DOI:
10.1039/C8RA06112B
(Paper)
RSC Adv., 2019,
9, 307-312
A novel 3D Cd(II) coordination polymer generated via in situ ligand synthesis involving C–O ester bond formation†
Received
19th July 2018
, Accepted 10th December 2018
First published on 2nd January 2019
Abstract
A novel 3D Cd(II) coordination polymer {[Cd(ddpa)(2,2′-bpy)]·H2O}n (1) (H2ddpa = 5,10-dioxo-5,10-dihydro-4,9-dioxapyrene-2,7-dicarboxylic acid, 2,2′-bpy = 2,2′-bipyridine) is hydrothermally synthesized in situ, and the influencing factors and mechanism for the in situ reaction are briefly discussed. The synthesis of 1 requires the formation of a new C–O ester bond. This current study confirms that metal ions and N-donor ligands play important roles in the domination of the in situ ligand from 6,6′-dinitro-2,2′,4,4′-biphenyltetracarboxylic acid (H4dbta). Furthermore, the structure, thermal stability and photoluminescent property of 1 are also investigated.
1. Introduction
Hydro(solvo)thermal in situ ligand syntheses are of great interest in coordination chemistry and organic chemistry for the preparation of unusual coordination compounds, discovery of new organic reactions and understanding their mechanisms.1–5 So far, a variety of in situ ligand syntheses have already been reported, which include the formation of C–C, C–N, S–S, C–O, –O–, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds,6–11 oxidation of a phenyl ring with a methyl group,12 formation of tetrazole,13–15 and decarboxylation of aromatic carboxylates.16 Since the hydro(solvo)thermal reaction is affected by several factors such as solvents, temperature, metal ions, organic ligands and anions,17–24 it is still a formidable challenge to elucidate the reaction mechanism of in situ ligand syntheses.
C bonds,6–11 oxidation of a phenyl ring with a methyl group,12 formation of tetrazole,13–15 and decarboxylation of aromatic carboxylates.16 Since the hydro(solvo)thermal reaction is affected by several factors such as solvents, temperature, metal ions, organic ligands and anions,17–24 it is still a formidable challenge to elucidate the reaction mechanism of in situ ligand syntheses.
It is well-known that the rational design and reasonable use of the organic multicarboxylate ligands are very important in the construction of desired coordination polymers. Although many studies have recently focused on the use of biphenyl-2,2′,4,4′-tetracarboxylic acid,25–35 the use of 6,6′-dinitro-2,2′,4,4′-biphenyltetracarboxylic acid (H4dbta) has not been adequately explored.36–38 Compared with biphenyl-2,2′,4,4′-tetracarboxylic acid, although the nitro groups of H4dbta might be unlikely to form coordination bonds with metal ions, they can influence the coordination and framework due to their electronegative and steric effects.39
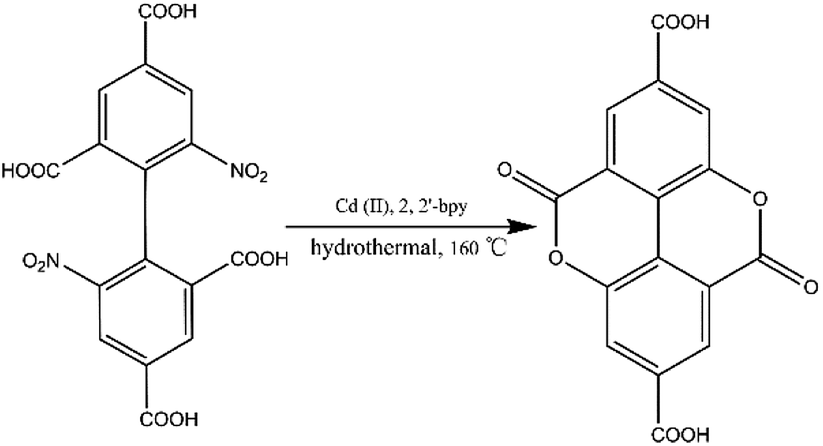
Prior to this study, we reported the synthesis of a 3D pillared bilayer Cd(II) coordination polymer based on H4dbta {[Cd2(dbta)(dpe)]n, H4dbta = 6,6′-dinitro-2,2′,4,4′-biphenyltetracarboxylic acid, dpe = 1,2-di(4-pyridyl)ethylene}.40 In a follow-up study, we found that the replacement of 1,2-di (4-pyridyl) ethylene (dpe) with 2,2′-bipyridine (2,2′-bpy) resulted in an in situ reaction containing a C–O ester bond in H4dbta molecule to form the ddpa2− ligand (ddpa2− = 5,10-dioxo-5,10-dihydro-4,9-dioxapyrene-2,7-dicarboxylate; its ring system is identical to that of the natural tetracyclic diester ellagic acid), which resulted in the synthesis of another three-dimensional (3D) coordination polymer {[Cd(ddpa)(2,2′-bpy)]·H2O}n (1). Although two coordination polymers of the ddpa2− ligand have been reported by Cheetham and Huang groups,41,42 the synthesis of the ddpa2− ligand includes the in situ reaction of 5-hydroxyisophthalic acid (OH–H2BDC) and Ni(II)/Ce(III) ions (Scheme 1). In this in situ synthesis, the formation of the C–O ester bond belongs to conventional esterifications, in which at least one –OH (hydroxyl) group of the carboxylic acids is replaced with an –O–alkyl (alkoxy) group. The most important difference between our work and the study reported by Cheetham and Huang groups is that we obtained the ddpa2− ligand by the in situ ligand synthesis of H4dbta, Cd(II) and 2,2′-bpy (Scheme 2). Thus, it is necessary to explore the influencing factors for the in situ reaction of H4dbta.
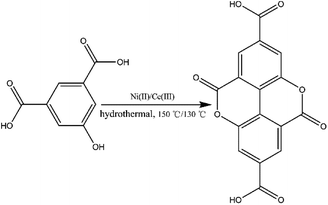 |
| | Scheme 1 In situ synthesis of the H2ddpa ligand from OH–H2bdc. | |
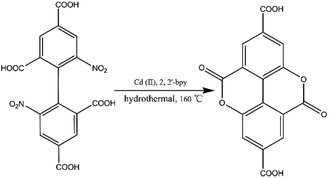 |
| | Scheme 2 In situ synthesis of the H2ddpa ligand from H4dbta. | |
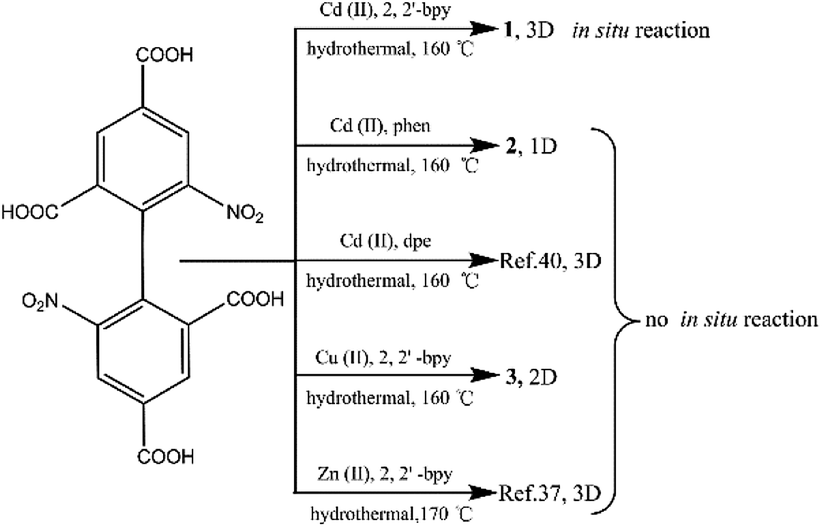
In this paper, the influencing factors and mechanism for the in situ generation of 1 are also briefly discussed. It is interesting that this in situ reaction is only observed when the metal ions and N-donor ligands are Cd(II) and 2,2′-bpy, respectively. If Cd(II) and 1,10-phenanthroline (phen)/dpe are used, 1D/3D structures of {[Cd2 (dbta) (phen) 2 (H2O)]·H2O}n (2) and [Cd2 (dbta) (dpe)]n40 are obtained by the hydrothermal reaction. In addition, the replacement of Cd(II) ions in 1 with Cu(II)/Zn(II) results in two different 2D/3D structures of {[Cu2 (dbta) (2,2′-bpy)2]·H2O}n (3) and [Zn2 (dbta) (2,2′-bpy)]n37 (Scheme 3). As far as we know, this is the first example of a coordination polymer obtained via in situ synthesis of the H4dbta ligand. Our study suggests that the synergetic effects of both 2,2′-bpy and Cd(II) ions are important factors in controlling the formation of the ddpa2− ligand. In this in situ synthesis, the formation of the C–O ester bond was not by conventional esterifications, which expands the conception of esterifications in organic chemistry. The result provides a promising route to design and construct a new organic ligand with special structures and properties. Furthermore, the thermal stability and photoluminescent properties of 1–3 are also investigated.
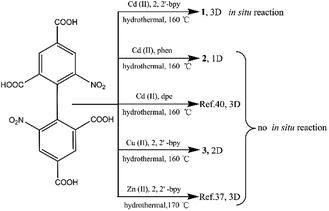 |
| | Scheme 3 The synthetic route for compounds 1–3, [Cd2(dbta)(dpe)]n (ref. 40) and [Zn2 (dbta) (2,2′-bpy)]n (ref. 37). | |
2. Experimental
2.1 Materials and methods
All reagents and solvents employed were commercially available and used without further purification. The C, H and N microanalyses were carried out with a PE 2400 Series II elemental analyzer. The IR spectra were recorded with a Shimadzu IR Affinity-1 spectrometer using the KBr pellet technique. Thermogravimetric analysis was performed on a NETZSCH STA 449F3 analyzer. The powder X-ray diffraction (PXRD) measurement was carried out using a Shimadzu XRD-7000 diffractometer with Cu Kα radiation (λ = 1.5418 Å). Photoluminescence spectra were obtained on a Hitachi F-4500 fluorescence spectrophotometer at room temperature.
2.2 Synthetic procedures
2.2.1 Synthesis of {[Cd (ddpa) (2,2′-bpy)]·H2O}n (1). A mixture of CdO (0.0064 g, 0.05 mmol), H4dbta (0.0210 g, 0.05 mmol), 2,2′-bpy (0.0078 g, 0.05 mmol) and 10 mL H2O was stirred for 30 min. The mixture was then placed in a 25 mL Teflon-lined stainless steel vessel and heated at 160 °C for 3 d. Yellow block crystals were obtained when the mixture was cooled to room temperature. Yield: ca. 56% based on Cd. Calcd. for C26H14CdN2O9 (%): C, 51.13; H, 2.31; N, 4.59. Found (%): C, 50.42; H, 2.38; N, 4.69. IR (KBr pellet, cm−1): 3488 m, 1743 s, 1625 m, 1593 m, 1549 s, 1372 s, 1294 s, 1186 s, 1091 s, 1014 m, 805 m, 766 m, 696 m, 652 w, 576 w, 525 w.
2.2.2 Synthesis of {[Cd2 (dbta) (phen)2 (H2O)]·H2O}n (2). The preparation of 2 was similar to that of 1 except that phen (0.0099 g, 0.05 mmol) was used instead of 2,2′-bpy (0.0078 g, 0.05 mmol). Yellow block crystals were obtained. Yield: ca. 62% based on Cd. Calcd. for C40H24Cd2N6O14 (%): C, 46.31; H, 2.33; N, 8.10. Found (%): C, 45.59; H, 2.35; N, 8.21. IR (KBr pellet, cm−1): 3488 w, 3068 w, 2362 w, 1599 w, 1561 w, 1523 m, 1434 m, 1389 m, 1338 s, 1097 w, 932 m, 925 w, 843 s, 728 s, 785 m, 716 s, 626 w, 544 w, 423 w.
2.2.3 Synthesis of {[Cu2 (dbta) (2,2′-bpy)2]·H2O}n (3). The preparation of 3 was similar to that of 1 except that CuO (0.0080 g, 0.1 mmol) was used instead of CdO (0.0064 g, 0.05 mmol). Blue plate block crystals were obtained. Yield: ca. 45% based on Cu. Calcd. for C36H22Cu2N6O13 (%): C, 49.49; H, 2.54; N, 9.62. Found (%): C, 49.71; H, 2.42; N, 9.21. IR (KBr pellet, cm−1): 3434 m, 1620 s, 1593 m, 1536 s, 1441 m, 1390 m, 1328 s, 1098 w, 761 m, 717 m, 627 w, 515 w, 414 w.
2.3 X-ray data collection and structure determination
Intensity data were collected on a Bruker Smart APEX II CCD diffractometer equipped with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) using ω–φ scan mode. A semi-empirical absorption correction was applied using the SADABS program.43 The structure was solved with direct methods and refined by full-matrix least-squares on F2 using the SHELXS-2014/7 and SHELXL-2014/7 programs, respectively.44,45 Non-hydrogen atoms were refined anisotropically and hydrogen atoms were placed in the geometrically calculated positions. The (dbta)4− anions and lattice water molecules in 3 were disordered and refined with different occupancy ratios. The hydrogen atoms of the disordered lattice water molecules in 3 were not added. The crystallographic data and selected bonds and angles for 1–3 are given in Tables S1 and S2,† respectively.
3. Results and discussion
3.1 Syntheses and characterization
The hydrothermal reactions of CdO and 6,6′-dinitro-2,2′,4,4′-biphenyltetracarboxylic acid (H4dbta) with 2,2′-bpy gave the 3D framework of 1. To explore the influencing factors and formation mechanism for the in situ reaction of H4dbta in 1, compounds 2 and 3 were synthesized under hydrothermal conditions (Scheme 3). When N-donor phen/dpe ligands were used instead of 2,2′-bpy in 1, a structurally different 1D chain/3D pillared bilayer framework was formed in 2 and [Cd2 (dbta) (dpe)]n40 under similar reaction conditions. Similarly, the replacement of Cd(II) ions in 1 by Cu(II)/Zn(II) resulted in two different 2D network/3D “brick-wall” frameworks.37 Apparently, the in situ reaction of H4dbta to H2ddpa did not occur for compounds 2 and 3, which suggests that the metal ions and N-donor ligands have great effect on the in situ reaction.
For compounds 1–3, the elemental analyses were consistent with their chemical formulae. PXRD showed that the observed patterns for 1 and 2 correlate well with the simulated patterns generated from the single-crystal X-ray diffraction data (Fig. S2 and S3,†). In the IR spectra of 1–3 (Fig. S4,†), the absence of any strong bands around 1700 cm−1 indicates that all the carboxylic groups are depronated. For 1, a sharp absorption band at 1743 cm−1 is observed, confirming the existence of the νCO group. The absorption bands at 1372 and 1186 cm−1 for 1 can be assigned to the vibrations of 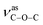 and
and 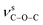 groups, respectively. Thermal stability studies (Fig. S5,†) demonstrate that 1 is thermally stable up to 250 °C; then, a quick weight loss (ca. 55%) from 250 to 360 °C is observed, which is caused by the decomposition of ligands. For 2, weight loss of about 3.6% is observed from 25 to 166 °C, which is ascribed to the release of lattice and coordinated water, followed by a quick weight loss of about 65.6% from 285 to 373 °C. For 3, weight loss of about 2.2% is observed from 25 to 117 °C, which may be ascribed to the release of lattice water; this is followed a quick weight loss of about 47.8% from 225 to 276 °C. The second step of the weight loss for 2 and 3 is caused by the decomposition of the ligands.
groups, respectively. Thermal stability studies (Fig. S5,†) demonstrate that 1 is thermally stable up to 250 °C; then, a quick weight loss (ca. 55%) from 250 to 360 °C is observed, which is caused by the decomposition of ligands. For 2, weight loss of about 3.6% is observed from 25 to 166 °C, which is ascribed to the release of lattice and coordinated water, followed by a quick weight loss of about 65.6% from 285 to 373 °C. For 3, weight loss of about 2.2% is observed from 25 to 117 °C, which may be ascribed to the release of lattice water; this is followed a quick weight loss of about 47.8% from 225 to 276 °C. The second step of the weight loss for 2 and 3 is caused by the decomposition of the ligands.
3.2 Structural descriptions
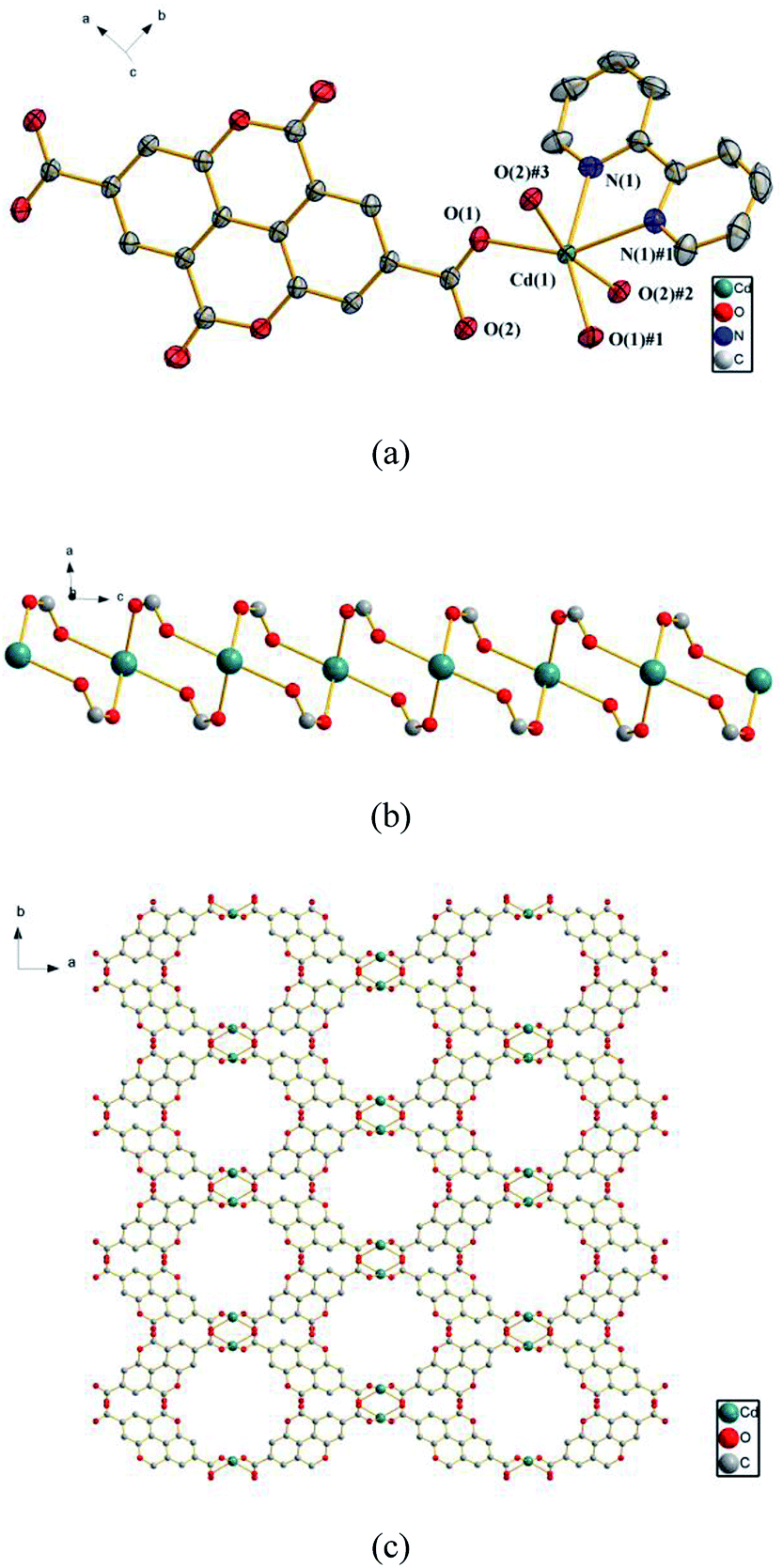
3.2.1 {[Cd (ddpa) (2,2′-bpy)] ·H2O}n (1). The X-ray analysis reveals that 1 crystallizes in the space group C2/c and has a 3D framework containing a 1D inorganic chain (–Cd–O–C–O–Cd–). As shown in Fig. 1a, each asymmetric unit of 1 contains one Cd(II) ion, one ddpa2− ligand, one 2,2′-bpy ligand and one lattice H2O molecule. The Cd(II) ion is coordinated by four oxygen atoms from four different ddpa2− ligands and two nitrogen atoms from one 2,2′-bpy ligand to furnish distorted octahedral coordination geometry. All the Cd–O and Cd–N bond lengths are in accordance with those reported in other related ref. 46 and 47 The two carboxyl groups in each ddpa2− ligand adopt a μ2-η1:η1 coordination mode to connect the adjacent Cd(II) ions and generate a 1D inorganic chain (–Cd–O–C–O–Cd–) (Fig. 1b). In the 1D chains formed by dinuclear rings, the Cd(II)⋯Cd(II) distance is 4.3658(2) Å. Along the c axis, such a chain is further linked by ddpa2− ligands to generate a 3D framework structure (Fig. 1c). The whole framework has octagonal-prism-like 1D channels filled with 2, 2′-bpy ligands and lattice H2O molecules, and the effective solvent accessible volume is 147 Å3 per unit cell (6.4% of the total cell volume calculated by the Platon program).
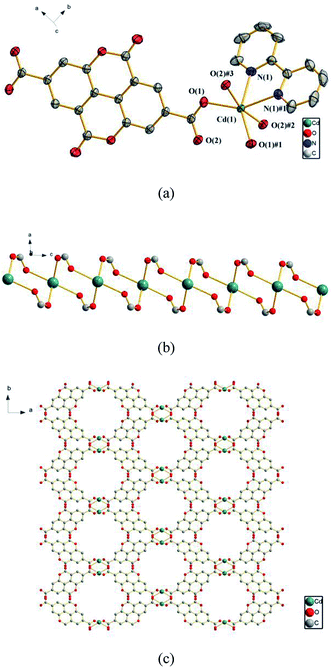 |
| | Fig. 1 (a) The coordination environments of Cd(II) ions in 1, symmetry codes: #1: −x, y, −z + 1/2; #2: −x, −y, −z + 1; #3: x, −y, z − 1/2. All hydrogen atoms are omitted for clarity. (b) The 1D chain (–Cd–O–C–O–Cd–) in 1. (c) The 3D framework assembled from the 1D chains. The 2,2′-bpy molecules, lattice water and hydrogen atoms are omitted for clarity. | |
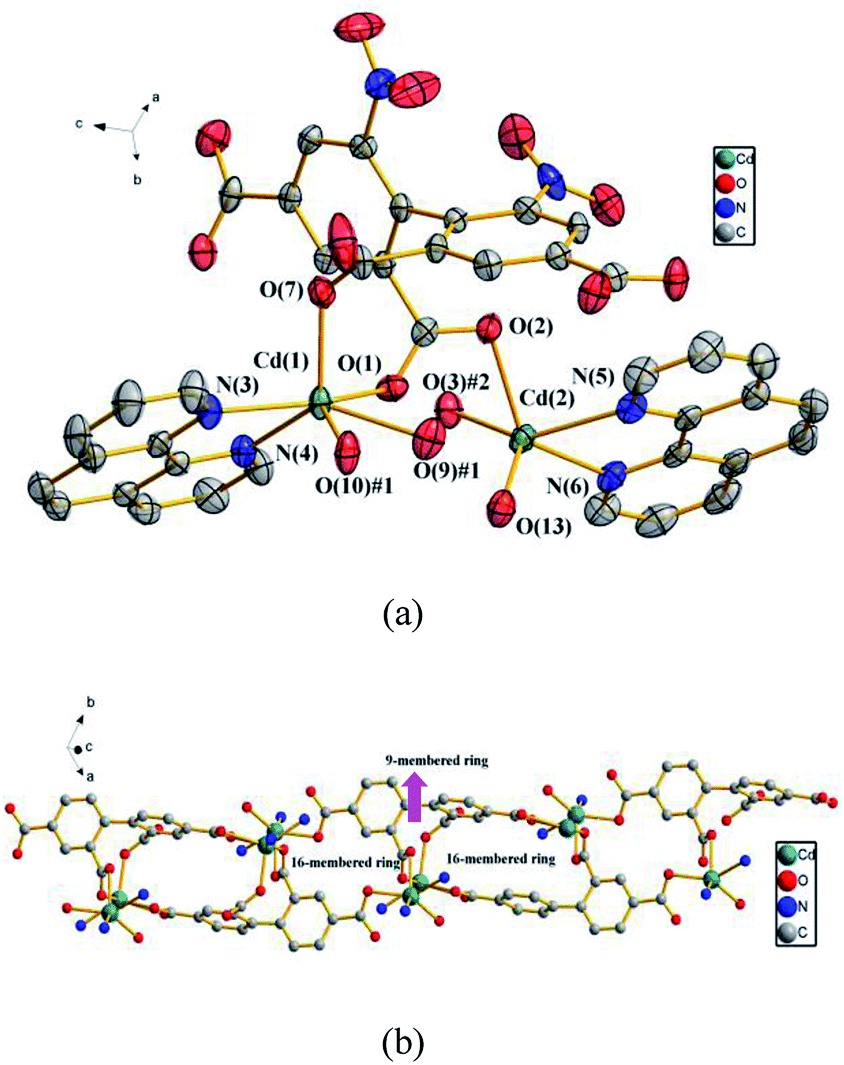
3.2.2 {[Cd2 (dbta) (phen)2 (H2O)]·H2O}n (2). To further examine the influence of the N-donor ligands on the structure of 1, a larger sized aromatic chelate ligand phen is used instead of 2,2′-bpy. Consequently, compound 2, which features a 1D looped chain, is obtained. Compound 2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . In the asymmetric unit, there exist two crystallographically unique Cd(II) ions, one dbta4− ligand, two phen ligands, one coordinated H2O molecule and one lattice H2O. As illustrated in Fig. 2a, the Cd (1) ion exhibits distorted octahedral geometry and is coordinated by two nitrogen atoms from a chelate phen and four oxygen atoms from three carboxylic groups of two dbta4− ligands. The Cd (2) ion is ligated by two nitrogen atoms from a chelate phen, one oxygen atom from one coordinated H2O molecule and two oxygen atoms from two dbta4− ligands to form distorted tetragonal pyramid geometry. These bond lengths are similar to those found in related dicadmium-tetracarboxylate polymers.37 The four carboxylate groups of the dbta4− ligands adopt three different coordination modes, i.e., monodentate, chelating bidentate and bridging bidentate to generate a 1D looped chain containing a 16-membered ring and a 9-membered ring (Fig. 2b).
. In the asymmetric unit, there exist two crystallographically unique Cd(II) ions, one dbta4− ligand, two phen ligands, one coordinated H2O molecule and one lattice H2O. As illustrated in Fig. 2a, the Cd (1) ion exhibits distorted octahedral geometry and is coordinated by two nitrogen atoms from a chelate phen and four oxygen atoms from three carboxylic groups of two dbta4− ligands. The Cd (2) ion is ligated by two nitrogen atoms from a chelate phen, one oxygen atom from one coordinated H2O molecule and two oxygen atoms from two dbta4− ligands to form distorted tetragonal pyramid geometry. These bond lengths are similar to those found in related dicadmium-tetracarboxylate polymers.37 The four carboxylate groups of the dbta4− ligands adopt three different coordination modes, i.e., monodentate, chelating bidentate and bridging bidentate to generate a 1D looped chain containing a 16-membered ring and a 9-membered ring (Fig. 2b).
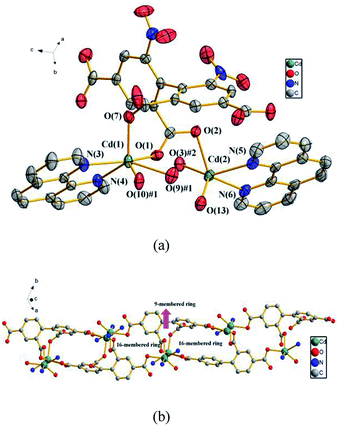 |
| | Fig. 2 (a) The coordination environments of Cd(II) ions in 2, symmetry codes: #1: −x, y, −z + 1/2; #2: −x, −y, −z + 1; #3: x, −y, z − 1/2. All hydrogen atoms are omitted for clarity. (b) The 1D looped chain in 2. The phen ligands, nitro groups and hydrogen atoms are omitted for clarity. | |
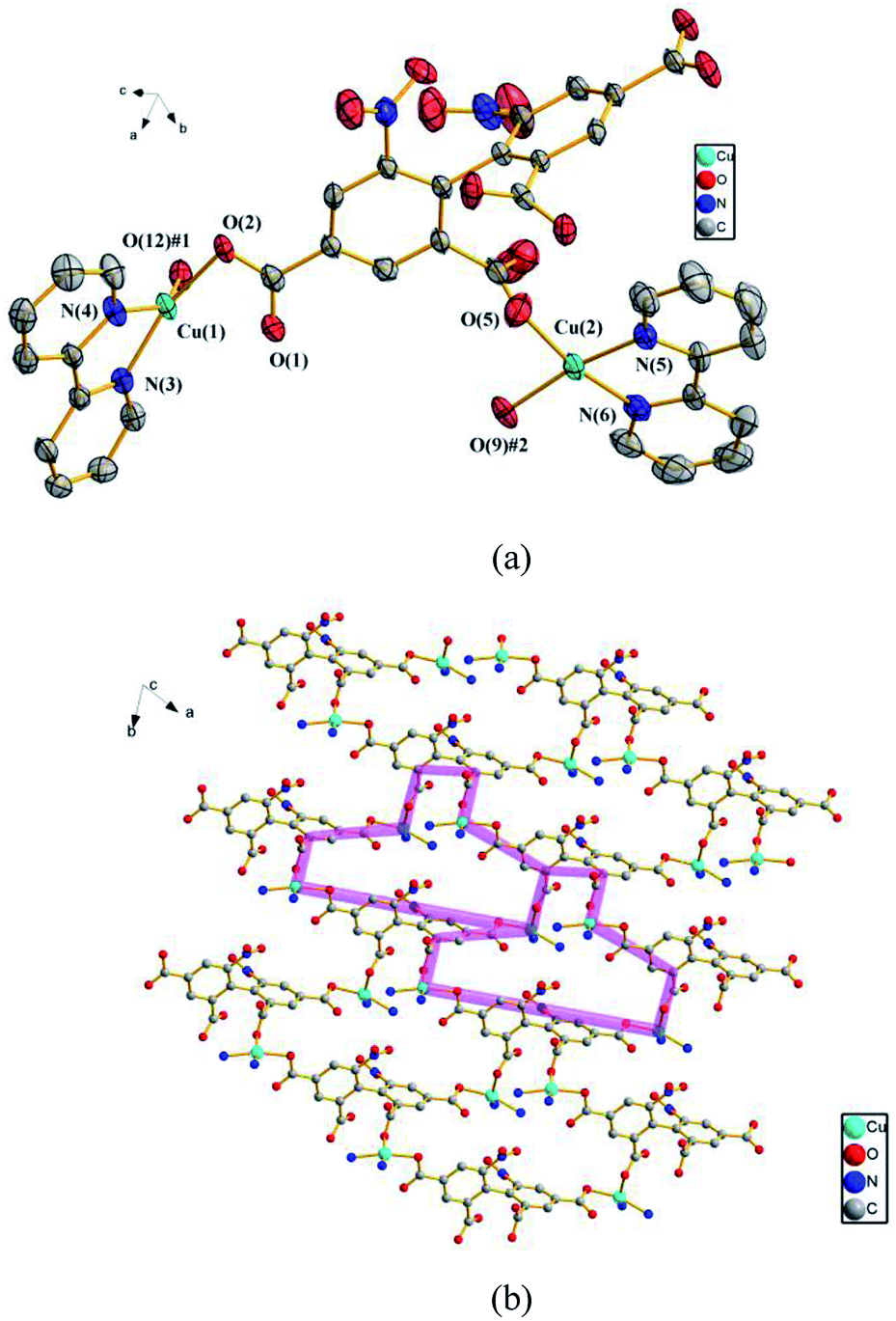
3.2.3 {[Cu2(dbta)(2,2'-bpy)2] ·H2O}n (3). To further explore the influence of metal ions on the structure of 1, Cu(II) ion is used instead of Cd(II) ion. Consequently, compound 3, which features a 2D network, is obtained. Compound 3 crystallizes in the triclinic space group P. In the asymmetric unit, there exist two crystallographically unique Cu(II) ions, one dbta4− ligand, two 2,2′-bpy ligands and one free H2O molecule. As illustrated in Fig. 3a, Cu (1) and Cu (2) ions exhibit distorted square geometry and are coordinated by two nitrogen atoms from a chelate 2,2′-bpy and two oxygen atoms from two carboxylic groups of two dbta4− ligands. The four carboxylate groups of dbta4− ligands adopt monodentate coordination modes. Each Cu(II) ion is bridged by dbta4− ligands to form a 2D convex-shaped network extending along the ab plane (Fig. 3b).
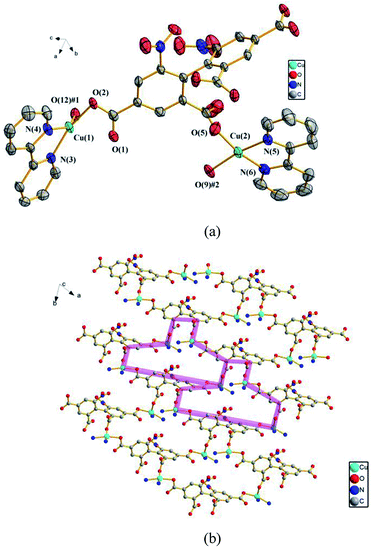 |
| | Fig. 3 (a) The coordination environments of Cu(II) ions in 3, symmetry codes: #1: −x, y, −z + 1/2; #2: −x, −y, −z + 1; #3: x, −y, z − 1/2. All hydrogen atoms are omitted for clarity. (b) The 2D convex-shaped network in 2. The 2,2′-bpy molecules and hydrogen atoms are omitted for clarity. | |
3.3 Influencing factors and mechanism for the in situ generation of ddpa2−
To evaluate whether both 2,2′-bpy and Cd(II) ions play crucial roles in the reaction process, after replacing 2,2′-bpy by phen/dpe ligands, the 1D looped chain/3D pillared bilayer framework was formed in 2 and [Cd2 (dbta) (dpe)]n40 under similar reaction conditions, respectively. Compound 2 and [Cd2 (dbta) (dpe)]n contain no ddpa2− ligand, suggesting that the in situ reaction may be prohibited. To further explore this interesting phenomenon, Cu(II) ion/Zn(II) is used instead of Cd(II) ion, followed by the same procedure as that for 1. The 2D network of 3 and the 3D “brick-wall” framework37 were obtained as new phases, indicating that Cu(II)/Zn(II) anions have the same negative effect on the in situ reaction as phen/dpe. Therefore, it can be concluded that the in situ reaction of the dbta4− ligand resulted from synergetic effects of both 2,2′-bpy and Cd(II) ions under hydrothermal conditions. This phenomenon is greatly different from the in situ generation of ddpa2− within the Ni(II)/Ce(III) reaction system.41,42
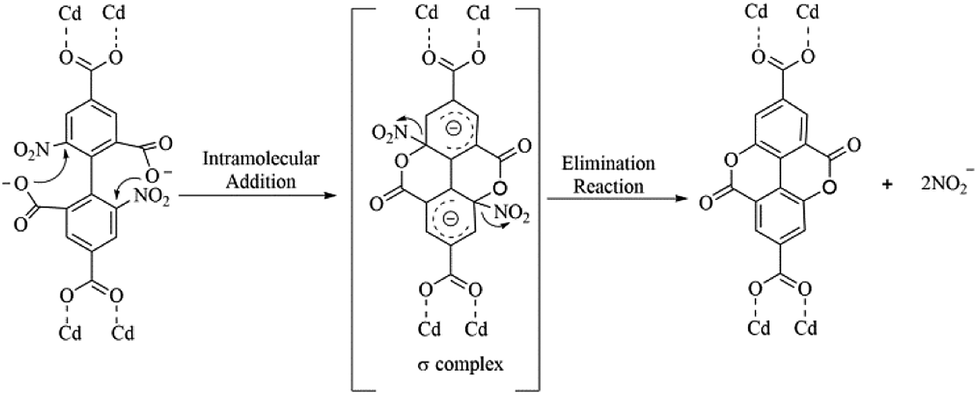
The possible mechanism for the in situ generation of ddpa2− was postulated and displayed in Scheme 4. An intramolecular nucleophilic aromatic substitution reaction (SNAr) may have occurred during hydrothermal synthesis processes. As shown in Scheme 4, the protonated carboxyl groups serve as nucleophilic agents to form the σ-complex intermediate (addition reaction), which can generate the cyclization products by eliminating the nitroso-group (elimination reaction). To illustrate the above plausible mechanism, several drops of AgNO3 solution (0.1 mol L−1) were added into the filtrate of the reaction mixture, and a large faint yellow precipitate was formed immediately, which revealed that AgNO2 might be formed. The clarification of the mechanism for internal esterifications as well as the origin of the synergy between 2, 2′-bpy and Cd(II) ion under hydrothermal conditions requires further studies, which are currently underway in our laboratory.
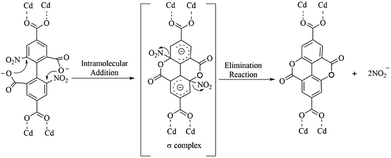 |
| | Scheme 4 The possible mechanism for in situ generation of ddpa2−. | |
3.4 Photoluminescent properties
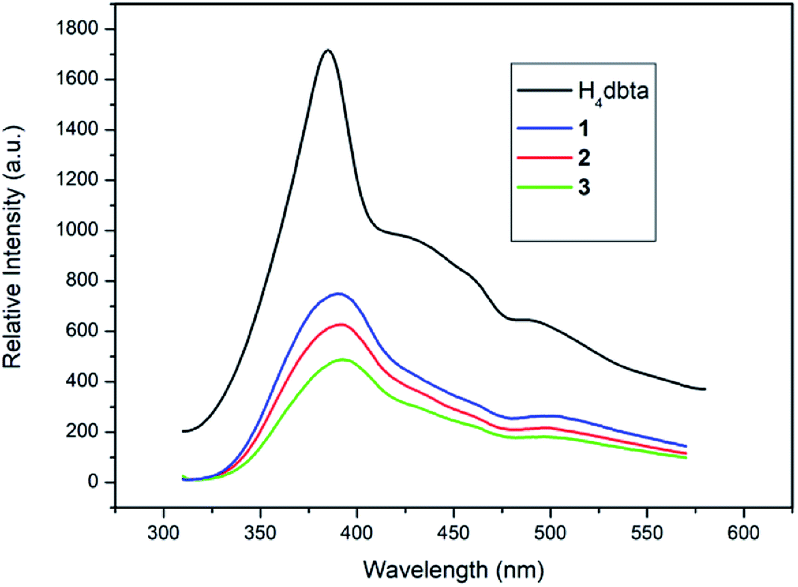
The solid state luminescence of free H4dbta and 1–3 was investigated at room temperature (Fig. 4). Upon excitation at 296 nm, the free H4dbta exhibits intense emission at 384 nm. Compounds 1–3 exhibited photoluminescence with emission maxima at 390 nm (1 and 2) and 393 nm (3), respectively. The three compounds exhibit a red-shift compared with the free H4dbta ligand. Compounds 1–3 exhibit similar emission peaks to the free H4dbta ligand. We tentatively assign them to intraligand photoluminescence.48,49 The different red-shifts and intensities of the emission of three compounds compared to that of the free H4dbta ligand may result from the differences in the coordination environments and different structures and in situ ligands.
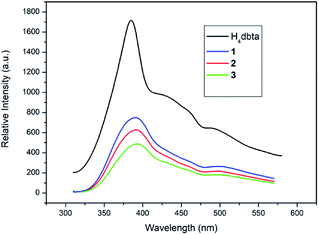 |
| | Fig. 4 The emission spectra of free H4dbta and 1–3 in the solid state at room temperature. | |
4. Conclusions
In summary, a novel 3D Cd(II) coordination polymer is hydrothermally synthesized in situ, and the influencing factors and mechanism for the in situ reaction are briefly discussed. The formation of 1 suggests that the in situ reaction for the dbta4− ligand resulted from synergetic effects of both 2,2′-bpy and Cd(II) ion under hydrothermal conditions. A study of the mechanism indicates that an intramolecular nucleophilic aromatic substitution reaction (SNAr) may have occurred during hydrothermal synthesis processes. The study also reveals that C–O ester bonds can also be formed in situ by H4dbta, which provides a promising route to design and construct a new organic ligand with special structures and properties. In the future, we will further clarify the mechanism for the internal esterifications as well as the origin of the synergy between 2,2′-bpy and Cd(II) ions under hydrothermal conditions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the NSF of China (No. 21373178, 21663031 and 21503183), which are gratefully acknowledged.
References
- X. M. Zhang, Coord. Chem. Rev., 2005, 249, 1201–1219 CrossRef CAS.
- X. M. Chen and M. L. Tong, Acc. Chem. Res., 2007, 40, 162–170 CrossRef CAS PubMed.
- Y. C. Chang and S. L. Wang, J. Am. Chem. Soc., 2012, 134, 9848–9851 CrossRef CAS PubMed.
- E. T. Nguyen, X. Zhao, D. Ta, P. L. Nguyen and X. H. Bu, Cryst. Growth Des., 2015, 15, 5939–5944 CrossRef CAS.
- T. Huang, Y. L. Wang, Q. Yin, B. Karadeniz, H. F. Li, J. Lü and R. Cao, CrystEngComm, 2016, 18, 2742–2747 RSC.
- G. B. Li, J. M. Liu, Z. Q. Yu, W. Wang and C. Y. Su, Inorg. Chem., 2009, 48, 8659–8661 CrossRef CAS.
- A. K. Ghosh, T. S. Mahapatra, R. Clérac, C. Mathonière, V. Bertolasi and D. Ray, Inorg. Chem., 2015, 54, 5136–5138 CrossRef CAS PubMed.
- C. E. Rowland, N. Belai, K. E. Knope and C. L. Cahill, Cryst. Growth Des., 2010, 15, 1390–1398 CrossRef.
- P. Kanoo, R. Matsuda, H. Sato, L. C. Li, H. J. Jeon and S. Kitagawa, Inorg. Chem., 2013, 52, 10735–10737 CrossRef CAS PubMed.
- X. M. Zhang, J. J. Hou and H. S. Wu, Dalton Trans., 2004, 3437–3439 RSC.
- F. L. Yang, J. Tao, R. B. Huang and L. S. Zheng, Inorg. Chem., 2011, 50, 911–917 CrossRef CAS PubMed.
- D. Wu, X. J. Bai, H. R. Tian, W. T. Yang, Z. W. Li, Q. Huang, S. Y. Du and Z. M. Sun, Inorg. Chem., 2015, 54, 8617–8624 CrossRef CAS PubMed.
- L. Sun, L. Ma, J. B. Cai, L. Liang and H. Deng, CrystEngComm, 2012, 14, 890–898 RSC.
- R. T. Dong, Z. Y. Ma, L. X. Chen, L. F. Huang, Q. H. Li, M. Y. Hu, M. Y. Shen, C. W. Li and H. Deng, CrystEngComm, 2015, 17, 5814–5831 RSC.
- C. C. Chang, Y. C. Huang, S. M. Huang, J. Y. Wu, Y. H. Liu and K. L. Lu, Cryst. Growth Des., 2012, 12, 3825–3828 CrossRef CAS.
- M. A. Nadeem, M. Bhadbhade, R. Bircher and J. A. Stride, Cryst. Growth Des., 2010, 10, 4060–4067 CrossRef CAS.
- W. Q. Zhang, W. Y. Zhang, R. D. Wang, C. Y. Ren, Q. Q. Li, Y. P. Fan, B. Liu, P. Liu and Y. Y. Wang, Cryst. Growth Des., 2017, 17, 517–526 CrossRef CAS.
- B. Liu, L. Wei, N. N. Li, W. P. Wu, H. Miao, Y. Y. Wang and Q. Z. Shi, Cryst. Growth Des., 2014, 14, 1110–1127 CrossRef CAS.
- Y. P. He, L. B. Yuan, H. Xu and J. Zhang, Cryst. Growth Des., 2017, 17, 290–294 CrossRef CAS.
- H. Wang, G. J. Xing, F. Chen, J. Sun and Y. H. Zhang, Chin. J. Struct. Chem., 2015, 34, 1113–1120 CAS.
- L. Qin, J. S. Hu, Y. Z. Li and H. G. Zheng, Cryst. Growth Des., 2012, 12, 403–413 CrossRef CAS.
- J. J. Liu, Y. F. Guan, M. J. Lin, C. C. Huang and W. X. Dai, Cryst. Growth Des., 2016, 16, 2836–2842 CrossRef CAS.
- E. Lee, H. Ju, S. Kim, K. M. Park and S. S Lee, Cryst. Growth Des., 2015, 15, 5427–5436 CrossRef CAS.
- B. Hu, T. Tao, Z. Y. Bin, Y. X. Peng, B. B. Ma and W. Huang, Cryst. Growth Des., 2014, 14, 300–309 CrossRef CAS.
- T. T. Jia, S. R. Zhu, M. Shao, Y. M. Zhao and M. X. Li, Inorg. Chem. Commun., 2008, 11, 1221–1223 CrossRef CAS.
- L. L. Wen, F. Wang, X. K. Leng, C. G. Wang, L. Y. Wang, J. M. Gong and D. F. Li, Cryst. Growth Des., 2010, 10, 2835–2838 CrossRef CAS.
- L. Cheng, S. H. Gou and J. Q. Wang, J. Mol. Struct., 2011, 991, 149–157 CrossRef CAS.
- J. J. Wang, T. T. Wang, L. Tang, X. Y. Hou, L. J. Gao, F. Fu and M. L. Zhang, J. Coord. Chem., 2013, 66, 3979–3988 CrossRef CAS.
- J. Jia, M. Shao, T. T. Jia, S. R. Zhu, Y. M. Zhao, F. F. Xing and M. X. Li, CrystEngComm, 2010, 12, 1548–1561 RSC.
- W. X. Chen, H. R. Xu, G. L. Zhuang, L. S. Long, R. B. Huang and L. S. Zheng, Chem. Commun., 2011, 47, 11933–11935 RSC.
- Y. H. Su, F. Luo, H. Li, Y. X. Che and J. M. Zheng, CrystEngComm, 2011, 13, 44–46 RSC.
- Z. R. Pan, J. Xu, Xi. Q. Yao, Y. Z. Li, Z. J. Guo and H. G. Zheng, CrystEngComm, 2011, 13, 1617–1624 RSC.
- H. Tian, K. Wang, Q. X. Jia, Q. Sun, Y. Ma and E. Q. Gao, Cryst. Growth Des., 2011, 11, 5167–5170 CrossRef CAS.
- F. Su, L. P. Lu and S. S. Feng, J. Mol.Struct., 2015, 1096, 38–42 CrossRef CAS.
- F. Su, L. P. Lu, S. S. Feng, M. L. Zhu, Z. Q. Gao and Y. H. Dong, Dalton Trans., 2015, 44, 7213–7222 RSC.
- C. Y. Ren, B. Liu, W. P. Wu, P. Liu, G. P. Yang, Y. F. Kang and Y. Y. Wang, Inorg. Chem. Commun., 2015, 53, 46–49 CrossRef CAS.
- Q. Q. Li, W. Q. Zhang, C. Y. Ren, Y. P. Fan, J. L. Li, P. Liu and Y. Y. Wang, CrystEngComm, 2016, 18, 3358–3371 RSC.
- W. Q. Zhang, W. Y. Zhang, R. D. Wang, C. Y. Ren, Q. Q. Li, Y. P. Fan, B. Liu, P. Liu and Y. Y. Wang, Cryst. Growth Des., 2017, 17, 517–526 CrossRef CAS.
- J. Y. Zhang, X. H. Jing, Y. Ma, A. L. Cheng and E. Q. Gao, Cryst. Growth Des., 2011, 11, 3681–3685 CrossRef CAS.
- J. W. Wang, Y. C. Su and J. J. Wang, Chin. J. Struct. Chem., 2015, 34, 1385–1390 CAS.
- R. K. Feller, P. M. Forster, F. Wudl and A. K. Cheetham, Inorg. Chem., 2007, 46, 8717–8721 CrossRef CAS PubMed.
- X. Y. Cao, L. Q. Yu and R. D. Huang, J. Solid State Chem., 2014, 210, 74–78 CrossRef CAS.
- G. M. Sheldrick, SADABS, A Program for Empirical Absorption Correction of Area detector Data, University of Göttingen, Germany 1997 Search PubMed.
- G. M. Sheldrick, SHELXS-2014/7, Program for Crystal Structure Solution, University of Göttingen, Germany 2014 Search PubMed.
- G. M. Sheldrick, SHELXL-2014/7, Program for Crystal Structure Refinement, University of Göttingen, Germany 2014 Search PubMed.
- J. J. Wang, M. L. Yang, H. M. Hua, G. L. Xue, D. S. Li and Q. Z. Shi, Z. Anorg. Allg. Chem., 2007, 633, 341–345 CrossRef CAS.
- L. L. Liu, C. X. Yu, Y. R. Li, J. J. Han, F. J. Ma and L. F. Ma, CrystEngComm, 2015, 17, 653–664 RSC.
- C. C. Du, X. F. Wang, S. B. Zhou, D. Z. Wang and D. Z. Jia, CrystEngComm, 2017, 19, 6758–6777 RSC.
- D. Chisca, L. Croitor, O. Petuhov, O. V. Kulikova, G. F. Volodina, E. B. Coropceanu, A. E. Masunov and M. S. Fonari, CrystEngComm, 2018, 20, 432–447 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Bond length/angle tables, figure of single-crystal, additional PXRD patterns, FT-IR, TGA. CCDC 1840510–1840512 for 1–3. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra06112b |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Zhuang Cao
*,
Zhuang Cao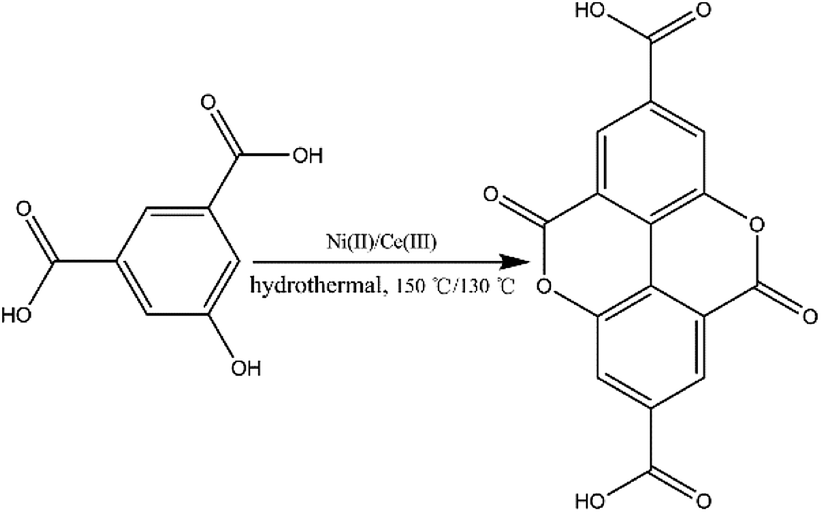
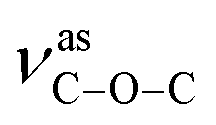 and
and 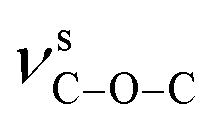 groups, respectively. Thermal stability studies (Fig. S5,†) demonstrate that 1 is thermally stable up to 250 °C; then, a quick weight loss (ca. 55%) from 250 to 360 °C is observed, which is caused by the decomposition of ligands. For 2, weight loss of about 3.6% is observed from 25 to 166 °C, which is ascribed to the release of lattice and coordinated water, followed by a quick weight loss of about 65.6% from 285 to 373 °C. For 3, weight loss of about 2.2% is observed from 25 to 117 °C, which may be ascribed to the release of lattice water; this is followed a quick weight loss of about 47.8% from 225 to 276 °C. The second step of the weight loss for 2 and 3 is caused by the decomposition of the ligands.
groups, respectively. Thermal stability studies (Fig. S5,†) demonstrate that 1 is thermally stable up to 250 °C; then, a quick weight loss (ca. 55%) from 250 to 360 °C is observed, which is caused by the decomposition of ligands. For 2, weight loss of about 3.6% is observed from 25 to 166 °C, which is ascribed to the release of lattice and coordinated water, followed by a quick weight loss of about 65.6% from 285 to 373 °C. For 3, weight loss of about 2.2% is observed from 25 to 117 °C, which may be ascribed to the release of lattice water; this is followed a quick weight loss of about 47.8% from 225 to 276 °C. The second step of the weight loss for 2 and 3 is caused by the decomposition of the ligands.