
Imprinting supramolecular chirality on silica from natural triterpenoid-regulated helical ribbons†
Yuxia
Gao
ab,
Jie
Hao
a,
Jinguo
Liu
a,
Yun
Liang
a,
Fengpei
Du
 b,
Jun
Hu
*cd and
Yong
Ju
*ad
b,
Jun
Hu
*cd and
Yong
Ju
*ad
aKey Laboratory of Bioorganic Phosphorus Chemistry and Chemical Biology, Ministry of Education, Department of Chemistry, Tsinghua University, Beijing 100084, China. E-mail: juyong@tsinghua.edu.cn
bDepartment of Applied Chemistry, College of Science, China Agricultural University, Beijing 100193, China
cBeijing Advanced Innovation Center for Soft Matter Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: jhu@mail.buct.edu.cn
dState Key Lab of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China
First published on 18th December 2018
Abstract
Supramolecular chirality has attracted significant attention because of its critical roles in the life and material sciences. In this study, a natural triterpenoid-tailored amphiphilic molecule C4-MOP was designed and synthesized, in which the pyridinium head group was modified on the skeleton of triterpenoid through an alkyl linker. The introduction of pyridinium not only offers a hydrophilic cation to promote the assembly process but also renders itself as the nucleation point to adsorb silica precursors. The results showed that by adjusting the solvent compositions and the concentration of C4-MOP, well-ordered helical nanoribbons with both right- and left-handedness were fabricated by the assembly of C4-MOP, where the hydrophilic pyridinium cations were helically displayed on the surface of ribbons. Subsequently, by taking advantage of electrostatic interactions between pyridinium and the silica precursor, the supramolecular chirality of C4-MOP was successfully imprinted onto the silica nanostructures using the gel–sol mineralization process. Our work provides a simple yet useful strategy to prepare chiral silica, which could promote the applications of chiral natural products in material science.
Introduction
Chirality is an intriguing phenomenon in organisms, where the chiral biomolecules largely direct the biological processes and the biofunctionality. To improve the understanding of the assembly mechanism of well-ordered hierarchical chiral structures in organisms, biomineralization, especially silica biomineralization, is widely used as a biomimetic “bottom-up” route to transcribe the intrinsic information from biomolecules to inorganic materials.1,2 Several classes of chiral biomolecules like DNA,3 amino acids4–8 and steroids9 have already been explored as templates for silica biomineralization, with imprinting of the unique supramolecular chirality on silica materials which have been utilized in recognition,10 chiroptical switch11 and enantioselective separation.4,6,12,13As one kind of central biomolecules in living organisms, natural triterpenoids have attracted immense attention for their prominent biological activities over the past decades.14 They are usually classified to acyclic linear type, mono-cyclic, and fused-polycyclic structures (bicyclic, tricyclic, tetracyclic, and pentacyclic) based on the carbon skeleton.15,16 Among them, pentacyclic triterpenoids are particularly appealing not only due to their significant contribution to pharmacology but also their intriguing performance in supramolecular chemistry. Recently, they were recognized as the ideal building blocks for creating supramolecular nano/micro-structures because of their inherent superiorities, such as the rigid hydrophobic skeleton, multiple functional groups, unique chiral nature, good biocompatibility, and biological activity.17,18 To date, a number of triterpenoid-derived functional molecules have been applied in emulsion,19 catalysis,20in situ heavy metal reduction,21 self-healing materials22etc. However, the study of triterpenoid-directed chiral silica mineralization replication has never been reported, which might be due to the difficulty in fabricating well-ordered chiral assemblies of triterpenoids as well as the weak interactions between the silica precursors and the triterpenoid skeletons.
Herein, we provided a simple yet useful strategy to prepare chiral silica material by imprinting triterpenoid-regulated helical ribbons. Oleanolic acid (OA), one of the typical pentacyclic triterpenoids, was selected as the central building block for constructing supramolecular chiral structures. OA features five fused structures consisting of six-member rings bearing hydroxyl and carboxylic groups at the C-3 position and C-28 position, respectively (chemical structure see Scheme S1, ESI†). The hydroxyl group was modified with a cationic hydrophilic pyridinium ion with an alkyl spacer allowing for an efficient interaction with anionic charges of the silica precursor, tetraethyl orthosilicate (TEOS).23,24 The carboxylic group was replaced by a methyl ester group to efficiently boost the hydrophobicity of OA framework. Such elaborate modification affords an ideal amphiphilic molecule C4-MOP, which owns a rigid hydrophobic skeleton and a flexible hydrophilic headgroup (Scheme S1, ESI†). The results showed that by adjusting the solvent compositions and concentration, C4-MOP could be regulated into uniform helical ribbons in methanol/water. Using them as a template, chiral silica materials have been generated successfully via a sol–gel silica mineralization process, driven by the electrostatic attractions between the positively charged pyridinium cation and the negatively charged TEOS (Scheme 1), thus realizing supramolecular chirality imprints on silica from natural chiral biomolecules.
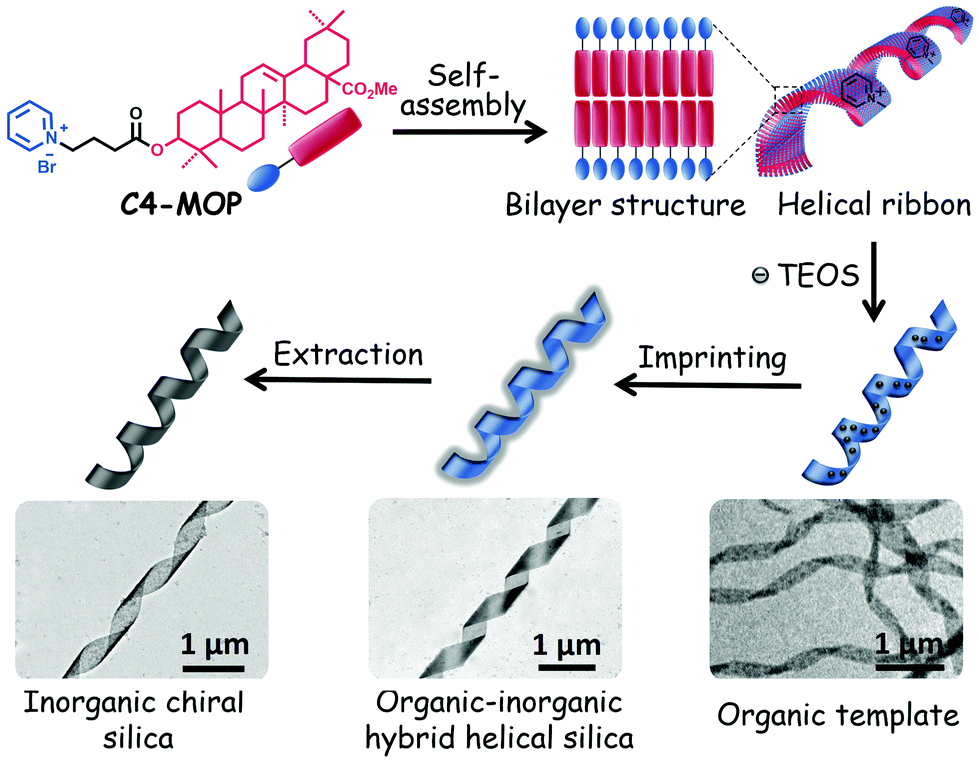 | ||
| Scheme 1 Schematic of the fabrication of helical ribbons assembled from C4-MOP and its chirality imprinting to inorganic silica. | ||
Results and discussion
Considering that a water-containing solvent is necessary for the hydrolysis of the silica precursor during the sol–gel process, the assembly of C4-MOP was investigated in methanol/water to appoint the organic chiral template. As can be seen from Table S1 (ESI†), under high concentrations of 5.0 mg mL−1, C4-MOP formed transparent gels when the percentage of water exceeded 50%, (entries 3 and 4), but the gel was not strong and tended to flow after long term inversion. As for low concentrations of 0.5 mg mL−1, C4-MOP required a higher water percentage (75%) to form the partial gel (entries 6 and 7). Evidently, the increase in either concentration or water content could accelerate the assembly process of C4-MOP.Transmission electron microscopy (TEM) was employed to investigate the gel morphologies of C4-MOP. As shown in Fig. 1a, abundant thin nanofibers with diameters around 20 nm occurred under the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. With the increase in water percentage, the nanofibers entangled with each other to generate robust ribbons with a certain helical sense (Fig. 1b, marked by arrows). When the water content reached 67%, well-ordered helical nanoribbons appeared, where the pitch was in the range of 0.8–2.0 μm, with the length at several micrometers and the width varied from 0.1 μm to 0.3 μm (Fig. 1c). It is noteworthy that both left-handed (M) and right-handed (P) nanoribbons existed in the system, as indicated by the arrows. After careful statistics, the P helices were numerically higher than the M helices. As the uneven helices could give rise to a CD signal, a strong negative Cotton effect at 264 nm was observed in Fig. 2a, which experimentally proved the excess of P helices. This hierarchical assembly process could also be verified by the morphology transformation of C4-MOP at a low concentration (0.5 mg mL−1). As shown in Fig. S1 (ESI†), the bundling and fusion process of the nanofibers into helical ribbons was clearly observed under both 1:2 and 1:3 ratio. On the basis of the amphiphilic nature of C4-MOP, the above morphology transformation was primarily attributed to the hydrophobic effects of the triterpenoid backbone, thus reducing the exposure of hydrophobic regions to the surrounding polar environment and making them energetically favourable.25,26 In this process, spontaneous symmetry breaking occurred and a particular symmetry was lost, consequently leading to the unequal numbers of mixed helices.27,28 It is well-known that the handedness of the ribbons depends not only on the molecular chirality but also the molecular orientation, which can be affected by the molecular geometries, solvents, ultrasound and ions.29 However, the factor that affected the tilt direction and symmetry breaking in our case is still unclear so far.
:1 ratio. With the increase in water percentage, the nanofibers entangled with each other to generate robust ribbons with a certain helical sense (Fig. 1b, marked by arrows). When the water content reached 67%, well-ordered helical nanoribbons appeared, where the pitch was in the range of 0.8–2.0 μm, with the length at several micrometers and the width varied from 0.1 μm to 0.3 μm (Fig. 1c). It is noteworthy that both left-handed (M) and right-handed (P) nanoribbons existed in the system, as indicated by the arrows. After careful statistics, the P helices were numerically higher than the M helices. As the uneven helices could give rise to a CD signal, a strong negative Cotton effect at 264 nm was observed in Fig. 2a, which experimentally proved the excess of P helices. This hierarchical assembly process could also be verified by the morphology transformation of C4-MOP at a low concentration (0.5 mg mL−1). As shown in Fig. S1 (ESI†), the bundling and fusion process of the nanofibers into helical ribbons was clearly observed under both 1:2 and 1:3 ratio. On the basis of the amphiphilic nature of C4-MOP, the above morphology transformation was primarily attributed to the hydrophobic effects of the triterpenoid backbone, thus reducing the exposure of hydrophobic regions to the surrounding polar environment and making them energetically favourable.25,26 In this process, spontaneous symmetry breaking occurred and a particular symmetry was lost, consequently leading to the unequal numbers of mixed helices.27,28 It is well-known that the handedness of the ribbons depends not only on the molecular chirality but also the molecular orientation, which can be affected by the molecular geometries, solvents, ultrasound and ions.29 However, the factor that affected the tilt direction and symmetry breaking in our case is still unclear so far.
 | ||
| Fig. 1 TEM images of C4-MOP assemblies (5.0 mg mL−1) in the mixed solvents of methanol and water with different volume ratios: (a) 2:1, (b) 1:1, and (c) 1:2. | ||
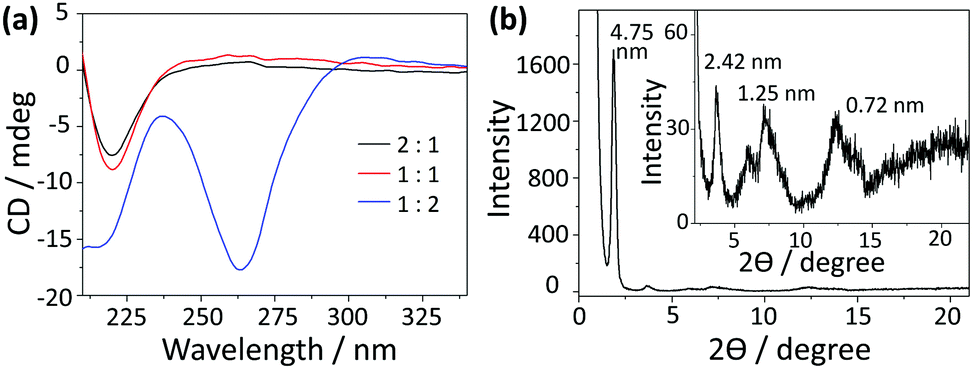 | ||
| Fig. 2 (a) CD spectra of C4-MOP assemblies in the mixed solvents of methanol and water with different volume ratios; (b) XRD pattern of helical ribbons assembled from C4-MOP (inset: the enlarged peaks between 2 degree and 22 degree). | ||
In addition, more chirality information about these assemblies is shown in Fig. 2a. The 2:1 and 1:1 systems exhibited similar CD signals, where the intensity of the negative Cotton effect at 220 nm increased slightly as the water content increased. It demonstrated that the assemblies in these systems had the similar chirality, which was consistent with the morphologies observed under TEM. Although a small percentage of helical ribbons appeared in the 1:1 system, nanofibers were still the dominant structures. Considering that the CD spectra reflected the comprehensive chirality information of the system, the chirality of the dominant morphology (nanofibers) would be detected as the final result. Thus, the 2:1 and 1:1 systems exhibited similar CD performance.30,31 The small enhancement at 220 nm was probably due to the rearrangement of C4-MOP into more ordered assemblies. It should be pointed out that the 1:2 system showed a markedly different CD spectrum, in which another negative Cotton effect at 264 nm was observed besides the one at 220 nm. This can be explained by the close chiral packing of the pyridinium groups. In comparison with the 2:1 and 1:1 systems, C4-MOP molecules in the 1:2 system were arranged in a more orderly manner on account of the increasing hydrophobic effect, leading to homogeneous helical ribbons. That is to say, the increase of water caused the differences in assemblies shapes and chiral arrays, which may affect differently the chromophores and their absorption maxima. Within the helical ribbons of the 1:2 system, pyridinium groups arranged in close proximity to each other in a chiral manner, resulting in an exciton-coupled absorption at 264 nm that was typically distinct from those nanofibers in the 2:1 and 1:1 systems.32 This phenomenon was further confirmed by the CD spectra of C4-MOP assemblies under the low concentration, in which a similar negative Cotton effect at 264 nm strengthened with water increasing (Fig. S2, ESI†). Apparently, the molecular chirality of C4-MOP has been translated into the supramolecular level via the self-assembly and amplified by the water-induced hydrophobic interactions.
To gain insight into the arrangement of C4-MOP within the helical ribbons, UV-vis, theoretical computational and X-ray diffraction (XRD) measurements were conducted. As shown in Fig. S3 (ESI†), there was no obvious change in the intensity or shape of the UV-vis spectra, and the slight enhancement in intensity at 260 nm was probably due to scattering by the larger assemblies as the water content increased.33 Moreover, four reflection peaks corresponding to d-spacing of 4.75 nm, 2.42 nm, 1.25 nm, 0.72 nm were observed in XRD patterns (Fig. 2b), which was close to the ratio of 1:1/2:1/4:1/6, verifying the lamellar structures in the helical ribbons. Since the d-spacing of 4.75 nm corresponded closely to twice that of the theoretical molecular length (2.21 nm in Fig. S4, ESI†), helical ribbons were considered to be fabricated by the basic bilayer structures with the rejection of the hydrophilic pyridinium cation on the outer surface (Scheme 1). Initially, the molecules assembled into bilayer structures under non-covalent interactions, the growth of which along the long axis resulted in fibrils; subsequently, these fibrils further stacked orderly and evolved into larger nanofibers (Fig. 1a). Driven by the increasing hydrophobic interactions, the nanofibers tend to form more stable structures to shield the hydrophobic part, thus bundling with each other to form wide ribbons (Fig. 1b), during which the intrinsic molecular chirality caused the emergence of asymmetric supramolecular morphologies (Fig. 1c). This hierarchical assembly process from the bilayer structures to fibrils, nanofibers, and helical ribbons realized the multi-step translation and magnification of chiral information.
Based on the above findings, C4-MOP showed excellent chiral assembly behaviors in methanol/water, i.e. well-ordered helical ribbons under both high concentration (5.0 mg mL−1, methanol/water = 1:2, v/v) and low concentration (0.5 mg mL−1, methanol/water = 1:3, v/v). It should be noted that the assembly under the low concentration was quite slow; hence, the samples needed to be incubated at room temperature for at least 2 weeks. Thus, the condition (5.0 mg mL−1, methanol/water = 1:2, v/v) was used to prepare the helical ribbons, which only needed 1 day before its mineralization replication from organic chirality to inorganic silica materials.
Having a negative charge, TEOS was selected as the precursor to generate silica on the surface of the helical ribbons, mainly promoted by electrostatic attractions. In general, TEOS was pre-hydrolyzed in water for 10 h and then placed in contact with helical ribbons at room temperature. Both the co-assembly and the pre-assembly methods were employed (Fig. S5 and S6, ESI†). TEM images in Fig. 3a and Fig. S7 (ESI†) showed that the chiral silica ribbons were produced successfully through both the co-assembly and the pre-assembly methods. The length of these hybrid helical ribbons was about several micrometers, the width ranged from 150 nm to 350 nm, and the pitch was around 1.7 μm, which was a duplicate of the triterpenoid-regulated helical ribbons. Therefore, it could be concluded that the hydrolysis products of TEOS did not disturb the assembly of C4-MOP and would further adsorbed on their surfaces through electrostatic attractions. Upon polymerization and mineralization, chiral nanostructures were successfully imprinted into the silica materials. In addition, the thickness of these hybrid ribbons increased with the aging time, a near 10 nm increase occurred after one week of incubation, as a result of the continuous sol–gel polymerization process (Fig. S8, ESI†). These hybrid helical structures were further verified by SEM and AFM images (Fig. 3b and c), based on which the thickness of the hybrid structures was measured as 25 nm. Notably, few spherical particles could be observed in these images, which may be caused by insufficient contact between the hydrolysis products of TEOS and the organic chiral templates.
 | ||
| Fig. 3 (a) TEM, (b) SEM, and (c) AFM images of organic–inorganic hybrid helical ribbons prepared by the pre-assembly method; HRTEM-EDS mapping images of organic–inorganic hybrid helical ribbons: (d) HRTEM image, (e) silicon mapping and (f) oxygen mapping. | ||
Besides morphologies, their elemental compositions were characterized by high-resolution transmission electron microscopy (HRTEM) images with mapping mode and energy dispersive spectroscopy (EDS). As shown in Fig. 3d–f, the silicon and oxygen elements, marked in green and yellow, respectively, showed a well-defined helical distribution. The EDS measurement further corroborated the formation of silica, where silicon and oxygen are the major constituents of the ribbons with a ratio close to 1:2 (Fig. S9, ESI†). It was unambiguous that the chiral information of the triterpenoid-regulated helical ribbons had been successfully imprinted to the inorganic silica materials.
In order to get the pure chiral inorganic silica, the C4-MOP template was removed by chemical extraction. As can be seen from Fig. 4a, the helical features still remained after the extraction, although the surface became a little rougher. The elimination of the template was further confirmed by FTIR spectra. As shown in Fig. 4b, both the C4-MOP solid and the organic–inorganic hybrid materials showed a stretching vibration at 1725 cm−1, which was attributed to a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group on C4-MOP. After extraction, the stretching vibration at 1725 cm−1 disappeared, while only the Si–O vibrations between 800 cm−1 and 1200 cm−1 were left, which was almost the same as the pure silica.34 This result strongly demonstrated the successful extraction of C4-MOP. Besides, thermogravimetric analysis (TGA) was also performed to demonstrate the elimination of C4-MOP. As shown in Fig. 4c, a 10% weight loss was observed for organic–inorganic hybrid silica and the template-removed silica materials around 50–120 °C, which may result from the adsorbed water and methanol molecules by the porous structures. Moreover, the C4-MOP solid and the organic–inorganic hybrid material exhibited a 98% and 10% weight loss between 200–520 °C, respectively, as a result of the degradation of C4-MOP. No other indication of weight loss was observed for the template-removed silica, revealing the almost complete removal of the templates.
O group on C4-MOP. After extraction, the stretching vibration at 1725 cm−1 disappeared, while only the Si–O vibrations between 800 cm−1 and 1200 cm−1 were left, which was almost the same as the pure silica.34 This result strongly demonstrated the successful extraction of C4-MOP. Besides, thermogravimetric analysis (TGA) was also performed to demonstrate the elimination of C4-MOP. As shown in Fig. 4c, a 10% weight loss was observed for organic–inorganic hybrid silica and the template-removed silica materials around 50–120 °C, which may result from the adsorbed water and methanol molecules by the porous structures. Moreover, the C4-MOP solid and the organic–inorganic hybrid material exhibited a 98% and 10% weight loss between 200–520 °C, respectively, as a result of the degradation of C4-MOP. No other indication of weight loss was observed for the template-removed silica, revealing the almost complete removal of the templates.
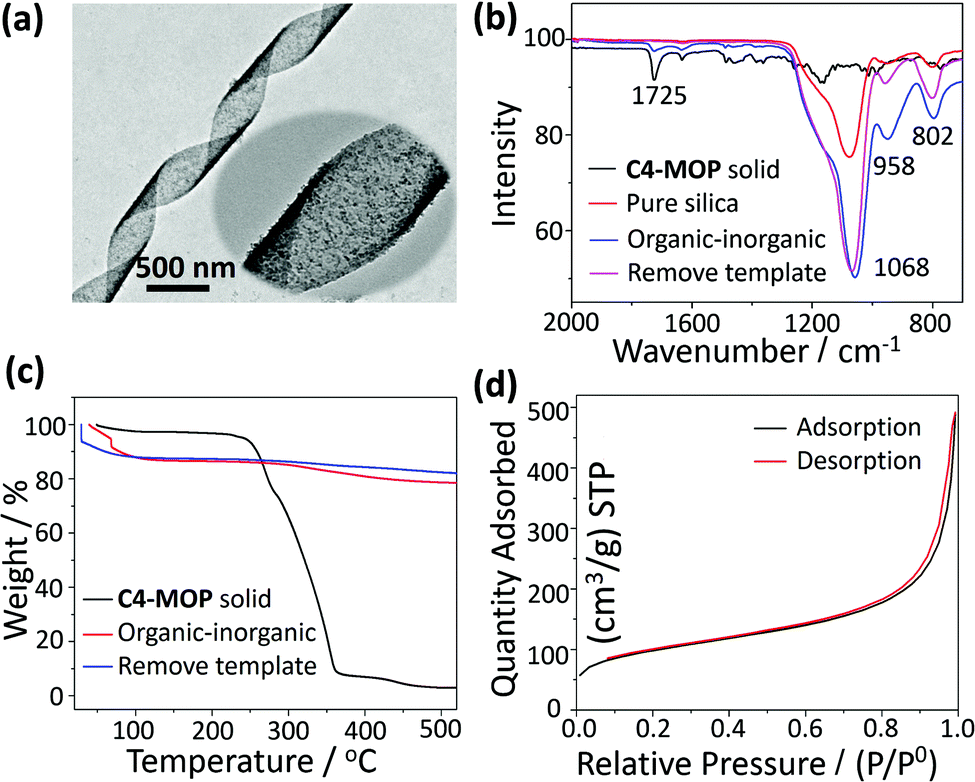 | ||
| Fig. 4 (a) TEM image of the template-removed helical silica (insert showed the enlarged view); (b) FTIR spectra of C4-MOP solid, pure silica, organic–inorganic hybrid silica and the template-removed helical silica; (c) TGA of the C4-MOP solid, organic–inorganic hybrid silica, and the template-removed helical silica; (d) nitrogen adsorption–desorption isotherms of the template-removed helical silica. | ||
Furthermore, the porosity of the template-removed chiral silica was characterized by Brunauer–Emmett–Teller (BET) surface-area measurement, during which the sample was first pre-degassed at 120 °C for 12 h, and then measured at the boiling temperature of nitrogen (77 K). As shown in Fig. 4d, nitrogen adsorption–desorption isotherms of the silica ribbons could be classified into type II, where the monolayer coverage was reached at a low relative pressure (P/P0 < 0.1) and unrestricted multilayer adsorption happened within the high P/P0 range from 0.9 to 1.0, revealing the macroporous structures of the helical silica.35,36 The wide pore-size distribution curve calculated by the Barrett–Joyner–Halenda (BJH) model further confirmed the porous nature of the silica with non-uniform pores, where most were in the range of 10–100 nm and few were around 3.80 nm (Fig. S10, ESI†). The BET surface area and pore volume were 336.85 m2 g−1 and 0.76 cm3 g−1, respectively.
A possible mechanism for the formation of helical silica ribbons is presented in Scheme 1. Initially, C4-MOP molecules assembled into the helical nanoribbons, mainly driven by hydrophobic interactions with embedding of the triterpenoid skeletons inside and projection of the pyridinium cations on the outside surface. Subsequently, these pyridinium cations served as the nucleation points to adsorb the hydrolysis products of TEOS, resulting in the polycondensation of TEOS on the surface of the helical ribbons through the electrostatic attractions. Consequently, the hydrolysis products of TEOS were aligned on the helical ribbons, imprinting the supramolecular chirality to the inorganic silica materials.
Conclusion
In summary, by introducing the pyridinium cation into the natural triterpenoid skeleton, an amphiphilic molecule C4-MOP was regulated into helical ribbons with both left- and right-handedness in methanol/water. Promoted by the electrostatic attractions, the pyridinium cations on the surface of the helical ribbons would serve as the nucleation points to adsorb the negatively charged hydrolysis products of TEOS. Upon polymerization and mineralization, uniform chiral silica materials were successfully constructed with imprinting of the supramolecular chirality on the silica from the natural triterpenoid-regulated organic helical ribbons. This work provides not only a simple and useful strategy to prepare chiral silica but can also promote the application of chiral natural products in material science.Experimental section
Materials
The following reagents were local commercial products and used as received: oleanolic acid (OA, >98.0%) was purchased from Shaanxi Huike Botanical Development Co., Ltd, Xi’an, China; 4-bromobutyric acid (98.0%), 4-dimethylaminopyridine (DMAP, 98.0%), dicyclohexylcarbodiimide (DCC, ≥99.0%), tetraethyl orthosilicate (TEOS, 99.0%), dimethylsulphoxide-d6 (DMSO-d6, >99.8%), and methanol (99.9%) were purchased from J&K Chemical Ltd, Beijing, China; methyl iodide (99.0%), pyridine (≥99.5%), dichloromethane (DCM, ≥99.5%), N,N-dimethylformamide (DMF, ≥99.0%) were supplied by the Beijing Chemical Works, Beijing, China. All organic solvents were dried and distilled before use.Instruments
1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a JOEL JNM-ECA 400 apparatus (JEOL Ltd, Tokyo, Japan); electrospray ionization mass spectrometry (ESI-MS) and high-resolution mass spectrometry (HRMS) were performed on a Bruker ESQUIRE-LC spectrometer (Bruker Daltonik, Bremen, Germany) and a quadrupole time-of-flight (Q-ToF) instrument, respectively.Scanning electron microscopy (SEM) images were taken on a SU-8010 instrument (Hitachi Ltd, Tokyo, Japan) with an accelerating voltage of 10 kV; transmission electron microscopy (TEM) images were obtained using a Hitachi H-7650B electron microscope (Hitachi Ltd, Tokyo, Japan), operating at the accelerating voltage of 80 kV; high resolution transmission electron microscopy (HRTEM) images were taken on JEM 2100F (JEOL Ltd, Tokyo, Japan) with an accelerating voltage of 120 kV; samples were prepared by drop-casting the sol on the carbon-coated copper grid, and allowed to stand for 1 min. Then, the excess sample solution was wiped away by the filter paper and dried in air; atomic force microscope (AFM) images were acquired continuously in the tapping mode using SPM-9600 (Shimadzu Co., Kyoto, Japan) under ambient conditions. Samples were placed on a freshly-cleaved mica plate and dried in air before the test.
UV-vis spectra were recorded on a TU-1901 instrument (Beijing Persee General Instrument Co. Ltd, Beijing, China); circular dichroism (CD) spectra were recorded on a Pistar π-180 instrument (Applied Photophysics Ltd, Leatherhead, UK) with a 150 W xenon lamp as the light source; X-ray diffraction (XRD) measurements were recorded on a Rigaku D/max 2500v X-ray diffractometer (Rigaku Co., Tokyo, Japan) with CuKα radiation (λ = 1.5406 Å), operating at a voltage of 45 kV and a current of 100 mA; FTIR spectra were recorded on a Perkin Elmer spectrum 100 FTIR spectrometer (Perkin Elmer, Waltham, MA, USA) in situ.
Energy dispersive spectroscopy (EDS) chemical element analysis and mapping were performed on a JEM 2100F (JEOL Ltd, Tokyo, Japan) with an accelerating voltage of 120 kV; thermogravimetric analysis (TGA) measurements were performed with a TA Instruments Q50 thermal gravimetric analyzer (TA Instruments, New Castle, USA) under the nitrogen atmosphere. 10 mg sample was placed in an aluminium crucible and heated from room temperature to 520 °C, at a heating rate of 10 °C min−1.
Nitrogen adsorption/desorption isotherms were recorded on a Micromeritics ASAP2020 instrument (Micromeritics Instrument Co., USA). The sample was predegassed at 120 °C for 12 h before the experiment, and then measured at the boiling temperature of nitrogen (77 K). The specific surface area was calculated according to the Brunauer–Emmett–Teller (BET) method, whereas the pore volume and pore size were estimated by the Barrett–Joyner–Halenda (BJH) method.
The theoretical calculation was realized using ChemBio 3D Ultra software (CambridgeSoft, USA).
C), 4.63 (t, 2H, J = 7.36 Hz, N–CH2), 4.38 (m, 1H, 3-CH–O–), 3.53 (s, 3H, OCH3), 2.45 (t, 2H, J = 8.72 Hz, OC–CH2), 2.21 (m, 2H, NCH2C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2), 1.10, 0.88, 0.80, 0.79, 0.65 (s, 7 × 3H, 7 × CH3); 13C NMR (100 MHz, CDCl3, ppm): 178.33 (O
2), 1.10, 0.88, 0.80, 0.79, 0.65 (s, 7 × 3H, 7 × CH3); 13C NMR (100 MHz, CDCl3, ppm): 178.33 (O![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) –OCH3), 172.2 (3-O–CO), 145.6 (CH–), 145.3, 143.9, 128.6 (5C, pyridinium-C), 122.2 (HC–), 81.9 (3-CH–O), 60.8, 55.3, 51.6, 47.6, 46.7, 45.9, 41.7, 41.3, 39.3, 38.1, 37.8, 36.9, 33.9, 33.1, 32.6, 32.4, 30.7, 30.6, 28.3, 27.7, 27.3, 26.0, 23.7, 23.5, 23.1, 18.3, 16.8, 15.4.
–OCH3), 172.2 (3-O–CO), 145.6 (CH–), 145.3, 143.9, 128.6 (5C, pyridinium-C), 122.2 (HC–), 81.9 (3-CH–O), 60.8, 55.3, 51.6, 47.6, 46.7, 45.9, 41.7, 41.3, 39.3, 38.1, 37.8, 36.9, 33.9, 33.1, 32.6, 32.4, 30.7, 30.6, 28.3, 27.7, 27.3, 26.0, 23.7, 23.5, 23.1, 18.3, 16.8, 15.4.
(i) Preparation of pre-hydrolyzed TEOS solutions. TEOS was placed in aqueous solution (pH 5.8) with the ratio of 15% (v/v) under constant agitation for 10 h before transcription experiments;
(ii) Sol–gel transcription. Two methods were used to transcribe the helical sense of the template to the silica: the co-assembly method and the pre-assembly method. The only difference between them is the sequence of replicating processes. As for the co-assembly method, the formation of organic helical ribbons and the sol–gel polymerization process happened simultaneously. That is, 5 mg C4-MOP was initially dissolved in 335 μL methanol to form a homogeneous solution, and then 500 μL pre-hydrolyzed silica precursor suspension and 165 μL DI-water were added. The volume ratio of methanol/water in this system should be in accordance with the condition, where chiral assemblies of C4-MOP formed (methanol/water = 1
:2, v/v). Upon incubation at room temperature for a week the mixture formed a uniform, translucent and stable gel. Moreover, for the pre-assembly method, the organic helical ribbons were formed initially, and then the sol–gel polymerization process of TEOS occurred on the surface of these ribbons. As follows: 1 mL chiral assemblies (5 mg mL−1, methanol/water = 1:2, v/v) were prepared, and subsequently 500 μL pre-hydrolyzed silica precursor suspension in DI-water was introduced. The mixture was vortexed (2000 rpm) for a few seconds to increase contact, and then left in static conditions for a week.
(iii) Extraction of the C4-MOP template. The organic template was removed by extracting with methanol and then dried in air. Before the extraction, the hybrid materials were aged at 80 °C for 24 h to make the structures more stable, and then dispersed into methanol with stirring at room temperature. This procedure was repeated at least five times to make sure that the C4-MOP templates were removed completely.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by NSFC (No. 21472108, 21604085, 21772112, 21802166), National Key R&D Program of China (No. 2017YFD0200302), the Fundamental Research Funds for the Central Universities (No. ZY1831), and Jilin Science Foundation for Youths (No. 20160520135JH). J. H. and Y. J. thank the support of State Key Laboratory of Polymer Physics and Chemistry, CIAC.Notes and references
- B. Liu, Y. Cao, Z. Huang, Y. Duan and S. Che, Adv. Mater., 2015, 27, 479–497 CrossRef CAS PubMed.
- C. J. Bueno-Alejo, L. A. Villaescusa and A. E. Garcia-Bennett, Angew. Chem., Int. Ed., 2014, 53, 12106–12110 CrossRef CAS PubMed.
- B. Liu, L. Han and S. Che, Angew. Chem., Int. Ed., 2012, 51, 923–927 CrossRef CAS PubMed.
- S. Lacasta, V. Sebastián, C. Casado, Á. Mayoral, P. Romero, Á. Larrea, E. Vispe, P. López-Ram-de-Viu, S. Uriel and J. Coronas, Chem. Mater., 2011, 23, 1280–1287 CrossRef CAS.
- H. Qiu and S. Che, Chem. Soc. Rev., 2011, 40, 1259–1268 RSC.
- P. Paik, A. Gedanken and Y. Mastai, ACS Appl. Mater. Interfaces, 2009, 1, 1834–1842 CrossRef CAS.
- T. Yokoi, K. Ogawa, D. Lu, J. N. Kondo, Y. Kubota and T. Tatsumi, Chem. Mater., 2011, 23, 2014–2016 CrossRef CAS.
- T. Kawasaki, Y. Araki, K. Hatase, K. Suzuki, A. Matsumoto, T. Yokoi, Y. Kubota, T. Tatsumi and K. Soai, Chem. Commun., 2015, 51, 8742–8744 RSC.
- J. H. Jung, H. Kobayashi, M. Masuda, T. Shimizu and S. Shinkai, J. Am. Chem. Soc., 2001, 123, 8785–8789 CrossRef CAS.
- H. Qiu, Y. Inoue and S. Che, Angew. Chem., Int. Ed., 2009, 48, 3069–3072 CrossRef CAS.
- J. Jiang, T. Wang and M. Liu, Chem. Commun., 2010, 46, 7178–7180 RSC.
- J. H. Jung, S. J. Moon, J. Ahn, J. Jaworski and S. Shinkai, ACS Nano, 2013, 7, 2595–2601 CrossRef CAS PubMed.
- T. Yokoi, S. Sato, Y. Ara, D. Lu, Y. Kubota and T. Tatsumi, Adsorption, 2010, 16, 577–586 CrossRef CAS.
- H. Kuzuhara, S. Nishiyama, N. Minowa and K. Sasaki, J. Nat. Med., 2006, 60, 113–120 CrossRef CAS.
- B. G. Bag, C. Garai, R. Majumdar and M. Laguerre, Struct. Chem., 2012, 23, 393–398 CrossRef CAS.
- R. A. Hill and J. D. Connolly, Nat. Prod. Rep., 2017, 34, 90–122 RSC.
- B. G. Bag and S. S. Dash, Nanoscale, 2011, 3, 4564–4566 RSC.
- B. G. Bag and S. S. Dash, Langmuir, 2015, 31, 13664–13672 CrossRef CAS PubMed.
- X. Chen, J. Wang, J. Guo, Z. Wan, S. Yin and X. Yang, Food Funct., 2017, 8, 823–831 RSC.
- A. Saha, J. Adamcik, S. Bolisetty, S. Handschin and R. Mezzenga, Angew. Chem., Int. Ed., 2015, 54, 5408–5412 CrossRef CAS PubMed.
- Y. Gao, J. Hao, Q. Yan, F. Du, Y. Ju and J. Hu, ACS Appl. Mater. Interfaces, 2018, 10, 17352–17358 CrossRef CAS.
- Y. Li, J. Li, X. Zhao, Q. Yan, Y. Gao, J. Hao, J. Hu and Y. Ju, Chem. – Eur. J., 2016, 22, 18435–18441 CrossRef CAS.
- C. Liu, Q. Jin, K. Lv, L. Zhang and M. Liu, Chem. Commun., 2014, 50, 3702–3705 RSC.
- X. Wu, S. Ji, Y. Li, B. Li, X. Zhu, K. Hanabusa and Y. Yang, J. Am. Chem. Soc., 2009, 131, 5986–5993 CrossRef CAS PubMed.
- I. A. Sedov, M. A. Stolov and B. N. Solomonov, J. Phys. Org. Chem., 2011, 24, 1088–1094 CrossRef CAS.
- L. Ziserman, H. Y. Lee, S. R. Raghavan, A. Mor and D. Danino, J. Am. Chem. Soc., 2011, 133, 2511–2517 CrossRef CAS PubMed.
- D. K. Smith, Chem. Soc. Rev., 2009, 38, 684–694 RSC.
- P. Duan, H. Cao, L. Zhang and M. Liu, Soft Matter, 2014, 10, 5428–5448 RSC.
- T. G. Barclay, K. Constantopoulos and J. Matisons, Chem. Rev., 2014, 114, 10217–10291 CrossRef CAS.
- Z. Shen, T. Wang and M. Liu, Angew. Chem., Int. Ed., 2014, 53, 13424–13428 CrossRef CAS PubMed.
- J. Li, K. Fan, X. Guan, Y. Yu and J. Song, Langmuir, 2014, 30, 13422–13429 CrossRef CAS PubMed.
- C. C. Lee, C. Grenier, E. W. Meijer and A. P. H. J. Schenning, Chem. Soc. Rev., 2009, 38, 671–683 RSC.
- K. V. Rao and S. J. George, Org. Lett., 2010, 12, 2656–2659 CrossRef CAS PubMed.
- Y. Okazaki, T. Buffeteau, E. Siurdyban, D. Talaga, N. Ryu, R. Yagi, E. Pouget, M. Takafuji, H. Ihara and R. Oda, Nano Lett., 2016, 16, 6411–6415 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- K. A. Cychosz, R. Guillet-Nicolas, J. García-Martínez and M. Thommes, Chem. Soc. Rev., 2017, 46, 389–414 RSC.
- Y. Gao, J. Hao, J. Wu, X. Zhang, J. Hu and Y. Ju, Nanoscale, 2015, 3, 13568–13575 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of C4-MOP; assembly behaviors of C4-MOP; TEM images, CD and UV-Vis spectra of C4-MOP assemblies; theoretical optimized structure of C4-MOP; photos of gels before and after imprinting; TEM image of hybrid helical ribbons; element analysis of hybrid helical ribbons by HRTEM/EDS; pore-size distribution curve of helical silica. See DOI: 10.1039/c8qm00498f |
| This journal is © the Partner Organisations 2019 |