
Thiacalixarene “knot” effect on protein binding by oligolactic acid particles†
Olga A.
Mostovaya
a,
Vladimir V.
Gorbachuk
a,
Olga B.
Bazanova
b,
Alexander V.
Gerasimov
 a,
Vladimir G.
Evtugyn
c,
Yury N.
Osin
c,
Viktor D.
Myakushev
d,
Ildar Kh.
Rizvanov
b and
Ivan I.
Stoikov
*a
a,
Vladimir G.
Evtugyn
c,
Yury N.
Osin
c,
Viktor D.
Myakushev
d,
Ildar Kh.
Rizvanov
b and
Ivan I.
Stoikov
*a
aKazan Federal University, A.M. Butlerov Chemistry Institute, Kremlevskaya Street, 18, Kazan, 420008, Russian Federation. E-mail: Ivan.Stoikov@mail.ru; Fax: +7-8432-752253; Tel: +7-8432-337463
bArbuzov Institute of Organic and Physical Chemistry, FRC Kazan Scientific Center of RAS, Kazan, Russian Federation
cInterdisciplinary Center for Analytical Microscopy of Kazan Federal University, 420008 Kazan, Russian Federation
dEnikolopov Institute of Synthetic Polymeric Materials, Russian Academy of Sciences, Moscow 117393, Russian Federation
First published on 11th December 2018
Abstract
Materials based on bioresorbable polymers and proteins have found wide ranging applications in biotechnology and in pharmaceutics to facillitate drug delivery. One of the key approaches to creating such systems is a bottom-up approach using a self-assembly method. In this article we report the effect of functionalization of oligolactic acid by a thiacalixarene “knot” on its self-organization and aggregation with proteins. Three carboxylic p-tert-butylthiacalix[4]arene derivatives were used in a copolycondensation with oligolactic acid, followed by a catalytic copolycondensation in the presence of tin(II) dioctoate. The most prominent increase in the oligolactide chain length was observed for the least sterically crowded macrocycle in the 1,3-alternate conformation. A slight decrease in the thermal stability of the (co)polyesters was also most prominently expressed for the oligolactic acid modified with a macrocycle in the 1,3-alternate conformation, which was established by simultaneous thermogravimetric and differential scanning calorimetry analysis. Linking the oligolactic acid fragments by using macrocyclic “knots” results in an increase in their affinity towards the proteins studied. Using fluorescent spectroscopy it was established that the most effective protein binding was observed for the oligolactic acids modified with the macrocyclic “knot” in the cone and partial cone conformations. A distinct feature of the binding is the formation of stable nanosized systems with transport proteins (bovine serum albumin and hemoglobin), while aggregation with lysozyme results in large precipitating structures.
Introduction
The attention of many researchers is now focused on the development of nanosystems capable of binding various biomolecules, such as, proteins and DNA, to utilize their recognition, separation and transport properties in medical applications.1,2 Proteins and colloidal systems based on these have found various applications in the area of drug delivery.3 Associations based on proteins and, in particular, enzymes, are interesting as building blocks for assembling biosensors.4 Systems based on hemoglobin were used for the development of artificial oxygen carriers.5,6 Various amphiphilic polymer materials have found wide ranging application as agents for biopolymer binding.7 Thus, polyesters were proposed for the encapsulation of proteins.8,9 Polylactides are one of the most promising materials among those mentioned owing to their low toxicity and fast biodegradation in comparison to polymers based on oil-refinery products.10 In this regard, it should be noted that not only is the polymer structure important for their medical use,11 but also its molecular weight.12,13 In particular, copolymerization of polylactic acid and its aqueous dispersion with some monomers, such as, D-gluconic acid, resorcin, calixarenes, and heparin, significantly changes the properties of the polymer.14–16 Moreover, oligolactic acid has been successfully implemented in hybrid materials, for example with magnesium hydroxide, and it exerted a significantly lower inflammatory response compared to common poly(L-lactide).17Modification of oligolactide (OLA) with a thiacalix[4]arene platform may result in the formation of new properties compared to the non-modified oligomer. Compounds based on this macrocycle have been proposed as extractants, transport agents, ionophores for ion-selective electrodes, agents for addressed drug delivery systems and so forth.18–26 The thiacalixarene platform can be rather easily functionalized with various fragments. Moreover, it favorably differs from the classic calixarene platform by allowing the possibility of selecting various spatial isomers by the pre-determined position of binding groups attached to the cyclophane platform using template synthesis.27 The relatively rigid spatial fixation of binding groups provides a high binding selectivity for various guest species. Combining all of the points mentioned above with the non-toxicity of the macrocycle28 offers wide ranging opportunities for the use of the copolymers described in various areas of industry, including use in pharmaceutical and fundamental science.
Previously, copolycondensation of the oligolactides (OLAs) with the acid based thiacalix[4]arene present in three spatial configurations (cone, partial cone and 1,3-alternate) has been described.29 The novel polymer materials obtained were utilized for modification of a glassy carbon electrode intended for the highly sensitive determination of tryptophan and thiocholine.30,31 The goal of this work is the synthesis of copolymers based on OLAs and p-tert-butylthiacalix[4]arene with a higher molecular weight and studying the influence of the macrocyclic “knot” and its structure (cone, partial cone, and 1,3-alternate) on their interaction with a number of model proteins at a physiological pH.
Experimental
Materials
Tin(II) 2-ethylhexanoate (92.5–100%) (Sigma), lysozyme from chicken egg white (Lys, Fluka Analytical), bovine serum albumin (BSA, Sigma), hemoglobin from bovine blood (Hb, Sigma), and KH2PO4 (HPLC grade) (Fisher Scientific) were used.The 1H and 13C NMR spectra (ESI,† Fig. S1 and S2) were recorded on a Bruker Avance 400 spectrometer (400.17 MHz for H-atoms) in 3–5% solutions in CDCl3 at 298 K. Residual solvent peaks were used as the internal standard.
Elemental analysis was performed on a PerkinElmer 2400 Series II instrument. The Fourier-transform infrared spectroscopy FTIR spectra (ESI,† Fig. S3) were recorded on a Spectrum 400 (PerkinElmer) infrared spectrometer.
The mass spectra (ESI,† Fig. S4–S7) were obtained on a Bruker Ultraflex III MALDI-TOF instrument. 2,5-Dihydroxybenzoic acid was used as the matrix.
Gel permeation chromatography (GPC) studies (ESI,† Fig. S8) were carried out on a GTsP chromatograph (Czech Republic) equipped with a refractometric detector and a 7.8 × 300 mm column. THF was used as the eluent. Phenogel 5 μm, pore size 10 Å (Phenomenex, USA), was utilized as a sorbent. Polystyrene standards were used for calibration.
The particle sizes and ζ-potentials were determined using a size analyzer of nanoparticles Zetasizer Nano ZS (Malvern) at 20 °C (ESI,† Fig. S9–S25). Dispersions of the polymers 5–8 (Scheme 1), obtained by self-assembly were left for 24 h after preparation and thermostated for 10 min before measurements were taken.
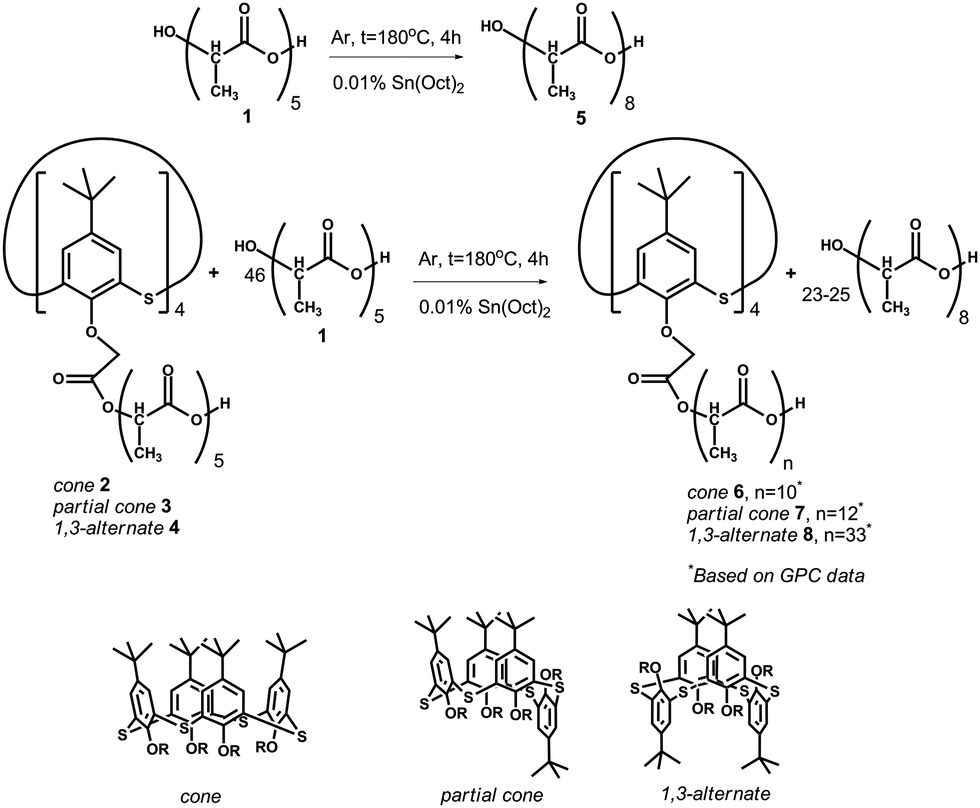 | ||
| Scheme 1 Copolycondensation of OLA with five monomer fragments (average size is given in Gorbachuk et al.29) in the presence of tin(II) dioctoate at 180 °C leads to elongation of the oligolactide chain by up to approximately eight fragments. | ||
Deionized water with a resistivity >18.0 MΩ cm was used for the preparation of solutions. It was obtained using a Millipore-Q purification system.
Transmission electron microscopy (TEM) imaging was carried out using a Hitachi HT7700 Exalens Microscope, 0.1% solutions were cast on a formvar-coated nickel grid.
Simultaneous thermogravimetry–differential scanning calorimetry (TG/DSC) was performed using a thermoanalyzer STA 449 F1 Jupiter (Netzsch) in an argon atmosphere with a total flow rate of 75 ml min−1 and a heating rate of 10 K min−1. In a typical experiment, 8.5–11.5 mg of the lactic acid oligomers synthesized were heated in a temperature range of 313–873 K in an Al crucible. The results were processed using the NETZSCH Proteus software.
Fluorescence spectra (ESI,† Fig. S26–S36) were recorded on a Fluorolog 3 luminescent spectrometer (Horiba Jobin Yvon) at an excitation wavelength of 285 nm and an emission scan range of 300–500 nm. Excitation and emission slits were equal to 3 nm for Lys, 2 nm for BSA and 4 nm for Hb. Quartz cuvettes with an optical path length of 10 mm were used. The cuvette was located in the front face position. Emission spectra were automatically corrected using the Fluorescence program. The fluorescence spectra were recorded with a 5 μM concentration of all of the proteins. The concentrations of the thiacalix[4]arene dispersions ranged from 0 to 0.5 mg ml−1 (for the compounds 6 and 7) and from 0 to 0.9 mg ml−1 (for the compounds 5 and 8). The molar concentrations of the thiacalix[4]arenes were calculated from their average molecular weights Mw (3909 Da for 6, 4251 Da for 7). The experiments were carried out at 20 °C. The temperature dependencies of the fluorescence were determined for all compounds at 5 and 35 °C.
Copolycondensation of OLA and tetraacid derivatives of p-tert-butylthiacalix[4]arene 2–4
OLA 1 or OLA 2–4 modified with derivatives of p-tert-butylthiacalix[4]arene29 (5 g) were placed into a 20 ml round-bottom flask, then a solution of 0.05 g of tin(II) dioctoate in 5 ml of chloroform was added and sonicated at room temperature in an Elmasonics 30 h ultrasonic bath (at 37 kHz, 100% of intensity) for 2 h until a homogeneous mixture was formed. The reaction mixture was then gradually heated up to 180 °C under bubbling with argon over 1 h and heated for 4 h.OLA 5 polycondensed in presence of tin dioctoate
1H NMR δH ppm: 1.48 (d, 3JHH = 6.8 Hz, CH3), 1.57 (d, 3JHH = 6.9 Hz, CH3), 4.36 (m, HO–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) (CH3)), 5.15 (m, C(O)–O–C(CH3), HOOC–C(CH3)). 13C NMR δC ppm: 16.91, 17.04, 17.16, 20.80, 20.91 (O–CH(
(CH3)), 5.15 (m, C(O)–O–C(CH3), HOOC–C(CH3)). 13C NMR δC ppm: 16.91, 17.04, 17.16, 20.80, 20.91 (O–CH(![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3)–C(O)O–), 65.92, 66.03, 66.19, 68.20, 68.70, 69.13, 69.24, 69.63 (–O–H(CH3)), 169.67, 170.17, 171.66, 174.49 (–(O)O–). MS (MALDI), m/z: calcd for [M + Na]+ 617.5, found 617.2. Elemental analysis: el. anal. calcd for lactic acid octamer (C24H34O17)%: C, 48.65; H, 5.75; found, %: C 48.71, H, 5.74. FTIR-ATR: 3508 (–OH stretch (free), lit. 3571), 3300 (OH stretch (bound)), 2996 (–CH stretch, asym.), 2946 (–CH stretch, sym., lit. 2944), 1748 (–C
H3)–C(O)O–), 65.92, 66.03, 66.19, 68.20, 68.70, 69.13, 69.24, 69.63 (–O–H(CH3)), 169.67, 170.17, 171.66, 174.49 (–(O)O–). MS (MALDI), m/z: calcd for [M + Na]+ 617.5, found 617.2. Elemental analysis: el. anal. calcd for lactic acid octamer (C24H34O17)%: C, 48.65; H, 5.75; found, %: C 48.71, H, 5.74. FTIR-ATR: 3508 (–OH stretch (free), lit. 3571), 3300 (OH stretch (bound)), 2996 (–CH stretch, asym.), 2946 (–CH stretch, sym., lit. 2944), 1748 (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch., lit. 1759), 1455 (–CH3 bend, lit. 1453), 1383, 1360 (–CH deformation including sym. and asym. bend, lit. 1382, 1362), 1293 (C–H + C–O–C), 1266 (–CO bend, lit. 1268), 1210 (rCH3 + νC–C), 1182, 1128, 1085 (C–O– stretch, lit. 1194, 1130, 1093), 1043 (C–CH3 s, C–O–C), 955, 871 (–C–C– stretch, lit. 926, 868, amorphous), 922, 823 (rCH3 + νC–C) 755, (C–C stretch, crystalline), 692 (δCO, lit. 695–715 cm−1).32 Literary values for the OLA absorption bands were taken from the review on polylactic acid.10
O stretch., lit. 1759), 1455 (–CH3 bend, lit. 1453), 1383, 1360 (–CH deformation including sym. and asym. bend, lit. 1382, 1362), 1293 (C–H + C–O–C), 1266 (–CO bend, lit. 1268), 1210 (rCH3 + νC–C), 1182, 1128, 1085 (C–O– stretch, lit. 1194, 1130, 1093), 1043 (C–CH3 s, C–O–C), 955, 871 (–C–C– stretch, lit. 926, 868, amorphous), 922, 823 (rCH3 + νC–C) 755, (C–C stretch, crystalline), 692 (δCO, lit. 695–715 cm−1).32 Literary values for the OLA absorption bands were taken from the review on polylactic acid.10
OLA 6 modified with a derivative of p-tert-butylthiacalix[4]arene in a cone conformation polycondensed in the presence of tin dioctoate
1H NMR δH ppm: 0.85–1.45 (m, C(CH3)3), 1.49 (d, 3JHH = 6.9 Hz, CH3), 1.57 (d, 3JHH = 7.1 Hz, CH3), 1.67 (d, 3JHH = 6.7 Hz, CH3), 4.36 (m, HO–C(CH3)), 5.04 (m, C(O)–O–C(CH3)–), 5.25 (m, C(O)–O–C(CH3)–, OC2CO), 7.0–7.9 (m, Ar-H). 13C NMR δC ppm: 16.91, 17.01, 20.80, 20.90 (O–CH(CH3)–C(O)O–), 66.04, 68.20, 68.71, 68.94, 69.13, 69.28, 69.50 69.93 (–O–CH(CH3)), 169.49, 169.55, 169.67, 169.75, 170.35 (–C(O)O–). FTIR-ATR (ν, cm−1): 3509 (–OH stretch (free), lit. 3571), 3300 (OH-bound), 2995 (–CH stretch, asym., lit. 2995), 2946 (–CH stretch, sym., lit. 2944), 2882 (νC–H), 1747 (–CO carbonyl stretch., lit. 1759), 1452 (–CH3 bend, lit. 1453), 1381, 1364 (–CH deformation including sym. and asym. bend, lit. 1382, 1362), 1267 (–CO bend, lit. 1268), 1182, 1127, 1083 (C–O– stretch, lit. 1194, 1130, 1093), 1043 (C–CH3 s, C–O–C), 955, 868 (–C–C– stretch, lit. 926, 868, amorphous), 829 (rCH3 + νC–C), 755 (C–C stretch, crystalline), 699 (δCO, lit. 695–715 cm−1).32 Literary values for the OLA absorption bands were taken from the review on polylactic acid.10
OLA 7 modified with a derivative of p-tert-butylthiacalix[4]arene in a partial cone conformation polycondensed in the presence of tin dioctoate
1H NMR δH ppm: 0.80–1.45 (m, C(CH3)3), 1.48 (d, 3JHH = 6.8 Hz, CH3), 1.57 (d, 3JHH = 7.0 Hz, CH3), 1.67 (d, 3JHH = 6.7 Hz, CH3), 4.19 (m, HO–C(CH3)), 4.36 (m, HO–C(CH3)), 5.04 (m, C(O)–O–C(CH3)–), 5.15 (m, C(O)–O–C(CH3)–, OC2CO), 6.8–8.0 (m, Ar-H). 13C NMR δC ppm: 16.92, 20.80, 20.91 (O–CH(H3)–C(O)O–), 65.94, 66.05, 68.19 68.70, 68.94, 69.12, 69.24, 69.39, 69.50, 69.63 (–O–H(CH3)), 169.65 (–C(O)O–). FTIR-ATR: 3509 (–OH stretch (free), lit. 3571), 3286 (OH-bound), 2996 (–CH stretch, asym., lit. 2995), 2946 (–CH stretch, sym.), 1754, 1748 (–CO carbonyl stretch., lit. 1759), 1455 (–CH3 bend, lit. 1453), 1383, 1360 (–CH deformation including sym. and asym. bend, lit. 1382, 1362), 1293 (δ2 CH), 1267 (–CO bend), 1210 (rCH3 + νC–C), 1182, 1129, 1085 (C–O– stretch, lit. 1194, 1130, 1093), 1043 (C–CH3 s, C–O–C), 956, 921, 871 (–C–C– stretch, amorphous), 829, (rCH3 + νC–C), 755 (C–C stretch, crystalline), 693 (δCO, lit. 695–715 cm−1).32 Literary values for the absorption bands of OLA were taken from the review on polylactic acid.10
OLA 8 modified with a derivative of p-tert-butylthiacalix[4]arene in a 1,3-alternate conformation polycondensed in the presence of tin dioctoate
1H NMR δH ppm: 0.82–1.45 (m, C(CH3)3), 1.48 (d, 3JHH = 6.9 Hz, CH3), 1.57 (d, 3JHH = 7.1 Hz, CH3), 4.36 (m, HO–C(CH3)), 5.25 (m, C(O)–O–C(CH3)–, OC2CO), 6.8–8.0 (m, Ar-H). 13C NMR δC ppm: 16.91, 17.17, 20.81 (O–CH(H3)–C(O)O–), 30.79 (t-Bu), 65.92, 66.04, 68.19, 68.58, 68.71, 68.97, 69.12, 69.24, 69.39, 69.50 (–O–H(CH3)), 169.36, 169.48, 169.67 (–C(O)O–). FTIR-ATR: 3509 (–OH stretch (free)), 3313 (–OH stretch (bound)), 2995 (–CH stretch, asym.), 2946 (–CH stretch, sym.), 2881 (νC–H), 1747 (–CO stretch), 1452 (–CH3 bend), 1381, 1364 (δ1 CH + δs CH3), 1267 (–CO bend, lit. 1268), 1182, 1127, 1082 (C–O– stretch, lit. 1194, 1130, 1093), 1043 (C–CH3 s, C–O–C), 956, 866 (–C–C– stretch, amorphous), 754 (δCO, C–C stretch, crystalline), 701 (δCO, lit. 695–715 cm−1).32 Literary values for the OLA absorption bands were taken from the review on polylactic acid.10
Preparation of the non-modified and modified OLAs dispersions
OLA 5 or OLAs modified with tetraacid derivatives of p-tert-butylthiacalix[4]arene 6–8 (11.4 mg) were placed into a plastic tube. Then, 10 ml of 50 mM phosphate buffer (pH = 7.4) were added and sonicated for 20 min with a Sonics VXW 450 ultrasonic microtip (power 90W, frequency 20 kHz). Dispersions were left unagitated for 24 h and then used to study the sizes and ζ-potentials of the particles or the interaction with proteins using fluorescent spectroscopy. The kinetic stability of the dispersions was determined by repetitive measurements of the sizes within 7 days of preparation.Results and discussion
Synthesis
Copolycondensation products of oligolactic acid 1 and the tetraacid derivatives of p-tert-butylthiacalix[4]arene 2–429 were introduced into a catalytic polycondensation reaction in the presence of tin(II) dioctoate.33 For homogenization, the reaction mixtures 1–4 were dissolved in chloroform. The reaction was conducted at 180 °C under argon to remove the water and solvent and to prevent the oxidation of OLA.The number of monomer fragments was estimated by end-group analysis based on the NMR 1H spectroscopy data. Two doublets at 1.57 and 1.48 ppm with 3JHH 6.9 and 6.8 Hz corresponded to the methyl group protons. The doublet at 1.48 ppm corresponds to the methyl group of the end fragment with a free hydroxyl group. The multiplet at 4.36 ppm was caused by the resonance of the methyne proton of the end-fragment with a free hydroxyl group. The other seven methyne protons of the ester and carboxylic groups appear in the spectrum as a multiplet at about 5.15 ppm. The ratio of the integral intensities of the signals in the areas of 4.36, 5.15, 1.57 and 1.48 ppm are equal to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7:3:21. This confirms the octameric structure of the chain which was further confirmed using MALDI mass spectrometry. The peak of the molecular ion with m/z 617.2, which corresponds to the octamer of lactic acid (molecular weight 594 Da) complexed with a sodium ion, is present in the mass spectrum (ESI,† Fig. S4). For the compounds 6–8, confirmation of the structure with the NMR 1H spectroscopy is troublesome owing to the low ratio of protons in the thiacalixarene fragment to those of the lactide groups making the quantitative estimation of their ratio difficult. For product 6 in the cone conformation, the whole set of signals in the range of 0.85–1.45 ppm corresponds to the tert-butyl groups. The aromatic proton signals resonate in the range 7.0–7.9 ppm. The quantity of the doublet signals caused by the protons of the methyl groups of the OLA fragment is larger than that of the unmodified OLA: 1.67 (3JHH 6.7 Hz), 1.57 (3JHH 7.1 Hz) and 1.49 ppm (3JHH 6.9 Hz). Their total intensity corresponds to 230 protons taken from the intensity of the signals in the range of 0.85–1.45 ppm, corresponding to 36 protons from the tert-butyl groups. A similar conclusion was drawn for the methyne protons of the OLA fragments. Signals for these protons in the NMR 1H spectrum are present as multiplets at 5.16 (58H) and 5.04 ppm (2H). Methylene fragments binding the OLA chains to the macrocyclic “knot” (ArOC2CO, 8H) resonate at 5.16 ppm. The multiplet with the chemical shift of 4.36 ppm corresponds to the methyne protons at the end of the lactide fragment with a free hydroxyl group present in the unreacted OLA. For the other synthesized compounds 7 and 8, the NMR 1H spectra are similar to each other. Unfortunately, the MALDI mass-spectrometry appears to not be informative for the macrocycle modified OLAs. Derivatives of p-tert-butylthiacalix[4]arene are not observed in the MALDI mass-spectra probably owing to their high molecular mass. In this regard, the study of the molecular mass of the products obtained was conducted by GPC analysis using polystyrene standards calibration. Three of the products, except for 8, have a similar polydispersity (2.27–2.67) calculated as the ratio of the weight-average to the number-average molecular weight. The lowest molecular weight and polydispersity were observed for the unmodified OLA 5 (Mn = 1060.95 Da, Mw = 2411 Da). It should be noted that the most intensive peak in the chromatogram, Mp, corresponded to the molecular weight of 2007 Da. This is in agreement with the weight-average molecular weight. The macrocyclic platform acts as a “knot” binding up to four oligolactide fragments in one molecule, therefore product 6 (cone) has a Mn = 1540, and Mw = 3909 Da (Mw/Mn = 2.54). Product 8 (1,3-alternate) has the highest molecular weight and polydispersity among the products obtained (Mn = 2570, Mw = 10532, Mw/Mn = 4.09). This can be easily explained by examining the difference in the spatial arrangement of the functional groups of the macrocyclic rim against the starting compounds. In the case of the 1,3-alternate, four carboxyl groups are at the maximal distance from each other. This allows the formation of derivatives with a higher molecular weight. This assumption was confirmed by the fact that the copolymer 7 (partial cone) with the substituents arranged in an intermediary configuration against 6 and 8 has also intermediate values for the molecular weight (Mn = 1589, Mw = 4251, Mw/Mn = 2.67). For all of the compounds studied, the Mp is closer to the weight-average molecular weight (Mp is equal to 3318 (6), 3386 (7) and 11073 Da (8)). The high value of the polydispersity and the underestimated number-average mass are caused by the unreacted OLA present in the products obtained. In addition, such a difference between the molecular weights of the unmodified OLA 5 derived from the GPC (calibration by the polystyrene standards) and the NMR 1H (end-group analysis) are usual for oligo- and polylactides.34 Although GPC leads to overestimation of the Mn and Mw values, it is doubtlessly a method that can be used to confirm the molecular weight increase pattern of the samples.
:7:3:21. This confirms the octameric structure of the chain which was further confirmed using MALDI mass spectrometry. The peak of the molecular ion with m/z 617.2, which corresponds to the octamer of lactic acid (molecular weight 594 Da) complexed with a sodium ion, is present in the mass spectrum (ESI,† Fig. S4). For the compounds 6–8, confirmation of the structure with the NMR 1H spectroscopy is troublesome owing to the low ratio of protons in the thiacalixarene fragment to those of the lactide groups making the quantitative estimation of their ratio difficult. For product 6 in the cone conformation, the whole set of signals in the range of 0.85–1.45 ppm corresponds to the tert-butyl groups. The aromatic proton signals resonate in the range 7.0–7.9 ppm. The quantity of the doublet signals caused by the protons of the methyl groups of the OLA fragment is larger than that of the unmodified OLA: 1.67 (3JHH 6.7 Hz), 1.57 (3JHH 7.1 Hz) and 1.49 ppm (3JHH 6.9 Hz). Their total intensity corresponds to 230 protons taken from the intensity of the signals in the range of 0.85–1.45 ppm, corresponding to 36 protons from the tert-butyl groups. A similar conclusion was drawn for the methyne protons of the OLA fragments. Signals for these protons in the NMR 1H spectrum are present as multiplets at 5.16 (58H) and 5.04 ppm (2H). Methylene fragments binding the OLA chains to the macrocyclic “knot” (ArOC2CO, 8H) resonate at 5.16 ppm. The multiplet with the chemical shift of 4.36 ppm corresponds to the methyne protons at the end of the lactide fragment with a free hydroxyl group present in the unreacted OLA. For the other synthesized compounds 7 and 8, the NMR 1H spectra are similar to each other. Unfortunately, the MALDI mass-spectrometry appears to not be informative for the macrocycle modified OLAs. Derivatives of p-tert-butylthiacalix[4]arene are not observed in the MALDI mass-spectra probably owing to their high molecular mass. In this regard, the study of the molecular mass of the products obtained was conducted by GPC analysis using polystyrene standards calibration. Three of the products, except for 8, have a similar polydispersity (2.27–2.67) calculated as the ratio of the weight-average to the number-average molecular weight. The lowest molecular weight and polydispersity were observed for the unmodified OLA 5 (Mn = 1060.95 Da, Mw = 2411 Da). It should be noted that the most intensive peak in the chromatogram, Mp, corresponded to the molecular weight of 2007 Da. This is in agreement with the weight-average molecular weight. The macrocyclic platform acts as a “knot” binding up to four oligolactide fragments in one molecule, therefore product 6 (cone) has a Mn = 1540, and Mw = 3909 Da (Mw/Mn = 2.54). Product 8 (1,3-alternate) has the highest molecular weight and polydispersity among the products obtained (Mn = 2570, Mw = 10532, Mw/Mn = 4.09). This can be easily explained by examining the difference in the spatial arrangement of the functional groups of the macrocyclic rim against the starting compounds. In the case of the 1,3-alternate, four carboxyl groups are at the maximal distance from each other. This allows the formation of derivatives with a higher molecular weight. This assumption was confirmed by the fact that the copolymer 7 (partial cone) with the substituents arranged in an intermediary configuration against 6 and 8 has also intermediate values for the molecular weight (Mn = 1589, Mw = 4251, Mw/Mn = 2.67). For all of the compounds studied, the Mp is closer to the weight-average molecular weight (Mp is equal to 3318 (6), 3386 (7) and 11073 Da (8)). The high value of the polydispersity and the underestimated number-average mass are caused by the unreacted OLA present in the products obtained. In addition, such a difference between the molecular weights of the unmodified OLA 5 derived from the GPC (calibration by the polystyrene standards) and the NMR 1H (end-group analysis) are usual for oligo- and polylactides.34 Although GPC leads to overestimation of the Mn and Mw values, it is doubtlessly a method that can be used to confirm the molecular weight increase pattern of the samples.
Thermal behavior of modified OLAs
The obtained OLA 5 and derivatives 6–8 were studied by TG/DSC. For all of the products studied, only the decomposition step was observed on the thermograms in the range of 256–329 °C. This corresponded to the decomposition of the OLA fragments of compound 5 and the products of the modification, 6–8. Based on the thermogravimetric analysis, the unmodified OLA 5 was found to be the most thermally stable. Its decomposition was observed in the range of 272–329 °C (Fig. 1). The product 8 was modified with the macrocycle in the 1,3-alternate conformation (256–308 °C) and was the most thermally unstable among the products studied. These results contradict the data previously obtained for OLAs with lower molecular weights, 1–4.29 Their thermal stability increased with the introduction of macrocyclic blocks. | ||
| Fig. 1 Results of the TG/DSC analysis of OLA 5 and the copolyesters of the OLAs with p-tert-butylthiacalix[4]arene in various conformations, 6–8, in a dynamic argon atmosphere of 75 ml min−1 in the temperature range of 40–600 °C. | ||
On the DSC curves, the endo-peaks have a complex shape that is most significantly expressed for compound 6 modified with the cone conformer. This two-stage DSC can be explained by the different kinetics for the chemical processes of the thermal degradation of the OLA of different molecular weight (first peak, 286.7 °C). In the case of the OLAs modified with the p-tert-butylthiacalix[4]arene derivatives in the partial cone and 1,3-alternate conformations, the average molecular weight is higher than that of 5 and 6, and only one peak (306.2 °C for 7 and 294.6 °C for 8) is present on the DSC. The values obtained correspond well to the middle of the thermal decomposition defined using the Marsh method (300.7 (5), 290.5 (6), 297.0 (7), and 282.7 °C (8)). Therefore, the thermal stability increases in the order 1,3-alternate8 < cone6 < partial cone7 < unmodified OLA 5.
The decrease in the thermal stability and the melting points observed with the introduction of the macrocyclic platform can probably be related to its disordering effect. It was shown previously that the unmodified OLA 1 of the lower molecular weight had the lowest decomposition temperature. The middle of the thermal decomposition defined using Marsh method was equal to 296 °C.29 The increased molecular weight, and hence the number of fragments, can increase the number and consecutively the energy of the inter- and intramolecular interactions (Van-der-Waals, hydrogen bonds).35 This results in the decomposition temperature of the unmodified OLA 5 increasing up to 301 °C. According to our hypothesis, the macrocyclic “knot” acts as an additional “disordering” factor reducing the interactions between the OLA chains. The above mentioned theory is confirmed by the significant effect of the thiacalix[4]arene platform on the conformations of the copolyesters and the resulting thermal properties observed.
Self-organization and interaction with model proteins
The 20 min ultrasonic treatment of the synthesized products 5–8 in 50 mM phosphate buffer (pH 7.4) resulted in the formation of dispersions that were stable for at least one week (concentration 0.19 mg ml−1). Study of the self-organization of the compounds 5–8 taken at the same concentration showed that OLA 6 formed particles of the lowest diameter (68 ± 3 nm, polydispersity index, PDI 0.28 ± 0.02). The modification of OLA by the macrocycle derivative 7 in the partial cone conformation leads to the formation of larger associates in the buffer (158 ± 8 nm, PDI 0.28 ± 0.02). The largest and most polydisperse self-associates were observed for the unmodified OLA 5 (1067 ± 187 nm, PDI 0.49 ± 0.02) while the size of OLA 8 was in the middle (418 ± 46 nm, PDI 0.30 ± 0.01). This is probably because such a significant difference in the size of the associate as observed for the compound 6 can be caused by the fundamental difference in its structure compared to that of the other compounds mentioned. In the case of 6, the polar oligolactide fragments were positioned on one side of the macrocycle. As a result, micelle-like structures are formed. Their core is composed of a hydrophobic macrocyclic fragment and an outer layer of more hydrophilic OLA fragments. For compounds 7 and 8, elongated vesicle-like structures are formed. In these compounds, the OLA chains are situated on the opposite sides of the macrocycle.36 Meanwhile, the unmodified OLA 5 forms associates by inter- and intramolecular interactions.35Determination of the ζ-potentials of the synthesized OLAs showed high values for the electrokinetic potentials that varied from −44 to −58 mV (Table 1) indicating the high stability of the appropriate colloidal systems.37 However, the stability of the systems was reduced in the presence of the proteins studied. Coagulation and fast sedimentation of Lys did not allow determination of the ζ-potentials of the system. It is interesting that both the associates formed in the presence of the Hb, which is uncharged (pI 7.1), and BSA, which is negatively charged (pI 4.8) under the reaction conditions, reduced their negative charge (Table 1). In the presence of Hb, there was a significant decrease in the ζ-potentials down to −10.8 mV for product 6 modified with a macrocycle in the cone conformation and hence a reduction of the colloidal stability was observed. The most significant changes in the particle sizes (coagulation) in the presence of the proteins studied were observed for associates with Lys (851 ± 152 nm, PDI 0.62 ± 0.05 for cone6, 2113 ± 1459 nm, PDI 0.43 ± 0.06 for partial cone7, 1970 ± 306 nm, PDI 0.39 ± 0.06 for 1,3-alternate8 and 460 ± 231 nm, PDI 0.65 ± 0.08 for unmodified OLA 5). This was expected as Lys was the only protein that was positively charged under the experimental conditions (pI 11.4). This promotes the electrostatic interaction with the negatively charged OLAs. It was unexpected that all of the synthesized compounds would interact with the negatively charged BSA (pI 4.8). In this case, an insignificant increase of the aggregate size took place (99 ± 1 nm, PDI 0.29 ± 0.01 for cone6, 150 ± 5 nm, PDI 0.26 ± 0.01 for partial cone7 and 422 ± 30 nm, PDI 0.36 ± 0.06 for 1,3-alternate8).
| Compound, 0.19 mg ml−1 | ζ-Potentials, mV | ||
|---|---|---|---|
| Self-associates | Associates with 10 μM of BSA | Associates with 10 μM of Hb | |
| OLA 5 | −44.9 ± 2.4 | −22.5 ± 1.3 | −12.1 ± 0.8 |
| Cone-OLA 6 | −44.4 ± 4.1 | −24.8 ± 0.9 | −10.8 ± 1.5 |
| Partial cone-OLA 7 | −47.4 ± 2.1 | −20.5 ± 0.7 | −11.6 ± 1.4 |
| 1,3-alternate-OLA 8 | −57.7 ± 2.5 | −20.6 ± 1.4 | −14.9 ± 1.2 |
The largest particles with the highest polydispersity were formed in the case of unmodified OLA 5 (2280 ± 194 nm, PDI 0.52 ± 0.08). It is likely that hydrophobic interactions prevail in the case of the BSA associates with modified OLAs.38 Another transport protein, neutrally charged Hb (pI 7.1), also associated with the OLAs obtained. For the cone6, particles of the size 121 ± 58 nm (79% by intensity) and a fraction of larger particles (1928 ± 1467 nm, PDI 0.60 ± 0.04) against 168 ± 13 nm, PDI 0.37 ± 0.01 for the partial cone7 were found. Particles formed by Hb, product 8 (1,3-alternate conformation) and the unmodified OLA 5 system remained polydisperse. Appropriate characteristics were found as follows: 477 ± 34 nm (PDI 0.41 ± 0.04) for 8 and 1568 ± 432 nm (76%) and 1698 ± 202 nm (PDI 0.56 ± 0.03) for 5. Therefore, stable systems of nanoscale sizes were formed from OLAs modified by the macrocyclic “knot” and the transport proteins (BSA and Hb). In the case of Lys, the systems tended to coagulate.
The results presented can be illustrated using the TEM data. The particles change their morphology in the samples 5–8. The particles of OLA 5 are amorphous and their polydispersity is quite high in accordance with the DLS data. The smallest particles with a spherical shape were observed for product 6. This agrees well with the DLS data. However, large plate-like particles were observed on the TEM images for compound 7 and particles that were close to a spherical shape were found for compound 8 based on an OLA modified with a derivative in the 1,3-alternate conformation (Fig. 2). The agreement between the DLS and TEM data observed for compound 6 is due to the macrocyclic structure. The cone conformation of 6 forms charged spherical particles (micelle-like) with hydrophobic fragments (thiacalixarene) pointed inwards and hydrophilic negatively charged fragments pointed outwards. After evaporation of the solutions in the TEM experiment, the charge on the micelles prevents their agglomeration. This leads to individual particles retaining a size similar to that in solution.
 | ||
| Fig. 2 TEM images of the particles of OLA 5 and OLAs functionalized with p-tert-butylthiacalix[4]arene derivatives 6–8. | ||
Similar behavior was observed for the unmodified OLA 5. In the case of the 1,3-alternate and partial cone (7 and 8) stereoisomers, the aggregates were found to have remarkably different structures. Hydrophilic fragments are arranged on both sides of the macrocycle. As a result, prolonged vesicles with a hydrophobic part encased between two layers of polar charged fragments were formed. Under TEM conditions, the particle agglomeration is governed by their hydrophobic interactions so that the particle size significantly increases and the aggregates visible on the TEM images are significantly larger than those in solution.39
Addition of the model proteins to the oligolactide mixtures changes the behavior, the BSA and Hb form spherical particles. In the case of Lys, micron-scale dendroid aggregates of spherical particles are formed (Fig. 3).
 | ||
| Fig. 3 TEM images of the aggregates formed by product 6 and the proteins (BSA, Hb and Lys). | ||
For final confirmation of the interaction with the model proteins, fluorescent spectroscopy was used. The protein fluorescence is caused by three amino acid residues (tyrosine, tryptophan and phenylalanine).40 Except for phenylalanine, the amino acids mentioned absorb in the range of 280–300 nm.
Therefore, tryptophan and tyrosine fluorescence can be separated by varying the excitation wavelength. At 285 nm, both residues are luminescent. This makes it possible to observe changes in the protein structure affected by the copolyesters studied. All of the proteins chosen showed significant fluorescence which decreased after addition of the synthesized products (Fig. 4). Significant changes were observed in the fluorescence spectra of all of the proteins in the presence of copolyesters 6 and 7 modified with the macrocycles in the cone and partial cone conformations, respectively. In the case of the 1,3-alternate8 and unmodified OLA 5, changes in the spectra observed in the concentration range studied appeared to be too low for their quantification. To specify the mechanism of quenching (dynamic vs. static one) the temperature dependence of fluorescence was used.40 For static quenching related to the complex formation, the constant decreases with increased temperature. Dynamic quenching is caused by collisions of the quencher molecules with a fluorophore, and this increases with the temperature. In appropriate experiments, a plot of the Stern–Volmer coordinates was used. The appropriate plot is linearized in the whole range studied and indicated a single mechanism of quenching. A decrease of the slope in the Stern–Volmer graph with increased temperature for Lys and Hb confirms the static character of the quenching. It is interesting to note that BSA showed dynamic quenching. Dynamic quenching has been reported in some articles considering BSA,41–44 but static quenching45,46 and a combination of the two quenching types47–52 prevails for this protein.
 | ||
| Fig. 4 Fluorescence spectra of BSA (5 μM) at different concentrations of compound 7 (0–0.5 mg ml−1) in phosphate buffer at pH 7.4. The inset shows the plots of F0/F versus concentration for compound 7 at different temperatures (5, 20 and 35 °C). λex = 285 nm; λem = 330 nm. | ||
Thus, association constants were determined for the model proteins with the OLAs obtained. Binding constants were determined from the analysis of the binding isotherms obtained by fluorescence spectroscopy and fitted to a 1:1 stoichiometry of binding.21,53–55 The Bindfit application developed for supramolecular systems56 was used for this purpose (ESI,† Fig. S37–S42). To confirm the proposed stoichiometry, titration results were also processed using the above mentioned binding model and for the host:guest ratio equal to 1:2 and 2:1. However, the constants were estimated in this case with much greater uncertainty. Changes in the emission spectra in the presence of oligolactides 5 and 8 for all of the proteins studied were too low to allow calculation of the constants. For the OLA modified with a macrocycle in a partial cone7 conformation, interaction with the model proteins was found to be more effective. The highest Ka was determined for 7 when it interacted with Hb (8680 M−1). The association constant with negatively charged BSA was slightly lower (Ka = 7340 M−1). The lowest association constant was obtained for the unstable system with Lys (4100 M−1). The same tendency also remained for compound 6 containing the macrocyclic fragment in the cone conformation (Ka = 6630 M−1 with Hb, 5340 M−1 with BSA and 3100 M−1 with Lys). Therefore, the OLAs modified with macrocycles in the cone and partial cone conformations bind all of the model proteins. We can conclude that the linking of the oligolactic acid fragments by a macrocyclic “knot” into a single structure leads to an increase of the affinity of the OLAs towards all of the proteins studied. Conformation of the macrocyclic fragment has a definitive impact on the protein binding ability. It is probable that decisive contribution to these interactions is provided by hydrophobic interactions between the macrocyclic fragments of the copolyesters and the protein binding sites.38
Conclusions
In the presence of tin(II) dioctoate, a catalytic copolycondensation of low-molecular weight OLA derivatives of p-tert-butylthilacalix[4]arene with the OLA increases the chain length of the OLA and its average molecular weight. The most significant increase of the chain length was observed for the macrocycle in a 1,3-alternate conformation, in which carboxylic end groups were arranged more flexibly. The structures and molecular weights of the products were determined using NMR 1H spectroscopy and GPC. All of the compounds obtained have similar thermal properties, however, an insignificant decrease in the thermal stability was observed for copolyesters that had a macrocyclic platform in their structure. All of the products formed stable dispersions in aqueous environments at a physiological pH close to that of blood (pH 7.4) and were able to interact with a number of model proteins (BSA, Lys, and Hb). This interaction was confirmed using DLS, TEM and fluorescent spectroscopy and is probably due to presence of the macrocyclic platform and its influence on the structure of the copolyester. The ability of the OLAs to bind proteins is determined by the macrocyclic “knot” conformation. All of the studied proteins were most effectively bound with the OLAs modified with a macrocycle in the partial cone conformation. The results obtained could find applications in non-toxic biodegradable systems for peptide binding and in biomaterials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Russian Science Foundation (grant no. 16-13-00005). Study of the compounds using NMR spectroscopy was funded by a subsidy of the Russian Government to support the Program of Competitive Growth of Kazan Federal University among the World's Leading Academic Centers.Notes and references
- D. K. Smith, Chem. Commun., 2018, 54, 4743 RSC.
- S. Raghav, R. Painuli and D. Kumar, Int. J. Pharmacol., 2017, 13(7), 890 Search PubMed.
- C. S. Kim, R. Mout, Y. Zhao, Y. C. Yeh, R. Tang, Y. Jeong and V. M. Rotello, Bioconjugate Chem., 2015, 26(5), 950 CrossRef CAS PubMed.
- S. A. Bhakta, E. Evans, T. E. Benavidez and C. D. Garcia, Anal. Chim. Acta, 2015, 872, 7 CrossRef CAS PubMed.
- Z. Tao and P. P. Ghoroghchian, Trends Biotechnol., 2014, 32(9), 466 CrossRef CAS PubMed.
- T. M. S. Chang, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2(4), 418 CAS.
- B. L. Banik, P. Fattahi and J. L. Brown, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 271 Search PubMed.
- M. Morales-Cruz, G. M. Flores-Fernández, M. Morales-Cruz, E. A. Orellano, J. A. Rodriguez-Martinez, M. Ruiz and K. Griebenow, Results Pharma Sci., 2012, 2, 79 CrossRef PubMed.
- S. Cohen, T. Yoshioka, M. Lucarelli, L. H. Hwang and R. Langer, Pharm. Res., 1991, 8(6), 713 CrossRef CAS.
- D. Garlotta, J. Polym. Environ., 2001, 9, 63 CrossRef CAS.
- S. Mao, C. Guo, Y. Shi and L. C. Li, Expert Opin. Drug Delivery, 2012, 9(9), 1161 CrossRef CAS PubMed.
- R. James, O. S. Manoukian and S. G. Kumbar, Adv. Drug Delivery Rev., 2016, 107, 277 CrossRef CAS PubMed.
- A. G. Andreopoulos, E. C. Hatzi and M. Doxastakis, Mater. Sci., 2000, 11(6), 393 CAS.
- K. Marcincinova-Benabdillah, M. Boustta, J. Coudane and M. Vert, Biomacromolecules, 2001, 2, 1279 CrossRef CAS PubMed.
- D. H. Go, Y. K. Joung, S. Y. Lee, M. C. Lee and K. D. Park, Macromol. Biosci., 2008, 8, 1152 CrossRef CAS PubMed.
- R. D. Dria, B. A. Goudy, K. A. Moga and P. S. Corbin, Polym. Chem., 2012, 3, 2070 RSC.
- Ch. H. Kum, Y. Cho, Y. Ki Joung, J. Choi, K. Park, S. Ho Seo, Y. S. Park, D. J. Ahn and D. K. Han, J. Mater. Chem. B, 2013, 1, 2764 RSC.
- O. A. Mostovaya, M. N. Agafonova, A. V. Galukhin, B. I. Khayrutdinov, D. Islamov, O. N. Kataeva, I. S. Antipin, A. I. Konovalov and I. I. Stoikov, J. Phys. Org. Chem., 2014, 27, 57 CrossRef CAS.
- P. L. Padnya, E. A. Andreyko, O. A. Mostovaya, I. K. Rizvanov and I. I. Stoikov, Org. Biomol. Chem., 2015, 13, 5894 RSC.
- R. R. Ibragimova, V. A. Burilov, A. R. Aimetdinov, D. A. Mironova, V. G. Evtugyn, Y. N. Osin, S. E. Solovieva and I. S. Antipin, Macroheterocycles, 2016, 9(4), 433 CrossRef CAS.
- O. A. Mostovaya, P. L. Padnya, A. A. Vavilova, D. N. Shurpik, B. I. Khairutdinov, T. A. Mukhametzyanov, A. A. Khannanov, M. P. Kutyreva and I. I. Stoikov, New J. Chem., 2018, 42, 177 RSC.
- P. Zlatušková, I. Stibor, M. Tkadlecová and P. Lhoták, Tetrahedron, 2004, 60, 11383 CrossRef.
- G. A. Evtugyn, R. V. Shamagsumova, P. L. Padnya, I. I. Stoikov and I. S. Antipin, Talanta, 2014, 127, 9 CrossRef CAS PubMed.
- E. A. Yushkova, I. I. Stoikov, J. B. Puplampu, I. S. Antipin and A. I. Konovalov, Langmuir, 2011, 27(23), 14053 CrossRef CAS PubMed.
- S. Kharchenko, A. Drapailo, S. Shishkina, O. Shishkin, M. Karavan, I. Smirnov, A. Ryabitskii and V. I. Kalchenko, Supramol. Chem., 2014, 26, 864 CrossRef CAS.
- J. Kroupa, I. Stibor, M. Pojarová, M. Tkadlecová and P. Lhoták, Tetrahedron, 2008, 64, 10075 CrossRef CAS.
- N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291 CrossRef CAS PubMed.
- F. Perret and A. W. Coleman, Chem. Commun., 2011, 47, 7303 RSC.
- V. V. Gorbachuk, O. A. Mostovaya, V. G. Evtugyn, Y. N. Osin, I. K. Rizvanov, A. V. Gerasimov and I. I. Stoikov, Macroheterocycles, 2017, 10(2), 174 CrossRef CAS.
- V. V. Gorbatchuk, A. V. Porfireva, V. B. Stepanova, Yu. I. Kuzin, V. G. Evtugyn, R. V. Shamagsumova, I. I. Stoikov and G. A. Evtugyn, Sens. Actuators, B, 2017, 246, 136 CrossRef CAS.
- A. V. Porifreva, V. V. Gorbatchuk, V. G. Evtugyn, I. I. Stoikov and G. A. Evtugyn, Electroanalysis, 2018, 30, 641 CrossRef CAS.
- G. Kister, G. Cassanas and M. Vert, Polymer, 1998, 39, 267 CrossRef CAS.
- H. R. Kricheldorf and A. Serra, Polym. Bull., 1985, 14, 497 CrossRef CAS.
- S. Rathi, J. P. Kalish, E. B. Coughlin and S. L. Hsu, Macromolecules, 2011, 44, 3410 CrossRef CAS.
- C. Funaki, S. Yamamoto, H. Hoshina, Y. Ozaki and H. Sato, Polymer, 2018, 137, 245 CrossRef CAS.
- O. A. Mostovaya, P. L. Padnya, D. N. Shurpik, A. A. Vavilova, V. G. Evtugyn, Yu. N. Osin and I. I. Stoikov, Macroheterocycles, 2017, 10, 154 CrossRef CAS.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337 CrossRef CAS PubMed.
- R. Abboud, C. Charcosset and H. Greige-Gerges, Chem. Phys. Lipids, 2017, 207, 260 CrossRef CAS PubMed.
- P. L. Padnya, I. A. Khripunova, O. A. Mostovaya, T. A. Mukhametzyanov, V. G. Evtugyn, V. V. Vorobev, Y. N. Osin and I. I. Stoikov, Beilstein J. Nanotechnol., 2017, 8, 1825 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, US, 2006, p. 954 Search PubMed.
- A. C. Bhowal and S. Kundu, Luminescence, 2018, 33, 267 CrossRef CAS PubMed.
- S. Panda, K. Kundu, A. Basaiahgari, A. P. Singh, S. Senapat and R. L. Gardas, New J. Chem., 2018, 42, 7105 RSC.
- M. B. Bolattin, S. T. Nandibewoor, S. D. Joshi, S. R. Dixit and S. A. Chimatadar, RSC Adv., 2016, 6, 63463 RSC.
- N. Kumar Das, L. Pawar, N. Kumar and S. Mukherjee, Chem. Phys. Lett., 2015, 635, 50 CrossRef.
- T. Janek, Z. Czyżnikowska, J. Łuczyński, E. J. Gudiña, L. R. Rodrigues and J. Gałęzowska, Colloids Surf., B, 2017, 159, 750 CrossRef CAS PubMed.
- V. Burilov, D. A. Mironova, R. R. Ibragimova, S. E. Solovieva and I. S. Antipin, J. Bionanosci., 2016, 6, 427 CrossRef.
- K. S. Adarsh, M. K. Singh, M. G. Kotresh, L. S. Inamdar, M. A. Shivkumar, B. N. Jagatap, B. G. Mulimani and S. R. Inamdar, Luminescence, 2017, 32, 35 CrossRef CAS PubMed.
- C. Hao, G. Xu, Y. Feng, L. Lu, W. Sun and R. Sun, Spectrochim. Acta, Part A, 2017, 184, 191 CrossRef CAS PubMed.
- S. K. Pawar, R. S. Naik and J. Seetharamappa, Anal. Bioanal. Chem., 2017, 409, 6325 CrossRef CAS PubMed.
- K. L. Zhou, D. Q. Pan, Y. Y. Lou and J. H. Shi, J. Mol. Recognit., 2018, e2716 CrossRef PubMed.
- P. Sengupta, P. S. Sardar, P. Roy, S. Dasgupta and A. Bose, J. Photochem. Photobiol., B, 2018, 183, 101 CrossRef CAS PubMed.
- S. Naveenraj, R. V. Solomon, R. V. Mangalaraja, P. Venuvanalingam, A. M. Asiri and S. Anandan, Spectrochim. Acta, Part A, 2018, 192, 34 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305 RSC.
- D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792 RSC.
- M. Tlustý, P. Slavík, M. Kohout, V. Eigner and P. Lhoták, Org. Lett., 2017, 19, 2933 CrossRef PubMed.
- Bindfit v0.5, Open Data Fit, 2016, http://supramolecular.org/bindfit/.
Footnote |
| † Electronic supplementary information (ESI) available: Full NMR, FTIR, MALDI, GPC, DLS plots, fluorescent spectra corresponding to protein titration with products synthesized. See DOI: 10.1039/c8qm00435h |
| This journal is © the Partner Organisations 2019 |