
Triazine based polyimide framework derived N-doped porous carbons: a study of their capacitive behaviour in aqueous acidic electrolyte†
Namrata
Deka
a,
Rajesh
Patidar
b,
S.
Kasthuri
c,
N.
Venkatramaiah
 *c and
Gitish K.
Dutta
*a
*c and
Gitish K.
Dutta
*a
aDepartment of Chemistry, National Institute of Technology Meghalaya, Bijni Complex, Laitumkhrah, Shillong-793003, Meghalaya, India. E-mail: gitish.dutta@nitm.ac.in
bAnalytical and Environmental Science Division & Centralized Instrument Facility, CSIR-Central Salt & Marine Chemicals Research Institute, Bhavnagar-364002, Gujarat, India
cDepartment of Chemistry, SRM Institute of Science and Technology (SRMIST), Chennai-603203, Tamil Nadu, India. E-mail: nvenkat83@gmail.com
First published on 22nd February 2019
Abstract
Nitrogen-doped porous carbon materials have been synthesized from nitrogen and oxygen rich triazine based polyimide (TPI-P/TPI-N) frameworks using ZnCl2 as an activating agent at different temperatures (600 and 700 °C) for electrochemical energy storage applications. The morphology and structural features of the materials were investigated using scanning electron microscopy (SEM), transmission electron microscopy (TEM), N2 adsorption/desorption isotherms, X-ray photoelectron spectroscopy (XPS) and Raman spectroscopic techniques. The resultant carbon materials possess large specific surface area and rich nitrogen contents. In particular, the material obtained at 700 °C (TPI-P-700) exhibits a surface area of up to 1650 m2 g−1 and a nitrogen content of up to 6.3%, and shows an excellent specific capacitance of 423 F g−1 in an aqueous acid electrolyte (1 M H2SO4) in a three electrode system. Moreover, the material also demonstrates nearly 100% capacitance retention up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 charge–discharge cycles. A symmetrical supercapacitor device assembled using TPI-P-700 as an active material delivered an energy density of 10.5 W h kg−1 at 0.5 A g−1.
000 charge–discharge cycles. A symmetrical supercapacitor device assembled using TPI-P-700 as an active material delivered an energy density of 10.5 W h kg−1 at 0.5 A g−1.
Introduction
With the global energy demands and inescapable depletion of fossil fuel based energy resources, the exploration of advanced and sustainable energy resources as well as efficient energy storage systems has attracted widespread attention in recent decades.1–3 Electrochemical energy storage devices, such as supercapacitors and batteries, are among the frontrunners for developing clean energy storage systems.4–7 Supercapacitors are superior to batteries in terms of rapid power delivery and quick charging due to their properties like faster charge–discharge rates, longer cyclability, reliable safety and high power density, but suffer from low energy density.8 In the context of their low energy density, efforts have been made to design better electrode materials with an aim to maximize the specific capacitance and/or potential window.9 Apart from metal oxide derived nanostructures10,11 and carbon–metal oxide composites,12 metal-free carbon based electrode materials like porous carbons,13 graphene,14,15 carbon nanostructures,16etc. are most commonly employed owing to their low cost, easy processability, good conductivity, high durability and high surface area.17 In spite of the many advantages of carbon based materials, their electrochemical performance is greatly hindered by their high pore tortuosity leading to poor ionic diffusion at high current densities.18 Therefore, rational design of 2-dimensional heteroatom-doped porous graphitic materials could effectively minimize the ion diffusion paths on the nanoscale.19,20 Among commonly incorporated heteroatom dopants (such as N, S, O, P, and B, etc.), nitrogen doping has been extensively studied and found to modify the charge distribution of nearby carbon atoms, introduce faradaic sites for pseudocapacitive charge storage and increase the wettability of the materials.21 Nitrogen doping has been accomplished by various post-treatment and pre-treatment methods including chemical vapor deposition (CVD), ammonia annealing, plasma treatment, etc.22 However, these methods fail to control the exact positioning of dopant sites. Therefore, controlled tailoring of nitrogen species into the carbon framework can allow us to monitor the structure–property relationship of these materials.23Recently, porous organic polymers (POPs) have been successfully applied as nitrogen containing precursors for the synthesis of carbon based materials for energy storage,24,25 catalysis,26 gas adsorption and storage,27 sensors,28etc. POPs consist of rigid aromatic building blocks covalently linked into a 2D or 3D backbone with a permanent porous structure and tunable heteroatom sites. The pore dimensions of the porous covalent skeleton are highly influenced by the choice of monomer unit and covalent linkage topology.29 In addition, surface chemical functionalization at precise locations plays an important role in controlling the electronic response of the materials.30 Subsequent carbonization of these precursors leads to the formation of electrically conductive heteroatom-doped carbon frameworks.31 For example, Wang et al. introduced sp-hybridized N atoms into pre-defined locations of a graphdiyne framework with an aim to enhance the electronic response of the material.32 Similar in situ incorporation of uniformly distributed heteroatoms into the carbon framework can enable the fine-tuning of the capacitive behavior of the materials.29 Also, carbonization of POPs based synthetic approaches enables the preparation of a thermally stable carbon framework without the use of harsh template removal steps. However, high temperature carbonization often results in partial collapse of pores leading to a decrease in surface area.33,34 Therefore, several activation strategies such as physical activation (using steam and CO2, etc.) or chemical activation (using KOH and NaOH, etc.) are used to control the porosity of the materials. Although KOH activation is a commonly reported method for the preparation of heteroatom doped porous carbons, certain drawbacks like low yield and extensive etching of heteroatoms need to be improved. Employing a “wet chemical” activation strategy, where inorganic molten salts like LiCl, ZnCl2, etc. react as a solvent at above their melting points and stabilize the porous framework against shrinkage, has been proven to have an overall positive effect on the yield and nitrogen content of porous carbons.35,36 In the context of organic polymer precursors, ZnCl2 also acts as a dehydrating agent, further facilitating the aromatization of carbon atoms.37
Antonietti and co-workers have demonstrated tuning of pore sizes in carbonaceous materials by varying the ZnCl2 activation conditions.38 Similarly, Xiong's group synthesized hierarchical porous carbon materials with substantial surface area (1203 m2 g−1) by ZnCl2 activation of Schiff-base networks and the resulting materials showed a specific capacitance (Cs) of 377 F g−1 at 0.2 A g−1.39 Porous carbons synthesized from a polyimide-based network have been studied for gas adsorptions and for supercapacitors.40,41 Simulation studies by Cooper and coworkers suggest that 2D polyimide networks lead to the formation of combined meso- and micro-porous structures depending on the linker moieties.42 On the other hand, nitrogen rich triazine moieties are a promising monomer choice owing to their high nitrogen content and ease of preparation. Hence, the rational design of triazine based polyimide frameworks as N-doped porous carbon precursors should result in porous carbon materials with inherent porosity and well-tailored dopant sites.
Herein, we report the synthesis of nitrogen-doped porous carbon materials derived from two TPI based microporous organic polymers using ZnCl2 as an activating agent at different temperatures (600 and 700 °C). It is found that the high specific surface area, the predominant microporous structure and the presence of high nitrogen content in the synthesized materials have resulted in a high specific capacitance of 423 F g−1. The charge storage kinetics of the materials was also investigated using Dunn's equation.43 Excellent cycling stability of the material further stresses on its extensive scope for practical supercapacitor applications.
Materials and methods
Materials
All reagents and solvents used were procured from Sigma Aldrich, Alfa Aesar, TCI Chemicals, HiMedia and Sisco Research Laboratories Pvt. Ltd. (SRL), India. Solvents were distilled prior to use. All the reactions were carried out under N2 atmosphere. Pyromellitic dianhydride (PMDA) and 1,4,5,8-naphthalenetetracarboxylic dianhydride (NTDA) were kept under vacuum and then under an inert atmosphere before use. 2,4,6-Tris(4-aminophenyl)-1,3,5-triazine (TAPT) and triazine based polyimide frameworks (TPI-P and TPI-N from PMDA and NTDA, respectively) were synthesized in high yields according to procedures described in the literature.44–46Synthesis
:7 weight ratio of polymer:ZnCl2. The mixture was dried at 80 °C overnight under vacuum to completely evaporate water, after which it was transferred into an alumina combustion boat and heated at high temperatures (600 °C and 700 °C) in a horizontal tube furnace (5 °C min−1) under a nitrogen atmosphere (a flow rate of ∼30 cc min−1) for 2 h. The black porous materials were immersed in 2 M HCl solution and then filtered. The filtered product was washed with distilled water until completely neutral and dried under vacuum to afford TPI-P-X and TPI-N-X (X = carbonization temperature; P = PMDA; and N = NTDA) with a 55–60% yield.
Characterization
The Metrohm Multi Autolab/M204 workstation was used to carry out all electrochemical measurements. FT-IR spectra were obtained using a PerkinElmer Spectrum Two Spectrometer (fitted with a UATR accessory). 13C CP-MAS solid-state NMR spectra were recorded on a JEOL ECX-400 spectrometer (400 MHz). Contact angles were measured under an air atmosphere on a KRUSS Drop Shape Analyser-DSA25. Nitrogen adsorption–desorption isotherms were measured at 77 K using a Micromeritics Instrument Corp., USA, model 3Flex. Surface areas of the samples were calculated by using the BET method and the pore size distributions were analyzed by NLDFT calculations. Field emission scanning electron microscopy, FE-SEM (JEOL JXA-8230) and transmission electron microscopy, TEM (JEOL JEM-2100) were used to analyze the surface morphology of the materials. Raman spectra of the samples were recorded using a Micro Raman Spectrometer: HORIBA, LABRAM HR Evolution with an excitation laser of wavelength 532 nm. X-ray photoelectron spectroscopy (XPS) was performed using an FEI Inc. PHI 5000 Versa Probe II. Powder X-ray diffraction studies were carried out in an Xpert3 PANalytical X-ray diffractometer with Cu Kα (1.54 Å) as an X-ray source.Electrochemical measurements
The electrochemical behavior of the porous carbon samples was initially tested in a typical three-electrode system using a 1 M H2SO4 electrolyte against Ag/AgCl as a reference electrode and platinum foil as a counter electrode. The active material (80%) was mixed with a PVDF binder (10%), acetylene black (10%) and NMP and coated on a 1 cm2 area of pre-cleaned flexible graphite sheet (current collector).47 The specific capacitance values were determined from cyclic voltammetry (CV) and galvanostatic charge–discharge (GCD) curves using reported equations.47,48 For a symmetrical cell, two identical working electrodes loaded with 1.1–1.2 mg of active materials were assembled using a glass fiber separator between them. The gravimetric capacitance of the symmetric cell and the electrode was calculated from GCD curves.49 Power density and energy density of symmetric cells were calculated as reported in the literature.19 Electrochemical impedance spectroscopy (EIS) was carried out on a Multi Autolab/M204 workstation coupled with an FRA32M module within a frequency limit of 100 kHz to 1 Hz at an amplitude of 10 mV.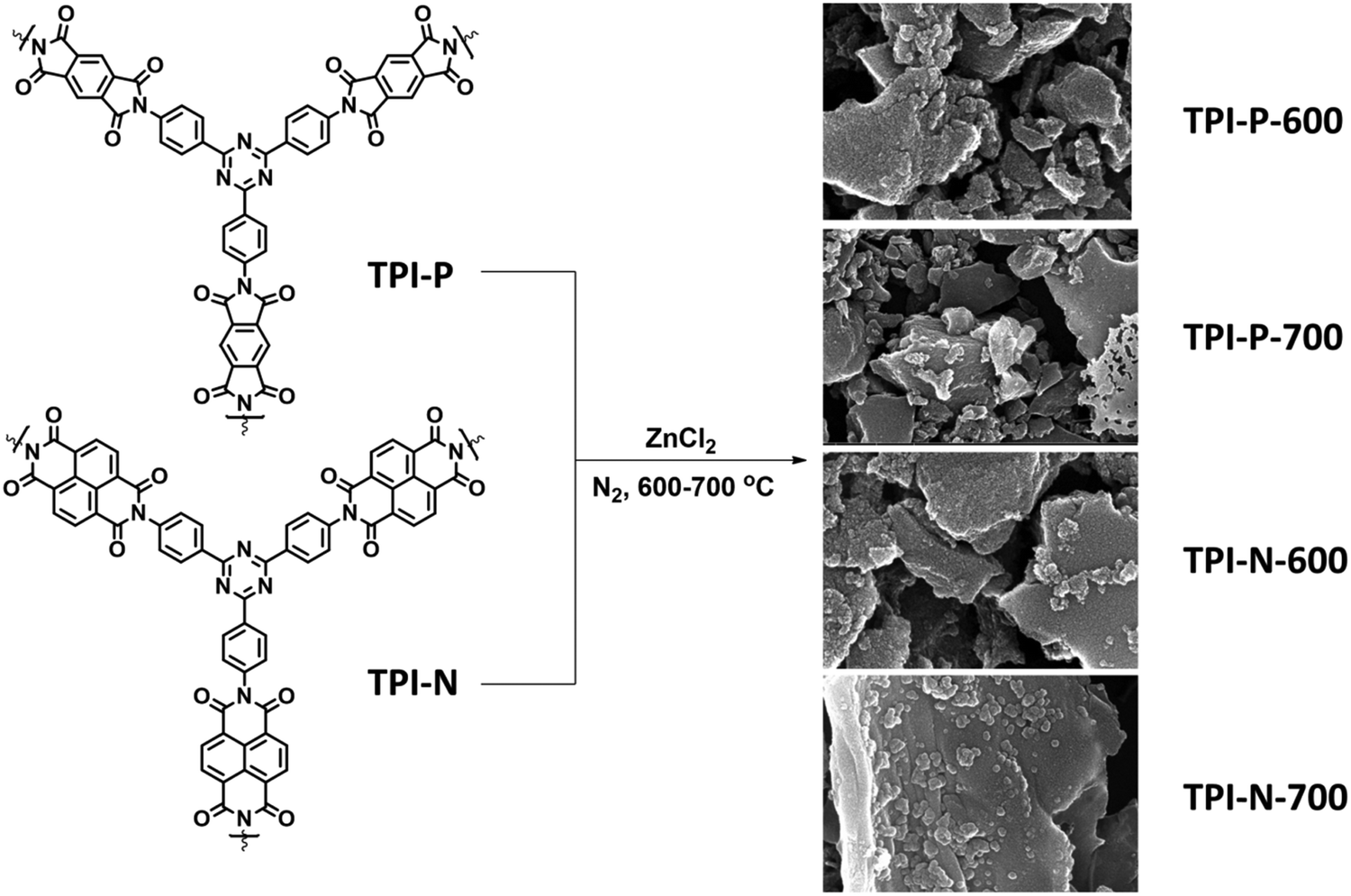 | ||
| Scheme 1 Synthetic scheme for N-doped porous carbons (TPI-P/TPI-N-X, X = carbonization temperature). | ||
Results and discussion
2,4,6-Tris(4-aminophenyl)-1,3,5-triazine (TAPT) was prepared by cyclotrimerization of 4-aminobenzonitrile in triflic acid (Scheme S1, ESI†). The polyimide based porous organic frameworks denoted as TPI-P and TPI-N (Scheme S1, ESI†) were synthesized by the reaction of TAPT and respective carboxylic dianhydride. The resultant materials were characterized by 13C CP-MAS spectra (Fig. S1, ESI†). Both the materials show triazine signals at ∼170 ppm and another signal at ∼160 ppm indicates the formation of polyimide rings.45,50 Successful formation of the polyimide network was also confirmed by FT-IR spectra (Fig. S1, ESI†). Characteristic peaks at 1670 and 1710 cm−1 represent the symmetric and asymmetric vibrations of imide carbonyls. The polyimide frameworks were carbonized using ZnCl2, which act as a template as well as a dehydrating agent during the activation process. The resulting porous carbon materials were denoted as TPI-P-X/TPI-N-X (X = carbonization temperature) and shown in Scheme 1. The amounts of ZnCl2 were optimized by preliminary electrochemical measurements (Table S1 and Fig. S2, ESI†) and the carbonization temperature was kept below its boiling point (i.e. 732 °C) to avoid evaporation loss. Also, the effect of ZnCl2 activation on the wettability of the materials has been evaluated by water contact angle measurements. ZnCl2 activation of the TPI precursors increases the wettability of the materials as shown in Fig. S3 (ESI†).51The surface morphologies of the materials were examined via field emission scanning electron microscopy (FE-SEM). Highly aggregated spherical morphology of the TPI-P and TPI-N precursors can be observed from FE-SEM images (Fig. S4a and b, ESI†). TPI-P carbonized without ZnCl2 activation shows no significant change in surface morphology (Fig. S4c, ESI†). On activation with ZnCl2, the spherical morphology is transformed into layered structures (Fig. S4d, ESI†) which may aid faster ion diffusion.19 In addition, elemental mapping in Fig. S5 (ESI†) reveals that nitrogen and oxygen functionalities evenly distributed over the surface of TPI-P-700. The FE-SEM images of all four porous carbon materials show similar irregularly shaped structures with a rough and uneven surface texture (Fig. 1a, b and Fig. S6, ESI†). Also, no detectable peaks for Zn or Cl were observed in the EDX spectra of TPI-P-700 (Fig. S8, ESI†) which implies that no traces of Zn or Cl ions remain in the materials after washing with HCl and deionized water. The transmission electron microscopy (TEM) images of TPI-P-700 as shown in Fig. 1c and d reveal the presence of random micropores and mesopores along with graphitic ribbons. The dark and light contrasts as shown in the TEM images correspond to the porous framework and the pore channels respectively.40 These images are consistent with the pore size distribution calculated using the non-local density functional theory (NLDFT) method and N2 isotherms.
 | ||
| Fig. 1 (a and b) FE-SEM images of TPI-P-700; (c and d) TEM images of TPI-P-700. | ||
Porous features and specific surface areas of the materials were analyzed by nitrogen adsorption–desorption isotherms at 77 K (Fig. 2a and b). All four materials display type I isotherms, typical of microporous solids.52 The sharp uptake at low relative pressures is a characteristic of abundant micropores present in the materials. The materials also show type-H4 hysteresis loops which are typically assigned to narrow slit-like mesopores present.53 The surface areas of TPI-P-600 (1651 m2 g−1) and TPI-P-700 (1650 m2 g−1), calculated by using a multipoint BET model, were almost identical, while TPI-N-600 and TPI-N-700 have comparatively lower surface areas (1057 m2 g−1 and 1237 m2 g−1, respectively). A decrease in surface area of TPI-N derived porous carbons could be attributed to higher volume occupied by the naphthalene moiety in the TPI framework cavity. Moreover, high torsional strain due to the greater dihedral angle of naphthalene-imide rings could be unfavorable for generating high porosity (Fig. S7, ESI†).46 Differential pore size distributions were calculated using the NLDFT method as depicted in Fig. 2c. TPI-P-600 and TPI-P-700 have similar total pore volume (VT = 0.77 cm3 g−1, Table 1), but TPI-P-700 contains a multitude of pores in the diameter range of 0.4–1 nm, which is appropriate for the adsorption of solvated electrolyte ions.54 TPI-P-700 also contains pores with diameters between 1.2–3 nm, which aid accessibility for electrolyte ion diffusion.55 On the other hand, an overall decrease was observed in VT of TPI-N-X (see Table 1) which may be attributed to the collapse of the TPI-N framework during thermal treatment. A well-balanced co-existence of micropores and mesopores in TPI-P-700 can contribute towards better electrochemical behavior.
 | ||
| Fig. 2 N2 adsorption–desorption isotherms of (a) TPI-P-X and (b) TPI-N-X, and (c) pore size distributions using the NLDFT method. | ||
| Sample | S BET (m2 g−1) | V T (cm3 g−1) | V Micro (cm3 g−1) | N (%) | O (%) | N-Species content (%) | |||
|---|---|---|---|---|---|---|---|---|---|
| N-6 | N-5 | N-Q | N-X | ||||||
| TPI-P-600 | 1651 | 0.77 | 0.60 | 7.1 | 10.6 | 10.8 | 64.5 | 9.5 | 15.1 |
| TPI-P-700 | 1650 | 0.77 | 0.67 | 6.3 | 12.7 | 20 | 58.4 | 13.2 | 8.4 |
| TPI-N-600 | 1057 | 0.54 | 0.45 | 11.4 | 9.4 | 6 | 62 | 31.7 | — |
| TPI-N-700 | 1237 | 0.65 | 0.46 | 3.0 | 8.7 | 32.9 | 45.4 | 9.5 | 12.1 |
Powder X-ray diffraction patterns for porous carbon samples were recorded to analyze their crystallinity (Fig. S9, ESI†). Two broad peaks centered around 25.5° and 43° reflect the (002) and (100) planes characteristic of graphitic layers of disordered carbon.56 The broad nature of the peaks indicates more amorphous nature of the materials which is also reflected in the Raman spectra of the materials. The degree of disorder or defects present in the materials was studied via Raman spectroscopy (Fig. 3a). The observed peaks at 1350 cm−1 (D band) and 1580 cm−1 (G band) are usually attributed to disordered carbon structures and in-plane vibrations of sp2 hybridized carbon, respectively.57 The ratio of intensities of the D to G band (ID/IG) of TPI-P-700, TPI-P-600, TPI-N-700 and TPI-N-600 was calculated to be 1.11, 0.97, 0.96 and 0.93 respectively. The ID/IG value increases with an increase in temperature indicating more disorganization in the graphitic lattice. At higher temperature, the decomposition of heteroatom functionalities in the frameworks can introduce more defects in the structure.58,59 Molten ZnCl2 impregnation in TPI-P precursors during carbonization also increases the number of defect structures in the carbon framework (Fig. S10, ESI†). The ID/IG value is greater for TPI-P-700 than in the precursor carbonized without ZnCl2 which has been reported to be beneficial for the electrochemical performance of carbon materials.60
 | ||
| Fig. 3 (a) Raman spectra of TPI-P-700 (black), TPI-P-600 (red), TPI-N-700 (blue), and TPI-N-600 (green); (b) full scan XPS spectra of TPI-P-700 (black), TPI-P-600 (red), TPI-N-700 (blue), and TPI-N-600 (green); (c) deconvoluted C 1s peak of TPI-P-700; and (d) deconvoluted N 1s peak of TPI-P-700. | ||
The presence of heteroatom surface functionalities was further investigated via X-ray photoelectron spectroscopy (XPS). Full scan XPS survey spectra show three peaks for carbon (285 eV, C 1s), nitrogen (398 eV, N 1s) and oxygen (533 eV, O 1s) for all four carbonaceous materials (Fig. 3b). The C 1s peak of TPI-P-700 can be deconvoluted into the following three peaks – 284 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 286 eV (C–C/C–N), and 288 eV (CO) (Fig. 3c). Activation by ZnCl2 has been previously reported to increase the oxygen content on the surface,61 whereas the concentration of nitrogen functionalities is dependent on the carbonization conditions. The nitrogen and oxygen surface contents from XPS analysis have been listed in Table 1. De-convolution of the N 1s spectra of TPI-P-700 reveals four peaks that can be assigned to pyridinic nitrogen (N-6, 398.4 eV), pyrrolic or pyridonic nitrogen (N-5, 400 eV), graphitic or quaternary nitrogen (N-Q, 402 eV) and oxidized nitrogen (N-X, 405 eV) as shown in Fig. 3d.61 Previous literature reports suggest that N-6 and N-5 species act as faradaic reaction sites in the material whereas N-Q and N-X species contribute towards enhanced electron transfer.62,63 From Table 1, it is evident that the carbonization of TPI-P/TPI-N frameworks at 700 °C results in a higher content of N-6 and N-5 species which is beneficial for higher specific capacitance of the materials.
C), 286 eV (C–C/C–N), and 288 eV (CO) (Fig. 3c). Activation by ZnCl2 has been previously reported to increase the oxygen content on the surface,61 whereas the concentration of nitrogen functionalities is dependent on the carbonization conditions. The nitrogen and oxygen surface contents from XPS analysis have been listed in Table 1. De-convolution of the N 1s spectra of TPI-P-700 reveals four peaks that can be assigned to pyridinic nitrogen (N-6, 398.4 eV), pyrrolic or pyridonic nitrogen (N-5, 400 eV), graphitic or quaternary nitrogen (N-Q, 402 eV) and oxidized nitrogen (N-X, 405 eV) as shown in Fig. 3d.61 Previous literature reports suggest that N-6 and N-5 species act as faradaic reaction sites in the material whereas N-Q and N-X species contribute towards enhanced electron transfer.62,63 From Table 1, it is evident that the carbonization of TPI-P/TPI-N frameworks at 700 °C results in a higher content of N-6 and N-5 species which is beneficial for higher specific capacitance of the materials.
The capacitive performance of the materials was initially tested in a three-electrode system, in a 1 M H2SO4 electrolyte. Cyclic voltammetry (CV) curves at 100 mV s−1 and galvanostatic charge–discharge (GCD) curves at 1 A g−1 for TPI-P-X/TPI-N-X materials are shown in Fig. 4a and b. The CV curves for all the materials display quasi-rectangular shapes, a characteristic of electrical double layer (EDLC) behavior. It is also noteworthy that broad redox-type humps are visible in the CV curves of the materials at low scan rates indicating significant pseudocapacitive contribution from heteroatom functionalities (Fig. 4c).63,64 By the comparison of the CV curves, it is revealed that TPI-P-700 covers the largest current density area. In addition, GCD curves of the materials are nearly symmetric in shape with a negligible IR drop which is normally observed in ideal capacitive materials with low internal resistance. The gravimetric specific capacitance (Cs) calculated at 1 A g−1 shows higher values for TPI-P-700 (423 F g−1) and TPI-N-700 (388 F g−1). On the other hand, materials afforded by carbonization at 600 °C showed lower Cs values. To understand the capacitive behavior of the materials, electrochemical impedance spectroscopy (EIS) measurements were carried out. The Nyquist plots (Fig. S12, ESI†) reveal that carbonization at 700 °C endows the materials with lower solution resistance (Rs = 2.63 Ω for TPI-P-700 and 2.66 Ω for TPI-N-700), whereas higher resistance was observed for TPI-P-600 (2.8 Ω) and TPI-N-600 (4.8 Ω). The low internal resistance of TPI-P-700 and TPI-N-700 as compared to TPI-P-600 and TPI-N-600 can contribute to their superior capacitive performance.65 Owing to its greater current response, high Cs value and low solution resistance, the supercapacitive behavior of TPI-P-700 was further studied at different scan-rates (Fig. 4c and Fig. S14, ESI†) and current densities (Fig. 4d). The redox peaks in the CV curves of TPI-P-700 become more prominent as the scan rate decreases from 100 mV s−1 to 5 mV s−1. However, the CV profiles of TPI-P-700 do not show much deviation even at scan rates as high as 100 mV s−1 (Fig. S14, ESI†) implying rapid ion transport into the porous framework.63 The rate capabilities of the electrode materials are shown in Fig. 4f by plotting the specific capacitance values against the corresponding current densities. TPI-P-700 displays better rate capability by exhibiting a high capacitance of 270 F g−1 at 20 A g−1 (65%). In addition, TPI-P-700 also exhibits an excellent capacitance retention of nearly 100% up to 10000 GCD cycles (Fig. 4e). Such superior electrochemical performance of TPI-P-700 can be associated with its optimal pore channels, high surface area and ideal heteroatom content. It should be mentioned that the specific capacitance of TPI-P-700 is higher than that of previously reported porous organic framework (POF) derived nitrogen-doped porous carbon materials (Table 2).
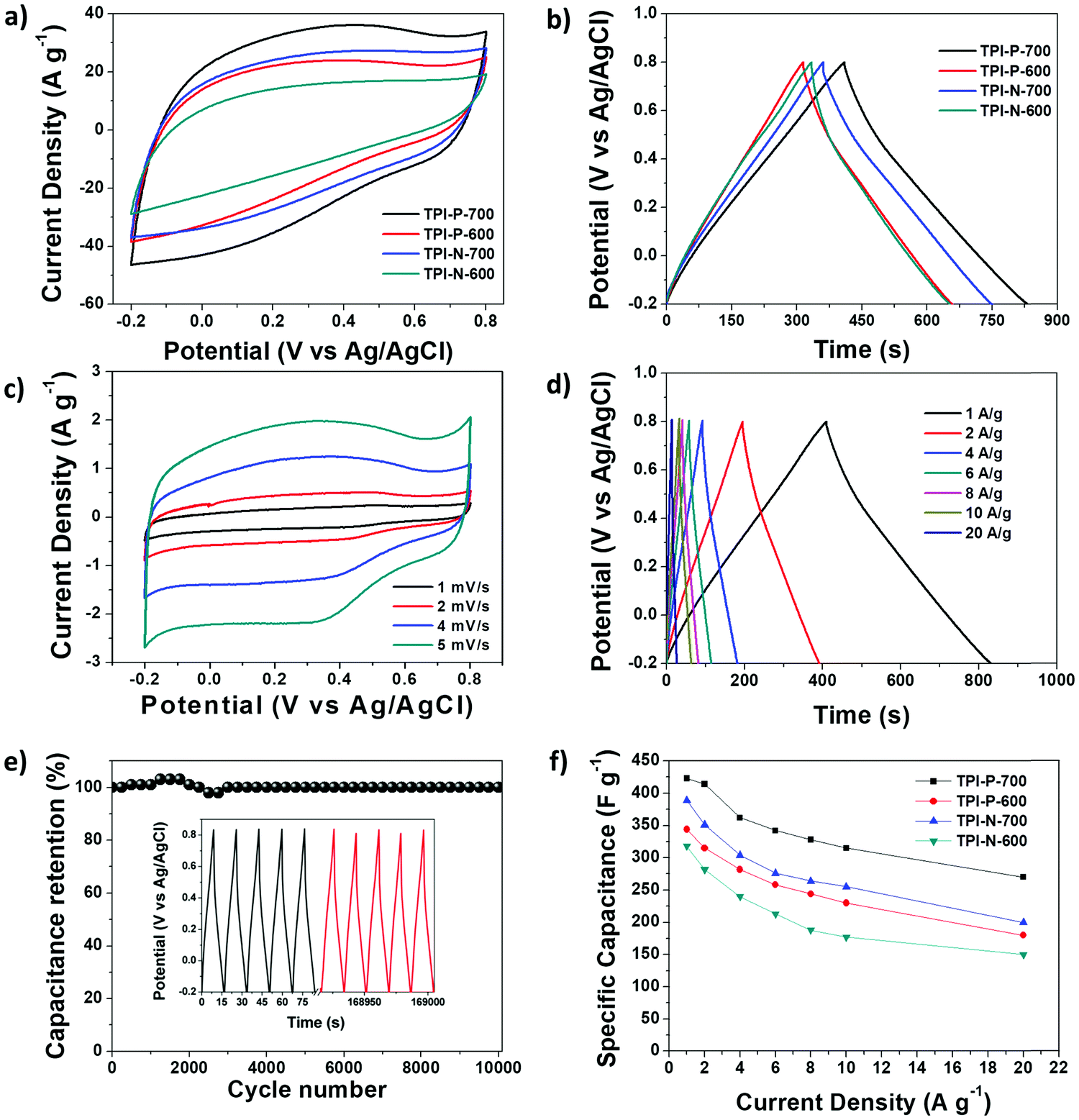 | ||
| Fig. 4 (a) CV curves of TPI-P-700 (black), TPI-P-600 (red), TPI-N-700 (blue), and TPI-N-600 (green) at 100 mV s−1; (b) GCD curves of TPI-P-700 (black), TPI-P-600 (red), TPI-N-700 (blue), and TPI-N-600 (green) at 1 A g−1; (c) CV curves of TPI-P-700 at different scan rates; (d) GCD curves of TPI-P-700 at different current densities; (e) cycling stability of TPI-P-700 at 30 A g−1; and (f) plot of specific capacitance vs. current densities. | ||
| Heteroatom-doped porous carbon precursor | C s (F g−1) | Current density (A g−1) | Potential range (V) | Electrolyte | Ref. |
|---|---|---|---|---|---|
| a Measurements in a two-electrode system. | |||||
| Schiff-base polymer sphere | 292 | 1 | −1 to 0 | 6 M KOH | 48 |
| Azine-linked COF | 234 | 1 | −1 to 0 | 6 M KOH | 70 |
| Schiff-base network | 377 | 0.2 | −0.2 to 0.8 | 1 M H2SO4 | 39 |
| Polyimide co-polymer | 157a | 0.05 | 0 to 1.5 | 1 M H2SO4 | 71 |
| Benzopyrrole-benzimidazole based polyimide | 179a | 1 | 0 to 1 | 1 M NaOH | 72 |
| Triazine based COF | 278 | 1 | −1 to 0 | 6 M KOH | 73 |
| Pyrene based conjugated microporous polymer | 301 | 1 | −0.2 to −1.1 | 6 M KOH | 74 |
| Hypercrosslinked microporous polymer | 374 | 0.1 | −0.1 to 0.8 | 1 M H2SO4 | 47 |
| N-rich microporous organic networks | 294a | 0.3 | 0 to 1 | 1 M H2SO4 | 75 |
| Triazine based polyimide derived framework | 423 | 1 | −0.2 to 0.8 | 1 M H2SO4 | This work |
The faradaic contributions in the materials may arise from both ion intercalation as well as redox reactions of the surface atoms. Surface redox processes and non-faradaic double layer contributions are capacitive processes, i.e. current (i) varies linearly with the scan rate (v). On the other hand, intercalation is a diffusion controlled process, i.e. i and v−1/2 have a linear relationship. To understand the capacitive behavior of TPI-P-700, the total capacitance can be differentiated into diffusive and capacitive contributions by applying eqn (1).43
 | (1) |
Fig. 5a displays the capacitive contribution (shaded area) to the total capacitance of TPI-P-700 at 5 mV s−1. Generally, diffusion controlled processes follow slow kinetics and are therefore more prominent at slower scan-rates (28% at 5 mV s−1) as evident from Fig. 5b. At higher scan-rates of 80 mV s−1, the contribution from diffusion controlled processes becomes negligible (9.7%).
 | ||
| Fig. 5 (a) Comparison of the capacitive contribution (the shaded region) to the total CV plots at 5 mV s−1. (b) Capacitive and diffusive contribution of TPI-P-700 with increasing scan-rates; (c) b value calculated by plotting logi against logv; (d) linear relationship of anodic/cathodic current with the scan rate. | ||
The charge storage mechanism of TPI-P-700 was further studied by applying the power law to the current (i)–scan rate (v) relation:66
| i = avb | (2) |
i vs. logv data plot yields b = 0.93 for TPI-P-700 in 1 M H2SO4, indicating a surface controlled process. The linear relationship between peak current (ip) and scan rate (v) is also demonstrated in Fig. 5d.
To evaluate the practical applications of the synthesized materials, a symmetric supercapacitor device was fabricated with TPI-P-700 in an aqueous acid electrolyte. The CV curves at scan rates from 10 to 100 mV s−1 are displayed in Fig. 6a. The gravimetric specific capacitance of the device and the electrode was calculated from the GCD curves (Fig. 6b) and found to be as high as 76 F g−1 and 304 F g−1 respectively at 0.5 A g−1.
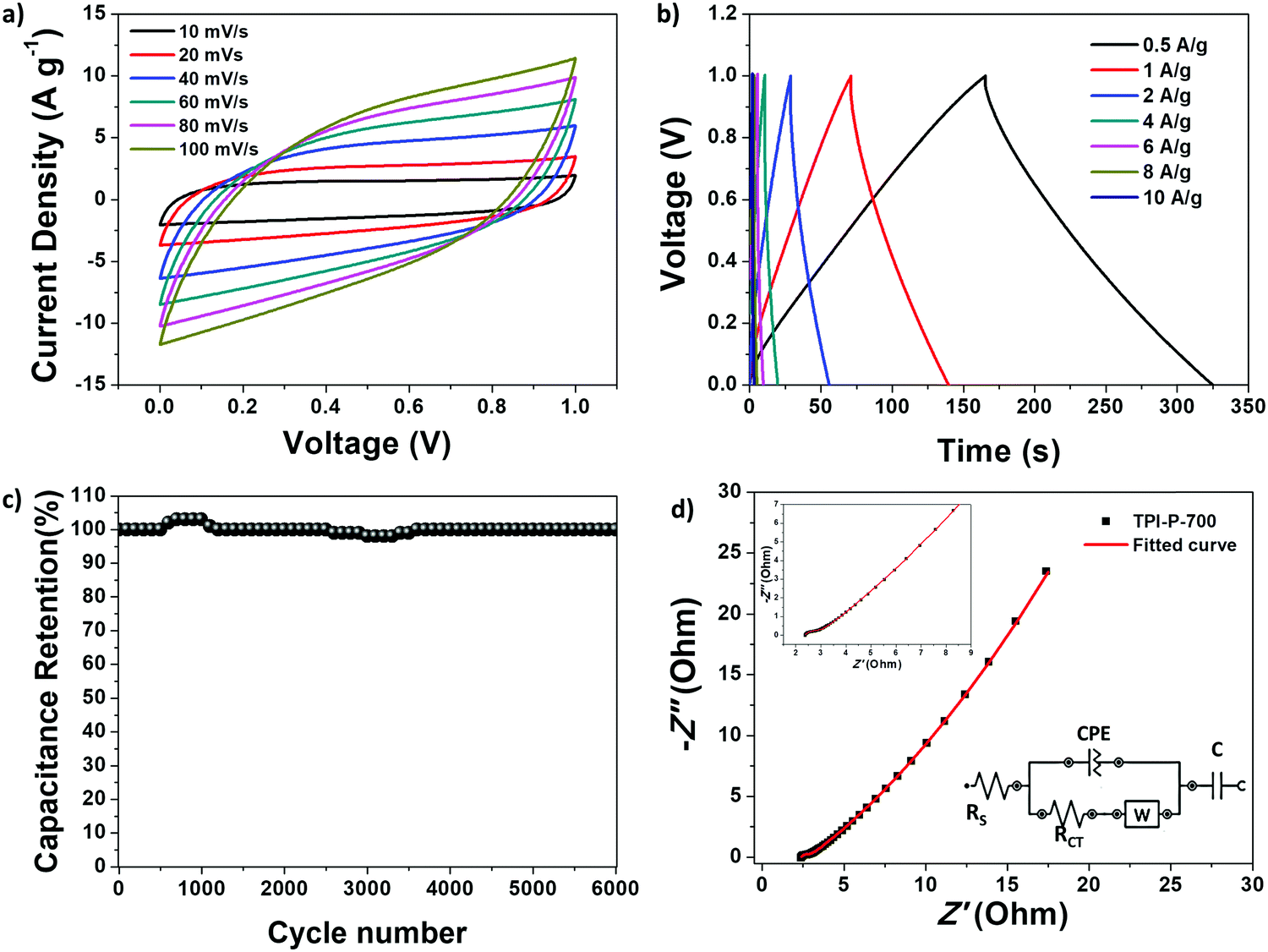 | ||
| Fig. 6 Electrochemical performance of TPI-P-700 based symmetrical supercapacitor device: (a) CV curves at different scan-rates; (b) GCD curves at different current densities; (c) cycling stability performance; (d) Nyquist plot with the equivalent circuit shown in the inset.68 | ||
The GCD curves also display symmetrical triangular shapes indicating good capacitive reversibility of the device. The scope of the TPI-P-700 based symmetrical supercapacitor device is further highlighted by its high energy density (10.5 W h kg−1 at 0.5 A g−1) and power density (5000 W kg−1 at 10 A g−1) as depicted by the Ragone plot in Fig. S15, ESI.† Long-term cycling stability of the device shows a complete retention of specific capacitance up to 6000 cycles (Fig. 6c). Also, the Nyquist plot from EIS measurements is depicted in Fig. 6d along with the appropriate equivalent circuit. The equivalent circuit consists of solution resistance, RS; charge-transfer resistance, RCT; Warburg impedance, W due to the diffusion of ions at the electrode interface from the bulk electrolyte; and the capacitive elements, C and CPE, arising from pseudocapacitive and double layer behavior. Small diameter of the semicircle in the high frequency region and a linear rise in the low frequency region are characteristic of rapid ion/electron diffusion and easy access of electrolyte ions into the porous material.69
Conclusion
Nitrogen-doped porous carbon materials were synthesized by carbonization of two triazine based polyimide (TPI) frameworks via ZnCl2 activation. The obtained materials exhibit high specific surface area, predominant microporosity and high nitrogen/oxygen contents. These structural features facilitate rapid ion adsorptions and energy storage to improve the supercapacitor performance. The optimal carbonized material (TPI-P-700) exhibits a high specific capacitance of 423 F g−1 (1 A g−1) and a good rate capability of 67% up to 20 A g−1 in a three-electrode system. TPI-P-700 also demonstrates excellent cycling stability (100%) up to 10000 cycles. In addition, the charge storage mechanism of the materials shows a surface controlled capacitive process in a sulphuric acid electrolyte. The symmetrical supercapacitor device using TPI-P-700 exhibits a superior gravimetric capacitance of 304 F g−1 at 0.5 A g−1, while maintaining 100% capacitance retention up to 6000 cycles. The symmetric device also delivers an energy density of 10.5 W h kg−1. These results demonstrate that nitrogen containing porous organic frameworks are suitable precursors to synthesize nitrogen-doped porous carbon materials for electrochemical energy storage devices.
Author contributions
The manuscript was written through contributions of all authors. All authors have given their approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the Science and Engineering Research Board (SERB), India (Grant no: SB/FT/CS-075/2014) for supporting this work financially. ND is grateful to the Ministry of Human Resource and Development (MHRD), India for a junior research fellowship. The authors would like to acknowledge the ACMS, IIT Kanpur; the Materials Science and Engineering Department, IIT Kanpur; SAIF, NEHU and SIF, Gauhati University for the analytical support. NVR acknowledges financial assistance from the Department of Science & Technology, Ministry of Science & Technology (DST-FIST) for the infrastructural development of SRMIST and SRM-SCIF for the Micro-Raman facility. The authors also thank Dr. Uttam Manna, IIT Guwahati for providing the contact angle measurement facility.References
- L. Dai, D. W. Chang, J.-B. Baek and W. Lu, Small, 2012, 8, 1130–1166 CrossRef CAS PubMed.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2014, 7, 19–29 CrossRef PubMed.
- J. Wang, H. Tang, L. Zhang, H. Ren, R. Yu, Q. Jin, J. Qi, D. Mao, M. Yang, Y. Wang, P. Liu, Y. Zhang, Y. Wen, L. Gu, G. Ma, Z. Su, Z. Tang, H. Zhao and D. Wang, Nat. Energy, 2016, 1, 16050 CrossRef CAS.
- Q. Yang, S. Cui, Y. Ge, Z. Tang, Z. Liu, H. Li, N. Li, H. Zhang, J. Liang and C. Zhi, Nano Energy, 2018, 50, 623–631 CrossRef CAS.
- X. Zhao, R. Yu, H. Tang, D. Mao, J. Qi, B. Wang, Y. Zhang, H. Zhao, W. Hu and D. Wang, Adv. Mater., 2017, 29, 1700550 CrossRef PubMed.
- D. Li, X. Zhao, R. Yu, B. Wang, H. Wang and D. Wang, Inorg. Chem. Front., 2018, 5, 535–540 RSC.
- J. Niu, R. Shao, M. Liu, J. Liang, Z. Zhang, M. Dou, Y. Huang and F. Wang, Energy Storage Mater., 2018, 12, 145–152 CrossRef.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- S. K. Balasingam, J. S. Lee and Y. Jun, Dalton Trans., 2015, 44, 15491–15498 RSC.
- M. Chen, J. Wang, H. Tang, Y. Yang, B. Wang, H. Zhao and D. Wang, Inorg. Chem. Front., 2016, 3, 1065–1070 RSC.
- J.-N. Zhang, P. Liu, C. Jin, L.-N. Jin, S.-W. Bian, Q. Zhu and B. Wang, Electrochim. Acta, 2017, 256, 90–99 CrossRef CAS.
- K. Xia, Q. Gao, J. Jiang and J. Hu, Carbon, 2008, 46, 1718–1726 CrossRef CAS.
- X. Wang, Y. Zhang, C. Zhi, X. Wang, D. Tang, Y. Xu, Q. Weng, X. Jiang, M. Mitome, D. Golberg and Y. Bando, Nat. Commun., 2013, 4, 2905 CrossRef PubMed.
- M. Cheng, Y. Meng, Q. Meng, L. Mao, M. Zhang, K. Amin, A. Ahmad, S. Wu and Z. Wei, Mater. Chem. Front., 2018, 2, 986–992 RSC.
- H. Jiang, P. S. Lee and C. Li, Energy Environ. Sci., 2013, 6, 41–53 RSC.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- X. Zheng, J. Luo, W. Lv, D.-W. Wang and Q.-H. Yang, Adv. Mater., 2015, 27, 5388–5395 CrossRef CAS PubMed.
- K. Yuan, T. Hu, Y. Xu, R. Graf, L. Shi, M. Forster, T. Pichler, T. Riedl, Y. Chen and U. Scherf, Mater. Chem. Front., 2017, 1, 278–285 RSC.
- M. Liu, J. Niu, Z. Zhang, M. Dou and F. Wang, Nano Energy, 2018, 51, 366–372 CrossRef CAS.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 RSC.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- H. M. Jeong, J. W. Lee, W. H. Shin, Y. J. Choi, H. J. Shin, J. K. Kang and J. W. Choi, Nano Lett., 2011, 11, 2472–2477 CrossRef CAS PubMed.
- Y. Kai, Z. Xiaodong, H. Ting, S. Lei, S. Stavroula, P. Ulrike, F. Michael, P. Thomas, R. Thomas, F. Xinliang, C. Yiwang and S. Ullrich, ChemElectroChem, 2017, 4, 709–715 CrossRef.
- S. Li, H. Yang, R. Xu, Y. Jiang, Y. Gong, L. Gu and Y. Yu, Mater. Chem. Front., 2018, 2, 1574–1582 RSC.
- L. Li, H. Zhao and R. Wang, ACS Catal., 2015, 5, 948–955 CrossRef CAS.
- N. B. McKeown and P. M. Budd, Chem. Soc. Rev., 2006, 35, 675–683 RSC.
- A. Palma-Cando, D. Woitassek, G. Brunklaus and U. Scherf, Mater. Chem. Front., 2017, 1, 1118–1124 RSC.
- F. Xu, D. Wu, R. Fu and B. Wei, Mater. Today, 2017, 20, 629–656 CrossRef CAS.
- Z. Xiang, D. Cao, L. Huang, J. Shui, M. Wang and L. Dai, Adv. Mater., 2014, 26, 3315–3320 CrossRef CAS PubMed.
- Y. Zheng and S.-Z. Qiao, Nat. Chem., 2018, 10, 900–902 CrossRef CAS PubMed.
- Y. Zhao, J. Wan, H. Yao, L. Zhang, K. Lin, L. Wang, N. Yang, D. Liu, L. Song, J. Zhu, L. Gu, L. Liu, H. Zhao, Y. Li and D. Wang, Nat. Chem., 2018, 10, 924–931 CrossRef CAS PubMed.
- K. Gayoung, Y. Jun, N. Naotoshi and S. Tomohiro, Chem. – Eur. J., 2017, 23, 17504–17510 CrossRef PubMed.
- T. Ben, Y. Li, L. Zhu, D. Zhang, D. Cao, Z. Xiang, X. Yao and S. Qiu, Energy Environ. Sci., 2012, 5, 8370–8376 RSC.
- Y.-B. Huang, P. Pachfule, J.-K. Sun and Q. Xu, J. Mater. Chem. A, 2016, 4, 4273–4279 RSC.
- X. Liu, N. Fechler and M. Antonietti, Chem. Soc. Rev., 2013, 42, 8237–8265 RSC.
- J. i. Hayashi, A. Kazehaya, K. Muroyama and A. P. Watkinson, Carbon, 2000, 38, 1873–1878 CrossRef CAS.
- K. Pierre, F. Aurélien, H. Jürgen, T. Arne and A. Markus, Adv. Mater., 2009, 21, 897–901 CrossRef.
- J.-S. Wei, H. Ding, Y.-G. Wang and H.-M. Xiong, ACS Appl. Mater. Interfaces, 2015, 7, 5811–5819 CrossRef CAS PubMed.
- J. Yan, B. Zhang and Z. Wang, J. Phys. Chem. C, 2017, 121, 22753–22761 CrossRef CAS.
- X. Zhixiao, Z. Xiaodong, Y. Chongqing, C. Jing, Y. Zhaoquan, T. Yanping, J. Jianzhong, W. Dongqing and F. Xinliang, Adv. Mater., 2016, 28, 1981–1987 CrossRef PubMed.
- A. Trewin and A. I. Cooper, CrystEngComm, 2009, 11, 1819–1822 RSC.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 RSC.
- R. Gomes, P. Bhanja and A. Bhaumik, Chem. Commun., 2015, 51, 10050–10053 RSC.
- M. R. Liebl and J. Senker, Chem. Mater., 2013, 25, 970–980 CrossRef CAS.
- Z. Wang, B. Zhang, H. Yu, L. Sun, C. Jiao and W. Liu, Chem. Commun., 2010, 46, 7730–7732 RSC.
- J.-S. M. Lee, M. E. Briggs, C.-C. Hu and A. I. Cooper, Nano Energy, 2018, 46, 277–289 CrossRef CAS.
- D. Zhu, J. Jiang, D. Sun, X. Qian, Y. Wang, L. Li, Z. Wang, X. Chai, L. Gan and M. Liu, J. Mater. Chem. A, 2018, 6, 12334–12343 RSC.
- C. Xue, L. Feng, Y. Hao, F. Yang, Q. Zhang, X. Ma and X. Hao, Green Chem., 2018, 20, 4983–4994 RSC.
- K. V. Rao, R. Haldar, C. Kulkarni, T. K. Maji and S. J. George, Chem. Mater., 2012, 24, 969–971 CrossRef CAS.
- G. Ma, F. Hua, K. Sun, Z. Zhang, E. Feng, H. Peng and Z. Lei, RSC Adv., 2016, 6, 103508–103516 RSC.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- K. A. Cychosz, R. Guillet-Nicolas, J. García-Martínez and M. Thommes, Chem. Soc. Rev., 2017, 46, 389–414 RSC.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- S. Kumagai, M. Sato and D. Tashima, Electrochim. Acta, 2013, 114, 617–626 CrossRef CAS.
- Z. Q. Li, C. J. Lu, Z. P. Xia, Y. Zhou and Z. Luo, Carbon, 2007, 45, 1686–1695 CrossRef CAS.
- J. Niu, R. Shao, J. Liang, M. Dou, Z. Li, Y. Huang and F. Wang, Nano Energy, 2017, 36, 322–330 CrossRef CAS.
- V. Datsyuk, M. Kalyva, K. Papagelis, J. Parthenios, D. Tasis, A. Siokou, I. Kallitsis and C. Galiotis, Carbon, 2008, 46, 833–840 CrossRef CAS.
- M. Demir, S. K. Saraswat and R. B. Gupta, RSC Adv., 2017, 7, 42430–42442 RSC.
- N. Wang, Y. Wang, X. Xu, T. Liao, Y. Du, Z. Bai and S. Dou, ACS Appl. Mater. Interfaces, 2018, 10, 9353–9361 CrossRef CAS PubMed.
- X. Xiang, E. Liu, Z. Huang, H. Shen, Y. Tian, C. Xiao, J. Yang and Z. Mao, J. Solid State Electrochem., 2011, 15, 2667–2674 CrossRef CAS.
- H.-J. Denisa, K. Masaya, S. Soshi, H. Hiroaki, Z. Z. Hua and L. G. Qing, Adv. Funct. Mater., 2009, 19, 1800–1809 CrossRef.
- T. Lin, I.-W. Chen, F. Liu, C. Yang, H. Bi, F. Xu and F. Huang, Science, 2015, 350, 1508–1513 CrossRef CAS PubMed.
- Y.-H. Lee, K.-H. Chang and C.-C. Hu, J. Power Sources, 2013, 227, 300–308 CrossRef CAS.
- L. Hao, B. Luo, X. Li, M. Jin, Y. Fang, Z. Tang, Y. Jia, M. Liang, A. Thomas, J. Yang and L. Zhi, Energy Environ. Sci., 2012, 5, 9747–9751 RSC.
- H. Lindström, S. Södergren, A. Solbrand, H. Rensmo, J. Hjelm, A. Hagfeldt and S.-E. Lindquist, J. Phys. Chem. B, 1997, 101, 7717–7722 CrossRef.
- V. Augustyn, J. Come, M. A. Lowe, J. W. Kim, P.-L. Taberna, S. H. Tolbert, H. D. Abruña, P. Simon and B. Dunn, Nat. Mater., 2013, 12, 518–522 CrossRef CAS PubMed.
- C.-S. Dai, P.-Y. Chien, J.-Y. Lin, S.-W. Chou, W.-K. Wu, P.-H. Li, K.-Y. Wu and T.-W. Lin, ACS Appl. Mater. Interfaces, 2013, 5, 12168–12174 CrossRef CAS PubMed.
- X. Hao, J. Wang, B. Ding, Z. Chang, Y. Wang, H. Dou and X. Zhang, ChemNanoMat, 2017, 3, 895–901 CrossRef CAS.
- G. Kim, J. Yang, N. Nakashima and T. Shiraki, Chem. – Eur. J., 2017, 23, 17504–17510 CrossRef CAS PubMed.
- C. Huang, A. M. Puziy, O. I. Poddubnaya, D. Hulicova-Jurcakova, M. Sobiesiak and B. Gawdzik, Electrochim. Acta, 2018, 270, 339–351 CrossRef CAS.
- Y. Li, J. Dong, J. Zhang, X. Zhao, P. Yu, L. Jin and Q. Zhang, Small, 2015, 11, 3476–3484 CrossRef CAS PubMed.
- M. Kim, P. Puthiaraj, Y. Qian, Y. Kim, S. Jang, S. Hwang, E. Na, W.-S. Ahn and S. E. Shim, Electrochim. Acta, 2018, 284, 98–107 CrossRef CAS.
- Y. Zhao, F. Xie, C. Zhang, R. Kong, S. Feng and J.-X. Jiang, Microporous Mesoporous Mater., 2017, 240, 73–79 CrossRef CAS.
- J. Lee, J. Choi, D. Kang, Y. Myung, S. M. Lee, H. J. Kim, Y.-J. Ko, S.-K. Kim and S. U. Son, ACS Sustainable Chem. Eng., 2018, 6, 3525–3532 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting information contains synthetic schemes, electrochemical investigation data, IR and 13C CP-MAS NMR spectra, water contact angle images, FESEM images and elemental mapping, powder X-ray diffraction, Raman spectra, deconvoluted XPS spectra and Nyquist plots. See DOI: 10.1039/c8qm00641e |
| This journal is © the Partner Organisations 2019 |