
Fluorophore core-engineered supramolecular discotic columnar liquid crystals with tunable fluorescent behavior†
Bin
Mu
 ,
Xuhong
Quan
,
Yu
Zhao
,
Xun
Li
and
Wei
Tian
*
,
Xuhong
Quan
,
Yu
Zhao
,
Xun
Li
and
Wei
Tian
*
MOE Key Laboratory of Material Physics and Chemistry under Extraordinary Conditions, Shanxi Key Laboratory of Macromolecular Science and Technology, Department of Applied Chemistry, School of Natural and Applied Sciences, Northwestern Polytechnical University, Xi’an, 710072, China. E-mail: happytw_3000@nwpu.edu.cn
First published on 12th June 2019
Abstract
Achieving hydrogen-bonded supramolecular discotic columnar liquid crystals with tunable fluorescent behavior, especially in the solid state, is still a substantial challenge due to the absence of suitable fluorophore components. To this end, we herein report a function-directing expansion based on a C3-symmetric tris(imidazole-2-yl)benzene skeletal structure to obtain various fluorophore cores, which are either fused with π-expanded benzene or naphthalene rings, or substituted with tolyl groups twisted out of the core plane. By means of complementary hydrogen-bonding interactions with alkylated benzoic acids, well-organized supramolecular discotic columnar liquid crystals can be formed and readily aligned in a homeotropic fashion. Efficient color-tunable fluorescent behavior, ranging from blue, cyan, green to yellow emission, is achieved with these liquid crystals even in the solid state, which manifest a close correlation with the different structural parameters shown by each of the designed cores, and also the assembled structures of the resultant complexes. Our study will lead to the design of new functional supramolecular liquid crystals that can potentially be applicable as anisotropic optoelectronic materials.
Introduction
The use of self-assembling blocks to develop functional supramolecular liquid crystals (LCs) through hydrogen bonding has been a subject of intense research, as it expands the scope of LCs that are often limited by tedious synthetic and purification procedures, and also endows them with dynamic functions.1–7 Among them, hydrogen-bonded discotic LCs, often with C3-symmetry, based on a heterocyclic template in combination with complementary hydrogen-bonding motifs have attracted ever-increasing interest.8–16 Such systems have a tendency to self-organize into columnar structures, which have great potential for use in a range of applications, such as ion-conductive materials,17,18 organic optoelectronics,19–23 chiral induction and amplification,24–26 particularly nanoporous cross-linked polymers with regular columnar channels.27–31 Moreover, when one of the components is intrinsically fluorescent, fluorescent supramolecular columnar LCs can be obtained as potential optoelectronic materials.32,33 Although such fluorescent columnar LCs obtained through hydrogen bonding in a multicomponent system have been reported,8,14–16 their applications are largely restricted in solution as they suffer from the problem of somewhat quenching in aggregated states, and tuning of the fluorescent behavior would not be allowed. Therefore, it is still highly desirable to develop new multicomponent hydrogen-bonded systems and obtain LC materials with efficient and finely tunable fluorescent behavior even in the solid state.To address the above-mentioned issues, it is of significance to consider elaborate designs and explore new fluorophore components consisting of hydrogen donor and acceptor moieties, which can form complementary systems. Thus, a useful strategy is to modify or extend the contents of existing hydrogen-bonded systems through the integration of certain functional motifs with the aim of constructing novel fluorescent supramolecular discotic columnar LCs and regulating their fluorescent properties. More specifically, the C3-symmetric tris(imidazole-2-yl)benzene (TIB) skeletal structure has 3 hydrogen-donors (–NH–) and 3 hydrogen-acceptors (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–), and its derivatives have been used as structure-directing agents for templated supramolecular self-assembly via hydrogen bonding with a variety of carboxylic acids.14,26,28–31 Therefore, we propose a function-directing expansion of such kind of TIB skeletal structure to obtain hydrogen-bonded supramolecular LCs with tunable fluorescence characteristics in both solution and solid states.
N–), and its derivatives have been used as structure-directing agents for templated supramolecular self-assembly via hydrogen bonding with a variety of carboxylic acids.14,26,28–31 Therefore, we propose a function-directing expansion of such kind of TIB skeletal structure to obtain hydrogen-bonded supramolecular LCs with tunable fluorescence characteristics in both solution and solid states.
Herein, various TIB cores including tris(4,5-dihydro-1H-imidazol-2-yl)benzene (HTIB), tris(1H-benzo[d]imidazol-2-yl)benzene (BTIB), and tris(1H-naphtho[2,3-d]imidazol-2-yl)benzene (NTIB), which have π-expanded benzene or naphthalene fused rings, and tris(4,5-di-p-tolyl-1H-imidazol-2-yl)benzene (TTIB), which has six tolyl substituents twisted out of the core plane (Fig. S15, ESI†), were designed and synthesized (Fig. 1). Then, these TIB-derived compounds served as complementary cores to afford discotic complexes (TIB–An) through hydrogen bonding with 3,4,5-trialkyloxybenzoic acid (An) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 stoichiometry. These carefully tailored supramolecular complexes self-organized into various columnar LC phases with broad mesomorphic ranges and high thermal stabilities. Interestingly, these columnar LCs exhibited color-tunable fluorescence in both solution and solid states; in particular, the twisted TTIB–An showed an unusual aggregation enhanced excimer emission characteristic. To the best of our knowledge, there have been virtually no attempts to construct hydrogen-bonded supramolecular discotic columnar LCs with color-tunable fluorescent behavior, especially in the solid state.
:3 stoichiometry. These carefully tailored supramolecular complexes self-organized into various columnar LC phases with broad mesomorphic ranges and high thermal stabilities. Interestingly, these columnar LCs exhibited color-tunable fluorescence in both solution and solid states; in particular, the twisted TTIB–An showed an unusual aggregation enhanced excimer emission characteristic. To the best of our knowledge, there have been virtually no attempts to construct hydrogen-bonded supramolecular discotic columnar LCs with color-tunable fluorescent behavior, especially in the solid state.
 | ||
| Fig. 1 Schematic illustration of the construction approach for the hydrogen-bonded supramolecular discotic complexes. Various TIB derivatives, including HTIB, BTIB and NTIB with increased π-conjugation length, and TTIB with a twisted geometry, were designed and served as rigid discotic cores, while 3,4,5-trialkyloxybenzoic acid An (n = 6, 10, and 14) was used to introduce flexible alkyl tails through complementary hydrogen bonding with the TIB cores in a 3:1 stoichiometry, leading to the formation of supramolecular discotic complexes TIB–An. | ||
Results and discussion
The detailed synthetic procedures for the TIB compounds and 3,4,5-trialkyloxybenzoic acid (An), and the preparation procedures for TIB–An complexes are provided in Section S2 of the ESI,† and all of the compounds or complexes have satisfactory molecular characterization data. The alkyl tail length of An was designed as n = 6, 10, and 14 to induce liquid crystallinity, by avoiding complete crystallization with shorter tails or the plasticity caused by too bulky alkyl segments. Although neither the TIB molecules nor the benzoic acids An are mesogenic (the crystalline melting points are listed in the Experimental section in ESI†), differential scanning calorimetry (DSC), polarized optical microscopy (POM) and X-ray diffraction (XRD) measurements confirm that the whole family of hydrogen-bonded TIB–An complexes can transform into birefringent, shearable LC mesophases within a very broad range of temperatures, including above 250 °C. All the detailed characterization data are available in Sections S3 and S4 of the ESI,† with a phase diagram depicted in Fig. 2a. | ||
| Fig. 2 Thermotropic LC behaviors of the hydrogen-bonded supramolecular discotic TIB–An complexes. (a) Phase diagram of the discotic TIB–An complexes determined by a combination of DSC, TGA, POM and XRD measurements. For DSC, the transition peak temperatures were used, while the temperature points of 5% mass loss were used in TGA. (b) Optical microscopy images of representative complexes, BTIB–A10 and HTIB–A10, after cooling from their isotropic melts at rates of 20 °C min−1 or 1 °C min−1, as indicated. (c) Representative XRD patterns of the discotic TIB–A10 complexes with proposed indexing at indicated temperatures, associated with plane symmetries, and proposed packing structures of the discotic columnar mesophases. | ||
In POM, the hydrogen-bonded supramolecular complexes of the NTIB and TTIB cores assembled with different benzoic acids An showed obvious birefringence but lacked typical LC textures (Fig. S14, ESI†) due to the unexpected occurrence of thermal decomposition before isotropization. Thus, the POM samples were prepared by direct manual pressing of the complexes in their LC states. However, the HTIB- and BTIB-based complexes exhibited obvious focal-conic fan-shaped textures after cooling from their isotropic melts at a rather fast rate of 20 °C min−1 (Fig. 2b), which is characteristic of the formation of a columnar mesophase that exhibits a randomly oriented multidomain morphology.22,23,32–34 In contrast, slowly cooling at a rate of 1 °C min−1 led to an almost completely dark field of view, typical for homeotropically aligned domains,29,31,34 for which the grain boundaries could be highlighted under 45° crossed polarizers (Fig. 2b). Thus, it would pave the way for high-performance optoelectronic applications because of their remarkable anisotropic properties.22,23,35–37
Variant temperature XRD experiments were then utilized to confirm the presence of LC mesophases for all the hydrogen-bonded supramolecular discotic TIB–An complexes (Fig. S16–S19, ESI†), with the typical diffraction patterns presented in Fig. 2c. A strong diffraction peak can obviously be seen at low angles, and a well-defined small shoulder superimposed upon a diffuse halo can be seen in the wide angle range, both of which are characteristic of columnar mesophases generated by discotic mesogens.32–34 The diffuse halo was approximately 14 nm−1 (∼0.45 nm) as a result of the liquid-like arrangement of peripheral aliphatic chains, on which the superimposed small diffraction shoulder indicated π-stacking of the rigid cores with a reciprocal spacing of about 0.35 nm within the columns. The strong peak associated with small diffractions at low angles was generated by the 2D periodic array of the columnar arrangement. The q ratio for all the complexes based on HTIB and BTIB, including TTIB–An (n = 10, 14) in the LC mesophase, and NTIB–An at low temperatures, was 1:√3:√4:√7, typical of a hexagonally arranged columnar mesophase (Colhex, p6mm). A ratio of 1:√2:√4 was observed for TTIB–A6, including NTIB–An in a higher temperature range, indicating the presence of a square columnar mesophase (Colsq, p4mm). Overall, all the complexes exhibited a single LC phase, except for the mesophase transition between Colhex and Colsq of NTIB–An, which could easily be realized by a 2D columnar rearrangement (Fig. S20, ESI†) as well as the accompanying variations in the intracolumnar correlations (Fig. 2c).
Based on the above results, nearly all the supramolecular complexes displayed ordered columnar structures with regular discotic stacking, but the rather weak π-stacking signal for the TTIB–An complexes indicated a lower intracolumnar correlation (Fig. 2c). This might result from the large steric hindrance of the outer six tolyl substituents of TTIB, which are twisted out of the core plane and do not permit regular stacking along the columnar axis (Fig. S15, ESI†). By an overall comparison to the TIB–An complexes with the same An, it was easily found that the diffraction peak derived from intracolumnar π-stacking order gradually diminished from HTIB to BTIB, NTIB, and TTIB based complexes (Fig. S16–S19, ESI†). Thus, it appeared that the dimension-dependent mobility of the rigid TIB cores rather than the π-conjugation length was the dominant factor influencing the intracolumnar order of such kind of hydrogen-bonded supramolecular discotic LC, as reorientation of a more bulky TIB core is known to show slower kinetics.
The lattice parameters deduced from the diffraction maxima at small angles clearly indicated that the diffraction units were comparable to the dimensions of the corresponding hydrogen-bonded 1:3 discotic complexes. The slightly lower experimental dimensions estimated from XRD analysis (Fig. 3 and Table S1, ESI†) than the theoretical values in an extended conformation (Fig. S21, ESI†) suggested that some degree of alkyl shrinkage or intercolumnar interdigitation was necessary to induce various mesophases and space filling of the aliphatic chains around the core.34,38,39 Owing to the same hydrogen-bonding characteristics of the benzoic acid An couple with the various TIB homologues (Fig. 1), the dimensions of the assembled columns or the average intercolumnar distances should be independent of the TIB cores and instead vary with the length of the peripheral alkyl chains of the benzoic acids An. There was no doubt that longer alkyl chains led to larger columnar diameters due to the increased thickness of the outer aliphatic shell layers (Fig. 3a). The average columnar diameters exhibited little variation or underwent very modest increases from HTIB- and BTIB- to NTIB-based complexes, but surprisingly, the diameters changed noticeably going from NTIB- to TTIB-based complexes with the same An (Fig. 3a). This phenomenon may result from the distinctly different steric hindrance generated by the TIB cores, which impacted the effective length of the hydrogen-bonded benzoic acids An. The alkyl tails in the homologous series of the HTIB-, BTIB- and NTIB-based complexes should suffer from similar influences, where relatively large void spaces exist between individual arms of the C3-symmetric TIB cores.40,41 The large void spaces may provide the hydrogen-bonded alkyl chains with a large degree of mobility; hence, they adopted a curved conformation (Fig. 3b) to minimize the energy, and interdigitation from different columns was permitted (Fig. 3c).38,39 In contrast, the crowded bulky tolyl substituents surrounding the core plane of TTIB may exert a certain amount of steric repulsion, forcing the hydrogen-bonded alkyl chains to adopt a more stretched conformation (Fig. 3b) and thus diminish the interdigitation from neighboring columns (Fig. 3c), which therefore resulted in relatively larger values for the average columnar diameters.
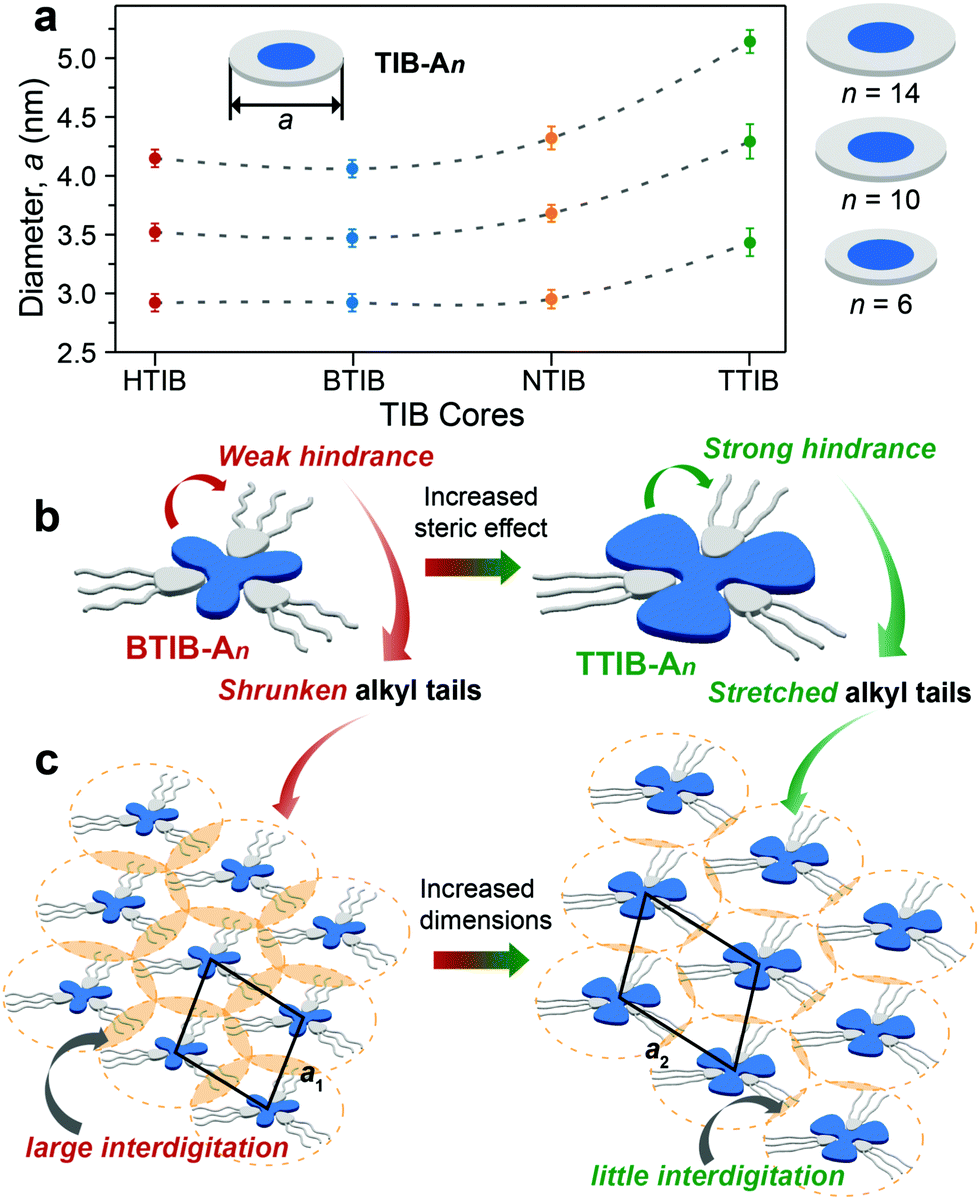 | ||
| Fig. 3 Illustration of the core influences on the assembled columnar LC structures. (a) Dependence of the average columnar diameter (a) in the LC mesophase determined by XRD analysis on the TIB core and corresponding alkyl tail length (n). (b) Proposed interaction mechanism for the increased steric hindrance from the representative BTIB to TTIB that forces conformational transformation of the hydrogen-bonded alkyl chains from shrunken and curved to their more stretched forms, resulting in (c) different packing arrangements of the BTIB–An complexes with relatively large intercolumnar interdigitation while smaller interdigitation is observed for the TTIB–An complexes. | ||
The spectroscopic properties of the supramolecular discotic complexes were investigated by ultraviolet visible (UV-vis) absorption and fluorescence emission for the representative TIB–A10 complexes in solution and in the solid state (Fig. 4), with the relevant data collected in Table S2, ESI.† In solution, by choosing the absorption maxima (Fig. 4a) as the excitation wavelength, progressively red-shifted fluorescence spectra with emission maxima changing from 343 to 381, 413 and 460 nm were observed for the HTIB, BTIB, TTIB and NTIB based complexes, respectively (Fig. 4b). This result appeared to be correlated with the π-conjugation lengths of the carefully designed TIB cores (Fig. 1 and Fig. S22, ESI†). Their corresponding fluorescence images taken under 365 nm UV light illumination, revealed a color change from no fluorescence to the naked eye, to slightly visible blue, and then to obvious cyan and green fluorescence (inset in Fig. 4b). Interestingly, an unexpected yellow fluorescence was observed for the TTIB–A10 complex in a concentrated solution such as 2 × 10−3 mol L−1, which was in sharp contrast to the cyan emission of the dilute solution of 2 × 10−6 mol L−1 (inset in Fig. 4c). The emission spectra were thereafter obtained and displayed a strong red-shift with the emission peak shifting from 413 nm to 510 nm (Fig. 4c), which was reminiscent of excimer emission resulting from the formation of nanoaggregates in a concentrated solution (Fig. S23, ESI†).42–46 To prove this, a series of solutions with progressively increasing concentrations were prepared, as excimer emission is known to be concentration dependent. Obviously, the peak at 413 nm in the 2 × 10−6 mol L−1 solution, corresponding to monomer emission, gradually decreased with increasing concentration, and the excimer band around 510 nm increased and became dominant until a concentration of 2 × 10−3 mol L−1 was reached (Fig. 4c), demonstrating that it truly originated from excimer emission.
 | ||
| Fig. 4 Spectroscopic properties of the hydrogen-bonded supramolecular discotic TIB–An complexes with various TIB cores. (a) UV-vis absorption and (b and c) fluorescence emission spectra of the representative discotic TIB–A10 complexes in chloroform at the indicated concentrations, with insets showing the corresponding fluorescence images taken under 365 nm UV light illumination. (d) UV-vis absorption and (e) fluorescence emission spectra of the representative TIB–A10 complexes in the solid state, with corresponding fluorescence images taken by adsorbing the complexes onto a thin-layer chromatography (TLC) plate. (f) Optimized geometries of the BTIB and TTIB based complexes, with simple benzoic acid instead of alkylated An for clarity. | ||
We further investigated the photophysical properties in the solid state. As shown in Fig. 4d and e, the HTIB–A10 complex, which only had a central π-conjugation benzene unit, showed similar absorption and emission spectra to those in solution, as well as similar fluorescence images under 365 nm illumination. The aggregated NTIB–A10 solid exhibited an obvious hypsochromic shift in both the solid-state absorption and emission spectra with respect to the monomer solution and displayed a weak cyan emission (Fig. 4d, e and Fig. S24, ESI†), all of which probably pointed to H-type aggregation in the solid state.47,48 Interestingly, for the BTIB- and TTIB-based complexes, in addition to their unstructured absorption spectra with respect to their monomer solutions, their solid-state fluorescence spectra revealed clear red-shifted emission for 130 nm and 148 nm, respectively (Fig. 4b and e). These results probably suggested that the distinct photophysical behaviors in the solid state resulted from the formation of excimers, resembling the emission spectrum of the TTIB–A10 nanoaggregates dispersed in a concentrated solution (Fig. 4c). Similar to traditional excimer systems, the BTIB–A10 complex exhibited a relatively weak green-yellow fluorescence in the solid state (inset in Fig. 4e) due to the aggregation of almost planar molecules (Fig. 4f), which caused partial fluorescence quenching via the nonradiative decay that resulted from the intermolecular π-stacking interactions (Fig. 2c). However, an exceptionally intense yellow fluorescence was observed for the TTIB–A10 solid (inset in Fig. 4e). This result may be attributed to the twisted geometry (Fig. 4f), which hindered intermolecular π-stacking (Fig. 2c) and restricted intramolecular motions in the aggregated state, thus blocking the nonradiative relaxations and inducing the aggregation-induced enhanced emission effect.49,50 It should be mentioned that the emissive excimer formation only for the BTIB- and TTIB-based complexes may be correlated with the different hydrogen-bonding manners between the An and various TIB cores (Fig. S10, ESI†).
To gain further insight into the underlying mechanism of the fluorescence emission of the BTIB–A10 and TTIB–A10 complexes that formed emissive excimer species, absolute fluorescence quantum yield (ΦF) and time-resolved fluorescence lifetime (τ) measurements in both solution and solid states were performed. In the isolated monomer state, the intramolecular motion, such as bond rotation, of both BTIB–A10 and TTIB–A10 allows the nonradiative deactivation process, but the hydrogen-bonding interactions tightly bind the TIB core and benzoic acid (An), limiting the intramolecular motion and activating the fluorescence emission. Therefore, the compromise between the two reverse pathways facilitated both complexes to produce reasonable fluorescence efficiencies with ΦF = 0.05 (τ = 4.1 ns) and ΦF = 0.02 (τ = 1.1 ns) for the BTIB–A10 and TTIB–A10 complexes, respectively, in dilute solution (Fig. 5a). However, the nonradiative rate constant (knr = 8.9 × 108 s−1) of TTIB–A10 was about 4 times larger than that of the almost planar BTIB–A10 complex (knr = 2.3 × 108 s−1), while the radiative rate constants (kr) were comparable to each other (Fig. 5b), which was due to the surrounding tolyl groups that were twisted out of the TTIB core plane inducing an additional relaxation deactivation process. In the solid state, although intramolecular motion was restricted by the π-stacking interactions and formation of ordered assembly structures to increase the fluorescence efficiency, the strong π-stacking in the BTIB–A10 complex provided a nonradiative deactivation pathway; therefore, a weak fluorescence was observed with radiative and nonradiative rate constants (kr = 0.8 × 107 s−1 and knr = 1.8 × 108 s−1) comparable to those of the isolated monomer solution (Fig. 5c and d). However, as for the TTIB–A10 solid, not only was the intramolecular motion that usually causes nonradiative relaxation restricted by the ordered molecular packing arrangement but also the twisted tolyl substituents provided steric hindrance to prevent the regular π-stacking between the rigid cores that usually leads to fluorescence quenching. As a consequence, a strong fluorescence with a high efficiency (ΦF = 0.13) and long emission lifetime (τ = 12.0 ns) was observed (Fig. 5c and d). When compared with the monomer solution, the nonradiative rate constant (knr = 7.2 × 107 s−1) was about one order of magnitude smaller than that in solution, while the radiative rate constant (kr = 1.1 × 107 s−1) remained comparable, accounting for the peculiar aggregation-enhanced excimer emission effect.
 | ||
| Fig. 5 Representation of the emission mechanism of the BTIB–A10 and TTIB–A10 complexes, which can form excimers. Fluorescence decay profiles (a) in a dilute solution and (c) in the solid state, with the best fitting curves shown as red lines. Schematic representations of (b) the isolated complex geometry and (d) columnar assembly concurrent with the intramolecular or intermolecular (= intracolumnar) actions related to the emission characteristics. The radiative rate constant kr = ΦF/τ, and the nonradiative rate constant knr = (1 − ΦF)/τ. | ||
Overall, the variations in the thermotropic behaviors and photophysical properties of the TIB–An complexes were well correlated with the different structural parameters shown by each TIB core and the corresponding An. The almost planar HTIB-, BTIB- and NTIB-based complexes were easily stacked into ordered columnar structures with regular discotic stacking due to the relatively strong intermolecular π-stacking interactions, whereas the twisted geometry of the TTIB–An complexes hindered regular stacking and thus exhibited low ordered columnar LCs with weak intracolumnar correlations. On the other hand, in dilute solutions, progressively red-shifted fluorescence spectra were observed going from the HTIB to BTIB, TTIB and NTIB based complexes, in great accordance with the extended π-conjugation lengths of the applied TIB cores. In the solid state, however, the NTIB-based complexes, which had the most extended π-conjugation length, exhibited H-type aggregation, probably resulting from the strong π-stacking interactions of the fused naphthalene rings, while the BTIB- and TTIB-based complexes, with similarly medium π-conjugation length, showed a red-shifted excimer emission. The almost planar BTIB–An complex displayed a weak fluorescence due to nonradiative relaxation deactivation via the regular π-stacking interactions. In contrast, the TTIB–An complex exhibited enhanced excimer emission arising from their twisted geometry that fulfills the structural requirements for well-known aggregation-induced enhanced emission. It is worth noting that although well documented for covalent LC compounds or polymers,51–55 aggregation-enhanced excimer emission characteristic is very rare in hydrogen-bonded supramolecular systems, especially for LC assemblies.
Conclusions
In summary, by function-directing expansion based on the TIB skeletal structure, HTIB, BTIB, NTIB and TTIB with different π-conjugation lengths or twisted geometry were prepared and then used to construct supramolecular discotic LCs via hydrogen-bonding motifs with An. These hydrogen-bonded supramolecular complexes self-organized into hexagonal or square columnar mesophases over very wide temperature ranges, for which homeotropic alignment could be easily realized via simple thermal treatment. Furthermore, the impact of the core on the intra- and inter-columnar correlations could be rationally explained based on the different dimensions, geometries and π-conjugation effects.On the other hand, a set of progressively red-shifted fluorescence spectra with emission wavelengths changing from 343 to 381, 413 and 460 nm were obtained for the HTIB-, BTIB-, TTIB- and NTIB-based complexes, respectively, in their isolated monomer solutions. These spectroscopic properties corresponded to ultraviolet, blue, cyan and green emissions. In aggregated solid, the NTIB-based complexes exhibited H-type aggregation and thereby emitted a weak cyan fluorescence, while the BTIB- and TTIB-based complexes showed a red-shifted excimer emission with a yellow color. In particular, the TTIB-based complex presented an unusual aggregation-enhanced excimer emission characteristic. Thus, color-tunable fluorescent behavior ranging from blue to cyan, green, and yellow was revealed for these tailor-made supramolecular LC complexes, which was closely correlated with their molecular architectures and assembled aggregate structures.
The present study provides insights into the structure–property relationships of hydrogen-bonded LCs based on a series of C3-symmetric homologous cores with variant π-conjugation lengths or twisted geometries. These complexes, exhibiting color-tunable fluorescent behavior, are expected to show great potential for application in various fields, such as anisotropic organic functional light emitting materials, even with color switching capability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21805228 and 21674086), the China Postdoctoral Science Foundation (2018M633563), the Natural Science Basic Research Plan in Shaanxi Province of China (2019JQ-414 and 2018JZ2003), the Postdoctoral Research Project Fund in Shaanxi Province of China (2018BSHYDZZ55), and the Fundamental Research Funds for the Central Universities (G2018KY0307 and 3102019PY003).Notes and references
- J. Goodby, P. J. Collings, T. Kato, C. Tschierske, H. Gleeson and P. Raynes, Handbook of Liquid Crystals, 2nd edn, Wiley-VCH, Weinheim, Germany, 2014 Search PubMed.
- T. Kato, J. Uchida, T. Ichikawa and T. Sakamoto, Angew. Chem., Int. Ed., 2018, 57, 4355–4371 CrossRef CAS PubMed.
- T. Kato, M. Yoshio, T. Ichikawa, B. Soberats, H. Ohno and F. Masahiro, Nat. Rev. Mater., 2017, 2, 17001 CrossRef.
- D. B. Amabilino, D. K. Smith and J. W. Steed, Chem. Soc. Rev., 2017, 46, 2404–2420 RSC.
- C. Tschierske, Angew. Chem., Int. Ed., 2013, 52, 8828–8878 CrossRef CAS PubMed.
- D. J. Broer, C. M. W. Bastiaansen, M. G. Debije and A. P. H. J. Schenning, Angew. Chem., Int. Ed., 2012, 51, 7102–7109 CrossRef CAS PubMed.
- K. Yano, Y. Itoh, F. Araoka, G. Watanabe, T. Hikima and T. Aida, Science, 2019, 363, 161–165 CrossRef CAS PubMed.
- M. H. Ryu, J. W. Choi, H. J. Kim, N. Park and B. K. Cho, Angew. Chem., Int. Ed., 2011, 50, 5737–5740 CrossRef CAS PubMed.
- L. X. Guo, Y. H. Liu, L. Wang, M. Wang, B. P. Lin and H. Yang, J. Mater. Chem. C, 2017, 5, 9165–9173 RSC.
- M. Lehmann and P. Maier, Angew. Chem., Int. Ed., 2015, 54, 9710–9714 CrossRef CAS PubMed.
- A. Kraft, A. Reichert and R. Kleppinger, Chem. Commun., 2000, 1015–1016 RSC.
- M. Pfletscher, S. Hölscher, C. Wölper, M. Mezger and M. Giese, Chem. Mater., 2017, 29, 8462–8471 CrossRef CAS.
- M. Pfletscher, C. Wölper, J. S. Gutmann, M. Mezger and M. Giese, Chem. Commun., 2016, 52, 8549–8552 RSC.
- J. F. Xiong, S. H. Luo, J. P. Huo, J. Y. Liu, S. X. Chen and Z. Y. Wang, J. Org. Chem., 2014, 79, 8366–8373 CrossRef CAS PubMed.
- I. Bala, S. P. Gupta, S. Kumar, H. Singh, J. De, N. Sharma, K. Kailasam and S. K. Pal, Soft Matter, 2018, 14, 6342–6352 RSC.
- A. A. Vieira, H. Gallardo, J. Barberá, P. Romero, J. L. Serrano and T. Sierra, J. Mater. Chem., 2011, 21, 5916–5922 RSC.
- B. Soberats, M. Yoshio, T. Ichikawa, X. Zeng, H. Ohno, G. Ungar and T. Kato, J. Am. Chem. Soc., 2015, 137, 13212–13215 CrossRef CAS PubMed.
- H. Shimura, M. Yoshio, K. Hoshino, T. Mukai, H. Ohno and T. Kato, J. Am. Chem. Soc., 2008, 130, 1759–1765 CrossRef CAS PubMed.
- B. Feringán, P. Romero, J. L. Serrano, C. L. Folcia, J. Etxebarria, J. Ortega, R. Termine, A. Golemme, R. Giménez and T. Sierra, J. Am. Chem. Soc., 2016, 138, 12511–12518 CrossRef PubMed.
- I. Paraschiv, M. Giesbers, B. Lagen, F. C. Grozema, R. D. Abellon, L. D. A. Siebbeles, A. T. M. Marcelis, H. Zuilhof and E. J. R. Sudhölter, Chem. Mater., 2006, 18, 968–1074 CrossRef CAS.
- I. Paraschiv, K. Lange, M. Giesbers, B. Lagen, F. C. Grozema, R. D. Abellon, L. D. A. Siebbeles, E. J. R. Sudhölter, H. Zuilhof and A. T. M. Marcelis, J. Mater. Chem., 2008, 18, 5475–5481 RSC.
- T. Wöhrle, I. Wurzbach, J. Kirres, A. Kostidou, N. Kapernaum, J. Litterscheidt, J. C. Haenle, P. Staffeld, A. Baro, F. Giesselmann and S. Laschat, Chem. Rev., 2016, 116, 1139–1241 CrossRef PubMed.
- S. Kumar, Chemistry of Discotic Liquid Crystals: From Monomers to Polymers, CRC, Boca Raton, FL, 2010 Search PubMed.
- F. Vera, J. Barberá, P. Romero, J. L. Serrano, M. B. Ros and T. Sierra, Angew. Chem., Int. Ed., 2010, 49, 4910–4914 CrossRef CAS PubMed.
- F. Vera, R. M. Tejedor, P. Romero, J. Barberá, M. B. Ros, J. L. Serrano and T. Sierra, Angew. Chem., Int. Ed., 2007, 46, 1873–1877 CrossRef CAS PubMed.
- P. Xing, S. Z. F. Phua, X. Wei and Y. Zhao, Adv. Mater., 2018, 30, 1805175 CrossRef PubMed.
- H. K. Lee, H. Lee, Y. H. Ko, Y. J. Chang, N. K. Oh, W. C. Zin and K. Kim, Angew. Chem., Int. Ed., 2001, 40, 2669–2671 CrossRef CAS PubMed.
- G. M. Bögels, J. A. M. Lugger, O. J. G. M. Goor and R. P. Sijbesma, Adv. Funct. Mater., 2016, 26, 8023–8030 CrossRef.
- J. A. M. Lugger, D. J. Mulder, S. Bhattacharjee and R. P. Sijbesma, ACS Nano, 2018, 12, 6714–6724 CrossRef CAS PubMed.
- S. Bhattacharjee, J. A. M. Lugger and R. P. Sijbesma, Chem. Commun., 2018, 54, 9521–9524 RSC.
- X. Feng, K. Kawabata, G. Kaufman, M. Elimelech and C. O. Osuji, ACS Nano, 2017, 11, 3911–3921 CrossRef CAS PubMed.
- B. Mu, B. Wu, S. Pan, J. L. Fang and D. Z. Chen, Macromolecules, 2015, 48, 2388–2398 CrossRef CAS.
- B. Mu, S. Pan, H. F. Bian, B. Wu, J. L. Fang and D. Z. Chen, Macromolecules, 2015, 48, 6768–6780 CrossRef CAS.
- B. Mu, X. T. Hao, J. Chen, Q. Li, C. X. Zhang and D. Z. Chen, Polym. Chem., 2017, 8, 3286–3293 RSC.
- C. Zou, J. Wang, M. Wang, Y. Wu, K. Gu, Z. Shen, G. Xiong, H. Yang, L. Jiang and T. Ikeda, Small, 2018, 14, 1800557 CrossRef PubMed.
- S. Sergeyev, W. Pisula and Y. H. Geerts, Chem. Soc. Rev., 2007, 36, 1902–1929 RSC.
- B. R. Kaafarani, Chem. Mater., 2011, 23, 378–396 CrossRef CAS.
- M. T. Allen, S. Diele, K. D. M. Harris, T. Hegmann, B. M. Kariuki, D. Lose, J. A. Preece and C. Tschierske, J. Mater. Chem., 2001, 11, 302–311 RSC.
- C. Krause, R. Zorn, F. Emmerling, J. Falkenhagen, B. Frick, P. Huber and A. Schönhals, Phys. Chem. Chem. Phys., 2014, 16, 7324–7333 RSC.
- M. Lehmann, Chem. – Eur. J., 2009, 15, 3638–3651 CrossRef CAS PubMed.
- T. Wöhrle, S. J. Beardsworth, C. Schilling, A. Baro, F. Giesselmann and S. Laschat, Soft Matter, 2016, 12, 3730–3736 RSC.
- M. Martínez-Abadía, R. Giménez and M. B. Ros, Adv. Mater., 2018, 30, 1704161 CrossRef PubMed.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed.
- B. Situ, M. Gao, X. He, S. Li, B. He, F. Guo, C. Kang, S. Liu, L. Yang, M. Jiang, Y. Hu, B. Z. Tang and L. Zheng, Mater. Horiz., 2019, 6, 546–553 RSC.
- Z. Zhao, S. Chen, J. W. Y. Lam, Z. Wang, P. Lu, F. Mahtab, H. H. Y. Sung, I. D. Williams, Y. Ma, H. S. Kwok and B. Z. Tang, J. Mater. Chem., 2011, 21, 7210–7216 RSC.
- Y. B. Gong, P. Zhang, Y. R. Gu, J. Q. Wang, M. M. Han, C. Chen, X. J. Zhan, Z. L. Xie, B. Zou, Q. Peng, Z. G. Chi and Z. Li, Adv. Opt. Mater., 2018, 6, 1800198 CrossRef.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- S. J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M. G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed.
- J. Mei, Y. Hong, J. W. Y. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- S. Yamane, Y. Sagara and T. Kato, Chem. Commun., 2013, 49, 3839–3841 RSC.
- A. S. Abd-El-Aziz, A. A. Abdelghani, B. D. Wagner and E. M. Abdelrehim, Polym. Chem., 2016, 7, 3277–3299 RSC.
- K. Sakajiri, T. Sugisaki and K. Moriya, Chem. Commun., 2008, 3447–3449 RSC.
- Y. Ren, W. H. Kan, M. A. Henderson, P. G. Bomben, C. P. Berlinguette, V. Thangadurai and T. Baumgartner, J. Am. Chem. Soc., 2011, 133, 17014–17026 CrossRef CAS PubMed.
- M. Martínez-Abadía, S. Varghese, P. Romero, J. Gierschner, R. Giménez and M. B. Ros, Adv. Opt. Mater., 2017, 5, 1600860 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and additional data regarding thermotropic behaviors and spectroscopy properties of the obtained complexes. See DOI: 10.1039/c9qm00337a |
| This journal is © the Partner Organisations 2019 |