
Chitosan based in situ and ex situ magnetic iron oxide nanoparticles for rapid endotoxin removal from protein solutions†
Achyut
Konwar
a,
Devasish
Chowdhury
 a and
Abhijit
Dan
*ab
a and
Abhijit
Dan
*ab
aPhysical Sciences Division, Institute of Advanced Study in Science and Technology, Paschim Boragaon, Garchuk, Guwahati – 781035, India
bDepartment of Chemistry & Centre for Advanced Studies in Chemistry, Panjab University – Chandigarh, Sector – 14, Chandigarh – 160014, India. E-mail: abhijit@pu.ac.in
First published on 8th March 2019
Abstract
Endotoxin removal from protein solutions remains a very challenging task for bioprocess engineers. In this study, we report a facile design and synthesis of chitosan-based iron oxide nanoparticles using two different approaches, namely in situ and ex situ, to address the current issue. In the in situ method, first chitosan hydrogel was prepared using glutaraldehyde (GA) as a cross-linker, and then the iron oxide nanoparticles were allowed to form inside the hydrogel matrix by a co-precipitation process, resulting in chitosan–iron oxide nanocomposites (in situ CHIO), while in the ex situ method, previously synthesized bare magnetic nanoparticles (MNP) were functionalized with chitosan to obtain chitosan coated iron oxide nanoparticles (ex situ CHIO). The characterization of the resulting materials using several physical techniques viz., Fourier Transform Infrared Spectra (FTIR), X-ray Diffraction (XRD), Thermal Gravimetric Analysis (TGA), Vibrating Sample Magnetometry (VSM) and Transmission Electron Microscopy (TEM), revealed that the characteristic features of the materials obtained by the two different approaches were remarkably different. We further explored the opportunity to employ the materials prepared by the two different methods to obtain their effectiveness in the clearance of a toxic bacterial component, namely, endotoxin, which could be removed from protein solutions by making use of the magnetic property of the prepared nanomaterials. A good endotoxin clearance with minimal protein loss was observed when the samples were treated with the iron oxide incorporated chitosan hydrogel nanocomposites as prepared by the in situ method. In comparison to the ex situ functional nanoparticles, the in situ hydrogel nanocomposites retained their high removal efficiency after five cycles, thus providing an excellent cost-effective material for endotoxin removal. The combination of remarkable removal efficiency, minimal protein loss and reusability of the in situ CHIO nanocomposites obtained by mineralization of the hydrogel offered a complete and versatile toolbox for rapid clearance of a biological toxic molecule in clinically relevant matrices.
Introduction
Endotoxin is the primary toxic constituent of the outer membrane of Gram-negative bacteria.1 It is a lipopolysaccharide (LPS) consisting of three structural components: lipid A, an anchor that secures the molecule within the membrane, a connecting core oligosaccharide, and a polysaccharide side chain that interacts with the external environment (Fig. 1a). Upon proliferation or death, Gram-negative bacteria shed endotoxin into the environment, thereby triggering severe physiological reactions in mammalian organisms, and thus leading to severe health conditions including death.1–12 Because LPS is an indicator of bacterial contamination, and host immune systems respond to LPS, detection, quantification and removal of LPS is critically important in a wide range of health-related contexts, including clinical and basic biomedical research,13–15 public and occupational health16–18 and device and agent safety.19–22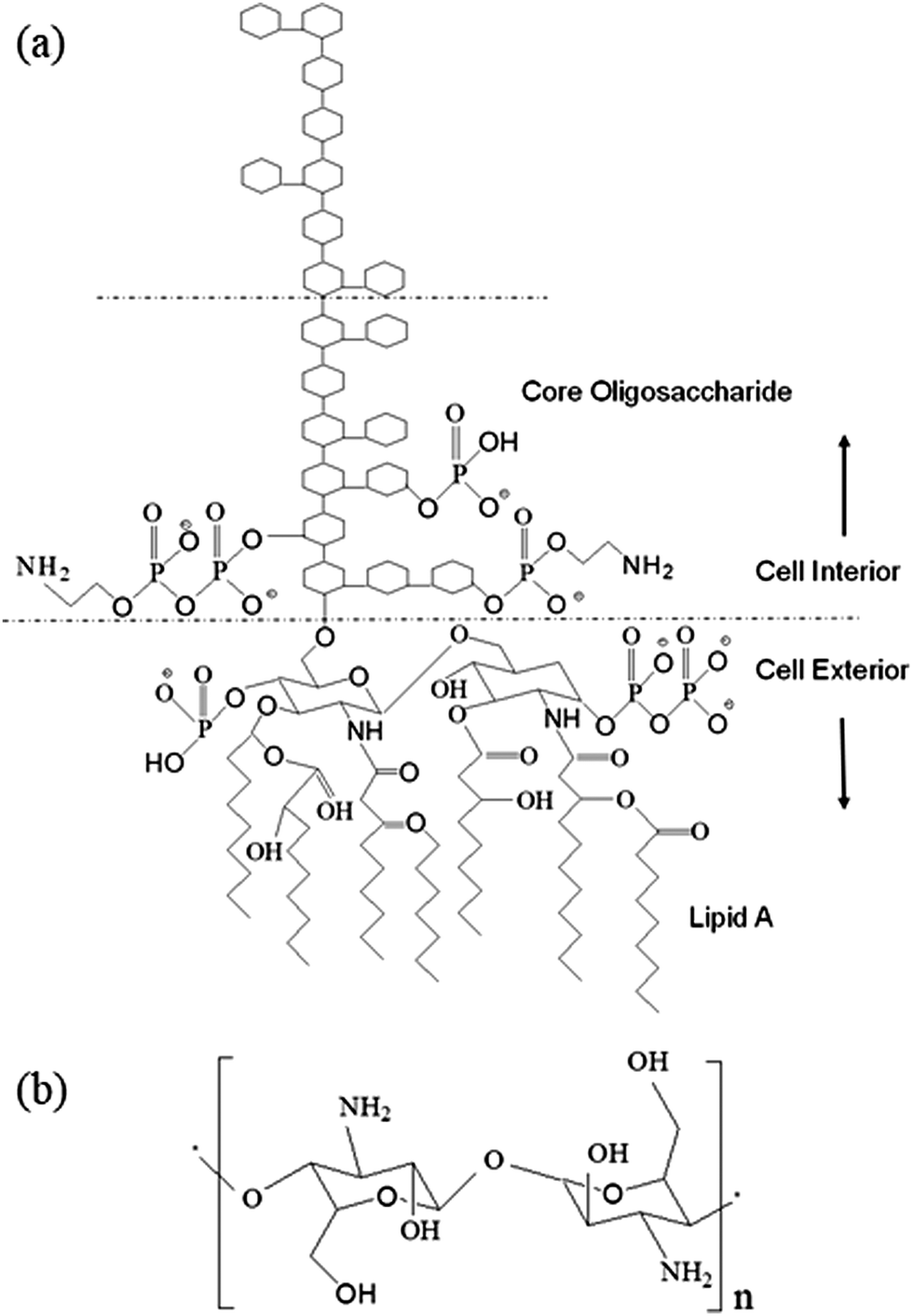 | ||
| Fig. 1 Schematic view of the chemical structure of (a) endotoxin from E. coli and (b) chitosan. | ||
Endotoxins are known to be frequent contaminants in protein solutions derived from biological preparations and their removal from such solutions remains a very challenging task for bioprocess engineers.23,24 Most commonly used methods such as ion exchange chromatography and cationic immobilized adsorbents viz., histamine, poly-L-lysine (PL), etc.,25,26 for endotoxin removal from protein solutions mostly exploit the electrostatic interactions between the negatively charged LPS and positively charged ligands. Caution must be taken when any type of ion exchange column is used to remove endotoxin, because (i) there may be significant ionic interactions between proteins and the resin, which often result in large losses in protein yield,27–29 and (ii) the endotoxin can be eluted from the column as it is tightly associated with the flow-through protein.29,30 Moreover, these methods are sensitive to pH and salt concentration, and are generally not suitable for acidic proteins.30 Methods using hydrophobic adsorbents are less effective as the endotoxin molecules shield their hydrophobic portion by aggregation or by associating with other protein molecules.30 Endotoxin removal may be robust over a wider range of conditions using biological affinity-based methods, but these methods generally impose high costs and risk of contamination by toxic affinity ligands.30–32 Ultra-filtration being a slow process requires high temperature for partial decomposition of endotoxin under acidic pH conditions, which may lead to the permeation of lipid A, the true toxic part of the molecule.33 Other methods such as detergent-based extraction are not suitable for protein solutions due to their limited effectiveness and the difficulty to remove residual detergents.30 Therefore, the development of a steady and efficient method for rapid clearance of endotoxin from protein solutions will be of great importance in bioprocess engineering, and such highly desirable studies have not been fully explored yet.
Numerous research efforts have been initiated towards the development of novel and specific strategies to rapidly remove such highly toxic endotoxins from contaminated protein solutions. In a study, Herrmann et al., describe a magnetic separation-based approach using polymyxin B functionalized nanomagnets to eliminate endotoxin from human blood in vitro.34 As reported by Gopal et al.,35 chitosan capped magnetic nanoparticles were used to investigate the separation of endotoxin from urine. More recently, a core–shell magnetic nanoparticle based abiotic system has been proposed that shows reversible capture and removal of LPS molecules in solution, thus offering a regenerable platform for endotoxin management.36 However, these methods are still not robust and efficient enough to meet the requirements for protein purification due to their complicated fabrication and limited biocompatibility.
In this work, we endeavored to fabricate chitosan–iron oxide nanoparticles (CHIO) by employing two different approaches, namely in situ and ex situ co-precipitation methods, to obtain a system that promoted effective as well as rapid clearance of endotoxin from protein solutions. Chitosan, the cationic (1–4)-2-amino-2-deoxy-β-D-glucan, contains –NH2 functional groups in the C-2 position of the D-glucosamine repeating unit, which make this polymer unique in its chemical behavior (Fig. 1b). It acquires positive charge due to the protonation of the –NH2 groups in acidic medium.37 This allows the negatively charged LPS to bind with the positively charged surfaces of materials by electrostatic interaction, which controls the endotoxin clearance from the contaminated solution. The removal of endotoxin could be monitored by quantitative estimation of LPS after treatment using a commercially available Limulus Amebocyte Lysate (LAL) assay.38
Here, we further compared the effectiveness of the magnetic hydrogel nanocomposite, in situ CHIO, obtained by in situ mineralization of the hydrogel in endotoxin removal with that of the chitosan coated iron oxide nanomaterials, ex situ CHIO, that were prepared by ex situ polymeric functionalization of iron oxide. A thorough experimental study was carried out to obtain a material that possesses the maximum chitosan loading on the iron oxide nanoparticles, which eventually measured the removal efficiency of the materials. In comparison to the ex situ chitosan coating of iron oxide nanoparticles, in situ synthesis of magnetic nanoparticles inside the hydrogel matrix turned out to be a promising approach for protein purification that allowed a good endotoxin removal with minimal loss of protein. Unlike the other techniques, this method did not allow retention or binding of protein molecules and hence provided an easy and selective separation of endotoxin. In addition, the in situ CHIO nanocomposite retained its high removal efficiency after five cycles, thus offering an excellent cost-effective material for rapid clearance of endotoxin from protein solutions.
Experimental
Materials
Biopolymer chitosan (M.W. ≃193![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 Da) was purchased from SRL, India. Lipopolysaccharides (LPS) from E. coli O55:B5 purified by ion-exchange chromatography, ferric chloride hexahydrate (FeCl3·6H2O) and ferrous chloride tetrahydrate (FeCl2·4H2O) were obtained from Sigma Aldrich, Germany. Other chemicals, viz., glutaraldehyde solution (GA, 25% in H2O), acetic acid, and sodium hydroxide (pellets) were purchased from Merck, India. All chemicals were of analytical grade reagents and used without further purification. A Limulus Amebocyte Lysate (LAL assay) kinetic-QCL kit (QCL-1000) for estimation of endotoxin concentration was purchased from LONZA, Switzerland.
400 Da) was purchased from SRL, India. Lipopolysaccharides (LPS) from E. coli O55:B5 purified by ion-exchange chromatography, ferric chloride hexahydrate (FeCl3·6H2O) and ferrous chloride tetrahydrate (FeCl2·4H2O) were obtained from Sigma Aldrich, Germany. Other chemicals, viz., glutaraldehyde solution (GA, 25% in H2O), acetic acid, and sodium hydroxide (pellets) were purchased from Merck, India. All chemicals were of analytical grade reagents and used without further purification. A Limulus Amebocyte Lysate (LAL assay) kinetic-QCL kit (QCL-1000) for estimation of endotoxin concentration was purchased from LONZA, Switzerland.
Preparation of chitosan hydrogel (CH-Gel)
A 20 mL chitosan solution (3% w/v) was prepared by dissolving chitosan in 2% (v/v) acetic acid with constant stirring for 2 hours at 60 °C. The solution was then allowed to cool to room temperature followed by dropwise addition of 120 μL GA (25% in H2O) into the solution with constant stirring. The solution was then poured into a Petridish and kept undisturbed for 24 hours. A soft cross-linked three dimensional network structure of chitosan hydrogel was formed and the solution turned light yellow to brownish yellow. The prepared chitosan hydrogel was then thoroughly washed with Milli-Q water several times to remove any unreacted monomer.In situ synthesis of magnetic iron oxide nanoparticles within the chitosan hydrogel matrix (in situ CHIO)
The prepared chitosan hydrogel was soaked in 1.5 M Fe3+ solution for 30 minutes. Then the Fe3+ loaded hydrogel was washed with Milli-Q water and soaked in 0.75 M Fe2+ solution for another 30 minutes. After that, the Fe3+/Fe2+ loaded hydrogel was subsequently washed with Milli-Q water. This cycle was repeated three times. Finally, the obtained hydrogel containing iron ions was soaked in 1 N NaOH solution for 12 hours with flow of nitrogen gas for the first few minutes. The reddish brown color gel turned black due to formation of iron oxide nanoparticles inside the hydrogel matrix, which was then washed with Milli-Q water to remove excess alkali until the pH of the washed water was close to 7 and dried at 60 °C in a hot air oven. These in situ synthesized nanoparticles using 3% (w/v) chitosan hydrogel are referred to as ‘In-3’ for their subsequent presentation in this paper.Synthesis of magnetic iron oxide nanoparticles (bare MNPs)
Typically, magnetic iron oxide nanoparticles were synthesized with the most commonly used co-precipitation technique as reported elsewhere.37,39 Briefly, a clear solution of Fe3+/Fe2+ ions was prepared in a 2:1 ratio by mixing 5 mL of 0.2 M Fe3+ and 5 mL of 0.1 M Fe2+ ion solutions at room temperature, to which a 50 mL solution of 1 M aqueous NH4OH was added. The solution was stirred at 400 rpm for 30 minutes with continuous supply of N2. The black precipitate obtained at the end was collected with the help of a strong magnet and washed several times with Milli-Q water until all the alkali was washed out.
Ex situ functionalization of iron oxide nanoparticles with chitosan (ex situ CHIO)
A dispersion of magnetic iron oxide of concentration 10 mg mL−1 was added into the chitosan solutions dissolved in 2% (v/v) acetic acid. The functionalization was carried out at various chitosan concentrations by varying the chitosan to iron oxide ratio in the mixture, viz., Ex 5:1, Ex 10:1 and Ex 20:1. The mixture was sonicated in a bath sonicator for 40 minutes followed by mechanical stirring at 60 °C for 2 hours. The prepared chitosan coated iron oxide nanoparticles were washed several times by magnetic decantation and collected after drying.
Characterization
The chitosan based magnetic iron oxide nanoparticles prepared by both in situ and ex situ methods were characterized by various physical techniques, viz., Fourier transform infrared spectra (FTIR), X-ray Diffraction (XRD), Vibrating Sample Magnetometry (VSM), Thermal Gravimetric Analysis (TGA), and Transmission Electron Microscopy (TEM). The FTIR spectra were obtained using a Nicolet 6700 FTIR instrument. Powder XRD spectra were collected on a Bruker D8 Advance diffractometer. The magnetic properties of the nanomaterials were studied with a Lake Shore model 7410 vibrating sample magnetometer (VSM). To analyze the thermo-stability of the composites, TGA thermograms were recorded with a PerkinElmer 4000 instrument, and the analysis was performed in the temperature range of 35–800 °C at a heating rate of 10 °C min−1 and with a nitrogen flow rate of 20 mL min−1. The samples were dried overnight in a vacuum oven before the TGA was performed. Transmission electron microscopy (TEM) was conducted on JEOL JEE 2100 operated at 200 kV.Endotoxin removal from protein solutions
Proteins samples (1 mg mL−1) were spiked with 2 μg mL−1 LPS. Subsequently, chitosan–iron oxide (CHIO) nanoparticles (20 mg mL−1) prepared by both in situ and ex situ methods were added and incubated for 2 hours at 37 °C. Then, the magnetic materials were separated from the solutions by applying an external magnetic field gradient using a magnetic separator. Following magnetic separation, the protein samples were collected for quantitative determination of endotoxin. A standard chromogenic Limulus Amebocyte Lysate (LAL) kinetic-QCL assay (QCL-1000, LONZA, Switzerland) was used to measure the concentration of bacterial endotoxin in the aqueous solution. The LAL reagent and chromogenic substrate were added to the diluted samples and incubated for 10 minutes. Gram-negative bacterial endotoxin catalyzed the activation of a proenzyme in the LAL. The initial rate of activation was measured by the concentration of endotoxin present. The activated enzyme then catalyzed the splitting of pNA from the colorless substrate Ac-Ile-Glu-Ala-Arg-pNA. The pNA released was determined photometrically at 405 nm continuously throughout the incubation period. The concentration of endotoxin in the samples was calculated from a standard curve. The endotoxin level of each sample was measured before and after treatment. Protein recovery was determined using a standard Bradford protein estimation assay (Sigma) at a wavelength of 595 nm.Reusability experiment
The LPS loaded materials were regenerated with 0.2 N NaOH in 20% ethanol for 2 hours. Then, they were washed with 2 M NaCl solution followed by endotoxin free water. These steps were repeated two additional times. Finally, the materials were collected from the solution by simple magnetic separation and equilibrated with endotoxin free buffer at pH 6–8 for further use in the succeeding cycles. The reusability experiments were conducted for five cycles. The concentration of LPS was subsequently measured by LAL assay after each cycle.Results and discussion
Chitosan based magnetic iron oxide nanoparticles (CHIO) were successfully prepared by both ex situ and in situ co-precipitation methods. While the ex situ method promoted the coating of the previously synthesized magnetic iron oxide nanoparticles (bare MNP) with chitosan, in the in situ method, first chitosan hydrogel was prepared using GA as a cross-linker, and then the iron oxide nanoparticles were allowed to form inside the hydrogel matrix. The resulting materials obtained by these two approaches, viz., in situ CHIO and ex situ CHIO were added to the LPS contaminated protein solutions to evaluate their effectiveness in endotoxin removal. A schematic representation of the protocols involving mechanistic steps followed for the formation of these nanoparticles and subsequent adsorption of endotoxin are illustrated in Scheme 1.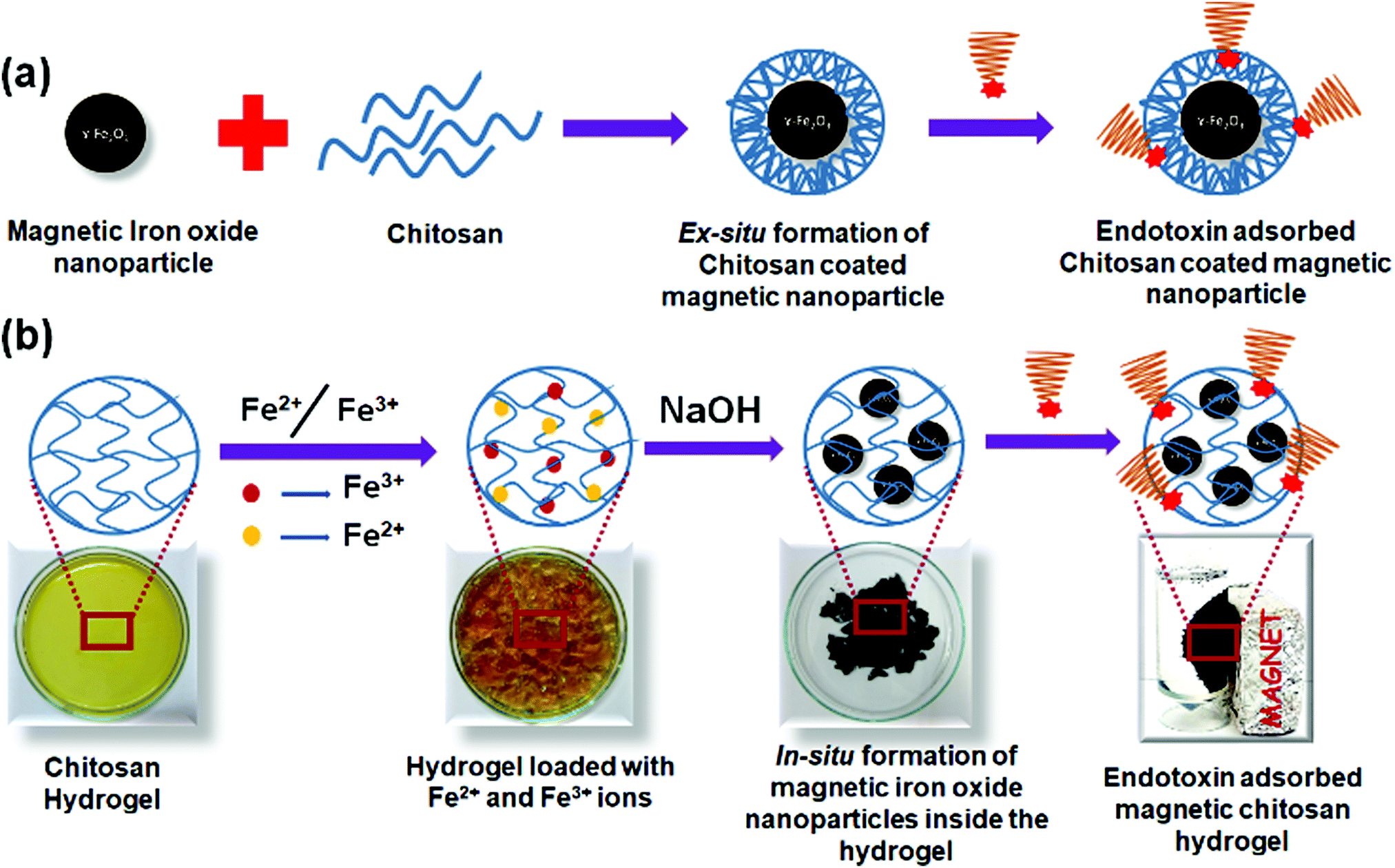 | ||
| Scheme 1 Schematic representation of the protocol followed for formation of (a) chitosan coated magnetic iron oxide nanoparticles by an ex situ method, and (b) an iron oxide incorporated chitosan hydrogel nanocomposite by in situ mineralization and their subsequent use in endotoxin removal. | ||
Formation of a GA cross-linked chitosan hydrogel was confirmed by the physical appearance as well as by the FTIR spectra (Fig. 2a), where the peak appearing at 3380 cm−1 for O–H stretching of chitosan powder broadened up after hydrogel formation due to increased hydrogen bonding. The peak at 1598 cm−1 associated with the N–H bending frequency also shifted to lower wavenumber, i.e., 1570 cm−1, suggesting that the –NH2 groups of chitosan were involved in some kind of secondary interactions. After formation of the iron oxide incorporated chitosan hydrogel by the in situ method, the color of the hydrogel system turned black. FTIR spectra of the hydrogel nanocomposite, in situ CHIO, showed that the peak at 3360 cm−1 associated with O–H stretching frequency broadened further and a new peak at 575 cm−1 appeared due to the Fe–O bending frequency, indicating successful formation of iron oxide nanoparticles inside the hydrogel matrix.
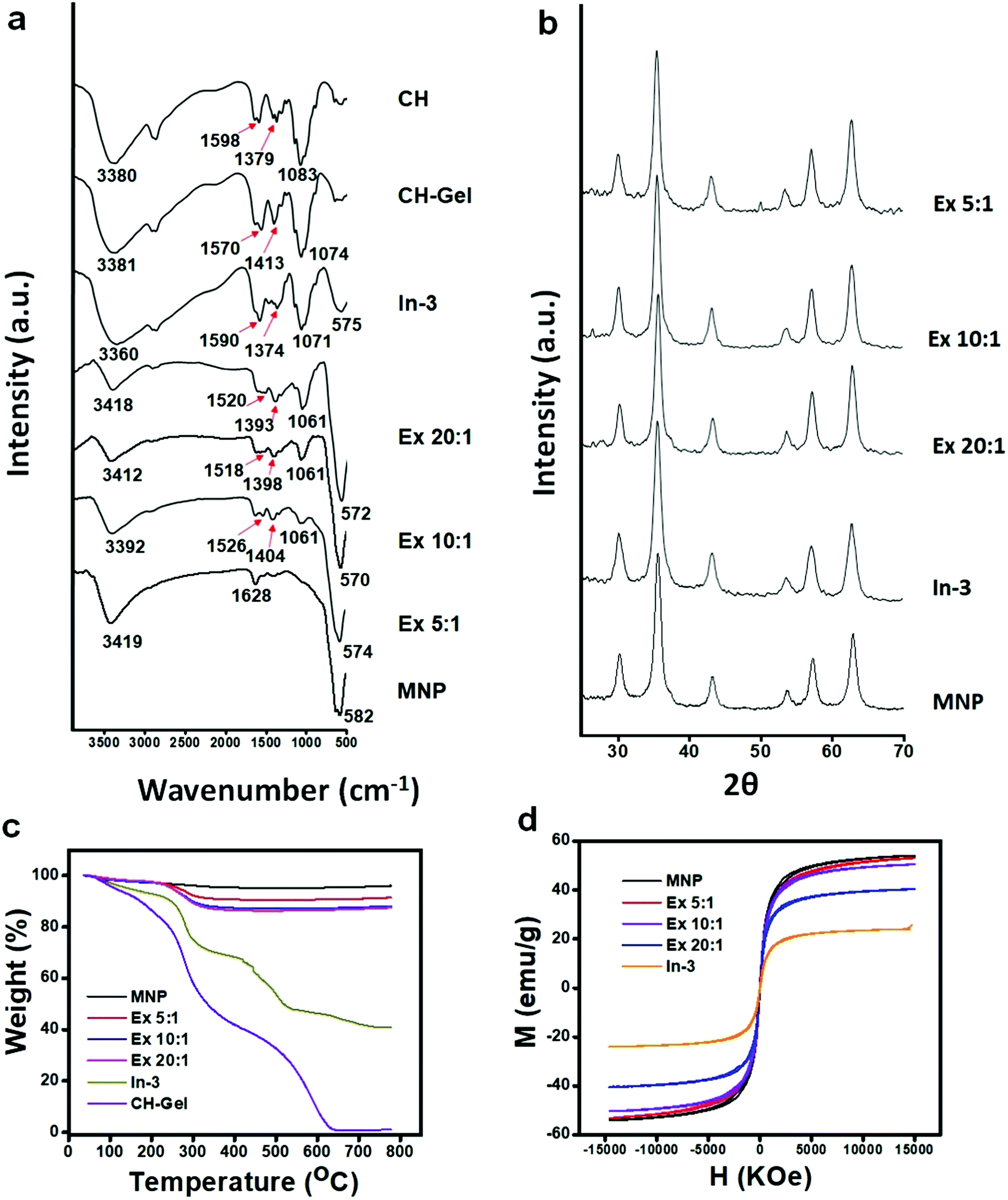 | ||
| Fig. 2 (a) FTIR, (b) XRD spectra, (c) TGA thermograms and (d) magnetization–hysteresis (M–H) loops of the CHIO nanoparticles prepared by in situ (In-3) and ex situ (Ex 20:1, Ex 10:1, Ex 5:1) methods. Note: CH = dry chitosan, CH-Gel = GA cross-linked chitosan hydrogel. | ||
The chitosan coated iron oxide nanoparticles, ex situ CHIO, were prepared at different mixing ratios and the resulting nanoparticles are represented as Ex x:y, where x:y refers to the weight ratio of chitosan to iron oxide. Chitosan loading to iron oxide increased with increasing chitosan content in the reaction mixture, which was evident by the increasing peak intensities associated with chitosan as observed in the FTIR spectra (Fig. 2a). The FTIR spectra of the bare MNP possessed two low intensity peaks arising at 3419 cm−1 and 1628 cm−1 due to the moisture absorbed by the iron oxide, and an intense peak at 582 cm−1 associated with the Fe–O bending frequency. New peaks at 1061 cm−1 (for C–O stretching of primary alcohol), in the range of 1518 to 1536 cm−1 (for N–H bending of amine) and 1393 to 1421 cm−1 (due to the –CH3 symmetrical deformation mode) appeared after chitosan coating with increasing intensity upon increase in the concentration of chitosan.
Here, we observed a remarkable difference in the FTIR spectra between the in situ and ex situ products. The peak arising at 1590 cm−1 for the N–H bending frequency of in situ CHIO moved to lower wavenumbers, i.e., between 1518–1526 cm−1 in the case of ex situ CHIO, which was attributed to the structural difference of these two products resulting from two different ways of their formation. In the in situ synthesis of the nanoparticles, hydrogel was formed by covalent crosslinking between the –NH2 groups of chitosan and the −CHO groups of GA.40 In contrast, during ex situ functionalization, the chitosan with protonated –NH2 groups was allowed to bind with the negatively charged surface of the iron oxide nanoparticles under acidic conditions. The peak positions for C–O stretching and –CH3 symmetrical deformation also changed significantly, which might be due to the change in hydrogen bonding caused by their structural differences. Interestingly, the peak appearing at 582 cm−1 for the Fe–O bending frequency of bare MNP shifted to lower wavenumbers (e.g., 575 cm−1 for In-3, 574 cm−1 for Ex 5:1, 570 cm−1 for Ex 10:1 and 572 cm−1 for Ex 10:1) upon chitosan loading, indicating an electrostatic interaction between chitosan and iron oxide nanoparticles.37,41
As shown in Fig. 2b, the XRD patterns further confirmed the existence of iron oxide nanoparticles in both types of CHIO nanomaterials. Six characteristic diffraction peaks for iron oxide, marked by their crystal planes (220), (311), (400), (422), (511), and (440), were observed in all the samples. The d-spacing values calculated from the peaks obtained in the XRD spectra and their intensities (see the ESI,† Table S1) revealed that both types of iron oxides, magnetite (Fe3O4) and maghemite (γ-Fe2O3), were obtained in different percentages with an inverse spinal structure inside the chitosan hydrogel matrix as well as for those used in ex situ chitosan coating.37 According to the Scherrer equation, all the iron oxide nanoparticles were highly crystalline with an average particle size of around 13.4–17 nm (see the ESI,† Table S2), which was consistent with the results obtained from TEM characterization as presented in the subsequent section.
The TGA spectrograms of the CHIO nanoparticles not only provided information about their thermostability but also determined the average mass content of chitosan in the materials prepared by both in situ and ex situ methods. As shown in Fig. 2c, about 3% weight loss of bare MNP was observed over the temperature range from 30 to 800 °C due to the loss of residual water in the sample. The TGA thermogram of the in situ sample (In-3) exhibited a two-stage degradation pattern, consistent with that obtained for a pure chitosan hydrogel (CH-Gel). The polysaccharide side chains of the pure CH-Gel started to degrade at around 180 °C, which shifted to 270 °C in the case of in situ CHIO. This tremendous improvement of thermostability for the in situ CHIO nanocomposites can be explained by the fact that the presence of strong interaction of iron oxide with the hydrogel matrix led to a decreased mobility of the chitosan side chains. The second-stage of weight loss was observed above 430 °C, which was mainly attributed to the degradation of aromatic moieties of chitosan. In the case of ex situ CHIO nanoparticles, the percentage weight loss at the first stage of degradation decreased and the second stage disappeared, indicating a lower amount of chitosan loading. The weight percentage of chitosan in the materials can be determined using the Peniche's formula.42
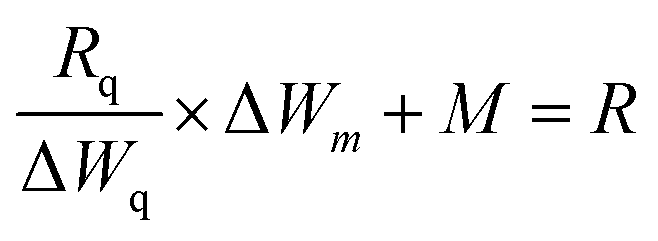 | (1) |
| Sl no. | Sample name | Percent chitosan content (wt%) |
|---|---|---|
| 1 | Ex 5:1 |
8.78 |
| 2 | Ex 10:1 |
12.18 |
| 3 | Ex 20:1 |
12.68 |
| 4 | In-3 | 60 |
The results demonstrated that the average mass content of chitosan in the ex situ CHIO nanoparticles increased to a small extent with the concentration of chitosan used in the synthesis, while a remarkable increase in the chitosan content (60 wt%) was found in the case of the in situ CHIO hydrogel nanocomposites. Thus, the hydrogel network system with increased percentage of chitosan in the in situ product made it more suitable for endotoxin removal from protein solutions.
The hysteresis loops (M–H) of the prepared materials measured at 300 K are shown in Fig. 2d. The M–H loops demonstrated that bare MNP as well as both in situ and ex situ CHIO nanomaterials exhibited a characteristic paramagnetic behavior with very minimal coercivity values (see the ESI,† Table S3). The saturated magnetization of bare MNP was found to be 54.15 emu g−1 and the values decreased with an increasing presence of chitosan in the materials – the saturation magnetization values for the Ex 5:1, Ex 10:1, Ex 20:1 and In-3 samples were 53.2, 50.5, 40.5 and 22.9 emu g−1, respectively. This decrease in saturation magnetization was attributed to the shielding of the polymeric loading resulting from the modification process. The in situ CHIO nanocomposites with low saturation magnetization value also possessed enough magnetic response to meet the need of magnetic separation.
The structural and morphological features of the prepared materials were characterized by TEM as shown in Fig. 3. It could be seen that the bare MNP (Fig. 3a) and ex situ functionalized iron oxide nanoparticles (Fig. 3c) consisted of a spherical structure with an average particle size of around 15 nm. In contrast, the in situ synthesis of iron oxide inside the chitosan hydrogel provided crystalline particles (Fig. 3b), the shapes of which were predominantly cubic with some spherical particles of 13 mm average diameter. The reason for this unique structural feature might be due to the fact that the iron ions inside the chitosan hydrogel became rigid owing to in situ mineralization that promoted agglomeration of the nanoparticles as they attempted to become stable. This occurred in a uniform and repeating pattern, resulting to the formation of nanocrystals. The TEM image of the in situ CHIO nanocomposites also clearly showed the existence of chitosan hydrogel matrix where the particles were dispersed uniformly, as indicated by the red arrows in Fig. 3b. The cumulative particle size distribution histograms of the respective nanomaterials are shown in Fig. 3(d)–(f). As compared to bare MNP and ex situ CHIO nanomaterials, a narrow size distribution of iron oxide nanoparticles was observed for the in situ CHIO hydrogel nanocomposites. The monodispersity of the size distribution resulted from the in situ mineralization of hydrogel by iron ions during the synthesis, which prevented the abnormal growth of magnetic grains.
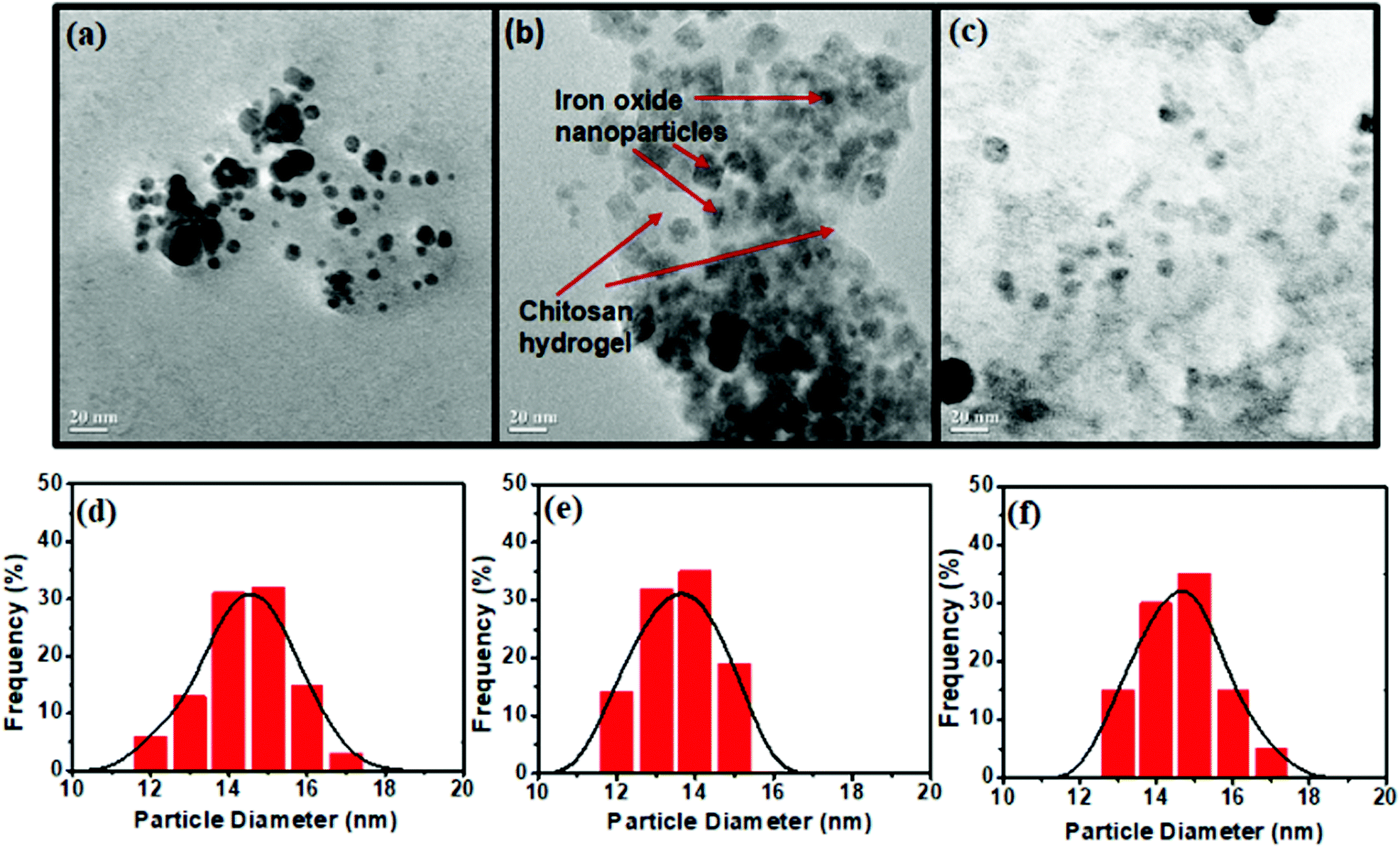 | ||
| Fig. 3 TEM images (a–c) and the particle size distribution (d–f) of bare MNP (a and d) and CHIO nanoparticles prepared by in situ (b and e) and ex situ methods (c and f). | ||
Removal of endotoxin
The feasibility of using CHIO nanoparticles to remove the bacterial endotoxin was exploited by treating the LPS contaminated samples with the prepared materials, the results of which are depicted in Fig. 4. Typically, the magnetic adsorbents (2 mg mL−1) were added to the LPS solutions of 2 μg mL−1 concentration and incubated for 2 hours at 37 °C with constant stirring. This allowed the negatively charged endotoxin to adsorb at the positively charged surface of the materials by electrostatic interaction. Long incubation time and increased temperature promoted LPS binding to chitosan at the surface of the adsorbents.43 The LPS loaded magnetic materials were subsequently removed by magnetic separation and the samples were collected for determination of LPS concentration using a commercially available Limulus Amoebocyte Lysate (LAL) assay. The essential steps for rapid clearance of endotoxin from contaminated samples are schematically presented in Fig. 4a. In order to evaluate the effectiveness of the materials as obtained by ex situ (Ex 5:1, Ex 10:1, Ex 20:1) as well as in situ (In-3) methods, the samples were treated with these materials under the same experimental conditions. As shown in Fig. 4b, the removal of LPS was strongly dependent on chitosan loading; among the ex situ CHIO nanomaterials, a maximum removal of around 88% was observed for the Ex 20:1 sample, while effective LPS removal (over 99%) could be easily achieved when in situ CHIO nanocomposites were used. Bare MNPs, used as controls, were added to account for unspecific adsorption of LPS. No significant removal was observed when the sample was treated with bare MNPs, which led us to conclude that the clearance of LPS was due to specific binding of LPS with chitosan controlled by electrostatic interaction.
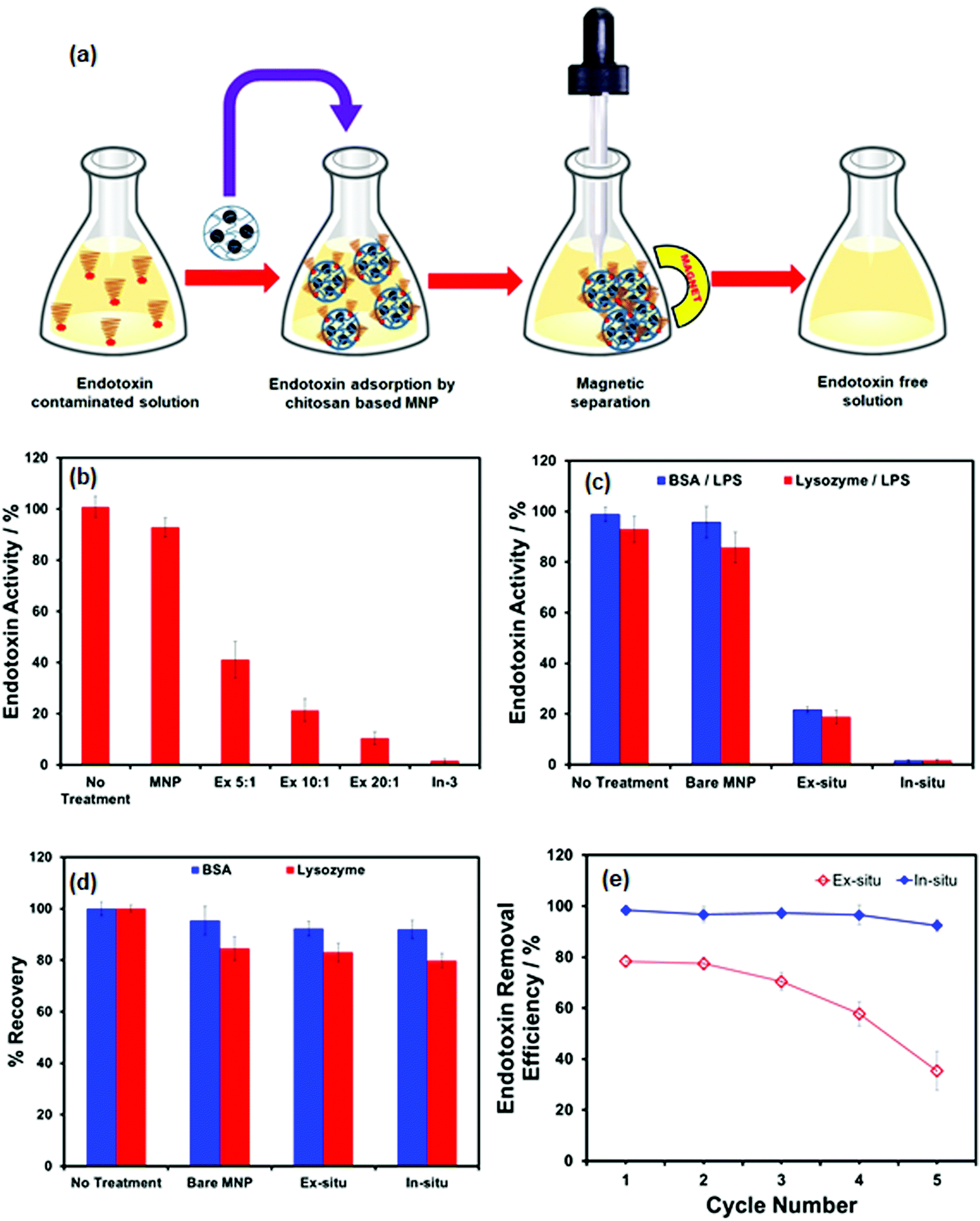 | ||
| Fig. 4 (a) Schematic illustration of magnetic separation based protein purification using CHIO nanomaterials, (b) effect of chitosan loading on endotoxin removal by ex situ chitosan coated iron oxide nanoparticles (Ex 5:1, Ex 10:1 & Ex 20:1) and in situ iron oxide incorporated chitosan hydrogel nanocomposites (In-3), (c) endotoxin removal from protein (BSA and lysozyme) solutions using as prepared bare MNP, and CHIO nanomaterials prepared by ex situ (Ex 20:1) and in situ (In-3) methods, (d) protein recovery for BSA and lysozyme (1 mg mL−1), (e) reusability test of the ex situ (Ex 20:1) and in situ (In-3) samples for five successive cycles. Note: the samples were spiked with 2 μg mL−1 LPS and treated with 2 mg mL−1 magnetic nanoparticles. | ||
Following this encouraging result, this approach was further used for a protein purification process. Endotoxin removal using CHIO nanomaterials was investigated for BSA and lysozyme of 1 mg mL−1 concentrations spiked with 2 μg mL−1 LPS. The contaminated protein solutions were treated with bare MNP, Ex 20:1 and In-3 samples for a comparative study. To evaluate the LPS removal efficiency, the endotoxin activity and protein recovery after treatment were determined by the LAL assay and the standard Bradford method, respectively and the results are presented in Fig. 4c and d, respectively. Bare MNPs were not able to eliminate LPS from the solutions at all, whereas around 80% LPS could be removed by using ex situ CHIO nanoparticles (Ex 20:1 sample with 12.68 wt% chitosan content) in the presence of both BSA and lysozyme. In contrast, when a similar batch experiment was conducted with in situ CHIO nanocomposites (In-3 sample), having a large amount of chitosan content (60 wt%), a good endotoxin reduction (over 99%) was obtained even in the presence of proteins. This suggested that the chitosan loading in the materials controlled the LPS clearance from the contaminated samples. Conversely, no obvious loss of the total proteins was observed during the process of LPS removal in both cases (Fig. 4d).
Reusability of the materials
Besides good removal performance, an excellent adsorbent should have benign reusability, which is crucial to provide an efficient, scalable, and economical system. To study the reusability of the prepared materials, NaOH solution in 20% ethanol was employed as a regeneration agent and an aqueous solution of NaCl was used as an eluent for endotoxin. The effect of five consecutive regeneration cycles was studied. As shown in Fig. 4e, the LPS removal efficiency decreased slowly after the second cycle with increasing number of cycles in the case of chitosan coated nanoparticles, ex situ CHIO, whereas the iron oxide incorporated chitosan hydrogel nanocomposites, in situ CHIO, could retain their high removal efficiency after five cycles, indicating that CHIO nanocomposite materials prepared by in situ mineralization of chitosan hydrogel had excellent regenerability and adsorptive properties. The decrease in removal efficiency of ex situ CHIO nanoparticles might arise from the slight loss of chitosan from the surface of the nanoparticles during the regeneration process. Thus, the results demonstrated that the as-prepared in situ CHIO hydrogel nanocomposites could act as an excellent cost-effective separation platform for endotoxin contaminated protein samples.Conclusions
We developed a simple method for reversible capture and removal of toxic bacterial LPS from aqueous protein solutions employing chitosan based magnetic iron oxide nanoparticles. Two different approaches were used to synthesize such materials: firstly, in situ mineralization of a chitosan hydrogel, in which the iron oxide nanoparticles were allowed to form, resulting in an iron oxide incorporated chitosan hydrogel nanocomposite, and secondly, ex situ functionalization of previously synthesized bare MNPs with chitosan to obtain chitosan coated iron oxide nanoparticles. The resulting materials obtained by two different approaches exhibited a remarkable difference in their characteristic features as investigated using several physical techniques such as FTIR, XRD, TGA, VSM, and TEM. The potential of the prepared materials for effective removal of bacterial endotoxin from protein solutions was investigated by conducting batch adsorption experiments, in line with a comparative study for different ways of material preparation. The endotoxins that were attached to the surface of the adsorbent materials could be separated from the protein solutions by making use of the magnetic property of the materials. The negatively charged LPS bound with the positively charged affinity ligand of the materials by ionic interaction, and thus the amount of chitosan loading to iron oxide nanoparticles measured the endotoxin removal efficiency of the materials. The characterization of these materials revealed that in situ mineralization of hydrogel possessed greater chitosan loading to the iron oxide nanoparticles than ex situ polymeric coating, and hence an effective LPS removal was achieved using the hydrogel nanocomposite materials, while no obvious loss of the total proteins was found during the process of endotoxin removal. As compared to ex situ CHIO nanomaterials, the in situ hydrogel nanocomposites showed regenerability and high adsorption efficiency after five cycles. In addition, the increased presence of chitosan made the in situ CHIO nanocomposites water soluble and biocompatible, and they could be used as a potential material for several biological applications such as endotoxin removal from protein solutions. Thus, iron oxide incorporated chitosan hydrogel nanocomposites prepared by in situ mineralization of hydrogel matrix could act as an excellent cost-effective separation platform for protein purification.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of this work by the Department of Science & Technology, Govt. of India under the Ramanujan Fellowship Scheme (SB/S2/RJN-119/2015), and University Grant Commission under a start up grant (F.4-5(244-FRP)/2015/BSR) is acknowledged.References
- C. R. H. Raetz and C. Whitfield, Lipopolysaccharide endotoxins, Annu. Rev. Biochem., 2002, 71, 635–700 CrossRef CAS PubMed.
- H. Brade, S. M. Opal, S. N. Vogel and D. C. Morrison, Endotoxin in health and disease, Marcel Decker Inc, New York, 1999, ISBN 0-8247-1944-1 Search PubMed.
- W. Su, M. Lin, H. Lee, M. Cho, W. S. Choe and Y. Lee, Determination of endotoxin through an aptamer-based impedance biosensor, Biosens. Bioelectron., 2012, 32, 32–36 CrossRef CAS PubMed.
- T. Y. Yeo, J. S. Choi, B. K. Lee, B. S. Kim, H. I. Yoon, H. Y. Lee and Y. W. Cho, Electrochemical endotoxin sensors based on TLR4/MD-2 complexes immobilized on gold electrodes, Biosens. Bioelectron., 2011, 28, 139–145 CrossRef CAS PubMed.
- K. Noda, H. Goto, Y. Murakami, A. B. F. Ahmed and A. Kuroda, Endotoxin assay by bioluminescence using mutant firefly luciferase, Anal. Biochem., 2010, 397, 152–155 CrossRef CAS PubMed.
- L. Mack, B. Brill, N. Delis and B. Groner, Endotoxin depletion of recombinant protein preparations through their preferential binding to histidine tags, Anal. Biochem., 2014, 466, 83–88 CrossRef CAS PubMed.
- S. E. Kim, W. Su, M. Cho, Y. Lee and W. S. Choe, Harnessing aptamers for electrochemical detection of endotoxin, Anal. Biochem., 2012, 424, 12–20 CrossRef CAS PubMed.
- J. Reich, H. Tamura, I. Nagaoka and H. Motschmann, Investigation of the kinetics and mechanism of low endotoxin recovery in a matrix for biopharmaceutical drug products, Biologicals, 2018, 53, 1–9 CrossRef CAS PubMed.
- M. Zandieh, S. N. Hosseini, M. Vossoughi, M. Khatami, S. Abbasian and A. Moshaii, Label-free and simple detection of endotoxins using a sensitive LSPR biosensor based on silver nanocolumns, Anal. Biochem., 2018, 548, 96–101 CrossRef CAS PubMed.
- J. Reich, P. Lang, H. Grallert and H. Motschmann, Masking of endotoxin in surfactant samples: effects on Limulus-based detection systems, Biologicals, 2016, 44, 417–422 CrossRef CAS PubMed.
- D. Petsch, T. C. Beeskow, F. B. Anspach and W.-D. Deckwer, Membrane adsorbers for selective removal of bacterial endotoxin, J. Chromatogr. B: Biomed. Sci. Appl., 1997, 693, 79–91 CrossRef CAS.
- R. Ma, J. Zhao, H.-C. Du, S. Tian and L.-W. Li, Removing endotoxin from plasmid samples by Triton X-114 isothermal extraction, Anal. Biochem., 2012, 424, 124–126 CrossRef CAS PubMed.
- M. Klevens, J. R. Edwards, C. L. Richards, T. C. Horan, R. P. Gaynes, D. A. Pollock and D. M. Cardo, Estimating health care-associated infections and deaths in U.S. Hospitals, Public Health Rep., 2007, 122, 160–166 CrossRef PubMed.
- W. B. Anderson, R. M. Slawson and C. I. Mayfield, A review of drinking-water-associated endotoxin, including potential routes of human exposure, Can. J. Microbiol., 2002, 48, 567–587 CrossRef CAS PubMed.
- R. Dales, D. Miller, K. Ruest, M. Guay and S. Judek, Airborne endotoxin is associated with respiratory illness in the first 2 years of life, Environ. Health Perspect., 2006, 114, 610–614 CrossRef CAS PubMed.
- H. Bisgaard, F. Buchvald, L. B. Halkjaer, K. Bønnelykke, M. Brasholt, A. Heltberg, N. Hawwa Vissing, S. V. Thorsen, M. Stage and C. B. Pipper, Childhood asthma after bacterial colonization of the airway in neonates, N. Engl. J. Med., 2007, 357, 1487–1495 CrossRef CAS PubMed.
- C. H. Lim, I. J. Yu, H. Y. Kim, S. B. Lee, M. G. Kang, D. R. Marshak and C. K. Moon, Inflammatory and immunological responses to sub chronic exposure to endotoxin-contaminated metalworking fluid aerosols in F344 rats, Environ. Toxicol., 2005, 20, 212–218 CrossRef CAS PubMed.
- S. R. Lane, P. J. Nicholls and R. D. E. Sewell, The Measurement and Health Impact of Endotoxin Contamination in Organic Dusts from Multiple Sources: Focus on the Cotton Industry, Inhalation Toxicol., 2004, 16, 217–229 CrossRef CAS PubMed.
- M. B. Gorbet and M. V. Sefton, Endotoxin: the uninvited guest, Biomaterials, 2005, 26, 6811–6817 CrossRef CAS PubMed.
- P. Malyala and M. Singh, Endotoxin limits in formulations for preclinical research, J. Pharm. Sci., 2008, 97, 2041–2044 CrossRef CAS PubMed.
- Y. Nakagawa, T. Murai, C. Hasegawa, M. Hirata, T. Tsuchiya, T. Yagami and Y. Haishima, Endotoxin contamination in wound dressings made of natural biomaterials, J. Biomed. Mater. Res., Part B, 2003, 66, 347–355 CrossRef CAS PubMed.
- M. A. Dobrovolskaia, B. W. Neun, J. D. Clogston, H. Ding, J. Ljubimova and S. E. McNeil, Ambiguities in applying traditional limulus amebocyte lysate tests to quantify endotoxin in nanoparticle formulations, Nanomedicine, 2010, 5, 555–562 CrossRef CAS PubMed.
- V. Vagenende, T.-J. Ching, R.-J. Chua, N. Thirumoorthi. and P. Gagnon, Amide-Mediated Hydrogen Bonding at Organic Crystal/Water Interfaces Enables Selective Endotoxin Binding with Picomolar Affinity, ACS Appl. Mater. Interfaces, 2013, 5, 4472–4478 CrossRef CAS PubMed.
- P. O. Magalhães, A. M. Lopes, P. G. Mazzola, C. Rangel-Yagui, T. C. V. Penna and A. Pessoa Jr, Methods of Endotoxin Removal from Biological Preparations: a Review, J. Pharm. Pharm. Sci., 2007, 10, 388–404 Search PubMed.
- J. L. Ding, Y. Zhu and B. Ho, High-performance affinity capture-removal of bacterial pyrogen from solutions, J. Chromatogr. B: Biomed. Sci. Appl., 2001, 759, 237–246 CrossRef CAS.
- C. M. Ongkudon, J. H. Chew, B. Liu and M. K. Danquah, Chromatographic removal of endotoxins: a bioprocess engineer's perspective, ISRN Chromatogr., 2012, 1–9 CrossRef , Article ID 649746.
- R. Ma, D.-D. Fan, W.-J. Xue, J.-Y. Xing, C.-H. Zhu and X. X. Ma, Endotoxin Removal during the Purification Process of Human-like Collagen, Sep. Sci. Technol., 2010, 45, 2400–2405 CrossRef CAS.
- B. D. Kelley, M. Switzer, P. Bastek, J. F. Kramarczyk, K. Molnar, T. Yu and J. Coffman, High-Throughput Screening of Chromatographic Separations: IV. Ion-Exchange, Biotechnol. Bioeng., 2008, 100, 950–963 CrossRef CAS PubMed.
- R. H. Chen, C. J. Huang, B. S. Newton, G. Ritter, L. J. Old and C. A. Batt, Factors affecting endotoxin removal from recombinant therapeutic proteins by anion exchange chromatography, Protein Expression Purif., 2009, 64, 76–81 CrossRef CAS PubMed.
- D. Petsch and F. B. Anspach, Endotoxin removal from protein solutions, J. Biotechnol., 2000, 76, 97–119 CrossRef CAS PubMed.
- J. Li, G. Shang, M. You, S. Peng, Z. Wang, H. Wu and G. Q. Chen, Endotoxin removing method based on lipopolysaccharide binding protein and polyhydroxyalkanoate binding protein PhaP, Biomacromolecules, 2011, 12, 602–608 CrossRef CAS PubMed.
- K. Shimokawa, R. Takakuwa, Y. Wada, N. Yamazaki and F. Ishii, Adsorption of various antimicrobial agents to endotoxin removal polymyxin-B immobilized fiber (Toraymyxin®). Part 2: Adsorption of two drugs to Toraymyxin PMX-20R cartridges, Colloids Surf., B, 2013, 101, 350–352 CrossRef CAS PubMed.
- P. O. Magalhães, A. M. Lopes, P. G. Mazzola, C. R. Yagui, T. C. V. Penna and A. Pessoa Jr., Methods of endotoxin removal from biological preparations: a review, J. Pharm. Pharm. Sci., 2007, 10, 388–404 Search PubMed.
- I. K. Herrmann, M. Urner, S. Graf, C. M. Schumacher, B. R. Z’graggen, M. Hasler, W. J. Stark and B. Beck-Schimmer, Endotoxin removal by magnetic separation-based blood purification, Adv. Healthcare Mater., 2013, 2, 829–835 CrossRef CAS PubMed.
- J. Gopal, H. N. Abdelhamid, P. Y. Huaa and H. F. Wu, Chitosan nanomagnets for effective extraction and sensitive mass spectrometric detection of pathogenic bacterial endotoxin from human urine, J. Mater. Chem. B, 2013, 1, 2463–2475 RSC.
- P. Prasad, S. Sachan, S. Suman, G. Swayambhu and S. Gupta, Regenerative core–shell nanoparticles for simultaneous removal and detection of endotoxins, Langmuir, 2018, 34, 7396–7403 CrossRef CAS PubMed.
- A. Konwar, S. Kalita, J. Kotoky and D. Chowdhury, Chitosan–iron oxide coated graphene oxide nanocomposite hydrogel: a robust and soft antimicrobial biofilm, ACS Appl. Mater. Interfaces, 2016, 8, 20625–20634 CrossRef CAS PubMed.
- A. P. Das, P. S. Kumar and S. Swain, Recent advances in biosensor based endotoxin detection, Biosens. Bioelectron., 2014, 51, 62–75 CrossRef CAS PubMed.
- A. Konwar, A. Gogoi and D. Chowdhury, Magnetic alginate–Fe3O4 hydrogel fiber capable of ciprofloxacin hydrochloride adsorption/separation in aqueous solution, RSC Adv., 2015, 5, 81573–81582 RSC.
- U. Filipkowska and T. Jóźwiak, Application of chemically-cross-linked chitosan for the removal of Reactive Black 5 and Reactive Yellow 84 dyes from aqueous solutions, J. Polym. Eng., 2013, 33, 735–747 CAS.
- A. Konwar, N. Gogoi, G. Majumdar and D. Chowdhury, Green chitosan–carbon dots nanocomposite hydrogel film with superior properties, Carbohydr. Polym., 2015, 115, 238–245 CrossRef CAS PubMed.
- H. Peniche, A. Osorio, N. Acosta, A. de Campa and C. Peniche, Preparation and characterization of superparamagnetic chitosan microspheres: application as a support for the immobilization of tyrosinase, J. Appl. Polym. Sci., 2005, 98, 651–657 CrossRef CAS.
- V. N. Davidova, I. M. Yermak, V. I. Gorbach and T. F. Solovieva, The effect of temperature on the interaction of Yersinia pseudotuberculosis lipopolysaccharide with chitosan, Membr. Cell Biol., 1999, 13, 49–58 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The data obtained from XRD and VSM analysis for the chitosan based iron oxide nanoparticles prepared by in situ (In-3) and ex situ (Ex 20:1, Ex 10:1, Ex 5:1) methods. See DOI: 10.1039/c8qm00668g |
| This journal is © the Partner Organisations 2019 |