-Y. Ye and P.-Q. Huang, Chem. – Eur. J., 2010, 16, 12792–12796 CrossRef CAS PubMed (CP); (o) Y.-H. Wang, W. Ou, L. Xie, J.-L. Ye and P.-Q. Huang, Asian J. Org. Chem., 2012, 1, 359–365 CrossRef CAS (CP); (p) E. B. Arevalo-Garcia, Heterocycl. Commun., 2014, 20, 47–50 CAS (CP); (q) Y. Natori, S. Kikuchi, T. Kondo, Y. Saito, Y. Yoshimura and H. Takahata, Org. Biomol. Chem., 2014, 12, 1983–1994 RSC (CP); (r) I. G. Rosset, R. M. P. Dias, V. D. Pinho and A. C. B. Burtoloso, J. Org. Chem., 2014, 79, 6748–6453 CrossRef CAS PubMed (rac); (s) Q.-R. Zhou, X.-Y. Wei, Y.-Q. Li, D. Huang and B.-G. Wei, Tetrahedron, 2014, 70, 4799–4808 CrossRef CAS [AUX to (−)-preussin]; (t) P.-Q. Huang, H. Geng, Y.-S. Tian, Q.-R. Peng and K.-J. Xiao, Sci. China: Chem., 2015, 58, 478–482 CrossRef CAS (CP); (u) M. Buchman, K. Csatayová, S. G. Davies, A. M. Fletcher, I. T. T. Houlsby, P. M. Roberts, S. M. Rowe and J. E. Thomson, J. Org. Chem., 2016, 81, 4907–4922 CrossRef CAS (AUX); (v) Y. Natori, T. Imahori and Y. Yoshimura, J. Synth. Org. Chem., Jpn., 2016, 74, 335–349 CrossRef CAS (KR); (w) C.-M. Si, L.-P. Shao, Z.-Y. Mao, W. Zhou and B.-G. Wie, Org. Biomol. Chem., 2017, 15, 649–661 RSC (KR); (x) H.-J. Rong, J.-J. Yao, J.-K. Li and J. Qu, J. Org. Chem., 2017, 82, 5557–5565 CrossRef CAS (rac); (y) H. Mao, H. Jeong, J. Yang, H.-J. Ha and J. W. Yang, Chem. – Eur. J., 2018, 24, 2370–2374 CrossRef CAS PubMed (AC).
- Typical examples: (a) S. Hoff, L. Brandsma and J. F. Arens, Recl. Trav. Chim. Pays-Bas, 1969, 88, 609–619 CrossRef CAS; (b) D. Gange and P. Magnus, J. Am. Chem. Soc., 1978, 100, 7746–7747 CrossRef CAS; (c) S. Hormuth and H.-U. Reissig, J. Org. Chem., 1994, 59, 67–73 CrossRef CAS; (d) O. Flögel and H.-U. Reissig, Eur. J. Org. Chem., 2004, 2797–2804 CrossRef; (e) M. Brasholz and H.-U. Reissig, Angew. Chem., 2007, 119, 1659–1662 ( Angew. Chem., Int. Ed. , 2007 , 46 , 1634–1637 ) CrossRef; (f) S. Cai, B. K. Gorityala, J. Ma, M. L. Leow and X.-W. Liu, Org. Lett., 2011, 13, 1072–1075 CrossRef CAS PubMed.
- Typical examples: (a) M. G. Okala Amombo, A. Hausherr and H.-U. Reissig, Synlett, 1999, 1871–1874 CrossRef; (b) O. Flögel and H.-U. Reissig, Synlett, 2004, 895–897 Search PubMed; (c) M. G. Okala Amombo, O. Flögel, S. Kord Daoroun Kalai, S. Schoder, U. Warzok and H.-U. Reissig, Eur. J. Org. Chem., 2017, 1965–1972 CrossRef; For a review of other allenyl amine cyclizations, see: (d) B. Alcaide and P. Almendros, Adv. Synth. Catal., 2011, 353, 2561–2576 CrossRef CAS.
- A recent computational study: F. Cumine, A. Young, H.-U. Reissig, T. Tuttle and J. A. Murphy, Eur. J. Org. Chem., 2017, 6867–6871 CrossRef CAS.
- (a) A. Hausherr, B. Orschel, S. Scherer and H.-U. Reissig, Synthesis, 2001, 1377–1385 CrossRef CAS; (b) A. Hausherr and H.-U. Reissig, Synthesis, 2018, 50, 2546–2554 CrossRef CAS.
- A. Hausherr and H.-U. Reissig, Eur. J. Org. Chem., 2018, 4071–4080 CrossRef CAS.
- A. Hausherr, R. Zimmer and H.-U. Reissig, Synthesis, 2018, 50 DOI:10.1055/s-0037-1609942.
- M. Haddad, F. Dahan, J.-P. Legros, L. Lopez, M.-T. Boisdon and J. Barrans, J. Chem. Soc., Perkin Trans. 2, 1992, 671–678 RSC.
- F. Chemla, V. Hebbe and J.-F. Normant, Synthesis, 2000, 75–77 CrossRef CAS.
- A. B. Smith III, T. A. Rano, N. Chida, A. G. Sulikowski and J. L. Wood, J. Am. Chem. Soc., 1992, 114, 8008–8022 CrossRef.
- See for instance: (a) T. Bach, H. Brummerhop and K. Harms, Chem. – Eur. J., 2000, 6, 3838–3848 CrossRef CAS PubMed.
- (a) H. M. Davies, J. J. Matasi, L. M. Hodges, N. J. S. Huby, C. Thornley, N. Kong and J. H. Houser, J. Org. Chem., 1997, 62, 1095–1105 CrossRef CAS; (b) R. Lin, J. Castells and H. Rapoport, J. Org. Chem., 1998, 63, 4069–4078 CrossRef CAS.
- Optical rotations reported for (−)-preussin: (a) −21.6 (c 1.0, CHCl3): W. Deng and L. E. Overman, J. Am. Chem. Soc., 1994, 116, 11241–11250 CrossRef CAS; (b) −28.8 (c 1.01 CHCl3) M. Okue, H. Watanabe, K. Kasahara, M. Yoshida, S. Horinouchi and T. Kitahara, Biosci., Biotechnol., Biochem., 2002, 66, 1093–1096 CrossRef CAS; (c) −34.7 (c 0.5, CHCl3) in ref. 16s.
- A. Kanazawa, S. Gillet, P. Delair and A. E. Greene, J. Org. Chem., 1998, 63, 4660–4663 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectra of all products. See DOI: 10.1039/c8ob02645a |
| This journal is © The Royal Society of Chemistry 2019 |